

Abstract
We demonstrate a complete nanotube electrophoresis system (nanotube radii in the range of 50 to 150 nm) based on lipid membranes, comprising DNA injection, single-molecule transport, and single-molecule detection. Using gel-capped electrodes, electrophoretic single-file transport of fluorescently labeled dsDNA molecules is observed inside nanotubes. The strong confinement to a channel of molecular dimensions ensures a detection efficiency close to unity and identification of DNA size from its linear relation to the integrated peak intensity. In addition to constituting a nanotechnological device for identification and quantification of single macromolecules or biopolymers, this system provides a method to study their conformational dynamics, reaction kinetics, and transport in cell-like environments.
Keywords: electrophoresis, lipid, conformation
Controlled transport, interrogation, and manipulation of single molecules in integrated nanoscale devices would provide new tools for fundamental studies of molecular properties, development of ultrasensitive biochemical assays, and new models for studies of transport and reaction phenomena in confined biological systems. For example, it was recently shown that lipid bilayer nanotubes ≈50-200 nm in diameter are involved in mediated transport of water-soluble and membrane-bound components between cells (1). Such observations do not only raise important questions about transport mechanisms for macromolecules and organelles in spaces comparable to the size of the cargo itself, but also about how such strong confinement affects diffusion, conformation, and chemical reactions of enclosed molecules and particles (2-4). Furthermore, along with previous understanding of sorting and routing of individual molecules, e.g., in the Golgi-endoplasmic reticulum network (5, 6), these observations point out clearly that what has been an engineering dream for decades, i.e., to create manmade devices that can operate with single molecules in a controlled fashion, is a reality in biology and therefore can be a reality in the world of engineering provided that we procure sufficient knowledge and tools to emulate these systems. However, experimental systems for controlled confinement and transport of materials dissolved in fluids approaching the theoretical size limit, i.e., where the ratio of channel inner diameter and dimensions of the cargo are close to unity, have been difficult to make in combination with transport control. This difficulty is mainly because these systems have been fabricated by using solid-state materials and processing technologies used in the computer industry that are limited in terms of the smallest accessible length scales, topologies, materials properties, complexity of fabrication, and their necessary integration to large-scale instrumentation to drive fluid flow. Nonetheless, powerful micro/nanofluidic protocols for polymer transport in solid-state nanochannels and pores (7-12) as well as in polydimethylsiloxane channels of a few micrometers in diameter (13, 14) have been developed.
Here, we demonstrate a complete nanotube electrophoresis system comprising DNA injection, single-molecule transport, and single-molecule detection, which by contrast is based on engineering of 2D fluids into nanotube networks. Such lipid-based networks can be prepared with designed connectivity, geometry, topology, and dimensionality, and their contents and surface properties can be controlled as described (15-21). These in vitro-fabricated networks are of the same amphiphilic and self-assembling nature as nanotubes found in vivo, and with similar dimensions regarding both radii (50-150 nm) (1) and lengths (up to tens of μm).
Materials and Methods
Liposome Preparation. The chemicals used for buffer solutions were of analytical grade and purchased from Sigma. Soybean polar lipid extract (Avanti Polar Lipids) was used to prepare giant unilamellar liposomes as described (16). The rehydration step was performed with our standard buffer (10 mM K3PO4/10 mM KH2PO4/5 mM Trizma base/90 mM KCl/1 mM MgSO4/0.5 mM EDTA, pH adjusted to 8.1 with KOH). A mixture of such unilamellar and multilamellar vesicles was used to form the vesicle-nanotube system, as described in Results and Discussion. The ζ-potential of the lipid membrane was measured by microelectrophoresis, applied to small unilamellar vesicles.
DNA Labeling. DNA was labeled with the LabelIT Fluorescein Nucleic Acid Labeling Kit (Mirus, Madison, WI) according to the standard labeling protocol, except that the reaction time was extended to 2 h at 37°C. The samples were purified by gel filtration on G50 Microspin columns (Amersham Pharmacia). Fifty-microliter fractions were collected and analyzed with gel electrophoresis for reagent impurities. The average labeling density was ≈1 fluorophore per 15 bp for all DNA sizes, determined by absorbance measurements. The pure fractions were then pooled and used in the experiments.
