

Significance
Biomass-burning organic aerosol(s) (BBOA) not only significantly impact Earth’s climate by absorbing solar radiation but also generate excited triplet states (3BBOA*) that react with other atmospheric molecules, known as photosensitization. Understanding the photosensitization processes is increasingly important due to the intensifying wildfires worldwide. In this study, we demonstrate the high reactivity of 3BBOA* in aerosol particles, indicated by the rapid sulfate formation via SO2 oxidation. Notably, the photosensitized sulfate formation in BBOA particles occurs much more rapidly than in diluted solutions, which is attributable to prominent interfacial reactions. These findings suggest that multiphase photosensitization in BBOA can drive the rapid formation of secondary pollutants and may exacerbate air quality degradation, particularly for regions prone to wildfires.
Keywords: air pollution, wildfire, multiphase chemistry, photosensitization, brown carbon
Abstract
Biomass-burning organic aerosol(s) (BBOA) are rich in brown carbon, which significantly absorbs solar irradiation and potentially accelerates global warming. Despite its importance, the multiphase photochemistry of BBOA after light absorption remains poorly understood due to challenges in determining the oxidant concentrations and the reaction kinetics within aerosol particles. In this study, we explored the photochemical reactivity of BBOA particles in multiphase S(IV) oxidation to sulfate. We found that sulfate formation in BBOA particles under light is predominantly driven by photosensitization involving the triplet excited states (3BBOA*) instead of iron, nitrate, and S(IV) photochemistry. Rates in BBOA particles are three orders of magnitude higher than those observed in the bulk solution, primarily due to the fast interfacial reactions. Our results highlight that the chemistry of 3BBOA* in particles can greatly contribute to the formation of sulfate, as an example of the secondary pollutants. Photosensitization of BBOA will likely become increasingly crucial due to the intensified global wildfires.
Organic aerosols resulting from wildfires and open biomass burning, collectively termed biomass-burning organic aerosol(s) (BBOA), represent the predominant source of global primary organic aerosols (POA). Annually, BBOA contributes about 60 to 85% of the total POA, with emissions estimated at approximately 31 to 36 Tg (1). These aerosols are rich in brown carbon (BrC) that can significantly absorb solar radiation, accelerating the warming of the Earth–atmosphere system (2).
The light-absorption properties and atmospheric persistence of BrC have garnered extensive research interest (3–7). It is critical to recognize that the fundamental process underlying light absorption involves the transition of electrons between energy states. Excited states with high energy will be generated upon the photon absorption by a BrC molecule. Recent studies indicate that BrC, upon excitation to singlet states (i.e., 1C*), can transition to longer-lived triplet states (i.e., 3C*) via intersystem crossing (ISC), besides undergoing photolysis and nonreactive relaxation processes such as fluorescence (Fig. 1). Triplets have significantly longer lifetimes than their singlet counterparts (8, 9), which enables reactions with various molecules (referred to as substrates) at appreciable rates to form secondary pollutants, known as photosensitization (10–15).
Fig. 1.

The photosensitization processes in BBOA. This diagram illustrates the key steps in the photosensitization process within BBOA. The black arrows indicate the self-cycling of photosensitizers (PS). The arrow from the photosensitizer to 1C∗ depicts the excitation of molecules from ground states to singlet excited states upon photon absorption. Subsequent arrows from 1C* to PS represent the relaxation of singlet states through both reactive (e.g., photolysis) and nonreactive (e.g., fluorescence and internal conversion) pathways. The arrow from 1C∗ to 3C∗ denotes intersystem crossing to triplet states, while the final arrow from 3C∗ to PS shows the relaxation pathways for triplet states, including phosphorescence and triplet–triplet annihilation. The red arrow linking condensed and gas phase precursors to 3C* illustrates the multiphase reactions driven by 3C* to form secondary particulate pollutants (i.e., photosensitization).
Sulfate aerosols, an important secondary air pollutant, are predominantly produced by multiphase SO2 oxidation during haze episodes (16). However, field measurements of sulfate formation rates often greatly exceed predictions from models based on conventional gas-phase and in-cloud reaction schemes. While multiphase SO2 oxidation has been proposed as a potential explanation for this discrepancy, the detailed mechanisms and kinetics remain less understood (17, 18). Previous studies have noted the presence of sulfate in BB particles, especially those transported through urban and industrial areas with elevated SO2 levels (19–21). Liu et al. (22) observed an excess of sulfate beyond that expected from chloride depletion during the aging of BB plumes, suggesting additional sulfate formation pathways. Notably, recent studies have reported enhanced sulfate production in both incense-burning particles and model biomass-burning photosensitizer (PS) particles under light illuminations, compared to those in the dark (23–25). The sulfate formation in illuminated photosensitizer particles prevails over that in photolyzing nitrate particles (25), a potentially important pathway for sulfate formation (26, 27). This highlights the role of photochemical processes initiated by the triplets of BBOA (i.e., 3BBOA*) beyond the direct climate impacts of BBOA, particularly as the frequency and intensity of wildfires are expected to rise globally due to climate change (28–30).
Despite these findings, the aerosol-phase reaction kinetics involving 3C* and atmospheric substrates, such as SO2, remain poorly understood. One of the main challenges is the transient nature of 3C* which complicates direct measurements (31). The decay of a chemical probe with known reaction rate constants with 3C* was the only existing approach to determine the steady-state [3C*] in diluted solutions. However, the probe decay is subjected to back electron donation and light screening in concentrated aerosols, known as inhibition, which causes great underestimation (32, 33). The kinetic data, largely derived from dilute solutions using single-model photosensitizers, may not accurately reflect the complex interplays within BBOA, which contains a diverse mix of photosensitizers and substrates (34, 35). Moreover, increasing evidence has shown that the kinetics for multiphase reactions in concentrated aerosols with high solute strength can differ greatly from those in the bulk solutions (16, 36–39). Excess quenchers are typically used to ascertain the potential importance of 3C*, through observation of suppressed reactant decay or product formation. However, the quencher can also react with other oxidants and generate by-products, introducing uncertainties (23, 40, 41). Overall, the lack of adequate kinetic evaluations has significantly limited our understanding of the capability of 3BBOA* to drive multiphase reactions and exacerbate air pollution.
Herein, we reported a kinetic study on the multiphase reactivity involving 3BBOA* in submicron aerosol particles, using an aerosol flow reactor (AFT) coupled with multiple offline analytical tools. In this work, we referred to the water extract of the BB particles as BBOA. We focused on the water extract due to its better compatibility with analytical tools for deeper insights into our study. The potential effects of water-insoluble components were also discussed. We focused on sulfate formation from SO2 oxidation as an indicator of the multiphase reaction kinetics. This choice was driven by the atmospheric significance of sulfate and the relatively straightforward mechanism compared to that of organic oxidations. We used chemical probes and transient absorption spectroscopy (TA) to determine the oxidant concentrations and reaction rate constants in diluted solutions and then extrapolate to aerosol conditions, enabling modeling of the sulfate formation rate and comparisons with the measurement results. Mass spectrometry provides identification of the potential photosensitizers in BBOA. In addition, quantum chemical calculation and electron paramagnetic resonance spectroscopy (EPR) connect the reactivities of 3BBOA* to photochemical parameters such as redox potentials, broadening our mechanistic understanding of BBOA-driven reactions. The overall goal of this work is to provide quantitative insights into the photosensitizing capabilities of BBOA particles and facilitate future parameterization.
Results
Sulfate Formation Kinetic in Illuminated BBOA Particles.
Fig. 2 shows the kinetics of sulfate formation in the BBOA particles (atomized from the water extract of BB particles) under different SO2 concentrations and light intensities. The sulfate concentration in BBOA linearly increases with reaction time under all conditions, with faster rises observed at higher SO2 concentrations and light intensities, suggesting photochemical oxidation of SO2 (Fig. 2 A and B).
Fig. 2.

(A) Sulfate concentrations in BBOA as a function of illumination time at different SO2 concentrations. The light intensity of 100% indicates the full flux from light tubes; (B) Sulfate concentrations in BBOA as a function of illumination time at different light intensities. The light intensities of 50% and 20% were achieved by using neutral density filters for light attenuation. The SO2 concentration is 40 ppb; (C) The dependence of sulfate formation rate on SO2 concentrations, the light intensity is 100%; (D) The dependence of sulfate formation rate on the light intensity, the SO2 concentration is 40 ppb. All experiments were under N2 conditions and at 80% RH. The initial sizes of BBOA particles are all 100 nm.
In contrast, experiments conducted in the dark with SO2 exposure showed negligible sulfate formation, indicating minimal contribution from nonphotochemical pathways (SI Appendix, Fig. S1). SO2 at the air–water interface could undergo forbidden excitation to its triplet state (i.e., 3SO2*), followed by hydrolysis to hydroxyl radical (i.e., OH•) capable of oxidizing SO2 (42, 43). To determine whether 3SO2* photochemistry contributes to the observed sulfate formation in BBOA under light, we quantified the sulfate formation in nonphotoactive organic buffer droplets (Sodium malonate/Sodium bimalonate = 1:1) at the same [SO2], light intensity, initial particle size, and comparable pH (around 4, will be discussed later). The liquid water content of the buffer droplet is higher than that of BBOA, which allows more S(IV) dissolution. Malonate is less reactive than phenolic compounds toward OH•, thus it is not expected to suppress the [OH•] in buffer droplets compared with BBOA. However, negligible sulfate formation in organic buffer droplets under illumination was found, suggesting that 3SO2* chemistry plays a minor role (SI Appendix, Fig. S1). The production of oxidants via photolysis of iron salts, iron organic complexes, and nitrate is possible, but their roles in the sulfate formation were also deemed minor, as doubling the iron and nitrate concentrations in BBOA only resulted in less than 5% increases in sulfate formation (SI Appendix, Fig. S2). Consequently, the dominant pathway for sulfate formation in illuminated BBOA is likely through the oxidation of S(IV) by 3BBOA*. In addition to SO2, effective photosensitized conversions of organic sulfur compounds to sulfate have been reported in the aquatic environments and likely sea-spray aerosols (44–46). A recent study by Rao et al. (47) suggested that such conversions can take place at the interface of microdroplets even in the absence of light. Nevertheless, no sulfate formation was observed in illuminated BBOA particles in the absence of SO2, indicating organic sulfur compounds, if present, were not a significant source of sulfate in BBOA particles (SI Appendix, Fig. S1).
