

Abstract
Recently, research into the oncogenic driver genes associated with non-small cell lung cancer (NSCLC) has advanced significantly, leading to the development and clinical application of an increasing number of approved therapeutic agents. Among these, small molecule inhibitors that target mesenchymal-epithelial transition (MET) have demonstrated successful application in clinical settings. Currently, three categories of small molecule MET inhibitors, characterized by distinct binding patterns to the MET kinase region, have been developed: types Ia/Ib, II, and III. This review thoroughly examines MET’s structure and its crucial role in NSCLC initiation and progression, explores discovery strategies for MET inhibitors, and discusses advancements in understanding resistance mechanisms. These insights are anticipated to enhance the development of a new generation of MET inhibitors characterized by high efficiency, selectivity, and low toxicity, thereby offering additional therapeutic alternatives for patients diagnosed with NSCLC.
Keywords: NSCLC, MET inhibitors, drug discovery, structure-activity relationships, targeted therapy
1 Introduction
According to data from the International Agency for Research on Cancer (IARC), the global incidence of newly diagnosed cancer cases reached 19.6 million in 2022. Concurrently, cancer-related mortality accounted for approximately 9.7 million deaths worldwide. These statistics underscore the emergence of cancer as a critical public health issue on a global scale (Bray et al., 2024). In 2022, lung cancer accounted for over 12% of all new cancer diagnoses, with an estimated 2.48 million new cases reported, and it was associated with approximately 1.81 million deaths globally (Abu Rous et al., 2023). As shown in Figure 1, Lung cancer is a diverse group of tumors mainly categorized into two types based on cell origin: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC). Notably, NSCLC represents the predominant variant, comprising approximately 85%–90% of all lung cancer cases. Due to the often insidious nature of the early clinical manifestations of NSCLC, clinical data suggests that approximately 75% of patients are diagnosed at an advanced stage of the disease. In patients with advanced NSCLC, surgical intervention is considered the primary treatment modality. However, approximately 40% of NSCLC patients experience recurrence and metastasis following complete tumor resection, which contributes to a diminished 5-year survival rate (Imyanitov et al., 2021). Given the restricted applicability of surgical intervention for patients with advanced NSCLC, clinical practice frequently resorts to chemotherapy and radiation therapy utilizing platinum-based agents as the primary treatment approach for this patient population. Nevertheless, the prognosis for NSCLC patients remains suboptimal due to constraints on radiation dosage, the adverse side effects associated with chemotherapy, and the emergence of drug resistance (Wang L. et al., 2023; Wang N. et al., 2023). Consequently, it is imperative to identify and investigate treatment strategies that exhibit enhanced selectivity and a diminished occurrence of adverse reactions. The advent of genetic testing technology is particularly promising, as it facilitates the identification of specific targets and the development of targeted therapies, thereby heralding a new era in the management of NSCLC.
FIGURE 1.

The categorization of lung cancer and the primary driver mutation types associated with NSCLC.
Targeted therapy for NSCLC serves as a prominent illustration of precision medicine, wherein distinct subtypes characterized by specific driver mutations inform the development of targeted treatment strategies (Salgia et al., 2021). The National Comprehensive Cancer Network (NCCN) guidelines delineate specific clinically relevant genes, including the epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK), c-ros oncogene 1 tyrosine kinase (ROS1), v-Raf murine sarcoma viral oncogene homolog B1 (BRAF), human epidermal growth factor receptor 2 (HER2), and mesenchymal-epithelial transition factor (MET), along with other established and frequently implicated driver genes (Figure 1) (Alexander et al., 2020). In the past decade, significant advancements have been made in the research of driver genes associated with NSCLC. The clinical application of a range of targeted therapies directed at key driver genes has resulted in improved survival outcomes for patients with NSCLC. The MET gene serves as a critical oncogenic driver in NSCLC and is recognized as a significant therapeutic target, following EGFR, ALK, and ROS1 (Duma et al., 2019). The advancement of pharmacological agents aimed at MET gene mutations has attracted considerable interest among medicinal chemists. In recent years, the FDA has granted approval for small molecule inhibitors, including capmatinib, savolitinib, and tepotinib, for the treatment of patients with metastatic NSCLC who possess MET exon 14 skipping mutations (Wu and Lin, 2022).
This review offers a thorough overview of the structure, biological functions, and dysregulation of the MET receptor. The focus is particularly on recent advancements in the development of MET inhibitors, in addition to elucidating the mechanisms underlying resistance to these inhibitors. Furthermore, the review discusses clinical strategies for combination therapies involving MET inhibitors and explores innovative treatment modalities aimed at targeting MET, including the use of PROTAC technology. A comprehensive understanding of the advancements in MET inhibitors for the treatment of NSCLC may enhance the discovery and development of novel, more effective, and less toxic inhibitors.
2 HGF/MET signaling pathway
The MET proto-oncogene, located on the long arm of chromosome 7 in humans (7q21-31), encodes a protein that extends over 120 kilobases and consists of 21 exons along with 20 introns. This protein functions as a transmembrane receptor and is classified within the receptor tyrosine kinase (RTK) family (Koch et al., 2020). Structurally, the MET receptor is composed of several distinct domains, including an extracellular domain, a transmembrane domain, and an intracellular region that encompasses a juxtamembrane domain, a catalytic kinase domain, and a carboxy-terminal tail (Figure 2). The extracellular domain of the MET receptor comprises three distinct structural domains: the N-terminal fragment, which adopts a conformation resembling a large signaling proten-like (SEMA) domain that encompasses the entirety of the alpha subunit and a portion of the beta subunit; the plexin-semaphorin-integrin (PSI) domain, situated downstream of the SEMA domain and characterized by the presence of four disulfide bonds; and the PSI domain, which is linked to four immunoglobulin-plexin-transcription factor (IPT) domains (Pothula et al., 2020).
FIGURE 2.

Schematic representation of the MET protein domain structure. Mechanisms of multiple cellular processes regulated by HGF/Met signaling.
Under physiological conditions, mature hepatocyte growth factor (HGF) demonstrates a high-affinity interaction with the MET receptor, specifically within the extracellular ligand-binding domain (Yin et al., 2019). This interaction initiates the phosphorylation of tyrosine residues Tyr1234 and Tyr1235 located in the tyrosine kinase domain. The phosphorylation of these residues induces a conformational change in the receptor, which subsequently facilitates the phosphorylation of two additional docking tyrosine residues, Tyr1349 and Tyr1356, at the carboxy terminus (Mulcahy et al., 2020). The activation of these residues enables the recruitment of various downstream signaling molecules, thereby contributing to a homologous signaling cascade. Upon the activation of the HGF/MET signaling pathway, the downstream signaling cascades, specifically the PI3K/AKT/mTOR and RAS/RAF pathways, are subsequently activated, thereby facilitating a range of physiological functions (Figure 2). These pathways are essential to a range of cellular processes, encompassing proliferation, migration, inhibition of apoptosis, and angiogenesis, and function within both cytoplasmic and nuclear compartments (Cheng and Guo, 2019).
3 MET dysregulation in NSCLC
The dysregulation of the HGF/MET signaling pathway, attributable to exon 14 skipping mutations, amplifications, overexpression, and fusion events, plays a significant role in the initiation and progression of various malignant tumors (Remon et al., 2023). The extracellular domain encoded by exon 14 of the MET gene serves as a crucial negative regulatory region, playing a significant role in the ubiquitination and subsequent degradation of the MET protein (Guo et al., 2020). The intronic region adjacent to exon 14, along with exon 14 itself, or a complete genomic deletion of exon 14, disrupts the splicing mechanism of MET gene transcription. This deletion inhibits the processes of ubiquitination and endocytosis, consequently impairing the degradation of the MET receptor protein (Drusbosky et al., 2021). Such impairment leads to excessive activation of MET-mediated signaling pathways, thereby promoting cellular proliferation and tumor growth (Figure 3). In NSCLC, the overall incidence rate of MET exon 14 skipping mutations varies between 2.5% and 6.0%. These mutations do not co-occur with other driver genes such as EGFR and ALK, suggesting that they function as independent oncogenic drivers. Nonetheless, MET exon 14 skipping mutations can coexist with MET gene amplification and protein overexpression (Huang et al., 2020).
FIGURE 3.