Electrode Preparation. A silver wire electro-deposited with AgCl(s) was inserted into a borosilicate micropipette filled with standard buffer, thus making an Ag/AgCl electrode. To suppress the electroosmotic flow, the charge density of the pipette wall was reduced by reaction with hexamethyl disilazane (Sigma). The borosilicate capillaries (Harvard Apparatus) were immersed in 10 mM hexamethyl disilazane dissolved in hexane for 10 min and then thoroughly rinsed with hexane. After drying in room temperature, pipettes were pulled by using a CO2-laser puller instrument (model P-2000, Sutter Instruments, Novato, CA). Furthermore, the pipette tip was hydrodynamically plugged by polymerizing acrylamide (20% wt/wt) and bis-acrylamide (1% wt/wt) (Bio-Rad). The cross-linked gel acts as a salt bridge, preventing bulk solvent flow, but allowing electric current to pass.
Single-Molecule Confocal Fluorescence Microscopy. The confocal setup is equipped with an avalanche photodiode SPCM-AQR-16 (EG & G Canada, Vaudreuil, Canada) and is described in detail elsewhere (15). The detector is coupled to a data acquisition card (NI PCI-6024E, National Instruments, Austin TX) on a personal computer running in-house-built labview 7.1 data collection software counting the number of incident photons with a bin size of 5 ms.
Results and Discussion
Construction and Characterization of the Nanotube Electrophoresis System. A nanotube electrophoresis system was constructed from a unilamellar vesicle coupled to a multilamellar liposome, which acts as a membrane reservoir so that the unilamellar vesicle can be expanded in size by fluid injection to arbitrary size, and the nanotube can be retracted, without increasing the membrane tension. The experiments were performed in the following manner. First, a controlled volume of the DNA solution of known concentration (0.03 mg/ml) was delivered to the unilamellar vesicle, using an electroinjection protocol (16). Two gel-capped electrodes were then electro-inserted into the DNA-filled unilamellar vesicle. Subsequently, one of the electrodes was withdrawn from the liposome, pulling a lipid nanotube 40 ± 1 μm long. The diameter of the nanotube is ≈300 nm (21). The nanotube was positioned across the confocal excitation/detection volume of the inverted microscope setup by moving the electrode by use of high-graduation 3D micromanipulators. An electric potential was then applied between the electrodes, with the nanotube-coupled electrode having positive potential (Fig. 1 a-d). At an applied voltage of 100 mV, the electric field strength in the nanotubes was estimated to be ≈15 V/cm by using the equivalence circuit for the system (see Fig. 1d legend).
Fig. 1.

Schematics describing nanotube-vesicle network (NVN) formation, geometry of the NVN system, and fluorescence detection used in the experiments and simulations of the field distribution and flow profile. (a) The electrodes are pressed against the membrane of the unilamellar liposome filled with a DNA solution, and an electric pulse is applied. The pulse opens pores in the membrane, allowing the tips of the electrodes to enter into the liposome. (b) The pores in the membrane close, and the membrane seals around the electrode. (c) One of the electrodes is moved out of the unilamellar liposome, pulling a lipid membrane nanotube. (d) Schematic picture of the NVN. The nanotube is aligned across the confocal excitation/detection spot of the confocal microscopy setup, and an electric potential is applied between the electrodes, with the nanotube-coupled electrode having positive potential. The fluorescence signal from the DNA molecules is detected as they pass the excitation/detection spot. V1 is the velocity of the DNA molecules, V2 is the velocity of the membrane, and V3 is the velocity of the electroosmotic flow. Because V2 and V3 cancel each other out (see text), no net liquid flow occurs in the nanotube. (Inset) The equivalence circuit of the electrophoretic system is shown; 1 and 5 represent the electrode resistance (50 MΩ), 2 and 3 represent the vesicle-electrode seal resistance (50 MΩ) and the nanotube-electrode seal resistance (100 MΩ), respectively, and 4 represents the nanotube resistance (>1GΩ), calculated from the buffer conductivity (0.5 S/m) and the nanotube dimensions (L = 40 μm and R = 150 nm). Evaluation of the equivalence circuit gives a potential drop of 60 mV over the nanotube. (e) A Nomarski differential interference contrast microscopy image of a fully formed NVN with electrodes. The unilamellar vesicle is filled with fluorescently labeled DNA molecules. The two electrodes are used both to create the NVN and drive the electric current through it. The length of the nanotube is 40 ± 1 μm. (f) Field strength distribution inside and outside the NVN, modeled with the finite element method program femlab 3.0.(g) Flow profile of axial velocity inside the nanotube, drawn from the nanotube center line to the membrane surface.
