

Abstract
Characterizing the dynamics of specific RNA levels requires real-time RNA profiling in a single cell. We show that the combination of a synthetic modular genetic system with fluorescence correlation spectroscopy allows us to directly measure in real time the activity of any specific promoter in prokaryotes. Using a simple inducible gene expression system, we found that induced RNA levels within a single bacterium of Escherichia coli exhibited a pulsating profile in response to a steady input of inducer. The genetic deletion of an efflux pump system, a key determinant of antibiotic resistance, altered the pulsating transcriptional dynamics and caused overexpression of induced RNA. In contrast with population measurements, real-time RNA profiling permits identifying relationships between genotypes and transcriptional dynamics that are accessible only at the level of the single cell.
Keywords: fluorescence correlation spectroscopy, multidrug efflux, transcription, cell cycle, noise
Nearly half of a century ago, the discovery of messenger RNA as “an unstable intermediate” established RNA dynamics as one of the key properties of molecular adaptation in nature (1). Given the transient character of RNA transcripts, it has proved technically challenging to monitor in real time the transcription activity of a promoter within an individual prokaryotic cell (2-4). There exist various powerful in vitro methods for measuring RNA levels, including Northern blots, RT-PCR, and microarrays. Expression levels of a specific RNA species, extracted from population measurements, generally come from cells that are in different cell cycle states and exhibit different behaviors due to the variations of their internal biochemical parameters (5). Because of these inherent differences among single cells within a population, the transient dynamics of transcriptional networks may only be correctly characterized by monitoring transcriptional activity as a function of time within a single cell (6). Therefore, we need simple, noninvasive, real-time approaches to study the relationship between structure and dynamics of intracellular transcriptional networks in a single living cell. To this end, we constructed an in vivo synthetic genetic system that allows us to monitor the dynamics of a specific RNA species as a function of time within a single bacterium.
Materials and Methods
Fluorescence Correlation Spectroscopy (FCS) Apparatus. The incident excitation from a blue laser beam (Sapphire 488 nm, 20 mW, Coherent, Santa Clara, CA) is focused with a ×100 microscope objective lens (numerical aperture = 1.3, Olympus, Melville, NY) onto a diffraction-limited spot in the bacterium. The emitted green fluorescence is collected in a confocal geometry and detected with an avalanche photo-diode (spcmaqr-16fc, PerkinElmer). The fluorescent signal is analyzed in real time with a fast correlator (5000EPP, ALV, Langen, Germany). We visualized the bacterium attached onto a glass coverslip by using a dark-field illumination. The temporal variations from the emitted light arise from the fluorescent molecules diffusing in and out of the confocal volume of detection. When the number of diffusing molecules is low, the fluctuations of fluorescence intensity about the mean signal are large. Because this technique relies on fluctuations and not on the absolute value of the fluorescence signal, the measure of concentrations and diffusion constants is self-calibrated. The mathematical analysis of the fluctuations of the fluorescence signal leads to the determination of concentrations and the diffusion coefficient of the associated fluorescent molecules. The amplitude of the autocorrelation function at the intercept with the vertical axis is inversely equal to the number of molecules (N) in the detection volume. This function corresponds to two-dimensional diffusion and is described by G(t) = 1/N[1/1+ (4Dt/ω2)], where D is the diffusion constant of the fluorescent molecules, t is the time variable, and ω is the radius of the detection volume in the experimental configuration.
Determination of RNA Concentration with FCS. In all experiments, the fraction of free and bound MS2-GFP was determined by fitting the autocorrelation (7) functions with
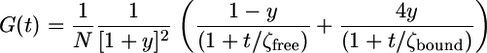 |
This formula takes into account the ratio in brightness between free homodimers and two homodimers bound to the two ms2-RNA-binding sites (see the supporting information, which is published on the PNAS web site). In this formula, N is the number of fluorescent molecules in the detection volume, y is the fraction of bound MS2-GFP, and ζfree and ζbound are diffusion half-times of free and fully bound MS2-GFP, respectively. We determined the RNA concentration in the detection volume (0.23 fl) by multiplying the fraction of bound MS2-GFP at any given time with the total number of MS2-GFP molecules N0 measured at the time point t = 0 s. Because of bleaching of GFP caused by successive laser excitations, the value of N is decreasing throughout the experiment. Therefore, we only use the initial concentration N0. The ratio y, the other fitting parameter, is insensitive to bleaching (see the supporting information). Estimation of ms2-RNA concentration is highly accurate when the fraction of bound MS2-GFP is between 20% and 60%. The errors in estimating RNA concentration are calculated by using the equation (δy/y + δN0/N0)[RNA], where δy and δN0 are the fitting errors of the fraction bound and of the MS2-GFP concentration inside the detection volume, respectively.