The involvement of MET exon 14 in NSCLC is illustrated through a schematic representation of the genomic regions adjacent to MET exon 14. The exclusion of MET exon 14 results in the upregulation of MET signaling pathways.
MET gene amplification is characterized by an increase in the copy number of the MET gene, encompassing both whole chromosome duplication and localized gene duplication (Tyler et al., 2022). Specifically, whole chromosome duplication refers to the presence of multiple copies of chromosome 7 within tumor cells. The amplification of the MET gene frequently occurs in conjunction with mutations in other oncogenes, including EGFR and KRAS (Yang et al., 2024). Current research indicates that MET amplification may not serve as a primary driver gene in NSCLC. Furthermore, a significant correlation exists between MET amplification and the activation of other driver genes, such as EGFR and KRAS, implying that MET amplification may represent one of the mechanisms contributing to acquired resistance in NSCLC characterized by EGFR mutations (Wang et al., 2019). Research has demonstrated that 15%–20% of patients with acquired resistance to EGFR inhibitors exhibit detectable amplification of the MET gene. Moreover, MET gene amplification is frequently associated with a poorer prognosis in patients with NSCLC (Shi et al., 2016).
Abnormal overexpression of either MET or HGF may result in prolonged activation of these pathways, potentially contributing to the initiation and progression of tumors (Chen et al., 2022). Research has demonstrated that the overexpression of HGF and its receptor, Met, in NSCLC is significantly correlated with the formation of lymphatic vessels (Kajiya et al., 2005). In pulmonary adenocarcinoma, the prevalence of MET overexpression can reach up to 65%; however, only 10% of cases exhibiting MET overexpression also present with mutations in the MET gene (Wang N. et al., 2024). Consequently, it is plausible that MET overexpression may not serve as the primary oncogenic driver, but rather as a secondary event arising from the activation of alternative driver genes, thereby facilitating tumor growth (Wang N. et al., 2023).
The advancement of genomics and proteomics has facilitated a transition in the treatment of NSCLC towards a personalized approach informed by the identification of driver genes (Bi et al., 2023). Over the past decade, numerous therapeutic strategies aimed at modulating aberrant HGF/MET signaling pathways have been documented, primarily encompassing three approaches: i) the utilization of biological agents or neutralizing antibodies to obstruct the extracellular interaction between MET and HGF. Especially, HGF antagonists, like liromalimumab and pemigatinib, competitively bind to the MET receptor, blocking its interaction with HGF without activating the receptor (Kim and Kim, 2017). The anti-MET monoclonal antibody Onartuzumab possesses the ability to inhibit the formation of the MET dimer, thereby attenuating downstream signaling and intracellular signal transduction, which ultimately contributes to the inhibition of tumor growth (Kim et al., 2021); ii) Small molecule inhibitors targeting MET kinase bind to the catalytic core region of MET, thereby inhibiting the phosphorylation of intracellular tyrosine kinases and disrupting the transmission of downstream signaling pathways (Spagnolo et al., 2023); iii) the inhibition of downstream signaling pathway proteins associated with HGF/MET to disrupt signal transduction (He et al., 2021). In recent decades, significant progress has been made in the development of small molecule inhibitors targeting the MET kinase, leading to the advancement of numerous candidates into clinical trials, several of which have subsequently received market approval (Ye et al., 2021). The following chapters will focus on the advancements in small molecule inhibitors specifically aimed at the tyrosine kinase domain of MET.
4 Small-molecule inhibitors of MET
To date, numerous MET inhibitors have been documented for the clinical management of NSCLC (Table 1) (Leonetti et al., 2019). Existing MET inhibitors can be categorized into three distinct classes based on their unique binding modes with biological target. Type I inhibitors interact with the active conformation of the kinase within the ATP binding pocket, further subdivided into non-selective type Ia and selective type Ib inhibitors. Type II inhibitors engage with the inactive conformation of the kinase within the ATP pocket. Finally, Type III inhibitors bind outside the ATP pocket, employing a non-ATP competitive allosteric mechanism (Mazieres et al., 2023). In light of the increasing prevalence of drug resistance, there has been a notable surge in research focused on novel MET inhibitors, complementing the previously discussed clinically available inhibitors (Jørgensen et al., 2024). The subsequent sections of this chapter will offer a comprehensive overview of the advancements in research and the structure-activity relationships associated with representative MET inhibitors from the three categories previously mentioned.