femlab simulations support this estimate. In the model, a spherical vesicle (radius 5 μm) connected to a nanotube (10 μm long, radius 150 nm) is immersed in electrolyte. The thickness of the lipid membrane is 5 nm. Both solvent and membrane are treated as conductors (22) with the conductivity of the electrolyte and membrane being set to 0.5 and 1 × 10-14 S/m, respectively. At the interface between lipid membrane and electrolyte, the boundary condition was set so that the normal component of the current density across the boundary is continuous. At the end of the nanotube a potential of 15 mV (reduced from the experimental potential difference of 60 mV because of the 4-fold shorter tube used in the simulation) was applied inside the membrane boundary, and the mesh element edges at the inside of the liposome boundary and opposite to the nanotubes entrance were given the potential 0 mV. Leakage currents passing through the seal between the pipette and the enclosing membrane and passing to the outside bulk electrolyte were included in the model by adding a current source in the bulk electrolyte outside the end of the nanotube and a corresponding current sink in the bulk electrolyte at the opposite end of the vesicle. The current densities were chosen so that a total current of 0.1 nA was obtained. The model was tested for membrane conductivities in the range of 10-9 to 10-14 S/m, and no noticeable differences in the field distribution were seen for these values. The field strength inside the nanotube is calculated to be 14.7 V/cm, and the field distribution is shown in Fig. 1f.
Membrane and Solvent Motion. The lipid mixture used gives a membrane that has a negative net charge with a zeta potential of ζ = -55 mV at pH 8.1. The membrane is therefore expected to be pulled by the electric field toward the positive electrode, and the transported lipid was indeed observed to enter the gel plug inside the electrode. In separate experiments we followed the electric field-driven transport of small unilamellar vesicles that were in direct contact with nanotube walls, and their velocity was measured to be ≈50 μm/s at 15 V/cm. We take this value as an estimate of the experimental membrane velocity, to be compared with theoretical predictions. In the simplest approach, electrophoretic theory (23, 24) predicts a velocity for lipid vesicles in free-solution given by the Smoluchowski formula νm = εEζ/η, where E is the applied electric field, ζ is the zeta potential, ε is the solvent dielectric constant, and η is the viscosity. At 15 V/cm in water this velocity is νm ≈ 55 μm/s, which is in fair agreement with our experimentally observed value for the velocity of the nanotube-confined vesicles.
However, the Smoluchowski formula is derived for spherical particles in free solution and does not necessarily apply to electrophoretic migration of the cylindrical nanotube wall in our system. The electric field is applied primarily inside the tube, and because of the low conductivity of the membrane, the field at the nanotube exterior surface is vanishingly small, as shown by the femlab simulations (Fig. 1f). We therefore solved the electrophoretic equations for the nanotube geometry with an electric field acting only on the interior side of the nanotube membrane wall. The concomitant solvent flow is given by the Stokes equation with an electric force density term, 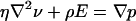 , where ρ is the density of mobile charges in solution and E is the electric field that will be taken to be the applied field (linear-response approximation). The mobile charge density is
, where ρ is the density of mobile charges in solution and E is the electric field that will be taken to be the applied field (linear-response approximation). The mobile charge density is 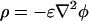 , where the electrostatic potential is given by the Poisson-Boltzmann equation
, where the electrostatic potential is given by the Poisson-Boltzmann equation 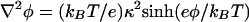 . Because the nanotube radius (R = 150 nm) is much larger than the Debye screening length under our conditions (κ-1 = 7.8 Å), the electrostatic potential (close to the membrane) can be approximated by that of a flat surface. The electric field inside the tube is constant and parallel to the tube axis; outside the tube the field is vanishingly small and the electric force on the exterior bilayer can be neglected. The cross-linked gel in the pipette connected to the nanotube has low water permeability; the net water flux in the tube is therefore set to zero.