Using TA, the quenching rate constant of 3BBOA* by S(IV) [k3BBOA∗, S(IV)] was determined to be 6.0 × 108, 5.1 × 108, and 3.2 × 108 M–1 s–1 at pH of 1, 4, and 7, respectively (SI Appendix, Fig. S3). Thermodynamic models suggested that the pH of BBOA particles ranges from 3 to 5 (48). Using a combined method of pH indicator paper and RGB analysis (SI Appendix, Fig. S4 and Text S1), we estimated the pH of BBOA particles to be around 4, which is consistent with the estimate based on the charge-conservation rule (49). At this pH, HSO3− dominates the dissolved S(IV) species (SI Appendix, Fig. S3). 3BBOA* may react with HSO3− via energy transfer, electron transfer, and hydrogen transfer. While the energy transfer from 3BBOA* to S(IV) can also lead to 3S(IV)* and ultimately OH•, probe experiments indicate that the OH• concentration would be too low (e.g., 10−18 M) to compete with 3BBOA* for oxidizing S(IV) (SI Appendix, Text S2), partially due to the low redox potential of 3BBOA* to abstract electrons from water to yield OH• (will be discussed later).
We propose the reactions between 3BBOA* and S(IV) via electron transfer and hydrogen transfer, illustrated as follows using HSO3− as an example:
| [R1] |
| [R2] |
Subsequent self-reactions of the sulfite radicals and reactions with HSO3− will lead to sulfate formation (50). The 3BBOA* concentration increases with light intensity. The observed slopes for the sulfate formation rate vs. SO2 concentration and light intensity are 1.12 and 1.02, respectively, on the log–log plots (Fig. 2 C and D), suggesting that the reaction kinetics between 3BBOA* and S(IV) could be considered as first order [R3] (8, 15).
| [R3] |
Despite the oxidative SO2 uptake to potentially acidify BBOA, prolonged illumination did not significantly reduce sulfate formation (Fig. 2 A and B).
The sulfate formation rate in BBOA can be given by
| [1] |
where Ka1 and Ka2 are the thermodynamic dissociation constants of H2SO3. HSO2 and PSO2 represent Henry’s law constant and partial pressure of SO2, respectively. The equilibrium constants are summarized in SI Appendix, Table S1.
The Significant Role of Interfacial Reactions.
We first model the sulfate formation kinetics using Eq. 1, based on data from diluted BBOA solutions mixed with S(IV) [BBOA to water mass ratio (BWR) of 10−3]. To compare results from solution and particle, we normalized the sulfate formation rate by the S(IV) concentration, prescribed in diluted solution and calculated by gas-particle partitioning, respectively. The pH of solution was adjusted to 4 to facilitate comparison. Fig. 3A shows that the normalized sulfate formation rate from modeling closely aligns with experimental measurements in the BBOA solution. This also suggested that the 3BBOA* quenching by S(IV) measured via TA represents the reaction well. However, a stark discrepancy arises with BBOA particles, where the modeled normalized sulfate formation rate is three orders of magnitudes lower than the measured rate. This divergence indicates potential underpredictions of the k3BBOA∗, S(IV) × [3BBOA*] in aerosols.
Fig. 3.

(A) The modeled and measured sulfate formation rate in bulk solution (purple), deposited droplets (red), and aerosol particles (pink), normalized by the concentration of S(IV). The sulfate formation rate in bulk solution was calculated up to 10% decay of S(IV), and the initial S(IV) concentration of 10 mM was used for normalization. The aerosol and deposited droplet experiments were conducted at 80% RH and 40 ppb SO2; (B) The sulfate formation rate as a function of the surface-to-volume ratio of the particles. The color and the symbol size show the RH and initial size of the BBOA particles, respectively. The equilibrated sizes at 70% and 40% RH are estimated by the size growth factor measured by H-TDMA; (C) The illustration for the distinct S(IV) oxidation kinetic between the interface and bulk phase of the BBOA particles; (D) The k3BBOA∗, S(IV) × [3BBOA*] ratio of the interface to the bulk phase of the BBOA particles (Left axis) as a function of relative volume of the interface for a 100 nm BBOA particle (Bottom axis). The corresponding interfacial thickness and k3BBOA∗, S(IV) × [3BBOA*] were shown on the Top and Right axes, respectively. The green column shows the typical thickness of the water monolayer. The BBOA particles were assumed spherical.
As the BWR increases, both the formation rate of 3BBOA* and sink rate by organic matters (OM) increase, resulting in modeled [3BBOA*] reaching a plateau (SI Appendix, Fig. S5). Despite the comparable modeled [3BBOA*] between solution at BWR of 10−3 and aerosol (e.g., BWR of 1), aerosol particles have a significantly higher surface-to-volume ratio (S/V) than bulk solutions. Our modeling based on Eq. 1 assumes identical environment for both the interface and the bulk of the particles. However, in reality, molecular behavior at the interface could differ significantly from that in bulk solutions (51). For example, the interfacial semisolvation reduces solvent rearrangement energy (52) and stabilizes excited state molecules (53), thereby increasing the reaction rate constant. In addition, photosensitizers and 3C*s can be concentrated at the interface in the presence of trace amount of surfactant molecules (e.g., a monolayer), facilitating interfacial reactions (54–57).
Recent work by Wang et al. (40) observed accelerated S(IV) oxidation in illuminated humic acid microdroplets that generate reactive triplets and OH•. While the electron localization fields for the photosensitizers are mostly identical between the air–water interface and the bulk, the interface stabilizes the radicals generated by electron transfer. This phenomenon is also found in our molecular dynamic simulations with 3,4-dimethoxy benzaldehyde (DMB, a known photosensitizer in BBOA), which could support a higher k3BBOA∗, S(IV) at the interface than bulk due to the reduced energy of the radical product (i.e., DMB•−) (SI Appendix, Figs. S6 and S7 and Text S3).
Using a flow cell, we exposed large deposited droplets (approximately 1 mm in diameter) to the same SO2 concentration, relative humidity (RH), and light intensity as we did for submicron aerosols in the AFT. Since the large droplets were fully equilibrated at 80% RH, their solute concentrations were expected to be the same as that of the submicron particles, with the only difference being the S/V. The sulfate formation rate in large BBOA droplets was slightly higher than that in diluted solutions but significantly lower than in submicron particles, further corroborating the importance of interfacial reactions (Fig. 3A). Additionally, an increase in sulfate formation rates was observed with higher S/V of submicron particles (Fig. 3B). This dependence holds even under two orders of magnitude higher viscosity at RHs (7, 58). Overall, the faster photosensitized sulfate formation in submicron aerosol particles compared to bulk solutions is attributed to prominent interfacial reactions rather than differences in pH or solute concentration.
We utilized a two-layer model comprising an interface layer and a bulk layer to illustrate the potential extent of acceleration for interfacial reactions over those in the bulk, as depicted in Fig. 3C. We assumed a fast equilibrium of S(IV) at the interface and the bulk, alongside a three times higher SO2 concentration at the air–water interface than that in the bulk predicted by Henry’s law (59). The normalized sulfate formation rate can be expressed as
| [2] |
The subscripts i and b represent interface and bulk. RV denotes relative volume. Using the same k3BBOA∗, S(IV), b and [3BBOA*]b of the aerosol as that derived from solution (i.e., 5 × 108 M−1 s−1 and 1.25 × 10−12 M), the k3BBOA∗, S(IV), i ×[3BBOA*]i across different RVi in the BBOA particle can be obtained from the Eq. 2 (Fig. 3D).
Unique photochemistry has been reported to occur within a monolayer at the interface (51, 56, 57). Assuming a typical interfacial water monolayer thickness (L) of 0.15 nm (51), the RVi is roughly 1% for 100 nm particles. Under these conditions, k3BBOA∗, S(IV), i × [3BBOA*]i reaches 102 s−1, which is five orders of magnitude higher than that for the bulk (Fig. 3D). While it is not possible to precisely quantify the size of the interfacial reaction “zone”, increased thickness of the interfacial layer would lead to lower k3BBOA∗, S(IV), i × [3BBOA*]i.
The mass accommodation coefficient (kMT) defines the fraction of collisions with a surface that lead to uptake of gas molecules to the surface, expressed as:
| [3] |
where Rp, Dg, α, and v are the radius of the particle, gas-phase molecular diffusion coefficient, mass accommodation coefficient, and mean molecular speed of SO2, respectively. Taking a typical α of 0.11 for SO2 (16), kMT would be approximately 5 × 108 s−1, which is much higher than the interfacial pseudo-first-order reaction rate constant with 3BBOA* (k3BBOA∗, S(IV), i × [3BBOA*]i = 102 s−1). This supported the fast equilibrium of S(IV) at the interface without mass-transfer limitation.