TABLE 1.
Overview of clinical approved MET inhibitors for the treatment of NSCLC.
| Types of MET inhibitors | Drugs | Chemical structures | Phase | Clinical trial number | Status |
|---|---|---|---|---|---|
| Ia | Crizotinib (1) |
|
III |
NCT02767804 NCT04632758 NCT03052608 NCT06569420 NCT02838420 NCT02075840 |
Active |
| I/II |
NCT01712217 NCT01970865 |
Completed | |||
| IV | NCT03672643 | Recruiting | |||
| II |
NCT01945021 NCT02183870 NCT03088930 NCT00932451 |
Completed | |||
| III |
NCT06140836 NCT06082635 NCT04603807 |
Recruiting | |||
| I/II | NCT05384626 | Recruiting | |||
| II | NCT01500824 | Withdrawn | |||
| IV | NCT05160922 | Active | |||
| I |
NCT01579994 NCT01998126 |
Completed | |||
| I/II | NCT02584634 | Terminated | |||
| II | NCT04322578 | Recruiting | |||
| III |
NCT02737501 NCT01639001 NCT00932893 NCT01828112 |
Completed | |||
| Ib | Capmatinib (2) |
|
II |
NCT04460729 NCT05567055 NCT03240393 |
Withdrawn |
| II |
NCT02414139 NCT03647488 NCT02750215 NCT01610336 NCT02276027 |
Completed | |||
| II | NCT04677595 | Active | |||
| IV | NCT05110196 | Recruiting | |||
| I/II | NCT05488314 | Active | |||
| III | NCT04427072 | Completed | |||
| III | NCT04816214 | Terminated | |||
| I |
NCT01911507 NCT01324479 |
Completed | |||
| II |
NCT05642572 NCT03040973 |
Recruiting | |||
| I | NCT02468661 | Terminated | |||
| Savolitinib (3) |
|
III | NCT04923945 | Recruiting | |
| II | NCT02117167 | Completed | |||
| II | NCT04322890 | Recruiting | |||
| II |
NCT03778229 NCT05163249 NCT03833440 NCT04606771 NCT03944772 |
Active | |||
| III | NCT05261399 | Recruiting | |||
| II | NCT04322578 | Recruiting | |||
| I | NCT02374645 | Completed | |||
| I | NCT02143466 | Active | |||
| SAR125844 (4) |
|
I | NCT01391533 | Completed | |
| I | NCT01657214 | Completed | |||
| II | NCT02435121 | Completed | |||
| Bozitinib (5) |
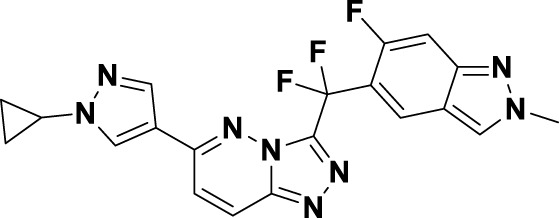
|
II | NCT03175224 | Recruiting | |
| I/II |
NCT04743505 NCT06343064 |
Recruiting | |||
| I | NCT02896231 | Completed | |||
| I/II | NCT03655613 | Terminated | |||
| Tepotinib (6) |
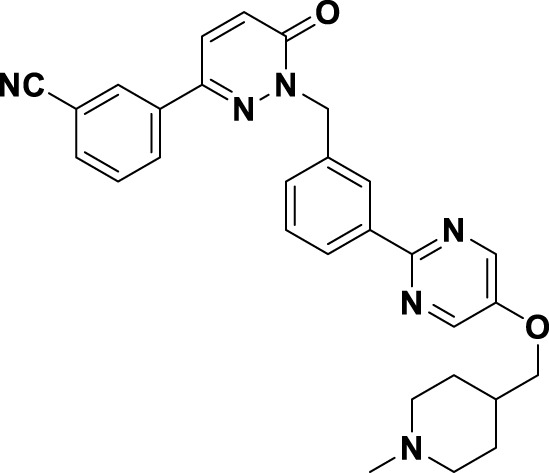
|
I/II | NCT04739358 | Terminated | |
| I/II | NCT06083857 | Recruiting | |||
| II |
NCT02864992 NCT03940703 |
Active | |||
| I/II | NCT01982955 | Completed | |||
| ABN401 (7) |
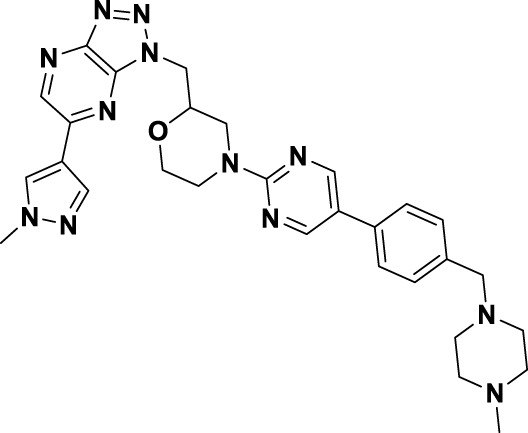
|
II | NCT05541822 | Recruiting | |
| I | NCT04052971 | Recruiting | |||
| I | NCT05248074 | Completed | |||
| Elzovantinib (8) |
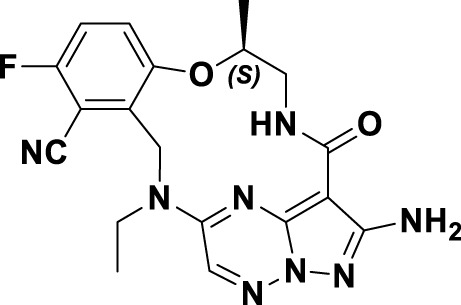
|
I/II | NCT03993873 | Active | |
| Glumetinib (9) |
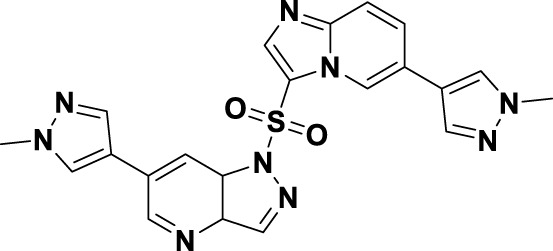
|
I/II | NCT04270591 | Unknown | |
| II | Cabozantinib (10) |
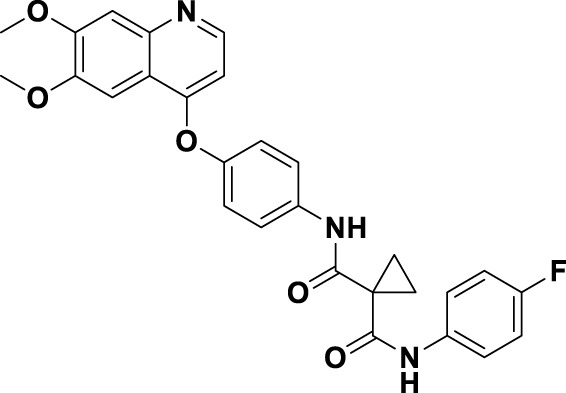
|
II | NCT02132598 | Terminated |
| I/II | NCT00596648 | Completed | |||
| I | NCT04173338 | Terminated | |||
| II |
NCT01639508 NCT05613413 |
Recruiting | |||
| II |
NCT00940225 NCT05200143 NCT03468985 NCT01866410 NCT02795156 |
Completed | |||
| III | NCT04471428 | Active | |||
| I/II | NCT04151563 | Withdrawn | |||
| II |
NCT04310007 NCT01708954 |
Active | |||
| Foretinib (11) |
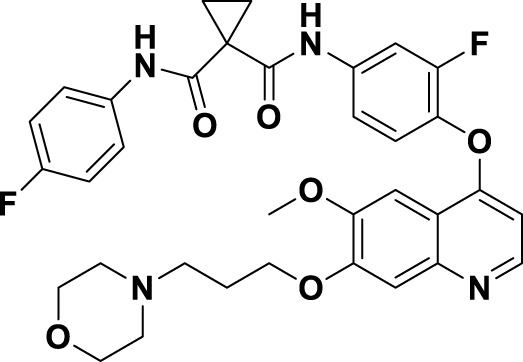
|
II | NCT02034097 | Withdrawn | |
| I/II | NCT01068587 | Completed | |||
| Glesatinib (12) |
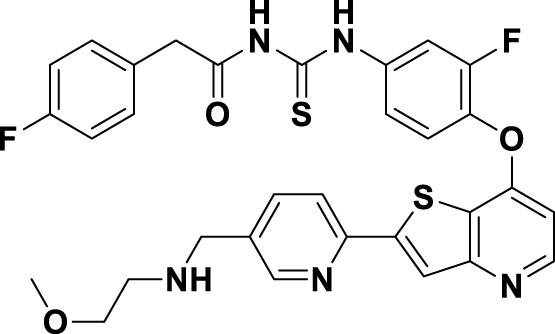
|
II | NCT02954991 | Terminated | |
| Merestinib (13) |
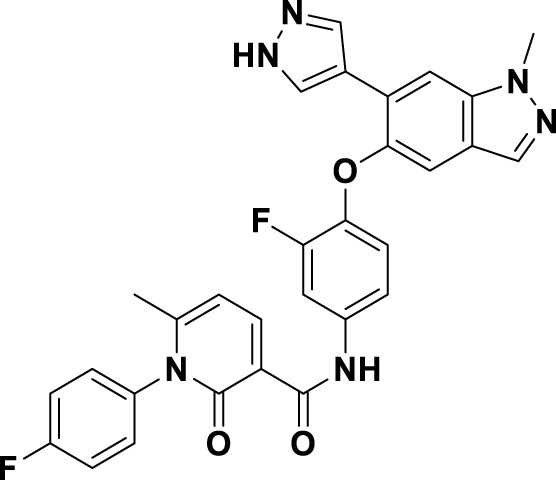
|
II | NCT02920996 | Terminated | |
| Ningetinib (14) |
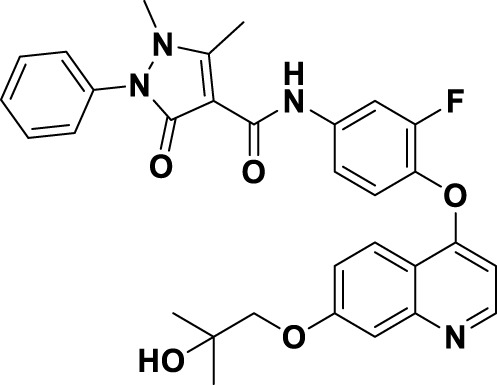
|
II | NCT03838848 | Terminated | |
| BMS-777607 (15) |
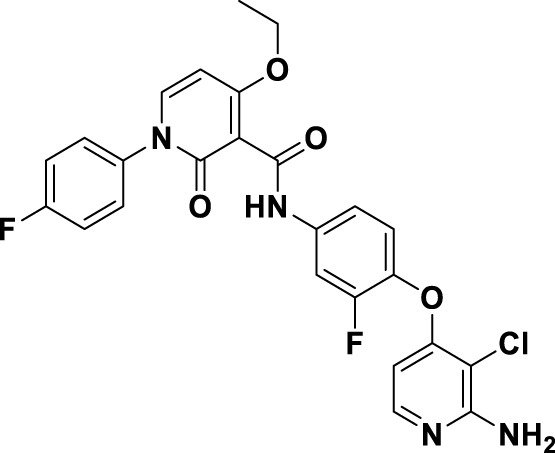
|
I/II | NCT00605618 | Completed | |
| I | NCT01721148 | Completed | |||
| III | Tivantinib (16) |
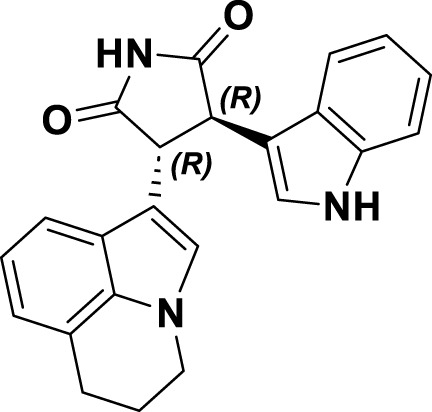
|
III | NCT01377376 | Terminated |
4.1 Type Ia inhibitors of MET
Type I small molecule inhibitors that target MET engage the active conformation of the protein’s ATP-binding pocket in a ‘U-shaped’ orientation, commonly referred to as ‘DFG-in’ (Sun et al., 2019). Type I inhibitors are further categorized into subclasses Ia and Ib, depending on their interaction with the G1163 residue, which is solvent-exposed. Notably, Type Ia inhibitors interact hydrophobically with the G1163 residue, whereas type Ib inhibitors do not (Zhang et al., 2019).