. Because the nanotube radius (R = 150 nm) is much larger than the Debye screening length under our conditions (κ-1 = 7.8 Å), the electrostatic potential (close to the membrane) can be approximated by that of a flat surface. The electric field inside the tube is constant and parallel to the tube axis; outside the tube the field is vanishingly small and the electric force on the exterior bilayer can be neglected. The cross-linked gel in the pipette connected to the nanotube has low water permeability; the net water flux in the tube is therefore set to zero.
Solving the above equations gives an electrophoretic membrane-wall velocity νm = εEζ/η(1 + ξ/8πη), where ξ = 4πη/log(L/R) is the friction coefficient per unit length of a moving cylinder of radius R. This velocity is ≈10% smaller than predicted by the Smoluchowski formula, because of surface friction at the exterior leaflet. Allowing for sliding of the two bilayers relative to each other did not change the resulting velocity, because the bilayer sliding friction is much larger than the hydrodynamics surface friction ξ of the nanotube.
Fig. 1g shows the calculated profile (axial velocity) of the solvent flow. There is an electroosmotic flow generated by the force on the positively charged Debye layer. However, because the membrane wall is also moving, the effective solvent velocity in the nanutube is much smaller than the velocity of the electroosmotic flow. The velocity at the center line is seen to be ≈10 times lower than the membrane electrophoretic velocity. The absence of any substantial liquid transport is in agreement with the fact that we do not observe any accumulation of fluid at the electrode tip. This behavior contrasts with the case of a solid, negatively charged capillary wall, where the electroosmotic flow is known to strongly reduce or even reverse the electrophoretic velocity of DNA.
DNA Transport. Fig. 2 shows representative electropherograms of the different DNA molecules used in this study. No peaks were observed in the absence of DNA in the vesicle or if the polarity was reversed compared with that in Fig. 1d, showing that the peaks correspond to DNA molecules that undergo unidirectional transport toward the positive electrode. The covalent labeling we used for the fluorescence-based DNA detection was found to reduce the background compared with reversibly intercalating dyes, e.g., YOYO-1, which dissociate with time and bind to the lipid membrane. Data collected at different occasions from different nanotube set-ups showed good reproducibility (<5% difference with respect to the average integrated peak area).
Fig. 2.

Electropherograms of DNA from φX174 (a), pΔ (b), T7/BsteII (c), T7 (noncleaved) (d), and T4 (e) (compare Table 1). The DNA molecules were transported through a 40-μm-long lipid membrane nanotube and detected at its midpoint. The effective field strength was ≈15 V/cm in all experiments.
dsDNA molecules of five different sizes were studied (Table 1). Exploiting the truly molecular dimensions of our microfluidic device, the set of DNA sizes were chosen to cover radii of gyration (Rg) that are either comparable or larger than the nanotube radius R. This size difference is expected to impose a strong confinement of the DNA, and consequently, the entrance and transport of the polymers is expected to require substantial perturbation of the coil or tube conformation for the largest DNA studied (25).
Table 1. Properties of DNA molecules used.
| DNA | N, bp* | Lc, μm† | Rg, nm‡ | Nb,§ | τ1, ms¶ | Δt, ms∥ |
|---|---|---|---|---|---|---|
| φX174** | 5,386 | 1.8 | 175 | 1.4 | 2.6 | 21 |
| pΔ | 8,000 | 2.7 | 213 | 2.0 | 4.8 | 21 |
| T7/BstEII | 20,065/19,871†† | 6.8 | 336 | 5.0 | 19 | 26 |
| T7 | 39,936 | 13.6 | 476 | 10.1 | 53 | 27 |
| T4 | 166,000 | 56.4 | 970 | 41.8 | 450 | 52 |
Degree of polymerization.