In Vitro Determination of RNA Concentration with FCS. To determine the diffusion times of free and bound ms2-RNA transcripts, we fit the autocorrelation functions with G(t) = 1/{N[(1 + (4Dt/ω2)]}. This function describes a two-dimension translational diffusion of either free or bound molecules. N is the number of fluorescent molecules in the detection volume, D = ω2/(2ζ) is the two-dimension diffusion constant of the fluorescent molecules (where ζ is the diffusion time), t is the time variable, and 2ω = 0.38 μm is the diameter of the detection volume. ms2-RNA was added to a final concentration of 0, 0.15, 0.30, 0.45, 0.60, 0.75, 0.90, 1.05, and 1.20 μM in 200 μl of PBS buffer containing 1.44 μM of purified MS2-GFP (Fig. 1B). FCS data were collected for increasing ms2-RNA concentrations. ms2-RNA concentrations determined with FCS were compared with RNA concentrations determined by absorbance at 260 nm. All MS2-GFP proteins became bound after 1.05 μM of ms2-RNA was added. The diffusion times of free and fully bound MS2-GFP to ms2-RNA were found to be 0.3 ± 0.03 ms and 1 ± 0.04 ms, respectively (Fig. 1 A). For accurate estimation of ms2-RNA concentration, the concentration of MS2-GFP protein should be at least 2-fold in excess of ms2-RNA concentration (Fig. 1B). MS2-GFP concentrations are given for homodimers that are the ms2-RNA-binding units. One ms2-RNA transcript binds to two MS2-GFP homodimers.
Fig. 1.

Quantifying RNA concentration with FCS. (A) Labeling ms2-RNA transcripts with MS2-GFP fusion protein in vitro. Purified MS2-GFP fusion proteins (1.44 μM) were titrated with ms2-RNA transcripts from 0 to 1.2 μM ([MS2-GFP] is given for homodimers). Binding of MS2-GFP proteins to ms2-RNA transcripts caused a shift in the diffusion constant of MS2-GFP. Autocorrelation functions (normalized) were obtained from free MS2-GFP proteins (black), a mixture of free and ms2-RNA-bound MS2-GFP proteins (gray), and fully bound MS2-GFP proteins to ms2-RNA transcripts ([ms2-RNA] from 0.9 to 1.2 μM) (light gray). (B) In vitro calibration curve of ms2-RNA concentration determined with absorbance at 260 nm (x axis) and with FCS (y axis). The calibration curve is linear for [MS2-GFP] = 1.44 μM and [ms2-RNA] from 0 to 0.8 μM. Error bars represent uncertainties in the fitting parameters of the autocorrelation function. The fraction of free and bound MS2-GFP molecules was determined by fitting (full lines) the autocorrelation functions with G(t) = (1/N)(1/[1 + y]2)(1 - y/(1 + t/ζfree) + 4y/(1 + t/ζbound)), where y (the only fitting parameter) is the fraction of bound MS2-GFP, and ζbound (1 ± 0.04 ms) and ζfree (0.3 ± 0.03 ms) are the intracellular diffusion times of bound and free MS2-GFP molecules. (C) Dual-plasmid reporting system for RNA expression in a living cell. Reporter plasmid (Left) carries an inducible lac promoter that regulates the expression of a mutant MS2 coat protein fused to GFP (12, 28). Expression plasmid (Right) carries an inducible tet promoter that regulates the expression of two ms2-RNA-binding sites (400 nt). (D) Monitoring RNA levels with FCS in living cells. Binding of MS2-GFP proteins to 70S ribosome-associated ms2-RNA transcripts causes a shift in the diffusion constant of MS2-GFP. Shown are typical normalized autocorrelation functions obtained from a single bacterium in which all MS2-GFP proteins are freely diffusing (black), mixture of free and bound MS2-GFP to ms2-RNA transcripts (gray), and all MS2-GFP proteins bound to ms2-RNA transcripts (light gray). Fitting curves (full lines) were plotted with ζbound (30 ± 3 ms) and ζfree (1.4 ± 0.5 ms).