Crizotinib (1), a prototypical type Ia inhibitor of MET, was identified from the precursor molecule SU5402 (17) via extensive structural modifications (Cui et al., 2011). Figure 4 demonstrate that strategically adding groups at the C5 position of the indolone and on the pyrrole ring of 17 can greatly improve the molecule’s inhibitory activity against the MET kinase, leading to the development of compounds SU11271 (18), SU11606 (19), and SU11274 (20). The substitution of the N-methylsulfonamide in compound 20 with a more flexible methylenesulfonyl group led to the synthesis of PHA-665752 (21), which demonstrated low nanomolar cellular antiproliferative and inhibitory activities against the MET kinase (Michaelides et al., 2023). The co-crystallization of 21 with the MET kinase (PDB ID: 2KWM) revealed that the benzylsulfonate moiety underwent self-folding to engage in interactions with the Y1230 residue, rather than directly associating with the hydrophobic region of the MET kinase. However, the poor pharmacokinetics of 21 hindered its clinical development, requiring structural optimization. Based on the binding mode analysis of 21 with MET, derivatives were identified by substituting its core with pyrido [2,3-b]indole or aminopyridine. Compound 22, featuring a 2-aminopyridine core, more effectively occupies the adenine pocket of the MET kinase and establishes a hydrogen bond network with residue M1160 compared to 21. Researchers found that the 2,6-dichlorophenoxy substituent significantly improved π-π interactions with the Y1230 residue in the A ring of the MET structure through detailed structure-activity relationship studies. Concurrently, the incorporation of a hydrophilic group at the tail region of the molecule further improved both the potency and pharmacokinetic properties, ultimately leading to the development of 1. Co-crystal analysis of compound 1 with MET kinase (PDB ID: 2WGJ) shows that the 2-aminopyridine forms hydrogen bonds with residues P1158 and M1160, while the 2,6-dichloro-3-fluorophenyl group interacts with Y1230 through a π-π interaction. The α-methyl group in the linkage between the 2,6-dichloro-3-fluorophenyl moiety and the parent nucleus facilitates interactions within the hydrophobic cavity constituted by residues V1092, L1157, K1110, and A1108. Additionally, it enhances the rigidity of the substituted phenyl group. This increased rigidity facilitates the establishment of additional electrostatic interactions between the substituted phenyl group and D1222. Furthermore, 1 shows a conserved hydrophobic interaction with M1211, potentially explaining its limited kinase selectivity.
FIGURE 4.

The identification and SAR studies of the pyrrole indolinone derivatives 17–21, 2-aminopyridine derivative 22, and crizotinib (1). Mauve dashed lines represent hydrogen bonds with key amino acids, and faint gray dashed lines indicate π-π interactions.
To date, numerous medicinal chemists have conducted comprehensive investigations into the structure-activity relationship of 1 (Yadav et al., 2024). The specific findings are as follows: i) Substituting 2-aminopyridine with alternative nitrogen-containing aromatic heterocycles, such as quinoline, pyridine, and 3-amino-pyrazine, results in a reduction of biological activity; ii) Substituting the oxygen atom in the linking region with either a sulfur atom or an amide bond leads to a decrease in potency. This reduction may be attributed to the alteration in the linking region, which impedes the formation of interactions between the substituted phenyl group and the methionine residue of the protein, thereby affecting the overall efficacy of the molecule; iii) The judicious substitution of hydrophilic groups can effectively sustain biological activity.
4.2 Type Ib inhibitors of MET
In contrast to type Ia inhibitors, type Ib inhibitors exhibit penetration into the solvent-accessible region adjacent to the residues D1222, Y1230, and R1228, thereby establishing a more substantial π-π interaction with Y1230 (Fujino et al., 2020). This interaction is instrumental in enhancing selectivity and increasing the inhibitory activity against the MET kinase. As shown in Table 1 (Rocco et al., 2024), several type Ib inhibitors targeting the MET kinase have been approved for clinical use, including capmatinib (2), savolitinib (3), SAR125844 (4), bozitinib (5), tepotinib (6), ABN401 (7), elzovantinib (8), and glumetinib (9).
Compound 2, recognized as the first highly selective MET inhibitor, has been granted approval for the treatment of metastatic NSCLC associated with MET exon 14 skipping mutations (Wolf et al., 2020). In vitro studies demonstrate that 2 displays considerable efficacy against MET (IC50 = 0.13 nM), as well as against MET-mediated signal transduction and tumor activity at low nanomolar concentrations. Unfortunately, the specific drug discovery pathway leading to the development of 2 has not been publicly disclosed. Recent clinical studies have indicated that the objective response rate (rwORR) of capmatinib as a first-line treatment is 73.4%, while the disease control rate (rwDCR) is 95%. Additionally, the median time to treatment discontinuation (mTTD) is reported to be 19.1 months. Although the median progression-free survival (rwPFS) and median overall survival (mOS) have not yet been reached, the 18-month rates for progression-free survival and overall survival are 68% and 92.6%, respectively. The research has demonstrated that capmatinib, when administered as a first-line treatment for NSCLC with MET exon 14 skipping mutations, yields a superior tumor response, a reduced risk of disease progression, and an extended survival rate compared to regimens based on chemotherapy and immunotherapy (Wu Y. et al., 2021; Dong et al., 2022). As shown in Figure 5, several MET inhibitors utilizing quinoline groups as ligands for binding to the hinge region of the MET kinase have been documented, including PF-04217903 (26) (Zou et al., 2012) and PF-04254644 (27) (Cui et al., 2013). Initially, compound 22 was recognized as a selective Ib-type MET inhibitor. Subsequently, through the methylation and modification of the core structure of 22, a more potent MET inhibitor, designated as compound 23, was identified, exhibiting an IC50 value of 1.3 nM. The co-crystal structure of 23 in complex with MET (PDB ID: 3ZZE) demonstrates that the phenolic group within 23 can interact with the M1160 residue located in the hinge region of MET, while the core moiety establishes a robust π-stacking interaction with the critical residue Y1230. Subsequently, compound 24, which incorporates a tetracyclic aromatic framework, was designed based on the coplanar conformation of 23. However, the non-pharmacological properties of 24 indicated the need for further structural optimization. Consequently, the tetracyclic framework was deconstructed into various bicyclic frameworks, resulting in the synthesis of compound 25. Structure-activity relationship studies indicate that the electron-deficient characteristics of aromatic heterocyclic parent nuclei, which contain multiple nitrogen atoms, are significantly correlated with the interaction strength between MET inhibitors and the Y1230 residues. This relationship is closely linked to the efficacy and specificity of small-molecule ligands that interact with the MET receptor. Compound 25 was further optimized through the substitution of the phenol group with a quinoline moiety and the replacement of the p-fluorophenyl group with a pyrazolyl ethanol group, resulting in the synthesis of compounds 26 and 27, which exhibited enhanced enzyme inhibitory and cellular inhibitory activities. The co-crystal structure of 26 with MET kinase (PDB ID: 3ZXZ) reveals that the quinoline nitrogen forms a hydrogen bond with M1160, while the triazolopyrazinyl moiety participates in a π-π stacking interaction with Y1230. Furthermore, the N atom and -OH group of the pyrazole moiety in 26 are capable of forming a water-mediated polyhydrogen bond network with the MET kinase (Cui et al., 2012). Currently, 26 has progressed to phase I clinical trials for the treatment of advanced solid tumors (Parikh and Ghate, 2018). In contrast, 27, which shares structural similarities with 26, has demonstrated cardiac-related toxicity attributed to its insufficient selectivity for the phosphodiesterase family, thereby hindering its advancement in clinical development (Knauf et al., 2018).
FIGURE 5.

The identification and SAR studies of quinoline-based Type Ib inhibitors 26 and 27 of Met are presented. The essential amino acid residues that mediate interactions between the small molecule and the MET protein are depicted. Mauve dashed lines represent hydrogen bonds with key amino acids, and faint gray dashed lines indicate π-π interactions.
