

Abstract
Essential genes are necessary to sustain the life of a species under adequate nutritional conditions. These genes have attracted significant attention for their potential as drug targets, especially in developing broad‐spectrum antibacterial drugs. However, studying essential genes remains challenging due to their variability in specific environmental conditions. In this study, the authors aim to develop a powerful prediction model for identifying essential genes in humans. The authors first obtained the essential gene data from human cancer cell lines and characterised gene sequences using 7 feature encoding methods such as Kmer, the Composition of K‐spaced Nucleic Acid Pairs, and Z‐curve. Subsequently, feature fusion and feature optimisation strategies were employed to select the impactful features. Finally, machine learning algorithms were applied to construct the prediction models and evaluate their performance. The single‐feature‐based model achieved the highest area under the Receiver Operating Characteristic curve (AUC) of 0.830. After fusing and filtering these features, the classical machine learning models achieved the highest AUC at 0.823 while the deep learning model reached 0.860. Results obtained by the authors show that compared to using individual features, feature fusion and feature optimisation strategies significantly improved model performance. Moreover, the study provided an advantageous method for essential gene identification compared to other methods.
Keywords: bioinformatics, essential gene, feature selection, neural nets
Essential genes are necessary to sustain the life of a species under adequate nutritional conditions. In this study, the authors aim to develop a powerful prediction model for identifying essential genes in humans. Results obtained by the authors show that compared to using individual features, feature fusion and feature optimisation strategies significantly improved model performance.
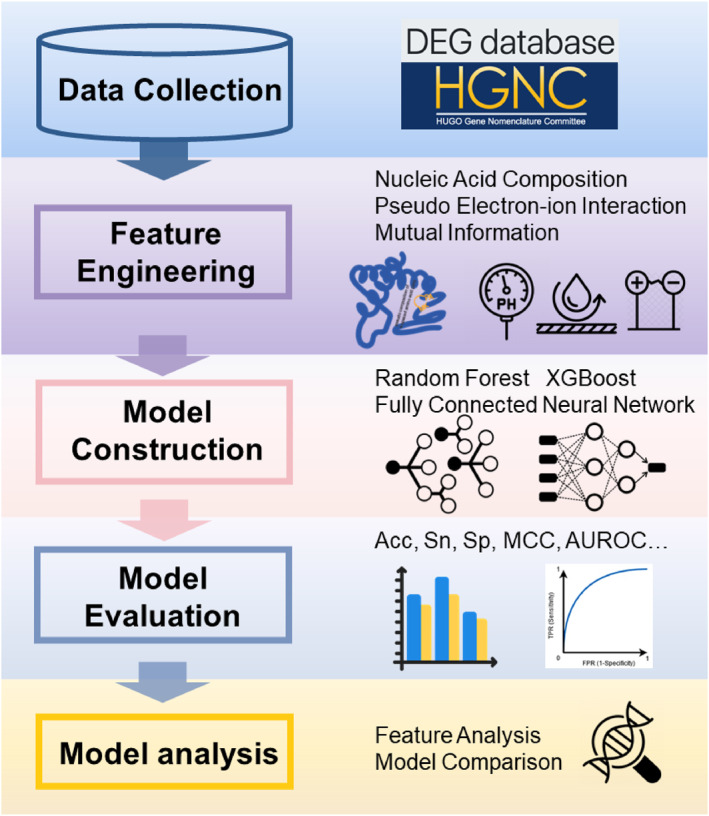
1. INTRODUCTION
A gene is a sequence of DNA that encodes functional products in an organism. Gene expression is highly regulated and can vary depending on the cell type, developmental stage, and physiological conditions. Essential genes constitute a specific class that is expressed at high levels and is indispensable for living organisms [1]. These genes are responsible for a range of basic life activities such as DNA replication, protein translation, growth and metabolism, and nutrient transport both in vivo and in vitro [2]. In unicellular organisms, the deletion or mutation of essential genes can directly result in death [3]. In higher eukaryotes, defects in these genes can lead to genetic disorders or birth defects [4]. Research has revealed that gene essentiality can differ across species, genetic backgrounds, and specific environments. For instance, the genes encoding enzymes, such as CYS3, were deemed essential in Σ1278b but not in S288c [5]. Therefore, the study of essential genes is crucial for understanding the biology of living organisms and identifying potential therapeutic targets for diseases [6, 7, 8, 9, 10, 11, 12, 13].
The identification of essential genes is a complex task demanding a comprehensive approach that integrates diverse methodologies and cutting‐edge technologies [14, 15]. In 1995, Itaya et al. [16] performed mutagenesis on 79 randomly selected genes from Bacillus subtilis. They identified six genes where mutations prevented colony formation, designating these as essential genes. In 2013, Sarmiento et al. [17] employed transposon mutagenesis for the first time to identify essential genes in archaea. The identification of essential genes has also been explored in higher eukaryotes. In 2015, three separate laboratories used mutagenesis or Clustered Regularly Interspaced Short Palindromic Repeats methods to report the essential characteristics of human genes, filling a research gap in human cells [18, 19, 20, 21]. Moreover, the use of computational methods for identifying essential genes is gaining popularity due to their ability to offer faster and more cost‐effective analyses. The strategies can be classified into two main categories: comparative genomics methods and machine learning methods. The core idea of comparative genomics methods is that essential genes have a higher level of evolutionary conservation. By conducting sequence alignment on the genome sequences of closely related species using tools such as BLAST [22], FASTA [23], PSI‐BLAST [24], HAlign [25, 26], and WMSA [27, 28] researchers can identify conserved genes. These identified genes can be prioritised for experimental validation as potential essential genes. In machine learning methods, the prediction of essential genes relies on models trained with experimental data [29, 30]. These models categorise genes into essential or non‐essential classes based on diverse features of genes including gene expression, sequence information, and functional annotations. Despite their low cost and quick implementation, continuous optimisation of computational methods for essential gene identification is motivated by their relatively lower accuracy compared to biological experiments.