Contour length; Lc = 0.34 nm·N.
Radius of gyration; Rg = (PLc/3)1/2 with a persistence length of P = 50 nm.
Number of DNA blobs in nanotube of radius R = 150 nm. Nb = N/n, where n = 3,970 is the number of bp per blob in a cylindrical tube, obtained from the condition that the blob radius-of-gyration equals the tube radius.
Longest relaxation time of unconfined polymer. Confinement will increase the relaxation time as described in the text.
Experimental average transit time across detection volume.
Linear form obtained by restriction cleavage of circular form by Eco 1471. The uncleaved sample contained >90% nicked circular form.
Two fragments in equimolar amounts obtained by restriction cleavage of T7 DNA by BstEII. Calculations and plots are based on the mean N.
Three parameters were extracted from the electropherograms: the maximum fluorescence signal detected from each DNA molecule (peak intensity Ip), the duration of the peak (transit time Δt), and the integrated fluorescence intensity (peak area AI). Fig. 3 a-c shows the distributions of these three parameters for four different DNA sizes. The distributions are all monomodal and can therefore be reasonably represented by an average value and a width. Whereas the transit time distributions do overlap for the two smallest sizes, it is clear that all three parameters have distributions that exhibit a significant trend toward higher values with increasing DNA size. By contrast, DNA topology has little effect on the distributions, because Fig. 3 d-f shows that they essentially overlap for the linear and relaxed circular forms of φX174 DNA for all three parameters.
Fig. 3.

Distributions of peak intensity, transit time, and integrated peak area for the different DNA species. Distributions for T4 DNA are not shown because of the small number of observed events (n = 12). (a-c) Distributions of peak intensity (a), transit time (b), and integrated peak area (c) for DNA from φX174, pΔ, T7/BsteII, and T7 (noncleaved) are shown. (d-f) Distributions of peak intensity (d), transit time (e), and integrated peak area (f) for the linear and relaxed circular forms of φX174 DNA. The distributions for T4 DNA are not shown because of the rarity of the detected peaks compared with the other DNA sizes (for which n > 100); only average values (n = 12) of the three parameters are given in Fig. 4.
The fact that the average peak area increases linearly with increasing DNA size (Fig. 4a) is consistent with a strong confinement inside the nanotube because it ensures that the entire molecule passes through the detection volume (26). This result is one of our most important, because it demonstrates that our system is capable of single-file transport and counting of DNA molecules. Notably, the average peak intensity is observed to increase linearly with DNA size as well (Fig. 4b). Finally, the transit time exhibits a linear dependence on DNA size, but in this case with an intercept with the ordinate axis at t0 = 20.0 ms (Fig. 4c). This result corresponds to the transit time for a small DNA molecule across the finite detection volume (0.8 μm in diameter), yielding a velocity of 40 μm/s toward the positive electrode (27). In fact, the velocity we measure is only somewhat lower than the free-solution velocity of DNA (51 μm/s at 15 V/cm) (28). This result is in agreement with our prediction that the solvent flow is weak (Fig. 1g). There is an electrostatic repulsion between DNA and the lipid wall that contributes to limit the time at least the small DNA molecules will experience the higher flow rates occurring close to the wall (within ≈2 Debye lengths).
Fig. 4.

Mean values of integrated peak area (AI) (a), peak intensity (IP) (b), and transit time (Δt) (c) vs. DNA size (N). Error bars correspond to the standard error. Where the error bars are not clearly visible, they are equal to or smaller than the data points.
DNA Conformation During Transport. The observed effect of DNA size on the three transport parameters has implications for the mode of DNA transport, as now will be discussed. The mechanism of transport of the larger DNA molecules is likely to depend on which conformation the strongly confined DNA coils attain in the tube and the strength of the repulsive interaction between DNA and membrane. Notably, the similar behavior of the linear and circular DNA forms (Fig. 3 d-f) indicates that a lack of free ends does not affect the migrational behavior of DNA. The DNA molecules are thus not forced to move end-on but can attain conformations where the polymer chain folds back on itself inside the tube, in agreement with the fact that the DNA persistence length (P = 50 nm) is smaller than the tube radius (≈150 nm).