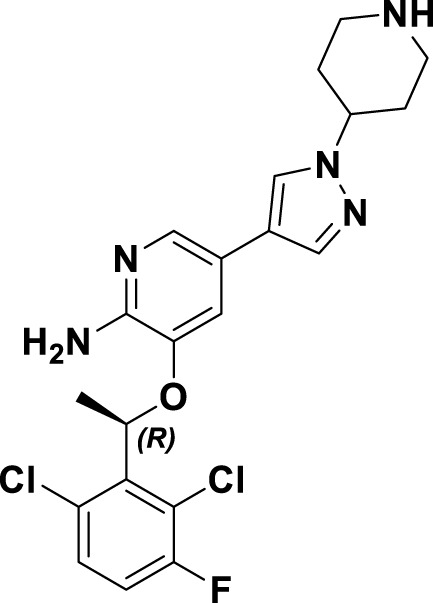
Savolitinib (3) is a strong MET kinase inhibitor belonging to the triazolopyrazine class. In 2021, marketing authorization has been approved in China for the therapeutic use in adult patients diagnosed with locally advanced or metastatic NSCLC exhibiting MET exon 14 skipping mutations (Pal et al., 2021). This authorization specifically pertains to individuals who have experienced disease progression following platinum-based chemotherapy or who are intolerant to standard platinum-based treatment regimens (Lu et al., 2021). At the molecular level, 3 demonstrates significant inhibitory activity against the MET kinase, exhibits exceptional kinase selectivity, and possesses a strong affinity for the kinase. In the realm of drug discovery, 3 was derived from compound 27 following multiple iterations of structural optimization (Figure 6). Notably, the phenolic group in compound 27 was substituted with a quinoline moiety, while the triazolopyridine group was replaced with a triazolopyrazine moiety, leading to the identification of 28. In comparison to 27, the enzyme inhibitory activity of 28 was significantly improved. Consequently, imidazopyridine derivatives were employed to optimize the quinoline moiety in 28, aiming to mitigate the potential toxicity and side effects associated with the quinoline group, which led to the identification of 29 (Baschnagel et al., 2021). The subsequent structural optimization process aimed to enhance cellular activity and pharmacokinetic properties by incorporating an (S)-methyl group at the methylene linkage of 29, ultimately yielding 3. The co-crystallization of 3 with MET (PDB code:6SDE) reveals that compound 3 adopts a U-shaped conformation within the binding cavity. The [1,2,3]triazolo [4,5-b]pyrazine core facilitates substantial π-stacking interactions with the critical residue Y1230. Additionally, the methyl-substituted pyrazole moiety engages in hydrogen bonding interactions with R1086, mediated by water molecules, while the imidazo [1,2-a]pyridine group forms hydrogen bonds with M1160. Additionally, the (S)-methyl group of 3 extends into the hydrophobic cavity created by the residues L1157, K1110, and V1092, thereby enhancing the molecule’s binding affinity to MET. In vivo pharmacokinetic studies demonstrated that 3 exhibited a satisfactory oral bioavailability (F% = 27.2) (Yoh et al., 2021).
FIGURE 6.

The identification and SAR studies of savolitinib (3) are presented. The essential amino acid residues that mediate interactions between the small molecule and the MET protein are depicted. Mauve dashed lines represent hydrogen bonds with key amino acids, and faint gray dashed lines indicate π-π interactions.
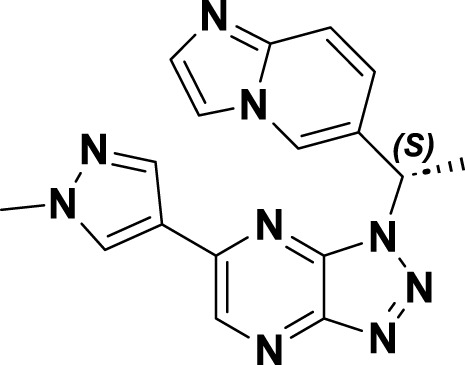
In 2023, glumetinib (9) received approval from the National Medical Products Administration (NMPA) for commercial distribution as a therapeutic agent for adult patients diagnosed with locally advanced or metastatic NSCLC characterized by MET exon 14 skipping mutations (Halder et al., 2024). Structurally, 9 functions as a MET inhibitor, incorporating a pyridine-pyrazole moiety that serves as the ligand for the hinge region of the MET kinase. This compound was developed from 30 through a series of iterative structural optimization processes (Figure 7) (Ai et al., 2018). Initially, 30 underwent structure-activity relationship studies, resulting in the development of 31, which demonstrated enhanced inhibitory efficacy against the MET kinase. The co-crystallization analysis of 31 with MET (PDB code: 2WD1) demonstrates that the presence of the sulfonyl group within the structure of 31 facilitates the interaction between the two aromatic ring systems, resulting in a U-shaped conformation. Consequently, due to the susceptibility of the pyridine-pyrrole moiety in the structure of 31 to hydroxylation at the nitrogen position in vivo, it has been substituted with a pyridine-pyrazole moiety to yield 32. To enhance the π-π interaction between the small-molecule ligand and the Y1230 residue of MET, the nitrobenzene moiety in 32 was substituted with a 6-phenyl-imidazo [3,2-a]pyridine group. This modification led to the identification of MET inhibitor 33, which demonstrated picomolar inhibitory activity. Subsequently, to enhance the druggability of the compound, the phenyl group in the structure of 33 was substituted with a 1-methyl-1H-pyrazole group, ultimately yielding 9. Simultaneously, other nitrogen-containing heterocycles, including thiazole and indazole, may also be utilized in the design of type Ib inhibitors, exemplified by SAR125844 (4) (Egile et al., 2015) and bozitinib (5) (Hu et al., 2023). As illustrated in Table 1, compounds 4 and 5 are presently undergoing clinical phase I/II trials and exhibit promising prospects for further development.
FIGURE 7.

The identification and SAR studies of glumetinib (9) are presented. The essential amino acid residues that mediate interactions between the small molecule and the MET protein are depicted. Mauve dashed lines represent hydrogen bonds with key amino acids, and faint gray dashed lines indicate π-π interactions. The chemical structures of candidate molecules SAR125844 (4) and bozitinib (5), currently undergoing clinical phase I/II studies, are presented.
Tepotinib (6), a highly selective oral MET inhibitor, is designed to target the oncogenic signaling of the MET receptor associated with variations in the MET gene (Albers et al., 2023; Wu Y. L. et al., 2021). In 2023, the NMPA of China granted marketing authorization for 6 for the treatment of NSCLC patients with MET exon 14 skipping mutations (Mazieres et al., 2023). Structurally, 6 is categorized as a small molecule inhibitor, distinguished by the presence of a single pyrimidine group that serves as the ligand for the hinge region of the MET. This compound was developed through several iterations of structural optimization derived from molecule 34 (Figure 8). In compound 34, the dimethoxy-substituted phenyl group and the thiadiazolone nucleus were replaced with a difluoro-substituted phenyl group and a pyridinone nucleus, respectively, resulting in the formation of compound 35. This modification resulted in enhanced therapeutic efficacy and reduced cardiotoxicity. However, amino acid methyl esters demonstrate potential metabolic toxicity in vivo; thus, further optimization of these compounds is warranted (Wu Z. et al., 2021). Consequently, building upon the original structure, a comprehensive investigation into the structure-activity relationship was undertaken. Compounds 6 and 36 were identified by substituting the amino acid methyl ester with a pyrimidine moiety that is modified with hydrophilic groups, and by replacing the difluoro-substituted phenyl group with a 3-cyanophenyl group. Research findings demonstrate that 6 exhibits low nanomolar enzymatic and cellular inhibitory activities, in addition to possessing favorable oral bioavailability (Jing et al., 2022). To date, the derivatization of 6 has primarily been documented with the aim of enhancing the in vivo metabolic stability of the drug, including derivatives 37, 38, 39, and ABN401 (7) (Kim et al., 2020). Notably, these derivatives demonstrate improved in vivo stability and pharmacokinetic profiles relative to 6. Among these derivatives, 7 is distinguished by its notable selectivity and advantageous pharmacokinetic properties. It is currently in Phase I/II clinical trials for the treatment of several solid tumors, including NSCLC (Wu Y. et al., 2020).