In this study, we present a new machine learning‐based model for identifying essential genes in humans. First, we collected essential gene data and extracted features from the DNA sequences using several feature extraction methods. Next, feature recombination and feature selection were performed to select predictive features. Maximal Information Coefficient (MIC) [31] and F‐score were adopted to identify well‐performing features. Then, machine learning algorithms were applied to build essential gene prediction models. These models were trained and evaluated using 10‐fold cross‐validation. Figure 1 depicts an overview of the various stages within the essential gene prediction workflow.
FIGURE 1.
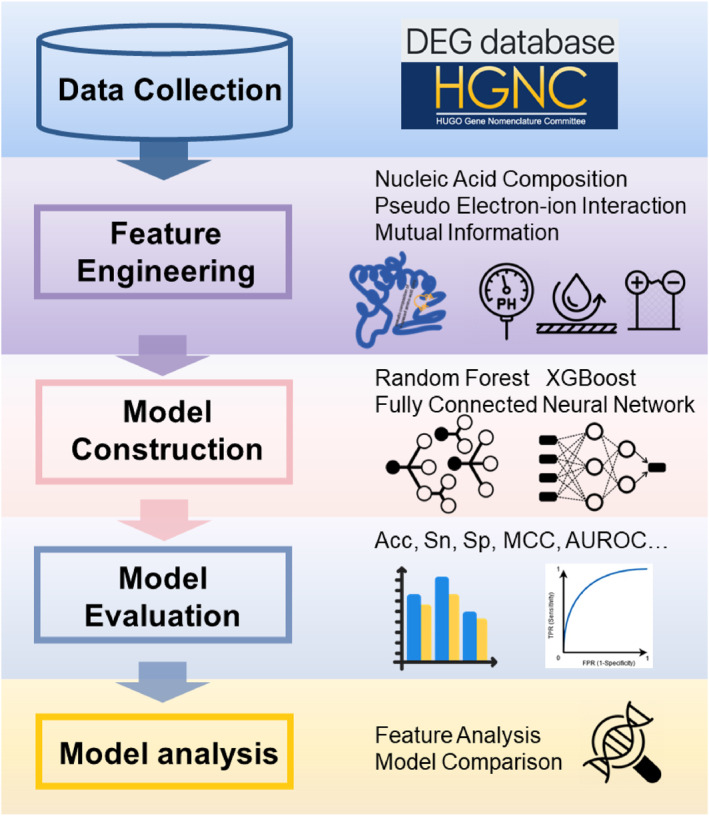
The flowchart of the essential gene identification.
2. MATERIALS AND METHODS
2.1. Data collection
The human essential gene datasets used in this study were sourced from Guo et al. [32] who compiled the information from the DEG database [33]. The dataset comprises 11 cell lines, which are KBM7, K562K, Raji, Jiyoye, A375, HAP1, DLD1, GBM, HCT116, HeLa, and rpel. The annotation information for protein‐coding genes was obtained from the HGNC database [34]. To ensure the robustness of our dataset, we defined essential genes as those showing essentiality in at least 6 of the 11 cell lines. Finally, we obtained 12,015 human genes including 1516 essential genes and 10,499 non‐essential genes. Essential genes are labelled as positive samples while non‐essential genes are labelled as negative samples.
2.2. Feature engineering
To transform and select raw sequences into informative features, we employed both feature extraction and feature selection techniques. Feature extraction aims to generate a set of features that can accurately represent the sequence data and facilitate subsequent machine learning analysis. In this study, we used seven methods to characterise the biological features of human genes: Nucleic acid composition (NAC) [35], K‐mer [36, 37, 38], reverse complement K‐mer (RCKmer) [39], the composition of K‐spaced nucleic acid pairs (CKSNAP) [40], Z‐curve [41], pseudo electron–ion interaction pseudopotentials (PseEIIP) [42], and multivariate mutual information (MMI) [43]. Nucleic acid composition, K‐mer, RCKmer, CKSNAP, and Z‐curve describe NAC. Pseudo electron–ion interaction pseudopotentials calculates the pseudo electron–ion interaction for each sequence. Multivariate mutual information calculates the mutual information for K‐mer. The dimension of the NAC, K‐mer, RCKmer, CKSNAP, Z‐curve, PseEIIP, and MMI feature sets are 4, 64, 32, 64, 9, 64, and 30, respectively. Feature extraction was performed using ilearnPlus [44]. To assess the impact of features on essential gene identification, we first developed separate machine learning models for each feature. The assessment of feature contribution was carried out by evaluating their performance in a 10‐fold cross‐validation. Next, we merged the features based on their individual performance and further refined the combined features using feature ranking scores calculated with F‐score and MIC [31]. Ultimately, we retained the top 200 features for subsequent model construction.
2.3. Model construction
The classification models in this study were trained using the random forest (RF) [45], XGBoost [46], and fully connected neural network (FCNN) [47] algorithms. Random forest is a widely used supervised learning algorithm known for its effectiveness in addressing classification problems. It achieves this by combining multiple weak classifiers which collectively vote to make the final decision. XGBoost is a gradient boosting algorithm proposed by Tianqi Chen. It introduces a regularisation term into the loss function and controls the model complexity to minimise the risk of overfitting [48]. The FCNN is a multilayer perceptron consisting of an input layer for receiving input information, hidden layers, and an output layer for generating the prediction probabilities. The FCNN model in this study comprises 3 hidden layers, with ReLU and softmax activation functions before and after the output layer, respectively. The CrossEntropyLoss and Adam optimiser were used for the model optimisation. The learning rate, epoch number, and batch data were set to 0.0001, 100, and 256, respectively.