The Blob Model. In thermal equilibrium, the confined DNA can be represented by a linear chain of subcoils (29). In this blob model, the DNA coil is divided into subcoils (blobs). Each blob contains a given contour length of DNA that is determined by the condition that the blob radius of gyration is equal to the tube radius R. The coil extension along the tube axis is 2R per blob. The smallest DNA segment used here has roughly the size of one blob, and the largest consists of ≈42 blobs (Table 1). The detection spot (0.8 μm in diameter) corresponds to between two and three blobs, and it would therefore be expected that the mean peak intensity should converge to a constant value for the three longest DNA molecules, whereas experimentally it increases linearly with size over the whole investigated range. This observation suggests that the DNA attains another, more axially compressed conformation.
The Compressed Globule Model. In a recent theoretical treatment of semiflexible polymers confined in soft tubes, Brochard-Wyart et al. (25) predict that if a polymer is large enough it deforms the tube and attains a globule-like conformation. The coil is spherical in shape, but compacted compared with the unconfined polymer coil because of forces arising from the wall of the expanded tube. In fact, the compression might be strong enough for even the largest DNA molecule to fit completely inside the detection volume, which would give a linear relation between the mean peak intensity and the number of base pairs. Given the membrane tension of ≈1 × 10-6 J/m2 in our system (15, 21), Brochard-Wyart et al. predict that the transition from the blob behavior (in an unperturbed channel) to the compressed globule occurs for a polymer size of ≈150 persistence lengths, or 22 kbp given that P = 150 bp at our ionic strength. Thus, the theory predicts that the two smallest DNA sizes we have studied should attain a blob-like conformation in an unperturbed tube, with φX174 and pΔ-DNA containing one and two blobs, respectively (Table 1), i.e., these coils are only slightly elongated. By contrast, the two largest T7 and T4 DNA coils are large enough to cause a deformation of the lipid nanotubes, and thereby be compressed into a globular conformation.
Nonequilibrium Aspects. The blob model as well as the compressed globule model of Brochard-Wyart et al. describe the equilibrium conformation of confined polymers, and it should be noted that during transport, nonequilibrium effects may affect the conformation of the DNA in the tube. The time span between the entry of a DNA molecule into the nanotube and its detection is ≈0.5 s, again assuming a velocity of 40 μm/s. By comparison, the conformational relaxation time for the T4 DNA molecule in free solution is τ1 ≈ 0.5 s (30), and this time is expected to increase by a factor of ≈10 because of the confinement within the nanotube (31). Therefore, it is plausible that the largest DNA molecules used in this study (Table 1) will not attain their equilibrium conformation by the time they pass the detection spot. Instead, they may retain a conformational memory caused by the entry into the nanotube. Because the relaxation time depends strongly on the molecular weight (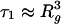 ) (32), the two shortest DNA chains used here have time to equilibrate and are expected to attain a blob-like conformation.
) (32), the two shortest DNA chains used here have time to equilibrate and are expected to attain a blob-like conformation.
Mechanisms Behind Distribution Widths. Polymers inherently exhibit a broad range of conformations, and single-molecule approaches have the potential to sample large conformational fluctuations, as observed experimentally in solution (33) and porous media (34). Such instantaneous conformational differences between individual DNA molecules at the detection moment may underlie the wide distributions of the parameters we measure (Fig. 3). Presently, we cannot rule out that system-specific sources affect the distribution widths. These may include fluctuations in the position of the flexible nanotube in the confocal detection volume, variations of the electric field strength inside the nanotube, and differences in the label density of different DNA molecules.