FIGURE 8.

The identification and SAR studies of tepotinib (6) are presented. The essential amino acid residues that mediate interactions between the small molecule and the MET protein are depicted. Mauve dashed lines represent hydrogen bonds with key amino acids, and faint gray dashed lines indicate π-π interactions. The chemical structures of tepotinib-based derivatives 37–39, as well as ABN401 (7), are depicted.
Macrocycles, defined as cyclic small molecules or peptides featuring ring structures that consist of 12 or more atoms, have gained recognition as promising chemical scaffolds in the domain of drug discovery (Garcia Jimenez et al., 2023). Their unique physicochemical properties, which include a high molecular weight and a significant number of hydrogen bond donors, distinguish them from linear analogues. In contrast to linear analogues, macrocyclic compounds exhibit a tendency to adopt pre-organized and constrained conformations, facilitating extensive interactions with target proteins (Cummings and Sekharan, 2019). As a result, these compounds are likely to exhibit enhanced binding affinity, improved selectivity, and superior pharmacological properties. Recently, the domain of macrocyclic pharmaceuticals has attracted considerable interest from medicinal chemists. Notably, over the past decade, there has been a substantial increase in the number of macrocyclic drugs approved by the FDA that were developed utilizing rational drug design methodologies (Acharya et al., 2023).
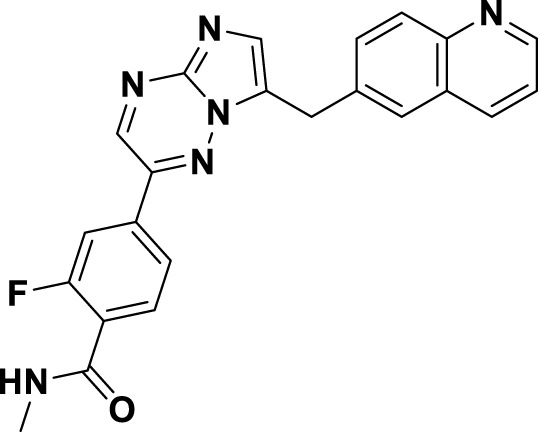
In 2019, a novel macrocyclic inhibitor targeting MET, SRC, and CSF1R, designated as elzovantinib (8), was reported, demonstrating picomolar-level inhibitory activity against these kinases. Currently, 8 is undergoing Phase I/II clinical trials in patients with NSCLC who have not previously received treatment with tyrosine kinase inhibitors (Qu et al., 2023). Preliminary results indicate that patients exhibit good tolerance to the treatment and show promising anti-tumor activity. It is noteworthy that the chirality of the methyl groups within the linker region significantly affects the cellular activity of 8. In comparison, the cellular activities of its enantiomer 40 and the deethylated derivative 41 are diminished by approximately 1,000-fold (Figure 9). This disparity may be ascribed to the influence of the spatial arrangement of atoms on the recognition of drug molecules by protein receptors. In particular, the (S)-methyl group within the structure of compound 8 appears to play a crucial role in constraining its conformation (Bouattour et al., 2018). In 2022, Lu and colleagues developed and synthesized a series of “U”-shaped MET inhibitors employing a pharmacophore hybridization strategy, guided by the chemical structures of NVP-BVU972 (41) and compound 6 (Figure 9) (Wang C. et al., 2022). Notably, 42 demonstrated significant inhibitory activity against the MET kinase and exhibited pronounced anti-proliferative effects on NSCLC cells. Subsequently, the research team implemented a macrocyclization strategy, which led to the discovery of several macrocyclic inhibitors of the MET kinase. Notably, 43 demonstrated the most promising enzymatic and cellular inhibitory activities, exhibiting an IC50 of 2.9 nM for the MET kinase and 0.7 nM for Hs746T cells. However, the pharmacokinetic properties of this compound are suboptimal, indicating the potential for structural refinement. Specifically, in the design of linker groups for macrocyclic inhibitors, it is imperative to consider not only the feasibility of synthesis but also the significant role these linker groups play in facilitating the interaction between the ligand and its target (Amrhein et al., 2021).
FIGURE 9.

The identification and SAR studies of compound 43 are presented. The essential amino acid residues that mediate interactions between the small molecule and the MET protein are depicted. Mauve dashed lines represent hydrogen bonds with key amino acids, and faint gray dashed lines indicate π-π interactions. The chemical structures of elzovantinib (8), along with its derivatives 40 and 41, are illustrated.
4.3 Type II inhibitors of MET
Structurally, the occupation of less conserved hydrophobic pockets surrounding the ATP-binding site by type II inhibitors facilitates the binding of small molecules to the protein, thereby enhancing selectivity (Zhao S. et al., 2021). Table 1 provides a summary of the representative type II MET inhibitors that have been incorporated into the clinical treatment of NSCLC.
Consider compound 44 as a representative example. As illustrated in Figure 10, most type II inhibitors of MET exhibit certain defining characteristics: (i) Various nitrogen-containing aromatic heterocycles (region I), including pyridine, pyrrolidine, and quinazoline, can be incorporated to establish hydrogen bonds with the amino acid residue Met1160 located in the hinge region of MET. Furthermore, the side chains of these aromatic heterocycles play a crucial role in modulating the affinity of the molecule for its target; (ii) Unsubstituted or mono-substituted pyridine and benzene rings (region II) possess the capacity to participate in π-π interactions with the residue F1223; (iii) The incorporation of a flexible chain or rigid ring structure, specifically a five-atom linking group, that includes one or more hydrogen bond donors or acceptors (designated as region III), significantly enhances the molecular potency. This region promotes the establishment of a hydrogen bond network with the amino acid residues of the MET kinase, including Asp1220, Lys1110, and Leu1245, among others; (iv) The region IV segment consists of six-membered aromatic heterocycles that may be unsubstituted, mono-substituted, or di-substituted, and this segment extends into the hydrophobic pocket of the MET kinase (Redmer et al., 2024).
FIGURE 10.

The chemical structure and binding mode characteristics of type II MET inhibitors in their interaction with the Met kinase were elucidated using compound 44 as a representative example. Mauve dashed lines represent hydrogen bonds with key amino acids, and faint gray dashed lines indicate π-π interactions. The strategy for the structural optimization of type II MET inhibitors has been elucidated.
The recently reported novel type II inhibitors of MET primarily focus on the optimization of regions I and III. In the context of region III, it is significant to note that one optimization strategy entails the identification of novel linkers that comply with the “5-atom rule” (Nan et al., 2020). This objective can be accomplished by introducing cyclopropyl and oxy groups at chemically unstable sites or by replacing these groups with alternative linear structures, such as ester groups, sulfone groups, and double bonds, as illustrated by compounds 10, 11, and 12 in Table 1 (Nan et al., 2023). Furthermore, an alternative optimization strategy entails the cyclization approach; however, it is crucial to verify that the region IV component can adequately extend into the hydrophobic pocket, as evidenced by compounds such as 13, 14, and 15. Macrocyclization can yield five-membered, six-membered, and bicyclic heterocycles. The macrocyclic structures described above have the ability to form hydrogen bond networks with the amino acid residues present in the kinase domain of the target protein (Bauder et al., 2021). Furthermore, region IV is highly likely to extend into the DFG-out conformation pocket, which comprises residues F1200, L1195, and F1134. Notably, the region IV of certain inhibitors may extend into the αC-helical pocket formed by the residues I1145, V1155, F1124, and E1127 (Figure 10). This pocket is infrequently observed in kinases other than the MET kinase, which consequently allows these inhibitors to demonstrate a high degree of selectivity for the MET kinase (Pievsky and Pyrsopoulos, 2016). In light of the significant advancements in research on type II inhibitors, several comprehensive reviews are currently available (Liu et al., 2022; Jin et al., 2024); consequently, a detailed discussion on this topic will not be undertaken in this context.