2.4. Data evaluation
To assess the effectiveness of the essential gene identification models, various evaluation metrics were employed including accuracy (Acc), sensitivity (Sn), specificity (Sp), Mathews correlation coefficient (MCC), recall, precision (Pre), F1‐score, and the area under the Receiver Operating Characteristic (ROC) curve (AUC) [49, 50, 51, 52, 53]. Acc measures the prediction accuracy across all samples. Sn and Sp evaluate the model's accuracy in predicting positive samples and negative samples, respectively. Mathews correlation coefficient assesses the correlation between the predicted labels and the true labels of the samples. Recall reflects the proportion of correctly predicted positive samples among the actual positive samples, while Pre represents the proportion of correctly classified positive samples relative to the total samples classified as positive. The F1‐score, which balances the influence of precision and recall, provides a more comprehensive evaluation of the classifier. Additionally, AUC is a widely used metric that offers a comprehensive measure of classifier performance [54, 55, 56, 57, 58]. ACC, Sn, Recall, Sp, Pre, MCC and F1‐score are formulated as follows:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
where TP (Ture positive) and TN (Ture negative) present the numbers of correctly identified essential genes and non‐essential genes, respectively. FP (False positive) and FN (False negative) denote the number of incorrectly non‐essential genes and essential genes, respectively.
3. RESULTS
3.1. Feature evaluation
To evaluate the effectiveness of seven feature extraction methods in characterising sequences, we constructed various machine learning models based on these features. For each feature set, we used RF, XGBoost, and FCNN algorithms to classify genes into essential and non‐essential categories. The assessment was carried out through a 10‐fold cross‐validation process. AUC values for different features using RF, XGBoost, and FCNN models were summarised in Table 1. The RF model achieved AUC values ranging from 0.636 to 0.779. The XGBoost model achieved AUC values ranging from 0.690 to 0.795. The FCNN models achieved AUC values ranging from 0.660 to 0.830. Notably, the RF and FCNN models using Kmer showed the highest ROC values of 0.779 and 0.830, respectively, while the XGBoost model constructed using PseEIIP had the highest ROC value of 0.795. Additionally, models based on CKSNAP and Z‐curve also demonstrated good performance.
TABLE 1.
10‐fold cross‐validation results of seven feature extraction methods using random forest (RF), XGBoost, and fully connected neural network (FCNN).
| Algorithm | Kmer | PseEIIP | Z‐curve | CKSNAP | MMI | RCKmer | NAC |
|---|---|---|---|---|---|---|---|
| RF | 0.779 | 0.778 | 0.773 | 0.769 | 0.761 | 0.726 | 0.636 |
| XGBoost | 0.789 | 0.795 | 0.777 | 0.788 | 0.766 | 0.736 | 0.689 |
| FCNN | 0.830 | 0.810 | 0.720 | 0.810 | 0.760 | 0.750 | 0.660 |
Note: Bold values represent the best statistically results.
Subsequently, we scored these features using the rank‐sum method based on the results from the three algorithms for a comprehensive evaluation. First, we sorted the features based on their AUC ranking. In the RF, XGBoost, and FCNN models, the ranked positions of K‐mer, PseEIIP, Z‐curve, CKSNAP, MMI, RCKmer, and NAC were [1, 2, 3, 4, 5, 6, 7], [1.5, 1.5, 4, 3, 5, 6, 7], and [1, 2, 6, 3, 4, 5, 7], respectively. We then calculated the rank sum ratio to determine the overall ranking of the features, resulting in the following order: K‐mer, PseEIIP, CKSNAP, Z‐curve, MMI, RCKmer, and NAC. Notably, the NAC feature performed poorly, likely due to its relatively small size, containing only four features. Given that the AUC values of the RF model were generally lower than those of the XGBoost model, we excluded the RF algorithm for subsequent modelling of fused features.
3.2. Fusion feature modelling results
To enhance the accuracy of gene classification we applied feature selection to various combinations of features. By combining Kmer, PseEIIP, Z‐curve, and CKSNAP features with MMI, RCKmer, and NAC features in turn, we employed F‐score and MIC to score feature contributions. The top 200 features from each combination were used to construct prediction models. For XGBoost and FCNN, eight models were created to distinguish essential and non‐essential genes, respectively.
The XGBoost‐based models achieved AUC values ranging from 0.818 to 0.825, all surpassing 0.8. Notably, the model constructed with Kmer, PseEIIP, Z‐curve, and CKSNAP using the MIC screening method achieved the highest AUC value of 0.825. This model reached Acc, Sn, Sp, and MCC of 88.18%, 13.52%, 98.96%, and 0.259, respectively. The performance results are detailed in Table 2. The ROC curves from 5‐fold cross‐validation are shown in Figure 2.
TABLE 2.
The 5‐fold cross‐validation results of XGBoost for different feature combinations.
| Feature | Feature filtering method | Acc (%) | Sn (%) | Sp (%) | MCC | AUC |
|---|---|---|---|---|---|---|
| Kmer + PseEIIP + Z‐curve + CKSNAP | F‐score | 88.22 | 16.03 | 98.65 | 0.277 | 0.823 |
| MIC | 88.18 | 13.52 | 98.96 | 0.259 | 0.825 | |
| Kmer + PseEIIP + Z‐curve + CKSNAP + MMI | F‐score | 88.20 | 17.15 | 98.46 | 0.282 | 0.820 |
| MIC | 88.15 | 14.98 | 98.71 | 0.265 | 0.823 | |
| Kmer + PseEIIP + Z‐curve + CKSNAP + MMI + RCKmer | F‐score | 88.15 | 16.09 | 98.55 | 0.272 | 0.819 |
| MIC | 88.13 | 13.92 | 98.85 | 0.258 | 0.821 | |
| Kmer + PseEIIP + Z‐curve + CKSNAP + MMI + RCKmer + NAC | F‐score | 88.12 | 15.17 | 98.66 | 0.265 | 0.819 |
| MIC | 88.14 | 18.54 | 98.19 | 0.286 | 0.818 |
Note: Bold values represent the best statistically results.