Taking the same diffusion coefficient (D) for S(IV) as that reported for ozone in the hydrophilic phase of BBOA particles (58), the characteristic time for S(IV) diffusion crossing interface () is 10−9 and 10−11 s at 40% and 80% RH, much shorter than the characteristic time for its interfacial reaction with 3BBOA* (10−2 s,). This implies that S(IV) is not confined to the interface. However, the interfacial reaction accounts for more than 99% of S(IV) oxidation in the BBOA. It should be noted that the rate constant for reactant diffusion and collision to form a precursor complex is typically 109 M−1 s−1 in aqueous solutions (60). If this represents the upper limit for k3BBOA∗, S(IV), i, then the [3BBOA*]i would reach 10−7 M, which was underestimated by five orders of magnitude using the extrapolated values from bulk solution measurements.
On the other hand, we observed that the decay of levoglucosan, a compound reported with no surface propensity and likely located in the BBOA bulk (61), is similar to the modeled results using the quenching rate constants and the predicted [3BBOA*] from diluted solution (SI Appendix, Fig. S8). This suggested that the k3BBOA∗ × [3BBOA*] may not be elevated for bulk phase reactions in the BBOA particles, assuming that the production of levoglucosan via back electron transfer to the oxidized levoglucosan is insignificant. Overall, an averaged enhancement factor (EF) of three orders of magnitude from the solution value can represent the k3BBOA∗, S(IV) × [3BBOA*] in BBOA particles for multiphase reactions.
The Photochemical Reactivity of the BBOA Photosensitizers.
Understanding how different photosensitizers in BBOA promote multiphase reactions is of great significance, given the expected variability of photosensitizer speciation in the ambient atmosphere. To simplify, we have treated the 3C* species in BBOA as a single entity in the above analysis (i.e., 3BBOA*, Eq. 1). Here, we consider the sulfate formation kinetics driven by N different 3C* species:
| [4] |
At steady state, the concentration of each 3C* species can be derived from its formation rate [the product of the light absorption rate (Rabs) and the intersystem crossing quantum yields (ФISC)] over the total sinking rates by OM and S(IV):
| [5] |
We identify the potential photosensitizers and characterize their light absorption in our BBOA sample. The two most prevalent chromophores at 300 to 400 nm were Sinapaldehyde (SinAld) and Coniferyl aldehyde (ConAld). Each of them accounts for more than 30% of the PDA absorption. Other known photosensitizers including DMB, syringaldehyde (SyrAld), and vanillin (VL) contributed moderately (2.9 to 9%) to PDA absorption. These major chromophores can be primarily categorized into aromatic propenals and aromatic aldehydes, with structures shown in Fig. 4A. The former compounds likely originated from lignin degradation and the subsequent oxidation forms the latter compounds (SI Appendix, Fig. S9). Their conjugated C = O moieties, not only enhance the light absorption by electron withdrawal from the aromatic ring (62) but also facilitate intersystem crossing through coupled n and π orbitals, making them potential photosensitizers (63).
Fig. 4.

(A) The structures of the major chromophores identified. The numbers in the blanket are the percentage contribution to the total PDA absorption; (B) The contribution of sulfate formation by different types of photosensitizers according to the enhanced sulfate formation by doubling the concentrations of the corresponding photosensitizer; (C) The map of energy and one-electron reduction potential for different 3C* of identified photosensitizers in BBOA. V. SHE indicates the volts relative to the standard hydrogen electrode. The data are summarized in SI Appendix, Table S2; (D) The EF of normalized sulfate formation in aerosol particles over bulk solution. Before EF calculation, the sulfate formation rates were normalized by S(IV) concentrations in surface and bulk, respectively. Both aerosol particles and bulk solutions contain 1 mM photosensitizers and around 3 M sodium bimalonate/sodium malonate=1:1 buffer.
By doubling the concentrations of each major chromophore and measuring the enhanced sulfate formation rate, we estimated their contributions to the sulfate formation, assuming a proportional increase in sulfate formation. In the BBOA collected from the AFT without illumination, the ratio of photosensitizers to K+ [used as an internal standard due to its stability toward photochemical reactions (23, 64)] decreased by less than 8% compared to the original solution for atomization. This indicates minimal evaporation of photosensitizers, consistent with their relatively high Log Kwa and Log Koa (SI Appendix, Table S2). Fig. 4B shows that these identified chromophores accounted for most of the sulfate formation under light, with contributions descending in order: DMB (29%) > ConAld (18%) > SinAld (15%) = SyrAld (15%) > VL (7%). Minor contributors such as Ferulic acid, Vanillylidene acetone, and 4-methoxy-cinnamaldehyde collectively accounted for 2% of the enhanced sulfate formation and are denoted as “others.”
Despite the lower contribution to PDA intensity than SinAld and ConAld, DMB was the greatest contributor to sulfate formation, which implies different reactivities and ISC quantum yield among 3C*s. We calculated the one-electron reduction potential (Eo) and triplet energy (ET) of the 3C* of these photosensitizers (SI Appendix, Text S4), which can reflect their reactivity via electron transfer and energy transfer, respectively, as displayed in Fig. 4C. It should be noted that we focused on the unprotonated 3C* for the ET and Eo map in Fig. 4C and that the protonation at low pH can increase the Eo of the 3C* to promote reactions (SI Appendix, Table S2) (11, 65). The aromatic propenals and aromatic aldehydes exhibited similar Eos of around 1 V. SHE, higher than the one-electron oxidation potential of S(IV) [0.63 to 0.84 V. SHE (66, 67)], indicating that electron transfer from S(IV) to 3BBOA* is thermodynamically favorable. The logarithm of the second-order quenching rate constants of 3C* by S(IV) does not increase with the difference between the one-electron reduction potential of 3C* and the oxidation potential of S(IV), suggesting that a diffusion-control regime has been approached (SI Appendix, Fig. S10). Furthermore, the DMPO-SO3 peaks detected in the illuminated BBOA and NaHSO3 mixture under N2 conditions support the electron transfer from S(IV) to 3BBOA* that yields SO3•− (SI Appendix, Fig. S11). The significant decay of TEMPO intensity in the EPR spectra of the illuminated BB and NaHSO3 mixture also serves as evidence for the electron-transfer reactions (68). The Eo of 3C*s in BBOA are mostly below the one-electron oxidation potential of water [e.g., 2.18 V. SHE (69)], rendering the electron transfer from water or OH− to form OH• unfavorable, consistent with the minimal OH• production. DMPO-OH was detected in the illuminated mixed solution of benzoquinone (BQ, Eo = 2.42 V.SHE) and DMPO, but not in BBOA, DMB, and VL solutions at the same mass concentration (SI Appendix, Fig. S12). This might explain the recent observation by Liu-Kang et al. (70), which suggested a minor role of OH• in secondary BrC formation in illuminated BBOA. For energy transfer reactions, the ETs of aromatic aldehydes are slightly higher than that of aromatic propenals, but still below the required value to excite S(IV) (e.g., 300 kJ mol−1) (71). Hence, sulfate formation from these photosensitizers is likely due to electron transfer rather than energy transfer reactions. Also, the similar ET and Eo suggested that the 3C* reactivities of aromatic propenals and aromatic aldehydes toward S(IV), and maybe OM, are likely close.
The ФISC of 3C*, another determinant of [3C*] besides the light absorption rate, plays a key role in their contributions to sulfate formation. Assuming the same 3C* reactivity, the enhanced sulfate formation rate after doubling the concentration of the photosensitizers is proportional to the Rabs and ФISC, simplified in Eq. 6:
| [6] |
Taking the PDA absorption intensity as Rabs and the reported ФISC for VL (0.21) and DMB (0.38) (8), we estimated the ФISC for SinAld (0.04), ConAld (0.05), and SyrAld (0.14) based on sulfate formation results. Aromatic aldehydes exhibited generally higher ФISC (24 ± 12%), making them more effective photosensitizers than aromatic propenals (ФISC = 5 ± 1%). Despite the low ФISC, aromatic propenals still contribute to the sulfate formation, benefiting from their around 2 to 3 folds higher molar absorptivity than aromatic aldehydes (11). The larger steric effects by vinyl groups in aromatic propenals may hinder electron transfer reactions, potentially leading to an underestimation of their ФISC.
To conclude, aromatic aldehydes possess higher ISC quantum yields and lead to higher [3C*] than aromatic propenals to drive multiphase oxidation. The relative abundance of aromatic aldehydes to aromatic propenals can vary across different types of BBOA, though our results align with those reported by the literature, which showed that the light absorption by aromatic propenals prevails over aromatic aldehydes (72, 73). More importantly, we found that for those identified major photosensitizers, the average EF of sulfate formation in aerosol particles over the bulk solution was (2.1 ± 0.3) ×103 (Fig. 4D), which is similar to the EF for BBOA (approximately 3 × 103).
Sulfate Formation by 3BBOA* Chemistry in Wildfire.
The transport of wildfire smoke in the atmosphere exhibits significant spatial and temporal complexities. Although it is currently not feasible to parameterize photosensitization reactions in BBOA in global models, the sulfate formation kinetics and behaviors of photosensitizers detailed in this study highlight the potential impact of multiphase BBOA photochemistry on air quality in wildfire-prone regions during fire seasons (74).
For comparative analysis, we calculated the sulfate formation through H2O2 oxidation, which has been reported to dominate global sulfate production (75), using local concentrations of SO2, H2O2, pH, and aerosol liquid water. We considered BBOA concentrations of 10 and 40 µg m−3, representing moderate and severe fire conditions, respectively. Details of these calculations are provided in SI Appendix, Table S3 and Text S5. We focused on the direct reactions between 3BBOA* and S(IV) for sulfate production. It is noteworthy that the presence of O2 will greatly complicate the chemistry. Specifically, 3BBOA* will react with the dissolved O2 via energy transfer to form 1O2* (76, 77). The HBBOA, formed via hydrogen abstraction from OM by 3BBOA*, can rapidly react with O2 to generate HO2 (78), which subsequently leads to H2O2 and OH• (79). The 1O2*, HO2/O2•−, H2O2, and OH• are collectively known as “secondary oxidants,” which can also contribute to the S(IV) oxidation (25). Sulfate formation in BBOA in air is around double of that in N2 (SI Appendix, Fig. S13). While resolving the concentrations of each secondary oxidant is beyond the focus of this work, we did not expect different [3C*] between air and N2 conditions since OM will be a predominant sink for 3C* in both cases, due to its much higher concentration (>30 M C) than dissolved oxygen (10−4 M) in BBOA particles. Moreover, we do not explicitly consider the emission of primary precursors (e.g., SO2) even for the severe fire scenario, which may also lead to underestimations.