4.4 Type III inhibitors of MET
Tivantinib (16) is classified as a type III inhibitor of Met. In vitro studies have demonstrated that 16 possesses an inhibitory constant (Ki) of approximately 355 nM, indicating a high degree of selectivity for the MET kinase when compared to 230 other human-derived kinases evaluated concurrently (Eathiraj et al., 2011). The co-crystal structure of compound 16 in complex with the unphosphorylated MET kinase (PDB ID: 3RHK) elucidates the formation of hydrogen bonds between the pyrrolidinylindolone fragment and the amino acid residues P1158 and M1160 (Figure 11). Additionally, the tricyclic moiety demonstrates a perpendicular orientation in relation to the indole ring within the binding site, subsequently extending into the hydrophobic cavity situated between the F1223 residues and the F1089 residues in MET kinase. The binding mechanism of 16 to the unphosphorylated MET receptor specifically stabilizes the “DFG-out” conformation, concurrently disrupting the salt bridge between residues E1127 and K1110 (Calles et al., 2015). Protein kinases demonstrate a significant degree of conservation in their three-dimensional structures, especially in the regions surrounding the ATP-binding site. Presently, most kinase inhibitors target this ATP-binding site, which often leads to challenges related to selectivity and off-target effects. Compound 16 is notable for its ability to occupy the hydrophobic pocket created by the displacement of the α-C helix in MET kinase. This modification alters the binding site’s conformation, transforming the polar ATP-binding pocket into a non-polar environment, making it unsuitable for ATP (Yoshioka et al., 2015).
FIGURE 11.

The chemical structure and binding mode characteristics of tivantinib (16) in their interaction with the unphosphorylated MET kinase domain were elucidated. Hydrogen bonds are represented by mauve dashed lines connecting to the key amino acids.
Preliminary clinical research suggests that the combination of 16 and erlotinib may significantly extend the survival duration of patients with refractory NSCLC. A Phase III clinical trial (NCT01377376) investigated the therapeutic efficacy of erlotinib in conjunction with tivantinib in an Asian cohort of patients diagnosed with stage IIIB/IV EGFR wild-type, non-squamous NSCLC (Han et al., 2024). The trial was prematurely terminated due to an increased incidence of interstitial lung disease (ILD) observed in the group treated with 16. Before its discontinuation, the clinical data demonstrated no statistically significant difference in median overall survival (OS) between the combination therapy group and the erlotinib monotherapy group. Despite the challenges encountered in the clinical development of 16, its unique mechanism of action has provided valuable insights for the subsequent identification of novel targeted therapeutic strategies aimed at the MET kinase in NSCLC.
5 Resistance mechanism of MET inhibitors
In recent years, several small molecule inhibitors that target the MET gene have been introduced to the market, offering survival advantages for numerous patients with NSCLC who possess MET gene mutations (Huang et al., 2019). However, although certain patients may initially exhibit therapeutic benefits, as indicated by tumor stabilization or reduction, these advantages are frequently temporary, resulting in subsequent tumor relapse and disease progression. Additionally, a subset of patients does not experience any therapeutic benefit from MET inhibitors. Specifically, drug resistance can be broadly classified into primary resistance and acquired resistance (Sun et al., 2022).
5.1 Primary resistance
Primary drug resistance is characterized by the inability of patients with MET alterations to exhibit an initial therapeutic response to MET inhibitors. Mechanistic investigations indicate that primary drug resistance is primarily attributable to the presence of activating mutations in bypass or downstream signaling pathways within tumor cells, particularly involving the activation of the RAS/MAPK pathway and alterations in the PI3K-AKT pathway (Figure 12) (Zhang Y. et al., 2021). Research has demonstrated that the co-occurrence of EGFR amplification, KRAS mutation, BRAF mutation, TP53 mutation, MDM2 amplification, PTEN protein loss, and PIK3CA mutation or amplification is prevalent in tumors exhibiting MET exon 14 skipping mutations. This coexistence of mutations is associated with primary resistance to MET inhibitors and a reduced duration of response to these therapies in affected patients (Zhou X. et al., 2021; Zhou J. et al., 2021). Preclinical investigations have demonstrated that the concurrent administration of MET inhibitors and PI3K inhibitors can effectively reestablish the sensitivity of drug-resistant tumor cells to MET inhibitors. Therefore, the concomitant use of MET inhibitor and PI3K inhibitors represents a viable approach to overcoming primary resistance to MET inhibitors (Fujino et al., 2019).
FIGURE 12.

Reported primary drug resistance (orange) and acquired drug resistance (brownish-red) mechanisms for MET inhibitors.
5.2 Secondary resistance
Following the successful initial treatment with a MET inhibitor, novel structural alterations in the MET domain may arise, leading to the development of secondary resistance. Secondary resistance is typically classified by its mechanisms into MET-dependent resistance, which accounts for about one-third of cases and includes secondary mutations and amplifications in the MET gene as well as HGF gene amplification, and non-MET-dependent resistance, characterized by aberrant activation of downstream signaling pathways related to MET and alternative bypass pathways (Recondo et al., 2020). Within the framework of MET-dependent secondary resistance, mutations at secondary sites within the MET kinase domain represent the most frequently observed alterations. Figure 12 provides a summary of the secondary MET mutation sites that have been documented in response to treatment with MET inhibitors. Specifically, the Y1230 and D1228 residues are key mutation sites for type I inhibitors, whereas L1195 and F1200 residues are mainly associated with type II inhibitors (Marona et al., 2019). Research in structural biology has elucidated that the interaction between Y1230 and type I molecules is pivotal for their efficacy, with mutations at the Y1230 position leading to a marked reduction in potency. Additionally, the conformation of residue Y1230 is closely linked to the salt bridge between D1228 and K1110. Consequently, the mutation at the D1228 site is highly likely to disrupt the integrity of the salt bridge, consequently altering the conformation of Y1230 and diminishing the binding affinity of the inhibitor to the target protein. Type II inhibitors interact with MET in a non-active “DFG-out” conformation, which does not depend on the π-π interaction with Y1230 (Sweeney et al., 2023). Consequently, these inhibitors retain their efficacy in patients with mutations at D1228 and Y1230. Residues L1195 and F1200 are crucial for type II MET inhibitors’ effectiveness due to their key positions in the hydrophobic pocket of region IV (Figure 10). In the “DFG-out” conformation, mutations at residues L1195 and F1200 will affect the sensitivity to type I inhibitors. Additionally, mutations at residue G1163, located within the solvent-exposed region, will hinder the binding of type Ia inhibitors within the hydrophobic pocket, thereby promoting drug resistance; however, these mutations will not influence the efficacy of type Ib and type II inhibitors of the MET kinase (Li et al., 2017). Clinical studies have demonstrated that secondary mutations arising from the administration of type I MET inhibitors can lead to acquired resistance, which may be effectively managed through the use of type II MET kinase inhibitors or by employing reverse strategies. This conclusion has been substantiated by clinical case analyses (Parsons et al., 2020). However, there exists a probability that the transition from type I to type II inhibitors consistently yields unsatisfactory outcomes for mutant variants. Especially, some patients harboring the METD1228N resistance mutation demonstrate insensitivity to type II inhibitors, including 10 (Xu et al., 2022). Consequently, following the emergence of resistance to MET inhibitors, elucidating the underlying resistance mechanisms through secondary biopsy and second-generation sequencing techniques is essential for informing the subsequent treatment strategy.
The resistance mechanism that operates independently of MET is predominantly mediated by aberrantly activated bypass signaling pathways linked to the ErbB family of proteins (Lei et al., 2023). Additionally, alterations in the RAS/RAF/MEK pathway and modifications in the PI3K/AKT/mTOR pathway contribute to this phenomenon (Figure 12). Combination therapy strategies have been shown to effectively counteract non-MET-dependent resistance mechanisms (Lombardi et al., 2024). Especially, tumors harboring KRAS gene mutations or amplifications may be effectively managed through a combination therapy approach that incorporates MET inhibitors. This strategy includes the concurrent administration of MET inhibitors with PI3K inhibitors, MEK inhibitors, or EGFR inhibitors.
6 Perspectives and conclusion
The MET gene assumes a vital role in the progression of NSCLC. MET inhibitors have manifested significant effectiveness in patients harboring MET gene mutations (Yang et al., 2021). Nevertheless, despite the considerable advancements achieved in the development of MET inhibitors to date, the future progression of these therapeutic agents continues to encounter numerous challenges.