FIGURE 2.
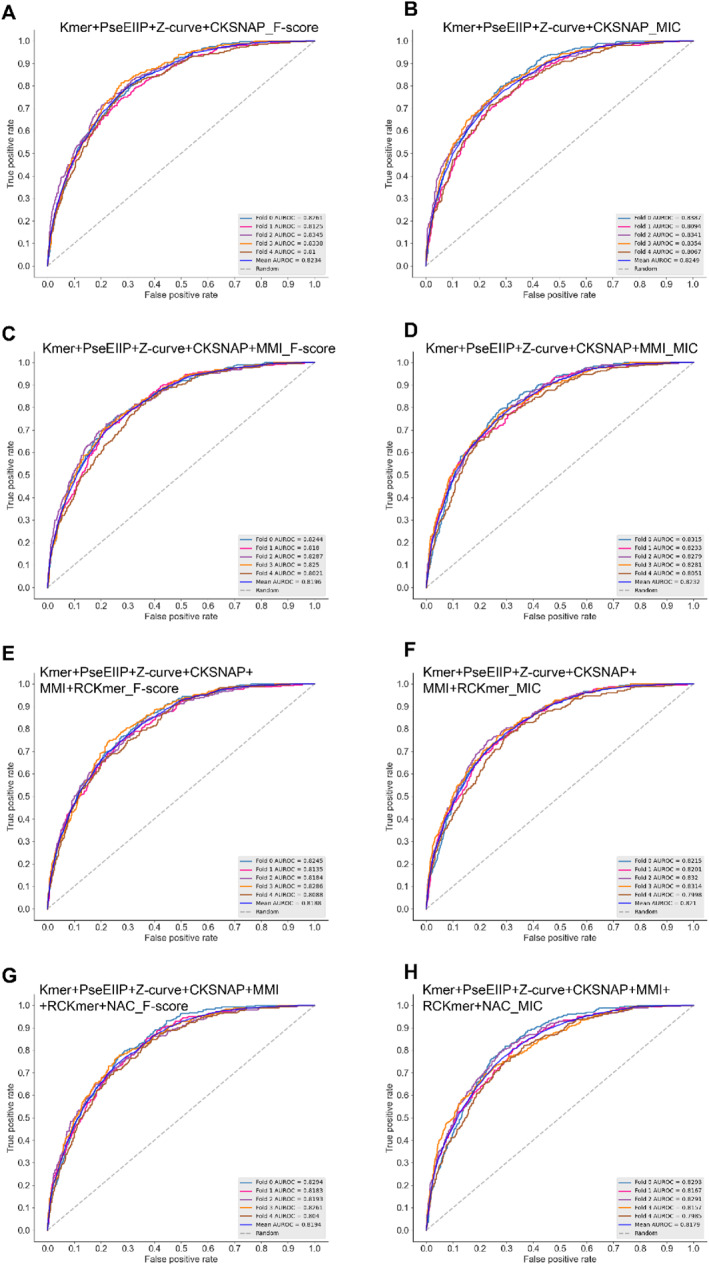
ROC curves of XGBoost models for different feature combinations. ROC, Receiver Operating Characteristic.
The FCNN‐based achieved AUC values ranging from 0.830 to 0.860. The model constructed with Kmer, PseEIIP, Z‐curve, and CKSNAP using the F‐score screening method achieved the highest AUC value of 0.860. Additionally, the values of Acc, Recall, Precision, and F1 achieved 79.57%, 75.58%, 82.13%, and 0.787, respectively. Detailed results are provided in Table 3. ROC curves and loss curves are depicted in Figures 3 and 4, respectively. The convergent loss curve indicates a well‐fitted model. Both the XGBoost and FCNN‐based models exhibit improved performance compared to models based on single features.
TABLE 3.
The results of fully connected neural network (FCNN) for different feature combinations.
| Feature | Feature filtering method | Acc (%) | Recall (%) | Precision (%) | F1 | AUC |
|---|---|---|---|---|---|---|
| Kmer + PseEIIP + Z‐curve + CKSNAP | F‐score | 79.57 | 75.58 | 82.13 | 0.787 | 0.860 |
| MIC | 76.48 | 69.70 | 80.65 | 0.748 | 0.840 | |
| Kmer + PseEIIP + Z‐curve + CKSNAP + MMI | F‐score | 76.43 | 70.93 | 79.70 | 0.751 | 0.840 |
| MIC | 75.54 | 70.78 | 78.10 | 0.743 | 0.840 | |
| Kmer + PseEIIP + Z‐curve + CKSNAP + MMI + RCKmer | F‐score | 76.68 | 69.85 | 80.82 | 0.750 | 0.840 |
| MIC | 77.88 | 72.44 | 81.35 | 0.766 | 0.850 | |
| Kmer + PseEIIP + Z‐curve + CKSNAP + MMI + RCKmer + NAC | F‐score | 76.35 | 70.13 | 80.02 | 0.748 | 0.840 |
| MIC | 75.49 | 69.02 | 79.29 | 0.738 | 0.830 |
Note: Bold values represent the best statistically results.
FIGURE 3.