According to the TUV model (SI Appendix, Text S5), an increase in BBOA loading from 10 to 40 µg m−3 attenuates the photon flux by 10 to 30%, which reduces the formation of 3BBOA* and partially offsets the increased sulfate formation (Fig. 5). Nonetheless, in regions like British Columbia, California, Amazonia, and Athens, the fractional contribution of 3BBOA* to sulfate formation increases significantly with fire severity to about 0.5 to 0.7, surpassing that by H2O2. In contrast, in Asian and African regions such as Guizhou and Leopoldville, the increase is more moderate at 0.04 and 0.09, respectively. It is important to note that we calculated the UV flux at the ground level to provide a conservative estimate of photosensitized sulfate formation, especially for severe fire scenarios.
Fig. 5.
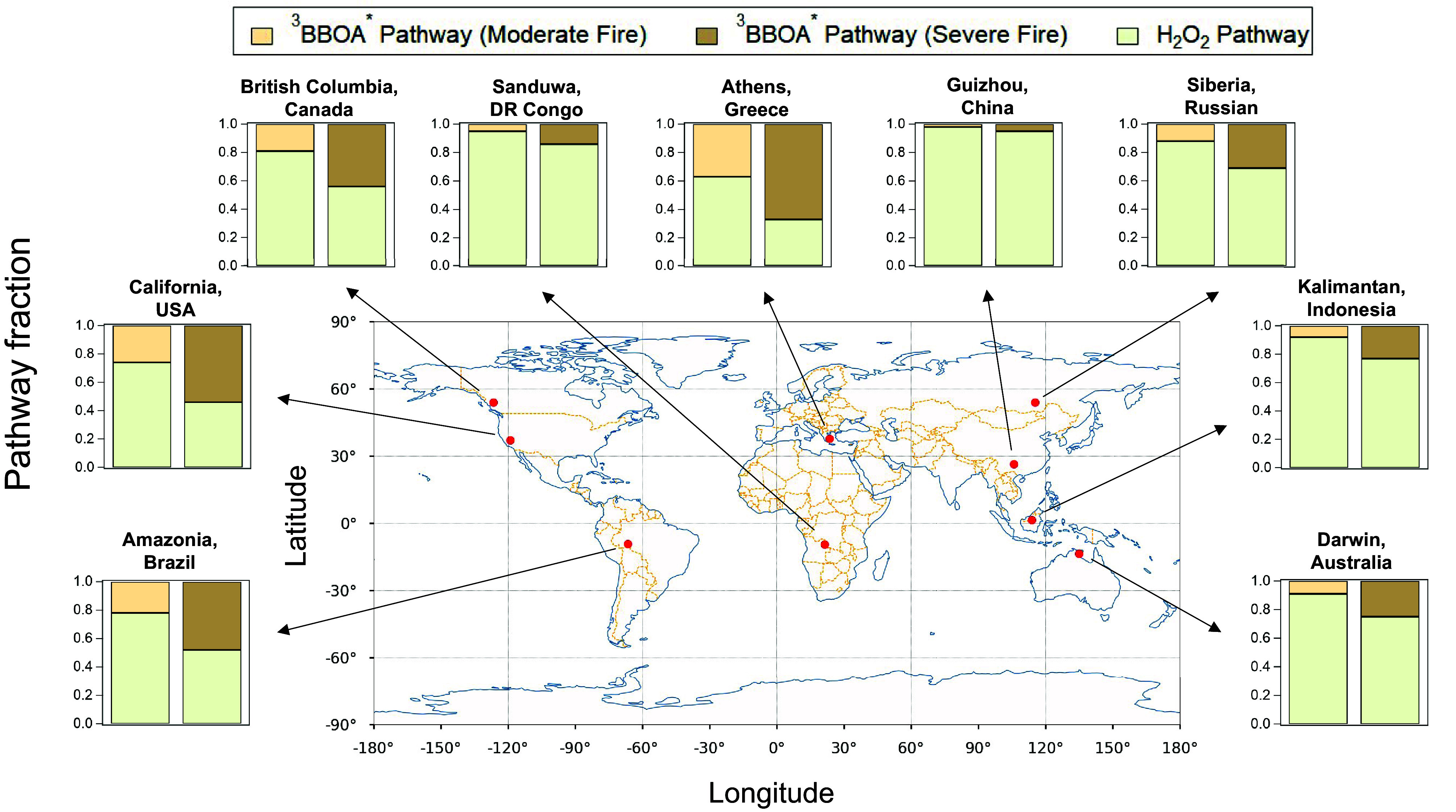
Sulfate formation in wildfire-prone regions. The histograms show the fractional contributions of the 3BBOA* and H2O2 oxidation pathways to the sulfate formation under different fire severity.
The increase in sulfate formation rate by 3BBOA* upon rising fire severity shows a negative correlation with aerosol pH but there is no clear correlation with SO2 or H2O2 concentrations (SI Appendix, Fig. S14). At pH above 2, where the increased SO2 solubility compensates for the reduced reaction rate constant, the sulfate formation rate by H2O2 oxidation is largely pH-independent (SI Appendix, Fig. S15) (16). At pH below 2, which applies to most cases in Fig. 5, the H2O2 sulfate formation rate decreases significantly due to a shift from HSO3− to less reactive H2SO3 as the dominant S(IV) species. On the other hand, the 3BBOA* sulfate formation is less sensitive to pH due to the comparable reactivity of 3BBOA* toward different S(IV) species. Therefore, enhanced sulfate formation driven by 3BBOA* in wildfires could be particularly significant in regions with lower aerosol acidity.
Discussion
Our results demonstrated that BBOA exhibited prominent photochemical reactivity, which can effectively drive multiphase reactions such as SO2 oxidation to sulfate. We further show that using concentrations and rate constants on photosensitization from bulk experiments can underestimate multiphase sulfate formation in aerosols by three orders of magnitude. Besides aromatic photosensitizers in BBOA, 2-imidazole carboxaldehyde (2-IC), a common atmospheric photosensitizer used in laboratory studies, gave comparable EF (SI Appendix, Fig. S16). While the direct determination of reaction rate constants and 3C* concentrations remains challenging, an EF of three orders of magnitude may be used to estimate the k3C∗ × [3C*] in BBOA particles for multiphase reactions, based on parameters derived from the bulk. For example, extrapolation from the solution data yields [3C*] of 10−13 M in ambient particles (SI Appendix, Fig. S17). Using this estimate and the reaction rate constant for ambient extract (bulk solution) (15), the sulfate formation rate of 10−4-10−2 µg m−3 h−1 can be obtained at pH of 4 to 6, as typical scenarios of Eastern Asian haze. After applying the EF, the sulfate formation rate in particles is estimated as 10−1 to 10 µg m−3 h−1, which aligns with the “missing” sulfate formation rate of 0.3 to 5 µg m−3 h−1 between modeling and field measurements.
The diverse speciation and reactivities of photosensitizers in BBOA have greatly complicated parameterization. As a step forward, we identified the potential photosensitizers in BBOA, profiled their ET and Eo, and correlated with their abilities in oxidizing S(IV) to sulfate. The results suggested that the tested BBOA photosensitizers may possess overall similar reactivities and varied ФISC by substitution. To our best knowledge, there is a limited number of identified photosensitizers in the atmosphere. Aromatic aldehydes, such as DMB, are commonly used as model BB photosensitizers in laboratory investigations, and our results support their validity (8).
With the photochemical properties of 3C*s, it could be feasible to parameterize photosensitized reactions in atmospheric waters containing a complex mixture of photosensitizers. Specifically, the reaction rate of a substrate (S) with 3C*s can be expressed as Eq. 7, by combining Eqs. 4 and 5:
| [7] |
k3C∗i, S, k3C∗i, OM, and k3C∗i, O2 denote the second-order quenching rate constant of 3C* by the substrate, OM (other than the substrate), and O2, which could be measured and estimated from the Eo and ET (e.g., using Rehm–Weller equation). In BBOA particles where the reactivities of 3C*s are potentially similar, and OM likely prevails over the other sinks, Eq. 7 can be simplified to Eq. 8:
| [8] |
Rabs can be derived from the concentration of the aromatic aldehydes and aromatic propenals and their molar absorptivity. With the corresponding ФISC, one can predict the substrate oxidation by 3BBOA* when the k3C∗i, S and k3C∗i, OM are available. An EF of 103 is recommended for multiphase reactions in aerosol particles. It is worth mentioning that a combined approach using ultrafast spectroscopy and chemical probes has been reported as effective for quantifying the quantum yield of short-lived oxidants in real time (80).