Monitoring Transcription in Single Living Cells with FCS. Plasmids. Vectors based on the pZ family (8) used were pZS12MS2-GFP (SC101 origin, 6-8 copies per cell, AmpR, PLlacO-1 promoter)/pZE31ms2 (ColE1 origin, 50-70 copies per cell, ChlR, PLtetO-1 promoter).
Cell strains. Strains used were as follows. DH5α PRO (Clontech): deoR, endA1, gyrA96, hsdR17(rk-mk+), recA1, relA1, supE44, thi-1, Δ(lacZYA-argF)U169, Φ80δlacZΔM15, F-, λ-, PN25/tetR, PlacIq/lacI, and SpR. Frag1A: F-, rha-, thi, gal, lacZam, ΔacrAB::kanR, PN25/tetR, PlacIq/lacI, and SpR. Frag1B: F-, rha-, thi, gal, lacZam, PN25/tetR, Placiq/lacI, and SpR. The PN25/tetR, Placiq/lacI, SpR cassette was transferred from DH5αPRO to Frag1 to generate Frag1B by P1 transduction. The ΔacrAB:kanR cassette was transferred from KZM120 to Frag1B to generate Frag1A.
Growth conditions. Cells carrying both reporter and expression plasmids were grown overnight at 30°C in M9 minimal salts (Qbiogene, Irvine, CA) supplemented with 0.1 mM CaCl2/2 mM MgSO4/0.4% glycerol/0.5% casamino acids/100 μg/ml ampicillin/34 μg/ml chloramphenicol/50 μg/ml spectinomycin/1 mM IPTG. Cells from overnight cultures were washed, diluted 20-fold, and regrown in fresh M9 media for an additional 2 h. Homogeneity and level of cellular MS2-GFP expression were checked with fluorescence microscopy.
Experimental conditions. First, cells were immobilized on a glass coverslip coated with flagellin antibodies as described in ref. 9. Second, cells were covered with 200 μl of 0.8% low-melting point agarose dissolved in M9 medium. We used a deletion mutant (FG loop deletion) of the coat protein of phage MS2 fused to GFP (denoted MS2-GFP), which binds a specific 23-nt sequence forming a bulged hairpin loop (MS2-binding site) (10). In our experiments, MS2-GFP was preexpressed from an inducible promoter controlled by LacI. Cells that express ≈25 μM MS2-GFP protein were selected for the assay. After the first division on the glass surface, a transcriptional inducer (anhydrotetracycline, aTc) was added to a final concentration of 400 ng/ml. Transcription of ms2-RNA was monitored through two generations by using the FCS setup. MS2-GFP protein concentration throughout measurements was at least 2-fold in excess of ms2-RNA concentration maximally expressed at 400 ng/ml aTc induction (≈3 μM). MS2-GFP concentrations are given for homodimers that are the ms2-RNA-binding units. One ms2-RNA transcript binds to two MS2-GFP homodimers.
In vivo determination of ζfree and ζbound. To determine in single cells the diffusion times of free and bound ms2-RNA transcripts, we use the same fit described for in vitro measurements. ζfree was determined to be 1.4 ± 0.5 ms by averaging the diffusion times of MS2-GFP in 100 cells carrying only the reporter plasmid pZS12MS2-GFP. Under this condition, all MS2-GFP molecules were free. Similarly, ζbound was determined by in vivo titration of the MS2-GFP molecules with an excess of ms2-RNA-binding sequences. Cells carrying the plasmids pZS12MS2-GFP/pZE31ms2 were induced overnight with 0.1 mM IPTG to express ≈2 μM MS2-GFP (see below). Overexpression of ms2-RNA was produced with 1 μg/ml aTc induction. Under this condition, the ms2-RNA transcripts were in excess, and all MS2-GFP molecules were bound. The maximum diffusion times of bound MS2-GFP from 30 cells were averaged to infer the value of ζbound (30 ± 3 ms).