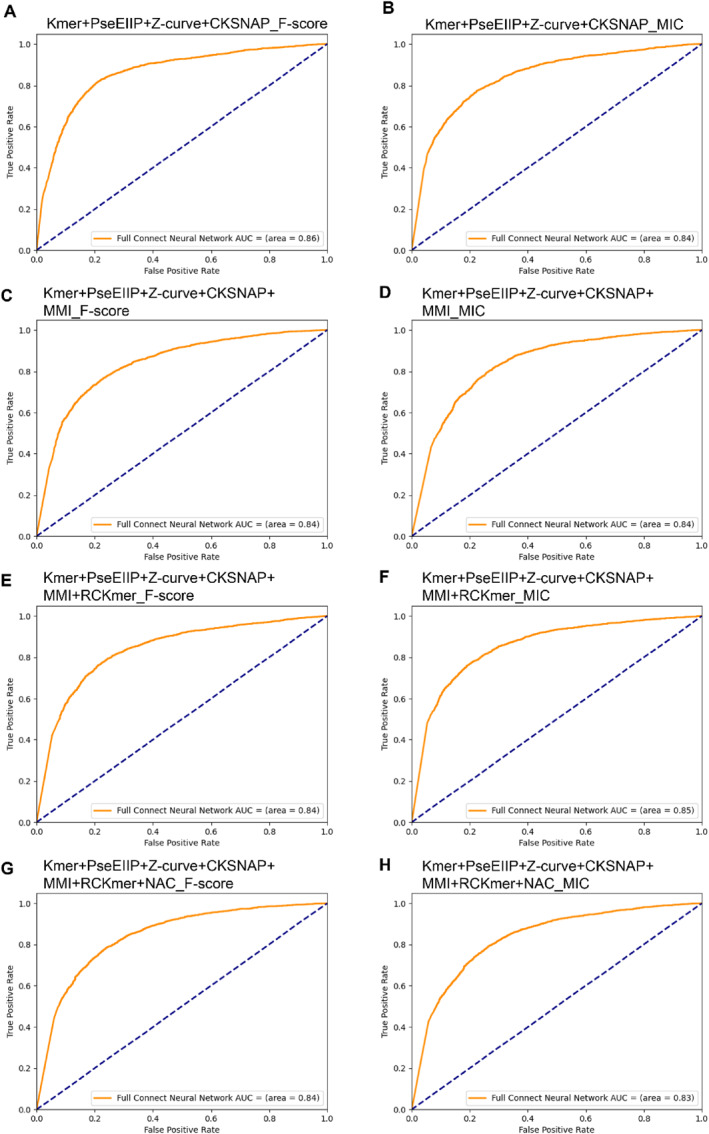
ROC curves of fully connected neural network (FCNN) models using fusion feature for different feature combinations. ROC, Receiver Operating Characteristic.
FIGURE 4.
Loss curves of fully connected neural network (FCNN) models for different feature combinations.
3.3. Feature contributions
To further explore the impact of individual features on the optimal classification model, the top 20 F‐score ranked features were listed in Table 4. Within this selection, 9 features originating from the Kmer constituted approximately 45% of the total, with 6 features from CKSNAP, 4 from PseEIIP, and one from Z‐curve. Three of the seven initial feature extraction methods did not make it to the top 20: MMI, RCKmer, and NAC. Overall, their dimensionality numbers were relatively low and performed poorly in the previous single‐feature modelling results. In addition, it was found that the models constructed by connecting the feature sets of MMI, RCKmer, and NAC were less effective than those constructed using fewer types of features. Combined with the fact that they did not appear in the top 20 features in terms of importance, it can be speculated that these feature extraction methods may not be able to distinguish human essential genes from non‐essential genes well. The results indicate the Kmer features play a relatively significant role in distinguishing essential genes from non‐essential genes.
TABLE 4.
The top 20 features of maximal information coefficient (MIC) feature assessment and their sources.
| Ranking of features | Feature | Feature source |
|---|---|---|
| 1 | f_50 | Kmer |
| 2 | f_8 | Kmer |
| 3 | f_199 | CKSNAP |
| 4 | f_87 | PseEIIP |
| 5 | f_192 | CKSNAP |
| 6 | f_151 | CKSNAP |
| 7 | f_7 | Kmer |
| 8 | f_18 | Kmer |
| 9 | f_56 | Kmer |
| 10 | f_24 | Kmer |
| 11 | f_66 | PseEIIP |
| 12 | f_130 | Z‐curve |
| 13 | f_99 | PseEIIP |
| 14 | f_163 | CKSNAP |
| 15 | f_2 | Kmer |
| 16 | f_29 | Kmer |
| 17 | f_23 | Kmer |
| 18 | f_198 | CKSNAP |
| 19 | f_96 | PseEIIP |
| 20 | f_160 | CKSNAP |
3.4. Compare with other methods
Machine learning techniques, known for their robust data learning capabilities, have been widely used to distinguish essential and nonessential genes. Since 2011, several models aimed at identifying essential genes have been proposed, primary focus on bacteria [59, 60]. In 2017, Guo et al. [32] introduced the first essential gene classification model, named Pheg. The authors represented nucleotide composition using λ‐interval Z‐curve and built a support vector machines (SVM)‐based classification model. Pheg achieved an AUC of 0.885. In 2023, iEsGene‐CSMOTE [61] addressed the issue of the imbalanced dataset by employing a clustering based synthetic minority oversampling technique. Then, the authors trained an SVM‐based model using the Z curve, and PseKNC feature for human essential genes identification. The model achieved an Acc of 83.36% and an AUC of 0.874. In 2024, Bingo [62] was developed for four organisms including human. Bingo used a large language model‐ and graph neural network (LLM–GNN)‐based approach to predict essential protein‐coding genes, achieving an AUC of 0.874 in the human dataset. These models show the promising ability of computational biological models in essential gene identification. The research on essential gene identification in humans using machine learning methods is summarised in Table 5.