In this study, we focus on the BBOA derived from water extracts, given that the BB plume directly from the furnace was not stable enough for kinetics and the organic solvent will interfere with probe experiments. It should be noted that the photosensitizers identified in the BBOA (water extract) still have much lower water solubilities (e.g., 10−2 M) than the atmospheric organics considered highly water-soluble, such as glyoxal (~2 M). GC×GC ToF MS analysis of the hexane extracts targeting the least water-soluble compounds showed that single-ring aromatics, led by aromatic propenals and aromatic aldehydes, are still predominant (SI Appendix, Fig. S18). This implies that the majority of photosensitizers in BB particles are likely included in water extracts. However, it should be noted some compounds with even lower water solubilities (e.g., <10−3 M) than single-ring aromatics, such as polyaromatic hydrocarbons may still be less recovered by water extraction (SI Appendix, Table S2) (72). In aerosol particles with limited liquid water, these poorly water-soluble compounds with high Log Koa values may form a hydrophobic shell of the particles, leaving the more soluble counterparts in the hydrophilic core (SI Appendix, Fig. S19) (58). These less soluble compounds generally exhibit stronger light-absorption capacities due to their planar and conjugated structures, making them potentially good photosensitizer candidates (81). Moreover, aromatic aldehydes and aromatic propenals can partition into the hydrophobic shell owing to their high Log Koa (around 10) and drive multiphase reactions after being excited to their triplet states. Though kinetic analysis was not possible, we found the enhanced sulfate formation in BB particles (directly from the furnace without undergoing extraction) over that in BBOA (water extract) under light, indicated by the mass ratio of SO42− to K+ (SI Appendix, Fig. S20).
The aging of BBOA during atmospheric transportation has been reported to show initial enhancements in light absorption, followed by photobleaching at slower rates (6, 70, 82). The typical BrC lifetime at ground levels is about 1 d or less (7, 83). However, when BBOA is elevated to higher altitudes, such as 5 km, through deep convection processes, the lower temperatures significantly increase its viscosity (84, 85). Such elevated viscosity could extend the lifetime of primary photosensitizers against oxidative bleaching by gaseous oxidants by 2 to 5 orders of magnitude, compared to conditions at 1 km altitude (7). Though increased viscosity hinders diffusion of gaseous precursors, the multiphase reactions triggered by interfacial photosensitization may still be effective. The rapid conversion of BBOA to oxidized organic aerosol (OOA) is well documented (86). However, OOA from biomass burning has also been identified as an important contributor to atmospheric 3C*(87). Despite the potentially limited diffusion of gas phase oxidants, photosensitization could serve as a source of “in-particle” oxidants to trigger particle aging and changes in physicochemical properties.
Beyond S(IV), the reactions between 3BBOA* and other atmospheric precursors, such as volatile organic compounds, warrant further quantitative investigation (88). The ET and Eo of 3BBOA* provide valuable references for inferring and parameterizing its reactivities toward various atmospheric precursors. Subsequent studies are also encouraged to explore how these photosensitization reactions depend on the combustion condition, environmental factors, and atmospheric aging of BBOA, to better understand their impacts on air quality and climate. Approaches for identifying the potential photosensitizers in diverse and complex particulate matrix will be invaluable.
Materials and Methods
Preparation of BBOA.
Biomass-burning (BB) particles were generated by smoldering untreated pine wood sourced from a Canadian hardware store. The apparatus for generating BB particles is shown in SI Appendix, Fig. S21. In brief, untreated pine wood was cut into strips (~20 × 2 × 2 mm) and positioned lengthwise inside a quartz flow tube furnace (inner diameter 36 mm, outer diameter 40 mm, length 1,400 mm). Zero air (Peak Scientific) was introduced through the tube at a flow rate of 2 L min−1 using a mass flow controller (Alicat). The heating temperature within the tube was calibrated and maintained at 400 °C, in the range of natural biomass burning (e.g., 300 to 800 °C) (6, 58). Aethalometer (USA) measurement shows that the black carbon concentration drops back to the baseline level after 1 min of heating, indicating a transition from the flaming phase to the smoldering phase. Therefore, the samplings were performed approximately two min after the temperature reached 400 °C. After dilution using a two-stage jet flow dilutor (Dekati), the BB particles from the smoldering phase were collected onto a 47 mm quartz filter (Pall) loaded on a stainless-steel filter holder. Prior to particle collections, the quartz filters were prebaked at 550 °C for 24 h to remove the organic residuals. BBOA was obtained by extracting the collected BB particles using ultrapure water, under sonication, followed by filtration through 0.22 µm Teflon filters to remove the insoluble components. The chemical composition of chromophores in the BBOA was characterized by high-performance liquid chromatography with a photodiode array and high-resolution orbitrap mass spectroscopy (HPLC-PDA-HRMS) (SI Appendix, Text S6). Chemical standards were used to identify the main chromophores. The light absorption pattern of the BBOA was determined by a UV-Vis spectrophotometer (Agilent) (SI Appendix, Fig. S22). Nonpolar components were extracted with hexane and characterized by two-dimensional gas chromatography coupled with time-of-flight mass spectrometry (GC×GC ToF MS), providing broad profiling of the particle constituents (SI Appendix, Text S7). Additional analyses, including Inductively Coupled Plasma Mass Spectrometry (ICP-MS, ThermoFisher), ion chromatogram (IC, ThermoFisher), and total organic carbon analyzer (TOC, Agilent) were used to quantify the ionic species and total organic carbon. All solvents used in the study are LC-MS grade from VWR. In addition, the BB particles were also deposited onto a glass slide coated with superhydrophobic and oleophobic material, using a PM2.5 impactor (TSI). An optical photothermal infrared microscope (O-PTIR, Photothermal Spectroscopy Corp.) was used to characterize the morphology and spatial distribution of the chemical components within the BB particles (SI Appendix, Text S8). A Hygroscopicity Tandem Differential Mobility Analyzer (BMI) was used to determine the size growth factor of the BBOA at different RHs, as detailed in our previous work (23) (SI Appendix, Fig. S23).
Flow Tube Experiments.
A stream of BBOA particles was generated using a constant output atomizer (TSI) and a flow of 0.1 L min−1 was first prehumified to reach equilibrium at 80% RH, then passed through a charcoal denuder to remove the potential volatile organic compounds (VOC). Subsequently, a differential mobility analyzer (DMA) selected the size of the BBOA particles, which were then introduced to an aerosol flow tube (AFT, 10 cm internal diameter, 120 cm length) made of quartz (SI Appendix, Fig. S24). The Reynold number in the AFT is around 11. A flow of SO2 carried by mixed dry-wet N2 (1.4 L min−1) was also introduced to the OFR to interact with the BBOA particles. The residence time of the BBOA particles in the OFR is approximately 380 s.
UVA light tubes (with a continuous emission spectrum over 300 to 420 nm) were evenly mounted along the length of the AFT on its outside (SI Appendix, Fig. S22). The photon flux to the AFT is comparable to the actinic flux at noon in the summer of British Columbia, Canada. The details for the determination of photon flux are available in SI Appendix, Text S9. At the exhaust of the AFT, an M170 digital sensor (Vaisala) was used to monitor the temperature and RH. The temperature for all experiments was around 293 K. The SO2 concentration and particle number concentrations were measured by an SO2 analyzer (Thermo Fisher) and a condensation particle counter (CPC, BMI). A noticeable increase in the particle number concentration was not observed when the light turned on, therefore, the new particle formation due to photochemistry was not considered.
We used 47 mm Teflon filters loaded in an in-line stainless steel filter holder to collect the aged particles (by SO2) at the exhaust. After that, the filters were extracted by ultrapure water and the sulfate moles in the aqueous extract were measured by IC. The sulfate concentration in the BBOA particles (M) can be quantified as
| [9] |
where mp is the mass loading of the particles measured by a calibrated microbalance (SI Appendix, Fig. S25 and Text S10). ρ is the density of BBOA assumed as 1.3 g cm−3 (89). A movable stainless-steel injector tube (inserted axially down the center of the flow tube) allowed variation in light exposure time, thus we can quantify the sulfate formed in BBOA particles as a function of illumination time and calculate the sulfate formation rate (SI Appendix, Table S4). We assumed no effects on particle size and density by considering the maximum abundance of the formed sulfate with the water it absorbs. To compare the sulfate formation initiated by individual photosensitizers between bulk solution and aerosol particles, we mixed the photosensitizers with organic buffer (sodium malonate/sodium bimalonate = 1:1), which provides aerosol liquid water to minimize the evaporation of photosensitizers.
Sulfate formation rates in particles and droplets with different surface-to-volume ratios (S/V) provide insight into the significance of interfacial reactions. To investigate this, we applied different voltages to the DMA for submicron particles of varying sizes (100 to 300 nm). We also conducted SO2 uptake experiments on large deposited BBOA droplets (diameter of approximately 1 mm after equilibrium) using a flow cell, as detailed in our previous works (90–92). In brief, 1 μL BBOA droplets were deposited onto hydrophobic substrates and equilibrated at 80% RH inside the flow cell. An optical microscope (Leica) equipped with a microruler was used to monitor the size change, ensuring equilibrium. Once equilibrium was achieved, pre-equilibrated SO2 steam was introduced into the flow cell, and the droplets were illuminated from the top–down using UVA light through the cell’s quartz window. The droplets were sampled at different time points, extracted, and the sulfate concentration was determined using IC for kinetic analysis.
Determination of the 3BBOA* Concentration.
Syringol (SYR) was used as an internal chemical probe to determine the production rate of the 3C* of BBOA (i.e., 3BBOA*) in solution upon illumination using a quartz cuvette reactor. The solution temperature was maintained at around 293 K using a temperature-controlled plate and cycling water bath. UVA light tubes were positioned above the reactor, simulating the photon flux conditions similar to those in the AFT.