In contrast with in vitro conditions, the change in the diffusion of MS2-GFP dimer bound to the RNA transcript is significantly larger (≈30-fold). It is hypothesized that the binding of the ribosome to the Shine-Dalgarno sequence within this RNA transcript dominates the observed diffusive process (see the supporting information). The concentration of ribosomes in an Escherichia coli cell is on the order of 10 μM, and the binding affinity of ribosome to a typical mRNA is 0.05-0.5 μM. Therefore, the binding of a single ribosome would increase the molecular mass of the MS2-GFP dimer (molecular mass ≈ 80 kDa)-bound RNA to ≈2,600 kDa (11) and would dominate the diffusion of the ms2-RNA transcript.
Results and Discussion
We used a RNA reporter system originally developed by Bertrand et al. (12), which employs the coat protein of phage MS2 fused to GFP. This fusion protein has a strong affinity for a specific RNA sequence: the ms2-binding site (13, 14) (ms2-RNA). Using FCS (9, 15), we first monitored the concentration of noncoding ms2-RNA transcripts from an in vitro transcription assay in the presence of purified MS2-GFP proteins. An important feature of the FCS technique is that it provides, in real time, concentrations and diffusion coefficients of fluorescent molecules either in vitro or within individual living cells (9, 16, 17). When the MS2-GFP fusion protein binds to the specific ms2-RNA, the RNA-protein complex diffuses through the detection volume slower than free MS2-GFP; the decay of the associated autocorrelation function is then delayed, reflecting the slower diffusion of the RNA target decorated with the MS2-GFP molecule (Fig. 1 A). The ratio of bound to free MS2-GFP proteins is then determined by adjusting the fitting parameters of a known mathematical function, which models the autocorrelation function associated with the diffusion of the fluorescent molecules (16, 18). Under these conditions, a 2-s acquisition is typically sufficient to measure concentration and diffusion constants at a given time point (see the supporting information). The FCS measurements showed that when the MS2-GFP fusion protein binds to the specific RNA sequence, the RNA-protein complex diffuses approximately three times slower through the detection volume than free MS2-GFP fusion molecules (Fig. 1 A). Measurements of RNA concentrations by FCS and by ultraviolet absorbance (260 nm) exhibit a one-to-one ratio. This one-to-one linear relationship demonstrates that if the MS2-GFP proteins are in excess, all of the target RNA molecules are bound to the MS2-GFP proteins (Fig. 1B).
Next, we monitored in real time and within a single E. coli bacterium the dynamics of transcriptional response when cells were exposed to an environmental stimulus: an inducer of gene transcription, aTc. E. coli strain DH5αPRO was transformed with two plasmids (see Fig. 1C and the supporting information). The MS2-GFP protein (19) was preexpressed from a LacI-controlled promoter. The inducible transcription of a short synthetic gene, encoding for two ms2-RNA-binding sites downstream of a ribosome binding site, was controlled by a tetR-regulated promoter (8). We determined that the diffusion time of MS2-GFP associated with the transcribed RNA was ≈30 times longer than free MS2-GFP (Fig. 1D). This large increase in diffusion time was mainly due to the interactions between the RNA transcripts and a cellular 70S ribosome (see the supporting information). The large difference in diffusion time between free and bound MS2-GFP was used to make RNA concentration measurements more sensitive. The diameter of the illumination spot defining the detection volume is approximately one-third of the cell body length, and we found that the measured ms2-RNA concentration was independent of the location of the spot along the cell body (see the supporting information).
Cells were mixed in a growth medium with low-melting point agarose and immobilized on the surface of microscope slides coated with an antibody raised against bacterial flagella. This attachment allows bacteria to grow and to divide. We monitored the variation of ms2-RNA concentration as a function of time within a single bacterium exposed to a steady level of inducer (Fig. 2A). Immediately after induction, the concentration of this specific RNA transcript rose sharply and peaked at ≈20 min. After this time point, the concentration of the ms2-RNA transcript unexpectedly decreased, and it eventually dropped to its initial preinduction level. Once the cell divided, another pulse of transcription coinciding with the cell cycle was observed. We induced the cells immediately after their first division to compare the transcriptional response from different cells at the same stage of their cell cycle. We found that the initial transcriptional responses to a fixed concentration of inducer ([aTc] = 400 ng/ml) were all synchronized among cells. However, as cells divided again, with asynchronous cell cycles, transcriptional responses occurred at different time points across the population (Fig. 2B).