TABLE 5.
Current status of research on machine learning methods to identify essential genes.
| Model | Feature | Algorithm | Accuracy (%) | AUC |
|---|---|---|---|---|
| Pheg [32] | λ‐interval form Z‐curve | SVM | / | 0.885 |
| iEsGene‐CSMOTE [61] | Z curve, PseKNC | SVM | 83.36 | 0.874 |
| Bingo [62] | ESM‐2 | LLM–GNN | / | 0.874 |
| Our model | Kmer + PseEIIP+ Z‐curve + CKSNAP | FCNN | 79.57 | 0.860 |
Abbreviations: FCNN, fully connected neural network; LLM–GNN, large language model‐and graph neural network; SVM, support vector machines.
4. DISCUSSION
Despite the availability of essential gene data in numerous databases, identifying these genes remains challenging due to their heterogeneity in certain circumstances. This study focuses on assessing the performance of various feature extraction methods in classifying essential genes. Initially, K‐mer, PseEIIP, Z‐curve, CKSNAP, MMI, RCKmer, and NAC were employed to characterise the biological properties of DNA sequences. Then, three machine learning algorithms including RF, XGBoost, and FCNN were used to train both single‐feature and fused‐feature prediction models. Subsequence analysis revealed the combination of K‐mer, PseEIIP, Z‐curve, and CKSNAP features demonstrated superior distinguishability in essential gene identification. These findings suggest that feature fusion and feature screening are effective in enhancing classifier performance.
However, our models still have room for improvement, primarily due to the imbalanced dataset. The imbalance makes the training model more inclined to predict test samples as negative. Additionally, we opted for a 10‐fold cross‐validation approach to evaluate the models without independent testing. To address the issue of an imbalanced dataset and enhance model accuracy, future work will involve considering resampling techniques. Besides, state‐of‐the‐art algorithms will be utilised to improve the performance of the model. Moreover, separate data will be used to further evaluate and improve the model's accuracy. Following the model optimisation, our future research aims to apply the improved prediction model to analyse the entire genome, identifying potential undiscovered essential genes. Additionally, we intend to develop a user‐friendly online tool for researchers. Users can input the DNA sequence of their target gene and our tool will provide an essentiality probability in response.
AUTHOR CONTRIBUTIONS
Zhao‐Yue Zhang and Yue‐Er Fan contributed to the study design, data analysis, and paper writing. Cheng‐Bing Huang and Meng‐Ze Du contributed to project oversight and paper revisiting. All authors read and agreed to publish the final version of the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no competing interests.
ETHICS APPROVAL STATEMENT
Not applicable.
PATIENT CONSENT STATEMENT
Not applicable.
ACKNOWLEDGEMENTS
This work was supported by the grant from the National Natural Science Foundation of China [62102067, 62172078].
Zhang, Z.‐Y. , et al.: Human essential gene identification based on feature fusion and feature screening. IET Syst. Biol. 18(6), 227–237 (2024). 10.1049/syb2.12105
Zhao‐Yue Zhang and Yue‐Er Fan contributed equally to this study.
Contributor Information
Zhao‐Yue Zhang, Email: zyzhang@uestc.edu.cn.
Cheng‐Bing Huang, Email: 20049607@abtu.edu.cn.
Meng‐Ze Du, Email: du_mengze@foxmail.com.
DATA AVAILABILITY STATEMENT
All data generated or analysed during this study are included in this published article.
REFERENCES
- 1. Bergmiller, T. , Ackermann, M. , Silander, O.K. : Patterns of evolutionary conservation of essential genes correlate with their compensability. PLoS Genet. 8(6), e1002803 (2012). 10.1371/journal.pgen.1002803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pal, C. , et al.: Chance and necessity in the evolution of minimal metabolic networks. Nature 440(7084), 667–670 (2006). 10.1038/nature04568 [DOI] [PubMed] [Google Scholar]
- 3. Juhas, M. , Eberl, L. , Church, G.M. : Essential genes as antimicrobial targets and cornerstones of synthetic biology. Trends Biotechnol. 30(11), 601–607 (2012). 10.1016/j.tibtech.2012.08.002 [DOI] [PubMed] [Google Scholar]
- 4. Georgi, B. , Voight, B.F. , Bucan, M. : From mouse to human: evolutionary genomics analysis of human orthologs of essential genes. PLoS Genet. 9(5), e1003484 (2013). 10.1371/journal.pgen.1003484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bosch‐Guiteras, N. , van Leeuwen, J. : Exploring conditional gene essentiality through systems genetics approaches in yeast. Curr. Opin. Genet. Dev. 76, 101963 (2022). 10.1016/j.gde.2022.101963 [DOI] [PubMed] [Google Scholar]
- 6. Cao, C. , et al.: Ravar: a curated repository for rare variant‐trait associations. Nucleic Acids Res. 52(D1), D990–D997 (2024). 10.1093/nar/gkad876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cao, C. , et al.: Webtwas: a resource for disease candidate susceptibility genes identified by transcriptome‐wide association study. Nucleic Acids Res. 50(D1), D1123–D1130 (2022). 10.1093/nar/gkab957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen, L. , Yu, L. , Gao, L. : Potent antibiotic design via guided search from antibacterial activity evaluations. Bioinformatics 39(2), btad059 (2023). 10.1093/bioinformatics/btad059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li, P. , et al.: Sparse regularized joint projection model for identifying associations of non‐coding rnas and human diseases. Knowl. Base Syst., 258 (2022) [Google Scholar]
- 10. Ai, C. , et al.: A multi‐layer multi‐kernel neural network for determining associations between non‐coding rnas and diseases. Neurocomputing 493, 91–105 (2022). 10.1016/j.neucom.2022.04.068 [DOI] [Google Scholar]