The rate constant for the SYR loss (k, s−1) under light illumination was determined. To obtain the pseudo-first-order SYR decay rate constant due to 3BBOA* (kSYR,3BBOA∗[3BBOA*], s−1), we subtracted k by the SYR direct photodegradation rate constant under UVA illumination (j, s−1) and pseudo-first-order oxidation rate constants by OH• (kSYR,OH•[OH•], s−1) and 1O2* (kSYR,1O2∗[1O2*], s−1):
| [10] |
The steady-state concentrations of these oxidants were determined by separately spiking benzoic acid (BA) and furfural alcohol (FFA) into the BBOA solution and monitoring the formation of p-hydroxybenzoic acid (p-HBA) and FFA decay via HPLC-PDA (78). The light screening effects on the probes were corrected (33).
kSYR,3BBOA∗, kSYR,OH•, and kSYR,1O2∗ are the bimolecular rate constants of SYR reacting with 3BBOA*, OH•, and 1O2*, respectively. We assumed that 3BBOA* shared the same second-order reaction rate constant as the 3DMB* with SYR [3.9 × 109 M−1 s−1 (93)] to calculate the steady-state [3BBOA*], based on Eq. 10.
Although SYR has been reported to undergo dark reactions that form BrC, particularly in the presence of Cl salts (94), we considered this pathway to be minor as negligible SYR decay was observed in the dark (SI Appendix, Fig. S26). It is also important to note that SYR has a high triplet energy [330 to 340 kJ mol−1 (95, 96)]. However, our analysis revealed that the major 3C* species in BBOA exhibit ET values below 300 kJ mol−1, making energy transfer from SYR to 3C* unlikely. On the other hand, the one-electron oxidation potential of SYR [approximately 0.7 V.SHE (97)] is lower than the Eo of the 3C* in BBOA. Therefore, we expect that the majority of 3C* were effectively quenched by SYR. The SYR decay could be subjected to back-electron donation of some electron-rich BBOA components and lead to underestimation of [3BBOA*], known as inhibition. The inhibition factor was quantified and used to correct [3BBOA*], according to Ma et al. (33) and Ma et al. (32). Overall, we considered the limitation of using SYR minor in this study.
The [3BBOA*] in concentrated aerosols cannot be directly measured, therefore we estimated it according to the formation rate (FT) and sink rates, derived from bulk measurements at a series of [OM] (SI Appendix, Table S5). At the steady state, the formation rate of 3BBOA* equals its removal rate by OM and O2:
| [11] |
where jabs is the light absorption rate constant, ФISC is the intersystem crossing quantum yield, f is the fraction of photosensitizers in the BBOA mixture, and k3BBOA∗, OM and k3BBOA∗, O2 are the second-order quenching rate constants of 3BBOA* by OM and O2, respectively.
At low concentrations of BBOA at which O2 is the dominant sink (SI Appendix, Fig. S5), FT can be determined by Eq. 12:
| [12] |
It should be noted diluted solution ([BBOA]/[H2O] = 10−5) was used here to derive FT specifically. For BBOA particles ([BBOA]/[H2O] = 1) discussed in the main text, O2 is a minor sink of 3BBOA* compared to OM. k3BBOA∗, O2 is determined as 1 × 109 M−1 s−1 based on transient absorption spectroscopy (will be introduced later), consistent with reported values for various aromatic photosensitizers (8). [O2] is the concentration of dissolved oxygen in the solution measured by a digital meter (Vio).
Rearrangement of Eq. 11 yields:
| [13] |
[3BBOA*] at a series of [OM] in bulk solution was used to fit Eq. 13 according to . k3BBOA∗, OM can be calculated according to b and k3BBOA∗, O2.
Finally, the [3BBOA*] in aerosol particles, in the presence of SO2 in N2 can be estimated by
| [14] |
Transient Absorption Spectroscopy (TA).
A laser flash photolysis system coupled with an excitation laser at 310 nm was used to measure the 3BBOA* quenching kinetics in the solution. A peristaltic pump was used to continuously supply the external BBOA solution to a quartz cuvette located inside the equipment, minimizing the effect of photobleaching on the kinetic measurements. For the 3BBOA* quenching experiment by S(IV) and levoglucosan (used as a control experiment for the reactions in the bulk phase of the particle), the solution was purged with ultrapure N2 before the measurements to minimize 3BBOA* quenching by O2.
3BBOA* was generated by a laser pump followed by decay. The decay of 3BBOA* was fitted with a single exponential term, to obtain the kobs. kobs for absorption peaks at different wavelengths were averaged to yield the overall observed 3BBOA* quenching rate constant (15):
| [15] |
The kobs were measured at a range of quenchers such as [S(IV)]. According to the Stern–Volmer equation (Eq. 16), the slope and intercept of the plot kobs vs. [S(IV)] yield the 3BBOA* quenching rate constant by S(IV) (k3BBOA∗, S(IV)) and the total natural deactivation rate of 3BBOA* via self-quenching, phosphorescence, etc., (kd), respectively.
| [16] |
Since the speciation of S(IV) is pH-dependent, we performed quenching experiments at pH = 1, 4, and 7 (by the addition of H2SO4 and NaOH), in which the SO2·H2O, HSO3−, and SO32− dominate (SI Appendix, Fig. S3), respectively. For the quenching kinetics between 3BBOA* and O2, we purged the solution using mixed N2 and O2 flow to achieve different O2 content, and the dissolved oxygen content was monitored.
Electro-Paramagnetic Resonance (EPR) Spectroscopy.
A continuous-wave EPR (CW-EPR) spectrometer (Bruker) coupled with a spin-trapping technique was used to measure the free radical formation in aqueous solutions under light. EPR peaks are generated by the absorption of microwave radiation by unpaired electrons in the presence of a magnetic field. The EPR peak intensity is directly proportional to the concentration of the radicals in the sample. We used a spin-trapping agent, 5,5-Dimethyl-1-pyrroline N-oxide (DMPO, Enzo), to convert the transient radicals such as OH to those with longer lifetimes such as DMPO-OH for measurements. The persistent 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO, Enzo) free radical was used to capture the dissolved electron during electron transfer from 3BBOA*. Specifically, the decay in TEMPO intensity indicates a reduction of TEMPO to the EPR-silent TEMPOH by dissolved electrons.
The spin-trap was spiked into the sample solution to make a final concentration of 0.1 M, and then 50 μL aliquot of the mixture was loaded into a 50 μL capillary tube and inserted in the resonator of the EPR spectrometer for temporal measurements over 90 min. An in situ UV irradiation system (Bruker) equipped with a 100 W Hg lamp was used with EPR. A safety shutter between the lamp and the resonator was used to control the start and stop of irradiation. The operating parameters for EPR measurements were as follows: a center field of 3,515.0 G, a sweep width of 100.0 G, a receiver gain of 25 dB, a modulation amplitude of 1.0 G, a scan number of 8, attenuation of 20 dB, a microwave power of 2 mW, a modulation frequency of 100 kHz, a microwave frequency of 9.86 GHz, and a conversion time and time constant of 5.12 ms. The Xenon software was used to calculate the peak area for the radical adducts.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We acknowledge the support from the baseline research fund (BAS/1/1432-01-01) and King Abdullah University of Science and Technology Center of Excellence for Smart Health fund (Award number 5932) received by C.K.C. We thank Dan Daniel and Marcus Lin for synthesizing the hydrophobic and oleophobic surfaces. Special acknowledgments go to Yuanlong Huang, Tengyu Liu, and Xinke Wang for fruitful discussions and the Instrumental Analysis and Research Center at Sun Yat-sen University for the laser flash photolysis system.
Author contributions
Z.L. and C.K.C. designed research; Z.L., L.Z., and Y.C. performed research; Z.L., L.Z., Y.C., Y.Q., and C.K.C. analyzed data; and Z.L., L.Z., Y.C., Y.Q., and C.K.C. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission. J.A. is a guest editor invited by the Editorial Board.