Fig. 2.

Real-time concentration profiles of specific RNA transcripts from an inducible tet promoter in a single living E. coli cell. (A) ms2-RNA concentration after induction as a function of time within a single dividing cell. Arrows indicate the cell followed during measurements. After the first division (0 min), the cell was exposed to a steady level of inducer (400 ng/ml aTc). Cell division is marked with a horizontal bar. (B) ms2-RNA concentration profiles from six individual cells induced as in A; uninduced cell profile is shown in gray. Cell division events are marked with horizontal bars. Error bars in A and B were obtained from the uncertainties in the fitting parameters of the autocorrelation function (see Materials and Methods). (C) Mean concentration profiles of ms2-RNA from 14 individual cells induced with 400 ng/ml aTc. Error bars represent the standard deviations from the ms2-RNA concentration distributions across a population of 14 cells at a given time point.
The standard assumption emerging from population measurements has been that cells exposed to steady levels of inducer would produce a steady transcription activity over a period much larger than the typical 20 min observed in our experiment (20). When 14 single-cell profiles were averaged together, after a transient peak of ≈20 min (due to the initial synchronization), the expected steady transcription activity established by population measurements was recovered (Fig. 2C). Conversely, real-time RNA profiling within single cells reveals that drug-inducible transcription exhibits an unexpected pulsating behavior coinciding with the cell cycle. Such transcriptional behavior could not be observed from ensemble measurements obtained from indiscriminately combining single-cell profiles. Interestingly, in a population of asynchronous cells, such a pulsating transcription observed at the single-cell level may also constitute an additional source of global noise in inducible genetic systems. Other sources of variability in simple transcriptional systems were recently identified by monitoring the expression level of GFP and its derivatives across a population of individual cells (21-26).
Because aTc is known to be a good substrate for efflux pumps (27-29), we also characterized the transcription dynamics from an efflux pump-deleted E. coli strain. Deleting efflux pumps modifies the effective permeability to aTc of the cell and therefore affects the effective intracellular concentration of inducer (28). We performed RNA profiling of the ΔAcrAB mutant strain, Frag1A (see Materials and Methods), in which the AcrAB-TolC multidrug efflux pump system, a key factor for multidrug resistance, was deleted (28, 29). This mutant strain was transformed with the dual plasmid system, and the transcription of the ms2-RNA gene in response to a steady level of aTc inducer was monitored. We found that after induction with aTc (400 ng/ml), the concentration of induced ms2-RNA transcripts from ΔAcrAB mutant cells exceeded the one obtained from the wild-type cells. Moreover, the concentration of induced ms2-RNA transcripts reached a steady-state value across generations (see Fig. 3A and the supporting information).
Fig. 3.

Inducible transcription in deleted acrAB efflux pump-mutant Frag1A cells. (A) Concentration profiles of ms2-RNA transcripts controlled from an inducible tet promoter in a dividing Frag1A mutant cell (ΔacrAB) without aTc (gray) and with 400 ng/ml aTc (black). Wild-type Frag1B cell with 400 ng/ml aTc is shown in dark gray. Cell division is marked with horizontal bars. Cells were induced immediately after their first division (time point 0 min). Error bars represent uncertainties in the fit parameters extracted from the autocorrelation function (see Materials and Methods). (B) Concentration of ms2-RNA transcripts expressed from an inducible tet promoter in a population of wild-type Frag1B cells (dark gray) and in a population of Frag1A (ΔacrAB) mutant cells (black). Cells were grown at constant density (A600 = 0.2) in M9 medium at 30°C. ms2-RNA concentration was determined with a primer extension assay. The error bars represent the distribution of ms2-RNA concentration in triplicate experiments.