- 11. Jin, J. , et al.: Idna‐abf: multi‐scale deep biological language learning model for the interpretable prediction of DNA methylations. Genome Biol. 23(1), 1–23 (2022). 10.1186/s13059-022-02780-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ning, L. , et al.: Development and application of ribonucleic acid therapy strategies against covid‐19. Int. J. Biol. Sci. 18(13), 5070–5085 (2022). 10.7150/ijbs.72706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ren, L. , et al.: Tcm2covid: a resource of anti‐covid‐19 traditional Chinese medicine with effects and mechanisms. iMETA 1(4), e42 (2022). 10.1002/imt2.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu, J. , et al.: Graph embedding and Gaussian mixture variational autoencoder network for end‐to‐end analysis of single‐cell rna sequencing data. Cell Reports Methods 3(1), 100382 (2023). 10.1016/j.crmeth.2022.100382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zou, Q. , et al.: Gene2vec: gene subsequence embedding for prediction of mammalian N(6)‐methyladenosine sites from mrna. RNA 25(2), 205–218 (2019). 10.1261/rna.069112.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Itaya, M. : An estimation of minimal genome size required for life. FEBS Lett. 362(3), 257–260 (1995). 10.1016/0014-5793(95)00233-y [DOI] [PubMed] [Google Scholar]
- 17. Sarmiento, F. , Mrazek, J. , Whitman, W.B. : Genome‐scale analysis of gene function in the hydrogenotrophic methanogenic archaeon methanococcus maripaludis. Proc. Natl. Acad. Sci. U. S. A. 110(12), 4726–4731 (2013). 10.1073/pnas.1220225110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Blomen, V.A. , et al.: Gene essentiality and synthetic lethality in haploid human cells. Science 350(6264), 1092–1096 (2015). 10.1126/science.aac7557 [DOI] [PubMed] [Google Scholar]
- 19. Hart, T. , et al.: High‐resolution crispr screens reveal fitness genes and genotype‐specific cancer liabilities. Cell 163(6), 1515–1526 (2015). 10.1016/j.cell.2015.11.015 [DOI] [PubMed] [Google Scholar]
- 20. Wang, T. , et al.: Identification and characterization of essential genes in the human genome. Science 350(6264), 1096–1101 (2015). 10.1126/science.aac7041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ren, L. , et al.: Metabolitecovid: a manually curated database of metabolite markers for covid‐19. Comput. Biol. Med. 167, 107661 (2023). 10.1016/j.compbiomed.2023.107661 [DOI] [PubMed] [Google Scholar]
- 22. Altschul, S.F. , et al.: Basic local alignment search tool. J. Mol. Biol. 215(3), 403–410 (1990). 10.1016/s0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 23. Pearson, W.R. : Searching protein sequence libraries: comparison of the sensitivity and selectivity of the smith‐waterman and fasta algorithms. Genomics 11(3), 635–650 (1991). 10.1016/0888-7543(91)90071-l [DOI] [PubMed] [Google Scholar]
- 24. Altschul, S.F. , et al.: Gapped blast and psi‐blast: a new generation of protein database search programs. Nucleic Acids Res. 25(17), 3389–3402 (1997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tang, F. , et al.: Halign 3: fast multiple alignment of ultra‐large numbers of similar DNA/rna sequences. Mol. Biol. Evol. 39(8), msac166 (2022). 10.1093/molbev/msac166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zou, Q. , et al.: Halign: fast multiple similar DNA/rna sequence alignment based on the centre star strategy. Bioinformatics 31(15), 2475–2481 (2015). 10.1093/bioinformatics/btv177 [DOI] [PubMed] [Google Scholar]
- 27. Wei, Y. , et al.: Wmsa: a novel method for multiple sequence alignment of DNA sequences. Bioinformatics 38(22), 5019–5025 (2022). 10.1093/bioinformatics/btac658 [DOI] [PubMed] [Google Scholar]
- 28. Chen, J. , et al.: Wmsa 2: a multiple DNA/rna sequence alignment tool implemented with accurate progressive mode and a fast win‐win mode combining the center star and progressive strategies. Briefings Bioinf. 24(4), bbad190 (2023). 10.1093/bib/bbad190 [DOI] [PubMed] [Google Scholar]
- 29. Zhang, Y. , et al.: Attention is all you need: utilizing attention in ai‐enabled drug discovery. Briefings Bioinf. 25(1), bbad467 (2024). 10.1093/bib/bbad467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang, Y. , et al.: P450rdb: a manually curated database of reactions catalyzed by cytochrome P450 enzymes. J. Adv. Res. 63, 35–42 (2023). 10.1016/j.jare.2023.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reshef, D.N. , et al.: Detecting novel associations in large data sets. Science 334(6062), 1518–1524 (2011). 10.1126/science.1205438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo, F.B. , et al.: Accurate prediction of human essential genes using only nucleotide composition and association information. Bioinformatics 33(12), 1758–1764 (2017). 10.1093/bioinformatics/btx055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang, R. , Ou, H.Y. , Zhang, C.T. : Deg: a database of essential genes. Nucleic Acids Res. 32(Database issue), D271–D272 (2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seal, R.L. , et al.: Genenames.Org: the hgnc resources in 2023. Nucleic Acids Res. 51(D1), D1003–D1009 (2023). 10.1093/nar/gkac888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dou, L. , et al.: Prediction of M5c modifications in rna sequences by combining multiple sequence features. Mol. Ther. Nucleic Acids 21, 332–342 (2020). 10.1016/j.omtn.2020.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang, Z.Y. , et al.: Design powerful predictor for mrna subcellular location prediction in Homo sapiens. Briefings Bioinf. 22(1), 526–535 (2021). 10.1093/bib/bbz177 [DOI] [PubMed] [Google Scholar]