Although PNAS asks authors to adhere to United Nations naming conventions for maps (https://www.un.org/geospatial/mapsgeo), our policy is to publish maps as provided by the authors.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Andreae M. O., Emission of trace gases and aerosols from biomass burning–An updated assessment. Atmos. Chem. Phys. 19, 8523–8546 (2019). [Google Scholar]
- 2.Chung C. E., Ramanathan V., Decremer D., Observationally constrained estimates of carbonaceous aerosol radiative forcing. Proc. Natl. Acad. Sci. U.S.A. 109, 11624–11629 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Q., et al. , Review of brown carbon aerosols in China: Pollution level, optical properties, and emissions. J. Geophys. Res. Atmos. 127, e2021JD035473 (2022). [Google Scholar]
- 4.Laskin A., Lin P., Laskin J., Fleming L. T., Nizkorodov S., “Molecular characterization of atmospheric brown carbon” in Multiphase Environmental Chemistry in the Atmosphere, Hunt S. W., Laskin A., Nizkorodov S. A., Eds. (ACS Publications, 2018), pp. 261–274. [Google Scholar]
- 5.Hems R. F., Schnitzler E. G., Liu-Kang C., Cappa C. D., Abbatt J. P., Aging of atmospheric brown carbon aerosol. ACS Earth and Space Chem. 5, 722–748 (2021). [Google Scholar]
- 6.Liu-Kang C., Gallimore P. J., Liu T., Abbatt J. P., Photoreaction of biomass burning brown carbon aerosol particles. Environ. Sci. Atmos. 2, 270–278 (2022). [Google Scholar]
- 7.Schnitzler E. G., et al. , Rate of atmospheric brown carbon whitening governed by environmental conditions. Proc. Natl. Acad. Sci. U.S.A. 119, e2205610119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Felber T., Schaefer T., He L., Herrmann H., Aromatic carbonyl and nitro compounds as photosensitizers and their photophysical properties in the tropospheric aqueous phase. J. Phys. Chem. A 125, 5078–5095 (2021). [DOI] [PubMed] [Google Scholar]
- 9.Felber T., Schaefer T., Herrmann H., Five-membered heterocycles as potential photosensitizers in the tropospheric aqueous phase: Photophysical properties of imidazole-2-carboxaldehyde, 2-furaldehyde, and 2-acetylfuran. J. Phys. Chem. A 124, 10029–10039 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Ma L., et al. , Kinetics and mass yields of aqueous secondary organic aerosol from highly substituted phenols reacting with a triplet excited state. Environ. Sci. Technol. 55, 5772–5781 (2021). [DOI] [PubMed] [Google Scholar]
- 11.Smith J. D., Kinney H., Anastasio C., Phenolic carbonyls undergo rapid aqueous photodegradation to form low-volatility, light-absorbing products. Atmos. Environ. 126, 36–44 (2016). [Google Scholar]
- 12.Li F., et al. , Aqueous photosensitization of syringaldehyde: Reactivity, effects of environmental factors, and formation of brown carbon products. ACS Earth Space Chem. 8, 1193–1203 (2024). [Google Scholar]
- 13.Mabato B. R. G., et al. , Aqueous secondary organic aerosol formation from the direct photosensitized oxidation of vanillin in the absence and presence of ammonium nitrate. Atmos. Chem. Phys. 22, 273–293 (2022). [Google Scholar]
- 14.Wang X., et al. , Naphthalene-derived secondary organic aerosols interfacial photosensitizing properties. Geophys. Res. Lett. 48, e2021GL093465 (2021). [Google Scholar]
- 15.Wang X., et al. , Atmospheric photosensitization: A new pathway for sulfate formation. Environ. Sci. Technol. 54, 3114–3120 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Liu T., Chan A. W., Abbatt J. P., Multiphase oxidation of sulfur dioxide in aerosol particles: Implications for sulfate formation in polluted environments. Environ. Sci. Technol. 55, 4227–4242 (2021). [DOI] [PubMed] [Google Scholar]
- 17.Zheng B., et al. , Heterogeneous chemistry: A mechanism missing in current models to explain secondary inorganic aerosol formation during the January 2013 haze episode in North China. Atmos. Chem. Phys. 15, 2031–2049 (2015). [Google Scholar]
- 18.Wang Y., et al. , Enhanced sulfate formation during China’s severe winter haze episode in January 2013 missing from current models. J. Geophys. Res. Atmos. 119, 10,425–410,440 (2014). [Google Scholar]
- 19.Diapouli E., et al. , Physicochemical characterization of aged biomass burning aerosol after long-range transport to Greece from large scale wildfires in Russia and surrounding regions, Summer 2010. Atmos. Environ. 96, 393–404 (2014). [Google Scholar]
- 20.Zauscher M. D., Wang Y., Moore M. J. K., Gaston C. J., Prather K. A., Air quality impact and physicochemical aging of biomass burning aerosols during the 2007 San Diego wildfires. Environ. Sci. Technol. 47, 7633–7643 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Hecobian A., et al. , Comparison of chemical characteristics of 495 biomass burning plumes intercepted by the NASA DC-8 aircraft during the ARCTAS/CARB-2008 field campaign. Atmos. Chem. Phys. 11, 13325–13337 (2011). [Google Scholar]
- 22.Liu X., Van Espen P., Adams F., Cafmeyer J., Maenhaut W., Biomass burning in southern Africa: Individual particle characterization of atmospheric aerosols and savanna fire samples. J. Atmos. Chem. 36, 135–155 (2000). [Google Scholar]
- 23.Liang Z., et al. , Sulfate formation in incense burning particles: A single-particle mass spectrometric study. Environ. Sci. Technol. Lett. 9, 718–725 (2022). [Google Scholar]
- 24.Tang R., et al. , Sulfate formation by photosensitization in mixed incense burning-sodium chloride particles: Effects of RH, light intensity, and aerosol aging. Environ. Sci. Technol. 57, 10295–10307 (2023). [DOI] [PubMed] [Google Scholar]
- 25.Zhou L., et al. , Sulfate formation via aerosol-phase SO2 oxidation by model biomass burning photosensitizers: 3, 4-dimethoxybenzaldehyde, vanillin and syringaldehyde using single-particle mixing-state analysis. Atmos. Chem. Phys. 23, 5251–5261 (2023). [Google Scholar]
- 26.Liu Y., et al. , Strong electric field force at the air/water interface drives fast sulfate production in the atmosphere. Chem 10, 330–351 (2024). [Google Scholar]
- 27.Gen M., Liang Z., Zhang R., Mabato B. R. G., Chan C. K., Particulate nitrate photolysis in the atmosphere. Environ. Sci. Atmos. 2, 111–127 (2022). [Google Scholar]
- 28.Westerling A. L., Hidalgo H. G., Cayan D. R., Swetnam T. W., Warming and earlier spring increase western US forest wildfire activity. Science 313, 940–943 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Abatzoglou J. T., Williams A. P., Impact of anthropogenic climate change on wildfire across western US forests. Proc. Natl. Acad. Sci. U.S.A. 113, 11770–11775 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balch J. K., et al. , Warming weakens the night-time barrier to global fire. Nature 602, 442–448 (2022). [DOI] [PubMed] [Google Scholar]
- 31.Woods E., et al. , Lifetime of triplet photosensitizers in aerosol using time-resolved photoelectric activity. ACS Earth Space Chem. 4, 1424–1434 (2020). [Google Scholar]
- 32.Ma L., Worland R., Tran T., Anastasio C., Evaluation of probes to measure oxidizing organic triplet excited states in aerosol liquid water. Environ. Sci. Technol. 57, 6052–6062 (2023). [DOI] [PubMed] [Google Scholar]
- 33.Ma L., et al. , Predicting photooxidant concentrations in aerosol liquid water based on laboratory extracts of ambient particles. EGUsphere 2023, 1–36 (2023). [Google Scholar]
- 34.Mabato B. R. G., Li Y. J., Huang D. D., Chan C. K., Aqueous-phase photoreactions of mixed aromatic carbonyl photosensitizers yield more oxygenated, oxidized, and less light-absorbing secondary organic aerosol (SOA) than single systems. Environ. Sci. Technol. 58, 7924–7936 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y., et al. , Co-photolysis of mixed chromophores affects atmospheric lifetimes of brown carbon. Environ. Sci. Atmos. 3, 1145–1158 (2023). [Google Scholar]
- 36.Liu T., Clegg S. L., Abbatt J. P., Fast oxidation of sulfur dioxide by hydrogen peroxide in deliquesced aerosol particles. Proc. Natl. Acad. Sci. U.S.A. 117, 1354–1359 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mekic M., Wang Y., Loisel G., Vione D., Gligorovski S., Ionic strength effect alters the heterogeneous ozone oxidation of methoxyphenols in going from cloud droplets to aerosol deliquescent particles. Environ. Sci. Technol. 54, 12898–12907 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Liu T., Abbatt J. P., Oxidation of sulfur dioxide by nitrogen dioxide accelerated at the interface of deliquesced aerosol particles. Nat. Chem. 13, 1173–1177 (2021). [DOI] [PubMed] [Google Scholar]
- 39.Liu T., Abbatt J. P. D., An experimental assessment of the importance of S(IV) oxidation by hypohalous acids in the marine atmosphere. Geophys. Res. Lett. 47, e2019GL086465 (2020). [Google Scholar]
- 40.Wang W., et al. , Significantly accelerated photosensitized formation of atmospheric sulfate at the air-water interface of microdroplets. J. Am. Chem. Soc. 146, 6580–6590 (2024). [DOI] [PubMed] [Google Scholar]
- 41.Wang N., et al. , Triplet-excited dissolved organic matter efficiently promoted atmospheric sulfate production: Kinetics and mechanisms. Separations 10, 335 (2023). [Google Scholar]
- 42.Zhong J., et al. , Interaction of SO2 with the surface of a water nanodroplet. J. Am. Chem. Soc. 139, 17168–17174 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Gong C., et al. , Fast sulfate formation initiated by the spin-forbidden excitation of SO2 at the air–water interface. J. Am. Chem. Soc. 144, 22302–22308 (2022). [DOI] [PubMed] [Google Scholar]
- 44.Ossola R., et al. , Photochemical production of sulfate and methanesulfonic acid from dissolved organic sulfur. Environ. Sci. Technol. 53, 13191–13200 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Ossola R., Clerc B., McNeill K., Mechanistic insights into dissolved organic sulfur photomineralization through the study of cysteine sulfinic acid. Environ. Sci. Technol. 54, 13066–13076 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Poulin B. A., Selective photochemical oxidation of reduced dissolved organic sulfur to inorganic sulfate. Environ. Sci. Technol. Lett. 10, 499–505 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rao Z., et al. , Spontaneous oxidation of thiols and thioether at the air-water interface of a sea spray microdroplet. J. Am. Chem. Soc. 145, 10839–10846 (2023). [DOI] [PubMed] [Google Scholar]