Conclusions
In conclusion, we have demonstrated single-file electrokinetic transport and detection of single-labeled dsDNA molecules in lipid membrane channels having radii of ≈50-150 nm. The single-photon burst characteristics can be used to measure the size of DNA molecules and interrogate their conformation during transport. In our soft-material device, the electroosmotic flow is cancelled by the membrane-driven liquid flow, in contrast to those with conventional solid channels. This soft-matter nanoscale device displays full-fledged capability for controlled single-molecule handling with potential applications for counting, characterization, manipulation, and modification of biopolymers. For further studies on conformational dynamics of confined DNA molecules and other polymers (e.g., RNA), nanotubes of different bore size, length, lipid composition, and mechanical properties (i.e., systems ranging from floppy and easily deformable tube walls to walls that are undeformable and solid state-like) can be used. Once the conformation of the transported DNA is deduced, it should then be feasible in a second step to control the conformation of DNA. Apart from pure physico-chemical characterization of polymer dynamics, such studies may shed light on intercellular traffic through biological surfactant nanotubes (1).
Another exciting possibility would be to carry out reactions on single DNA molecules in nanotube networks. We have previously shown (18) that topologically complex networks can be made. Hence the possibility exists that DNA can be routed to different nodes in a network where each of the nodes constitutes a discrete reaction container enclosing chemical species that react with an incoming DNA molecule. Such networks can then, theoretically, be used for e.g., hybridization of DNA, sequencing of DNA, synthesis of DNA, and studies of DNA-protein interactions. Of course, routing and transport of DNA in large networks requires a more complex electrophoresis system than we have presented here.
Acknowledgments
We thank Ken Cole of the National Institute of Standards and Technology for samples of pΔ DNA. This work was funded by the Swedish Research Council, the Swedish Foundation for Strategic Research, the Göran Gustafsson Foundation, and the Royal Swedish Academy of Sciences. P.D. acknowledges the receipt of a Marie Curie fellowship.
Author contributions: M.T., B.\A., and O.O. designed research; M.T. performed research; M.T., B.\A., J.O., J.-F.J., P.D., and O.O. analyzed data; M.T., B.\A., J.O., J.-F.J., P.D., and O.O. wrote the paper; and J.O. performed simulations.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: NVN, nanotube-vesicle network.
References
- 1.Rustom, A., Saffrich, R., Markovic, I., Walther, P. & Gerdes, H. H. (2004) Science 303, 1007-1010. [DOI] [PubMed] [Google Scholar]
- 2.Stange, P., Zanette, D., Mikhailov, A. & Hess, B. (1998) Biophys. Chem. 72, 73-85. [DOI] [PubMed] [Google Scholar]
- 3.Hess, B. & Mikhailov, A. (1996) Biophys. Chem. 58, 365-368. [DOI] [PubMed] [Google Scholar]
- 4.Khairutdinov, R. F. & Serpone, N. (1996) Progr. Reaction Kinetics 21, 1-68. [Google Scholar]
- 5.Sesso, A., Defaria, F. P., Iwamura, E. S. M. & Correa, H. (1994) J. Cell Sci. 107, 517-528. [PubMed] [Google Scholar]
- 6.Godi, A., Di Campli, A., Konstantakopoulos, A., Di Tullio, G., Alessi, D. R., Kular, G. S., Daniele, T., Marra, P., Lucocq, J. M. & De Matteis, M. A. (2004) Nat. Cell Biol. 6, 393-404. [DOI] [PubMed] [Google Scholar]