- 37. Zhu, W. , et al.: A first computational frame for recognizing heparin‐binding protein. Diagnostics 13(14), 2465 (2023). 10.3390/diagnostics13142465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang, R. , et al.: Deepbio: an automated and interpretable deep‐learning platform for high‐throughput biological sequence prediction, functional annotation and visualization analysis. Nucleic Acids Res. 51(7), 3017–3029 (2023). 10.1093/nar/gkad055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bi, Y. , et al.: Clarion is a multi‐label problem transformation method for identifying mrna subcellular localizations. Briefings Bioinf. 23(6) (2022). 10.1093/bib/bbac467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zulfiqar, H. , et al.: Identification of cyclin protein using gradient boost decision tree algorithm. Comput. Struct. Biotechnol. J. 19, 4123–4131 (2021). 10.1016/j.csbj.2021.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang, R. , Zhang, C.T. : A brief review: the Z‐curve theory and its application in genome analysis. Curr. Genom. 15(2), 78–94 (2014). 10.2174/1389202915999140328162433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. He, W. , et al.: 70propred: a predictor for discovering Sigma70 promoters based on combining multiple features. BMC Syst. Biol. 12((Suppl 4)), 44 (2018). 10.1186/s12918-018-0570-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Meng, L. , et al.: Mini‐review: recent advances in post‐translational modification site prediction based on deep learning. Comput. Struct. Biotechnol. J. 20, 3522–3532 (2022). 10.1016/j.csbj.2022.06.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen, Z. , et al.: Ilearnplus: a comprehensive and automated machine‐learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res. 49(10), e60 (2021). 10.1093/nar/gkab122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Breiman, L. : Random forests. Mach. Learn. 45(1), 5–32 (2001) [Google Scholar]
- 46. Chen, T.Q. , Guestrin, C. : Xgboost: a scalable tree boosting system. In: Kdd'16: Proceedings of the 22nd Acm Sigkdd International Conference on Knowledge Discovery and Data Mining,, pp. 785–794 (2016) [Google Scholar]
- 47. Hopfield, J.J. : Neural networks and physical systems with emergent collective computational abilities. P Natl Acad Sci‐Biol 79(8), 2554–2558 (1982). 10.1073/pnas.79.8.2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hasan, M.M. , et al.: Meta‐I6ma: an interspecies predictor for identifying DNA N6‐methyladenine sites of plant genomes by exploiting informative features in an integrative machine‐learning framework. Briefings Bioinf. 22(3) (2021). 10.1093/bib/bbaa202 [DOI] [PubMed] [Google Scholar]
- 49. Lai, H.Y. , et al.: Iproep: a computational predictor for predicting promoter. Mol. Ther. Nucleic Acids 17, 337–346 (2019). 10.1016/j.omtn.2019.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zulfiqar, H. , et al.: Deep‐stp: a deep learning‐based approach to predict snake toxin proteins by using word embeddings. Front. Med. 10 (2024). 10.3389/fmed.2023.1291352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zou, X. , et al.: Accurately identifying hemagglutinin using sequence information and machine learning methods. Front. Med. 10, 1281880 (2023). 10.3389/fmed.2023.1281880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhu, H. , Hao, H. , Yu, L. : Identifying disease‐related microbes based on multi‐scale variational graph autoencoder embedding wasserstein distance. BMC Biol. 21(1), 294 (2023). 10.1186/s12915-023-01796-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li, H. , Pang, Y. , Liu, B. : Bioseq‐blm: a platform for analyzing DNA, rna, and protein sequences based on biological language models. Nucleic Acids Res. 49(22), e129 (2021). 10.1093/nar/gkab829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang, H. , et al.: A gender specific risk assessment of coronary heart disease based on physical examination data. NPJ digital medicine 6(1), 136 (2023). 10.1038/s41746-023-00887-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dao, F.Y. , et al.: Accurate identification of DNA replication origin by fusing epigenomics and chromatin interaction information. Research 2022, 9780293 (2022). 10.34133/2022/9780293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu, M. , et al.: Geometric deep learning for drug discovery. Expert Syst. Appl. 240, 122498 (2023). 10.1016/j.eswa.2023.122498 [DOI] [Google Scholar]
- 57. Tang, Y. , Pang, Y. , Liu, B. : Idp‐Seq2seq: identification of intrinsically disordered regions based on sequence to sequence learning. Bioinformatics 36(21), 5177–5186 (2021). 10.1093/bioinformatics/btaa667 [DOI] [PubMed] [Google Scholar]
- 58. Li, H. , Liu, B. : Bioseq‐diabolo: biological sequence similarity analysis using diabolo. PLoS Comput. Biol. 19(6), e1011214 (2023). 10.1371/journal.pcbi.1011214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wen, Q.F. , et al.: Geptop 2.0: an updated, more precise, and faster geptop server for identification of prokaryotic essential genes. Front. Microbiol. 10, 1236 (2019). 10.3389/fmicb.2019.01236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wei, W. , et al.: Geptop: a gene essentiality prediction tool for sequenced bacterial genomes based on orthology and phylogeny. PLoS One 8(8), e72343 (2013). 10.1371/journal.pone.0072343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shi, H. , et al.: Identify essential genes based on clustering based synthetic minority oversampling technique. Comput. Biol. Med. 153, 106523 (2023). 10.1016/j.compbiomed.2022.106523 [DOI] [PubMed] [Google Scholar]
- 62. Ma, J. , et al.: Bingo'‐a large language model‐ and graph neural network‐based workflow for the prediction of essential genes from protein data. Briefings Bioinf. 25(1) (2023). 10.1093/bib/bbad472 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analysed during this study are included in this published article.