- 48.Pye H. O. T., et al. , The acidity of atmospheric particles and clouds. Atmos. Chem. Phys. 20, 4809–4888 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Y., et al. , Unveiling the role of carbonate radical anions in dust-driven SO2 oxidation. J. Geophys. Res. Atmos. 129, e2023JD040017 (2024). [Google Scholar]
- 50.Mottley C., Trice T. B., Mason R., Direct detection of the sulfur trioxide radical anion during the horseradish peroxidase-hydrogen peroxide oxidation of sulfite (aqueous sulfur dioxide). Mol. Pharmacol. 22, 732–737 (1982). [PubMed] [Google Scholar]
- 51.Ruiz-Lopez M. F., Francisco J. S., Martins-Costa M. T., Anglada J. M., Molecular reactions at aqueous interfaces. Nat. Rev. Chem. 4, 459–475 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Nowakowski P. J., Woods D. A., Verlet J. R. R., Charge transfer to solvent dynamics at the ambient water/air interface. J. Phys. Chem. Lett. 7, 4079–4085 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Kusaka R., Ishiyama T., Nihonyanagi S., Morita A., Tahara T., Structure at the air/water interface in the presence of phenol: A study using heterodyne-detected vibrational sum frequency generation and molecular dynamics simulation. Phys. Chem. Chem. Phys. 20, 3002–3009 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Martins-Costa M. T., Anglada J. M., Francisco J. S., Ruiz-López M. F., Photosensitization mechanisms at the air–water interface of aqueous aerosols. Chem. Sci. 13, 2624–2631 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tinel L., et al. , Mechanistic insights on the photosensitized chemistry of a fatty acid at the air/water interface. Environ. Sci. Technol. 50, 11041–11048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fu H., et al. , Photosensitized production of atmospherically reactive organic compounds at the air/aqueous interface. J. Am. Chem. Soc. 137, 8348–8351 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rossignol S., et al. , Atmospheric photochemistry at a fatty acid–coated air-water interface. Science 353, 699–702 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Gregson F. K. A., et al. , Phase behavior and viscosity in biomass burning organic aerosol and climatic impacts. Environ. Sci. Technol. 57, 14548–14557 (2023). [DOI] [PubMed] [Google Scholar]
- 59.Martins-Costa M. T. C., Anglada J. M., Francisco J. S., Ruiz-López M. F., Photochemistry of SO2 at the air-water interface: A source of OH and HOSO radicals. J. Am. Chem. Soc. 140, 12341–12344 (2018). [DOI] [PubMed] [Google Scholar]
- 60.Canonica S., Hellrung B., Wirz J., Oxidation of phenols by triplet aromatic ketones in aqueous solution. J. Phys. Chem. A 104, 1226–1232 (2000). [Google Scholar]
- 61.Devémy J., Dequidt A., Renard P., Deguillaume L., Malfreyt P., Energetic description of the liquid–vapor interface of water with organic coating molecules. RSC Appl. Interfaces 1, 800–811 (2024). [Google Scholar]
- 62.Chen K., et al. , Contribution of carbonyl chromophores in secondary brown carbon from nighttime oxidation of unsaturated heterocyclic volatile organic compounds. Environ. Sci. Technol. 57, 20085–20096 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rajagopal S. K., Mallia A. R., Hariharan M., Enhanced intersystem crossing in carbonylpyrenes. Phys. Chem. Chem. Phys. 19, 28225–28231 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Liang Z., et al. , Secondary aerosol formation in incense burning particles by O3 and OH oxidation via single particle mixing state analysis. Sci. Total Environ. 894, 164942 (2023). [DOI] [PubMed] [Google Scholar]
- 65.Smith J. D., Sio V., Yu L., Zhang Q., Anastasio C., Secondary organic aerosol production from aqueous reactions of atmospheric phenols with an organic triplet excited state. Environ. Sci. Technol. 48, 1049–1057 (2014). [DOI] [PubMed] [Google Scholar]
- 66.Jovanovic S. V., Harriman A., Simic M. G., Electron-transfer reactions of tryptophan and tyrosine derivatives. J. Phys. Chem. 90, 1935–1939 (1986). [Google Scholar]
- 67.Sarala R., Islam M. A., Rabin S. B., Stanburg D. M., Aromatic sulfonation by sulfite and the reduction potential of the sulfite radical: Oxidation of sulfite by the tetraammine (phenanthroline) ruthenium (II) cation. Inorganic Chem. 29, 1133–1142 (1990). [Google Scholar]
- 68.Cao Y., et al. , Photoactivation of chlorine and its catalytic role in the formation of sulfate aerosols. J. Am. Chem. Soc. 146, 1467–1475 (2024). [DOI] [PubMed] [Google Scholar]
- 69.McNeill K., Canonica S., Triplet state dissolved organic matter in aquatic photochemistry: Reaction mechanisms, substrate scope, and photophysical properties. Environ. Sci. Process. Impacts 18, 1381–1399 (2016). [DOI] [PubMed] [Google Scholar]
- 70.Liu-Kang C., et al. , Light exposure of wood smoke aerosol: Connecting optical properties, oxidation, radical formation, and chemical composition. ACS EST Air 1, 273–282 (2024). [Google Scholar]
- 71.Donaldson D. J., Kroll J. A., Vaida V., Gas-phase hydrolysis of triplet SO2: A possible direct route to atmospheric acid formation. Sci. Rep. 6, 30000 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang R.-J., et al. , Chromophoric fingerprinting of brown carbon from residential biomass burning. Environ. Sci. Technol. Lett. 9, 102–111 (2022). [Google Scholar]
- 73.Fleming L. T., et al. , Molecular composition and photochemical lifetimes of brown carbon chromophores in biomass burning organic aerosol. Atmos. Chem. Phys. 20, 1105–1129 (2020). [Google Scholar]
- 74.Shi P., Kasperson R., World Atlas of Natural Disaster Risk (Springer, 2015). [Google Scholar]
- 75.Gao J., et al. , Hydrogen peroxide serves as pivotal fountainhead for aerosol aqueous sulfate formation from a global perspective. Nat. Commun. 15, 4625 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Manfrin A., et al. , Reactive oxygen species production from secondary organic aerosols: The importance of singlet oxygen. Environ. Sci. Technol. 53, 8553–8562 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Bogler S., et al. , Singlet oxygen seasonality in aqueous PM10 is driven by biomass burning and anthropogenic secondary organic aerosol. Environ. Sci. Technol. 56, 15389–15397 (2022). [DOI] [PubMed] [Google Scholar]
- 78.Corral Arroyo P., et al. , Particle-phase photosensitized radical production and aerosol aging. Environ. Sci. Technol. 52, 7680–7688 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Wang Y., et al. , Photochemistry of imidazole-2-carbaldehyde in droplets as a potential source of H2O2 and its oxidation of SO2. Environ. Sci. Technol. 58, 11096–11104 (2024). [DOI] [PubMed] [Google Scholar]
- 80.Yan Y., et al. , Direct determination of absolute radical quantum yields in hydroxyl and sulfate radical-based treatment processes. Environ. Sci. Technol. 58, 8966–8975 (2024). [DOI] [PubMed] [Google Scholar]
- 81.Li J., et al. , Optical properties and molecular compositions of water-soluble and water-insoluble brown carbon (BrC) aerosols in northwest China. Atmos. Chem. Phys. 20, 4889–4904 (2020). [Google Scholar]
- 82.Hems R. F., et al. , Aqueous photoreactions of wood smoke brown carbon. ACS Earth Space Chem. 4, 1149–1160 (2020). [Google Scholar]
- 83.Forrister H., et al. , Evolution of brown carbon in wildfire plumes. Geophys. Res. Lett. 42, 4623–4630 (2015). [Google Scholar]
- 84.Ye X., et al. , Assessing vertical allocation of wildfire smoke emissions using observational constraints from airborne lidar in the Western US. J. Geophys. Res. Atmos. 127, e2022JD036808 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang Y., et al. , Top-of-atmosphere radiative forcing affected by brown carbon in the upper troposphere. Nat. Geosci. 10, 486–489 (2017). [Google Scholar]
- 86.Vasilakopoulou C. N., et al. , Rapid transformation of wildfire emissions to harmful background aerosol. npj Clim. Atmos. Sci. 6, 218 (2023). [Google Scholar]
- 87.Jiang W., Ma L., Niedek C., Anastasio C., Zhang Q., Chemical and light-absorption properties of water-soluble organic aerosols in Northern California and photooxidant production by brown carbon components. ACS Earth Space Chem. 7, 1107–1119 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li W.-Y., et al. , Photoactivated production of secondary organic species from isoprene in aqueous systems. J. Phys. Chem. A 120, 9042–9048 (2016). [DOI] [PubMed] [Google Scholar]
- 89.Lee A. K., et al. , Single-particle characterization of biomass burning organic aerosol (BBOA): Evidence for non-uniform mixing of high molecular weight organics and potassium. Atmos. Chem. Phys. 16, 5561–5572 (2016). [Google Scholar]
- 90.Liang Z., Zhang R., Gen M., Chu Y., Chan C. K., Nitrate photolysis in mixed sucrose–nitrate–sulfate particles at different relative humidities. J. Phys. Chem. A 125, 3739–3747 (2021). [DOI] [PubMed] [Google Scholar]
- 91.Liang Z., Cheng Z., Zhang R., Qin Y., Chan C. K., Distinct photochemistry in glycine particles mixed with different atmospheric nitrate salts. Atmos. Chem. Phys. 23, 9585–9595 (2023). [Google Scholar]
- 92.Liang Z., et al. , Inactivation of Escherichia coli in droplets at different ambient relative humidities: Effects of phase transition, solute and cell concentrations. Atmos. Environ. 280, 119066 (2022). [Google Scholar]
- 93.Smith J. D., Kinney H., Anastasio C., Aqueous benzene-diols react with an organic triplet excited state and hydroxyl radical to form secondary organic aerosol. Phys. Chem. Chem. Phys. 17, 10227–10237 (2015). [DOI] [PubMed] [Google Scholar]
- 94.Xu J., et al. , Aerosol brown carbon from dark reactions of syringol in aqueous aerosol mimics. ACS Earth Space Chem. 2, 608–617 (2018). [Google Scholar]
- 95.Canonica S., Jans U., Stemmler K., Hoigne J., Transformation kinetics of phenols in water: Photosensitization by dissolved natural organic material and aromatic ketones. Environ. Sci. Technol. 29, 1822–1831 (1995). [DOI] [PubMed] [Google Scholar]
- 96.Kaur R., Anastasio C., First measurements of organic triplet excited states in atmospheric waters. Environ. Sci. Technol. 52, 5218–5226 (2018). [DOI] [PubMed] [Google Scholar]
- 97.Silva D., Sousa A. C., Robalo M. P., Martins L. O., A wide array of lignin-related phenolics are oxidized by an evolved bacterial dye-decolourising peroxidase. New Biotechnol. 77, 176–184 (2023). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.