- 7.Turner, S. W. P., Cabodi, M. & Craighead, H. G. (2002) Phys. Rev. Lett. 88, 128103. [DOI] [PubMed] [Google Scholar]
- 8.Woods, L. A., Roddy, T. P., Paxon, T. L. & Ewing, A. G. (2001) Anal. Chem. 73, 3687-3690. [DOI] [PubMed] [Google Scholar]
- 9.Lyon, W. A. & Nie, S. M. (1997) Anal. Chem. 69, 3400-3405. [Google Scholar]
- 10.Cao, H., Yu, Z. N., Wang, J., Tegenfeldt, J. O., Austin, R. H., Chen, E., Wu, W. & Chou, S. Y. (2002) Appl. Phys. Lett. 81, 174-176. [Google Scholar]
- 11.Ito, T., Sun, L. & Crooks, R. M. (2003) Chem. Commun. 1482-1483. [DOI] [PubMed]
- 12.Turner, S. W., Perez, A. M., Lopez, A. & Craighead, H. G. (1998) J. Vacuum Sci. Technol. B 16, 3835-3840. [Google Scholar]
- 13.Chou, H. P., Spence, C., Scherer, A. & Quake, S. (1999) Proc. Natl. Acad. Sci. USA 96, 11-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu, A. Y., Spence, C., Scherer, A., Arnold, F. H. & Quake, S. R. (1999) Nat. Biotechnol. 17, 1109-1111. [DOI] [PubMed] [Google Scholar]
- 15.Karlsson, A., Karlsson, M., Karlsson, R., Sott, K., Lundqvist, A., Tokarz, M. & Orwar, O. (2003) Anal. Chem. 75, 2529-2537. [DOI] [PubMed] [Google Scholar]
- 16.Karlsson, M., Nolkrantz, K., Davidson, M. J., Stromberg, A., Ryttsen, F., Akerman, B. & Orwar, O. (2000) Anal. Chem. 72, 5857-5862. [DOI] [PubMed] [Google Scholar]
- 17.Cans, A. S., Wittenberg, N., Karlsson, R., Sombers, L., Karlsson, M., Orwar, O. & Ewing, A. (2003) Proc. Natl. Acad. Sci. USA 100, 400-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karlsson, M., Sott, K., Davidson, M., Cans, A. S., Linderholm, P., Chiu, D. & Orwar, O. (2002) Proc. Natl. Acad. Sci. USA 99, 11573-11578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karlsson, A., Karlsson, R., Karlsson, M., Cans, A. S., Stromberg, A., Ryttsen, F. & Orwar, O. (2001) Nature 409, 150-152. [DOI] [PubMed] [Google Scholar]
- 20.Karlsson, M., Sott, K., Cans, A. S., Karlsson, A., Karlsson, R. & Orwar, O. (2001) Langmuir 17, 6754-6758. [Google Scholar]
- 21.Karlsson, R., Karlsson, M., Karlsson, A., Cans, A. S., Bergenholtz, J., Akerman, B., Ewing, A. G., Voinova, M. & Orwar, O. (2002) Langmuir 18, 4186-4190. [Google Scholar]
- 22.Kummrow, M. & Helfrich, W. (1991) Phys. Rev. A 44, 8356-8360. [DOI] [PubMed] [Google Scholar]
- 23.Hiemenz, P. C. (1986) Principles of Colloid and Surface Chemistry (Dekker, New York).
- 24.Henry, D. C. (1931) Proc. R. Soc. London Ser. A 133, 106-129. [Google Scholar]
- 25.Brochard-Wyart, F., Tanaka, T., Borghi, N. & de Gennes, P.-G. (2005) Langmuir 21, 4144-4148. [DOI] [PubMed] [Google Scholar]
- 26.Filippova, E. M., Monteleone, D. C., Trunk, J. G., Sutherland, B. M., Quake, S. R. & Sutherland, J. C. (2003) Biophys. J. 84, 1281-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foquet, M., Korlach, J., Zipfel, W., Webb, W. W. & Craighead, H. G. (2002) Anal. Chem. 74, 1415-1422. [DOI] [PubMed] [Google Scholar]
- 28.Stellwagen, N. C., Gelfi, C. & Righetti, P. G. (1997) Biopolymers 42, 687-703. [DOI] [PubMed] [Google Scholar]
- 29.de Gennes, P.-G. (1979) Scaling Concepts in Polymer Physics (Cornell Univ. Press, Ithaca, NY).
- 30.Callis, P. R. & Davidson, N. (1969) Biopolymers 8, 379-390. [Google Scholar]
- 31.Bakajin, O. B., Duke, T. A. J., Chou, C. F., Chan, S. S., Austin, R. H. & Cox, E. C. (1998) Phys. Rev. Lett. 80, 2737-2740. [DOI] [PubMed] [Google Scholar]
- 32.Doi, M. & Edwards, S. F. (1988) The Theory of Polymer Dynamics (Clarendon, Oxford).
- 33.Perkins, T. T., Smith, D. E. & Chu, S. (1997) Science 276, 2016-2021. [DOI] [PubMed] [Google Scholar]
- 34.Larsson, A. & Akerman, B. (1995) Macromolecules 28, 4441-4454. [Google Scholar]