The fact that the induced RNA concentration reaches a steady plateau in mutant cells indicates that the AcrAB-TolC multidrug efflux system contributes to the observed pulsating dynamics of the inducible tet promoter in wild-type cells (30). It is also conceivable that the observed pulsating behavior in wild-type cells may be caused by periodic RNA degradation. However, in the ΔAcrAB mutant study, the RNA levels reached a steady plateau, suggesting that RNA degradation was constant throughout the cell cycle. Real-time RNA profiling in a single bacterium shows that the genetic deletion of multidrug resistance determinants, such as efflux mechanisms, can alter the dynamics of drug-inducible transcriptional circuits in response to environmental stimuli (31). In contrast, using primer extension, we monitored as a function of time the RNA concentration of transcripts from a population of ΔAcrAB mutant and wild-type cells grown at constant cell density (see the supporting information). We found that at the population level, ΔAcrAB mutant cells express twice the level of ms2-RNA transcripts compared with wild-type cells but that both ΔAcrAB mutant and wild-type cells exhibit similar transcriptional dynamics that leveled off to a steady plateau (Fig. 3B).
This study demonstrates that the combination of a FCS-based apparatus with a synthetic modular reporter system constitutes a noninvasive and efficient means by which to profile the transcriptional activity of a specific promoter as a function of time in a single prokaryotic cell. In particular, real-time RNA profiling in single cells becomes necessary to characterize genetic modifications that affect the dynamics of transcriptional responses not accessible to ensemble measurements or to single-cell sorting experiments. Labeling ms2-RNA with MS2-GFP fusion protein is a general framework that can be extended to other specific RNA-binding proteins. The use of other RNA-binding proteins, such as the spliceosomal U1A protein (32, 33), fused to fluorescent proteins of various colors would allow us to simultaneously measure transcriptional dynamics from more than one genetic element within the same cell. Most importantly, this plasmid system can be used in prokaryotes to report in real time the activity of any promoter controlling the expression of the recognition RNA sequence for RNA-binding proteins fused to GFP. As GFP made possible noninvasive studies of protein concentration in individual living cells (9), the reporter system presented in this work provides a noninvasive tool with which to measure the dynamics of various RNA species in vivo at the level of a single cell. Real-time RNA profiling should be applicable for a wide range of quantitative single-cell studies of transcriptional networks and noncoding RNAs.
Supplementary Material
Acknowledgments
We thank Y. L. Chan for help and advice with the ribosome association assays, W. Epstein (University of Chicago) for the gifts of the Frag1A and Frag1B mutant strains, T. J. Silhavy for suggesting the use of phage coat proteins, H. Nikaido (University of California, Berkeley) for the KZM120 strain, K. Bloom (University of North Carolina, Chapel Hill) for the pCP-GFP and pIIIMS2-2 plasmids, D. Peabody (University of New Mexico, Albuquerque) for the pCT119 dlFG plasmid, and T. Griggs for helpful comments on the manuscript. This work was supported by a National Institutes of Health training grant (to T.T.L.) and by the Materials Research Science and Engineering Center and Institute for Biophysical Dynamics seed fund (to S.H.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: FCS, fluorescence correlation spectroscopy; aTc, anhydrotetracycline.
References
- 1.Brenner, S., Meselson, M. & Jacob, F. (1961) Nature 190, 576-585. [DOI] [PubMed] [Google Scholar]
- 2.Eberwine, J., Yeh, H., Miyashiro, K., Cao, Y., Nair, S., Finnell, R., Zettel, M. & Coleman, P. (1992) Proc. Natl. Acad. Sci. USA 89, 3010-3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janicki, S. M., Tsukamoto, T., Salghetti, S. E., Tansey, W. P., Sachidanandam, R., Prasanth, K. V., Ried, T., Shav-Tal, Y., Bertrand, E., Singer, R. H. & Spector, D. L. (2004) Cell 116, 683-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Golding, I. & Cox, E. C. (2004) Proc. Natl. Acad. Sci. USA 101, 11310-11315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Korobkova, E., Emonet, T., Vilar, J. M., Shimizu, T. S. & Cluzel, P. (2004) Nature 428, 574-578. [DOI] [PubMed] [Google Scholar]
- 6.Paulsson, J. (2004) Nature 427, 415-418. [DOI] [PubMed] [Google Scholar]
- 7.Rauer, B., Neumann, E., Widengren, J. & Rigler, R. (1996) Biophys. Chem. 58, 3-12. [DOI] [PubMed] [Google Scholar]
- 8.Lutz, R. & Bujard, H. (1997) Nucleic Acids Res. 25, 1203-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cluzel, P., Surette, M. & Leibler, S. (2000) Science 287, 1652-1655. [DOI] [PubMed] [Google Scholar]
- 10.Peabody, D. S. & Ely, K. R. (1992) Nucleic Acids Res. 20, 1649-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore, P. B. (1998) Annu. Rev. Biophys. Biomol. Struct. 27, 35-58. [DOI] [PubMed] [Google Scholar]
- 12.Bertrand, E., Chartrand, P., Schaefer, M., Shenoy, S. M., Singer, R. H. & Long, R. M. (1998) Mol. Cell 2, 437-445. [DOI] [PubMed] [Google Scholar]
- 13.Ni, C. Z., Syed, R., Kodandapani, R., Wickersham, J., Peabody, D. S. & Ely, K. R. (1995) Structure 3, 255-263. [DOI] [PubMed] [Google Scholar]
- 14.Peabody, D. S. & Al-Bitar, L. (2001) Nucleic Acids Res. 29, E113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Magde, D., Elson, E. L. & Webb, W. W. (1974) Biopolymers 13, 29-61. [DOI] [PubMed] [Google Scholar]
- 16.Webb, W. W. (2001) Appl. Opt. 40, 3969-3983. [DOI] [PubMed] [Google Scholar]
- 17.Rigler, R. (1995) J. Biotechnol. 41, 177-186. [DOI] [PubMed] [Google Scholar]
- 18.Eigen, M. & Rigler, R. (1994) Proc. Natl. Acad. Sci. USA 91, 5740-5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beach, D. L., Salmon, E. D. & Bloom, K. (1999) Curr. Biol. 9, 569-578. [DOI] [PubMed] [Google Scholar]
- 20.Jacob, F. & Monod, J. (1961) J. Mol. Biol. 3, 318-356. [DOI] [PubMed] [Google Scholar]
- 21.Elowitz, M. B., Levine, A. J., Siggia, E. D. & Swain, P. S. (2002) Science 297, 1183-1186. [DOI] [PubMed] [Google Scholar]
- 22.Ozbudak, E. M., Thattai, M., Kurtser, I., Grossman, A. D. & van Oudenaarden, A. (2002) Nat. Genet. 31, 69-73. [DOI] [PubMed] [Google Scholar]
- 23.Blake, W. J., Kaern, M., Cantor, C. R. & Collins, J. J. (2003) Nature 422, 633-637. [DOI] [PubMed] [Google Scholar]
- 24.Raser, J. M. & O'Shea, E. K. (2004) Science 304, 1811-1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pedraza, J. M. & van Oudenaarden, A. (2005) Science 307, 1965-1969. [DOI] [PubMed] [Google Scholar]
- 26.Rosenfeld, N., Young, J. W., Alon, U., Swain, P. S. & Elowitz, M. B. (2005) Science 307, 1962-1965. [DOI] [PubMed] [Google Scholar]
- 27.Okusu, H., Ma, D. & Nikaido, H. (1996) J. Bacteriol. 178, 306-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li, X. Z. & Nikaido, H. (2004) Drugs 64, 159-204. [DOI] [PubMed] [Google Scholar]
- 29.Yu, E. W., McDermott, G., Zgurskaya, H. I., Nikaido, H. & Koshland, D. E., Jr. (2003) Science 300, 976-980. [DOI] [PubMed] [Google Scholar]
- 30.Barbosa, T. M. & Levy, S. B. (2000) J. Bacteriol. 182, 3467-3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guet, C. C., Elowitz, M. B., Hsing, W. & Leibler, S. (2002) Science 296, 1466-1470. [DOI] [PubMed] [Google Scholar]
- 32.De Guzman, R. N., Turner, R. B. & Summers, M. F. (1998) Biopolymers 48, 181-195. [DOI] [PubMed] [Google Scholar]
- 33.Oubridge, C., Ito, N., Evans, P. R., Teo, C. H. & Nagai, K. (1994) Nature 372, 432-438. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.