Firstly, the prevalent adverse reactions associated with MET-targeted therapies include peripheral edema, nausea and vomiting, and hepatotoxicity. Mechanistic investigations suggest that MET inhibitors disrupt the HGF/MET signaling pathway, which may compromise the permeability balance of vascular endothelial cells, consequently leading to tissue edema (Ayoub et al., 2021). Furthermore, the activity of metabolic enzymes in vivo may facilitate the conversion of MET inhibitors into active metabolites, which can subsequently interact with biological macromolecules within the organism. This interaction may lead to structural alterations and functional modifications of these macromolecules, potentially resulting in toxic effects and adverse reactions. Therefore, in clinical practice, the mechanisms underlying the toxicity induced by Met inhibitors are adjusted to minimize organ damage.
Secondly, the persistent challenge of drug resistance necessitates ongoing scrutiny. Especially, Mutations at residues Y1230 and D1228 disrupt the π−π interactions between the small-molecule ligand and the MET kinase, consequently abrogating the inhibitory effects of the ligand. To address drug resistance caused by kinase domain mutations, the common clinical practice is to alternate between type I and type II inhibitors, though this method is not always successful. For example, certain patients exhibiting the D1228N mutation demonstrate insensitivity to type II inhibitors, including cabozantinib (10). Consequently, there exists an urgent necessity to develop novel MET inhibitors to address the issue of targeted drug resistance in clinical practice (Lu et al., 2020). The following outlines feasible strategies for overcoming resistance to MET-targeted therapy:
(i) Research indicates that rational drug design focused on targeting mutant sites constitutes a pivotal strategy for identifying inhibitors that can effectively overcome MET resistance (Ferng et al., 2022). For example, the substitution of large-volume hydrophilic groups, such as 4-piperidylmorpholine, with small hydrophilic groups, including five-membered nitrogen heterocycles like pyrazole, isoxazole, furan, and thiophene, mitigates steric hindrance associated with solvent-exposed site mutations and may facilitate the formation of additional interactions. Moreover, the conformational constraints imposed by macrocyclization can mitigate entropy loss during ligand binding, enhance inhibitory activity, and simultaneously minimize steric hindrance in the solvent front region.
(ii) The D1228N mutation is commonly identified in patients undergoing treatment with type I MET inhibitors, such as crizotinib, capmatinib, and tepotinib; however, these patients exhibit insensitivity to type II MET inhibitors. Mechanistic studies suggest that the mutation at this site alters the electrostatic potential of the solvent-exposed region. In light of this alteration, and in accordance with the design strategy employed for the FLT3 inhibitor FF-10101, a small molecule inhibitor specifically targeting METD1228N was identified through a covalent drug design approach (Ma et al., 2024). Among these inhibitors, electrophilic groups (including acrylamide, epoxides, and halocarbonyl group) are present, which can form covalent bonds with nucleophilic residues such as cysteine or lysine at the active site (Huang et al., 2023). Unfortunately, no covalent inhibitors targeting MET have been reported to date.
(iii) The design of inhibitors targeting allosteric sites, or pockets, located in non-active regions of proteins is undertaken. In contrast to ATP-binding sites, these allosteric pockets exhibit a lower degree of conservation, suggesting that inhibitors directed at them may possess enhanced selectivity (Yan et al., 2016). For example, tivantinib (16) occupies the hydrophobic pocket created by the translocation of the α-C helix of Met, thereby functioning as an allosteric binding site.
(iv) Targeted protein degradation (TPD) represents an innovative approach to protein degradation that has garnered significant attention within the realms of chemical biology and drug discovery. Unlike the “occupation-driven” mechanisms characteristic of conventional small molecule inhibitors, TPD operates through an “event-driven” pharmacological mechanism, facilitating the degradation of target proteins via endogenous protein clearance pathways. This distinctive strategy offers several advantages, including its efficacy with minimal catalytic doses, the ability to circumvent established drug resistance mechanisms and off-target effects, the degradation of “undruggable” targets, and the capacity to target the entire protein framework to modulate both catalytic and non-catalytic functions of proteins. Furthermore, it has been successfully utilized in the development of novel anti-tumor therapeutics. Recently, Min et al. developed a series of MET-targeting degraders utilizing hydrophobic tag degradation technology, informed by the structural characteristics of 6 (Min et al., 2024). Their research demonstrated that the candidate molecules effectively inhibited tumor growth both in vivo and in vitro by facilitating the degradation of MET. In the Ba/F3-TPR-METD1228N and Ba/F3-TPR-METY1230H drug-resistant cell lines, the candidate molecule exhibited enhanced potency relative to 6. This study further substantiates the efficacy of targeted protein degradation strategies in addressing MET resistance.
(v) In consideration of the complex resistance mechanisms associated with MET inhibitors, which encompass a range of factors and signaling pathways, the strategic selection and implementation of diverse combination therapies may enhance the potential to overcome drug resistance. As shown in Table 1, numerous clinical trials have reported the utilization of MET inhibitors, either alone or in combination with other tumor-targeted therapies, for the treatment of various types of cancer. A substantial body of preclinical and clinical evidence has demonstrated that the combination of MET inhibitors with various tumor-targeted agents, including epigenetic therapies, immunotherapeutics, and other RTK inhibitors, exhibits synergistic effects. However, in the context of combination therapy, it is imperative to take into account several factors, including the cumulative dosage of the medications, the complex pharmacokinetic properties of the two drugs, and the potential for toxicities and adverse effects (Zhou X. et al., 2021). Alternatively, pharmacological agents that target dual or multiple pathways may reduce the risk of drug-related adverse reactions, demonstrate enhanced pharmacokinetic profiles, and improve overall safety.
Collectively, this article provides a thorough review of recent advancements in research concerning MET inhibitors for the treatment of NSCLC. It offers an in-depth analysis of the pathogenesis of MET in NSCLC, as well as the current progress in the development of MET inhibitors. These findings are anticipated to facilitate the research and development of future MET inhibitors that exhibit high efficacy, selectivity, and low toxicity.
Acknowledgments
We extend our sincere gratitude to Dr. Yang Li of the Changchun University of Chinese Medicine for his invaluable contributions to the refinement of this article.
Funding Statement
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Author contributions
DZ: Visualization, Writing–original draft, Writing–review and editing. WZ: Visualization, Writing–original draft. HL: Visualization, Writing–original draft. PL: Visualization, Writing–original draft. CL: Visualization, Writing–original draft. YL: Visualization, Writing–original draft. JH: Writing–original draft, Writing–review and editing. GZ: Writing–original draft, Writing–review and editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ALK, anaplastic lymphoma kinase; BRAF, oncogene homolog B1; EGFR, epidermal growth factor receptor; HER2, human epidermal growth factor receptor 2; HGF, hepatocyte growth factor; IPT domain, immunoglobulin-plexin-transcription factor domain; IRAC, the International Agency for Research on Cancer; MET, mesenchymal-epithelial transition factor; NCCN, The National Comprehensive Cancer Network; NSCLC, non-small cell lung cancer; PSI domain, the plexin-semaphorin-integrin domain; ROS1, c-ros oncogene 1 tyrosine kinase; RTK, receptor tyrosine kinase; SCLC, small cell lung cancer; TPD, targeted protein degradation.
References
- Abu Rous F., Singhi E. K., Sridhar A., Faisal M. S., Desai A. (2023). Lung cancer treatment advances in 2022. Cancer investig. 41 (1), 12–24. 10.1080/07357907.2022.2119479 [DOI] [PubMed] [Google Scholar]
- Acharya B., Saha D., Armstrong D., Jabali B., Hanafi M., Herrera-Rueda A., et al. (2023). Kinase inhibitor macrocycles: a perspective on limiting conformational flexibility when targeting the kinome with small molecules. RSC Med. Chem. 15 (2), 399–415. 10.1039/d3md00457k [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai J., Chen Y., Peng X., Ji Y., Xi Y., Shen Y., et al. (2018). Preclinical evaluation of SCC244 (Glumetinib), a novel, potent, and highly selective inhibitor of c-Met in MET-dependent cancer models. Mol. Cancer Ther. 17 (4), 751–762. 10.1158/1535-7163.MCT-17-0368 [DOI] [PubMed] [Google Scholar]
- Albers J., Friese-Hamim M., Clark A., Schadt O., Walter-Bausch G., Stroh C., et al. (2023). The preclinical pharmacology of tepotinib-a highly selective MET inhibitor with activity in tumors harboring MET alterations. Mol. Cancer Ther. 22 (7), 833–843. 10.1158/1535-7163.MCT-22-0537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander M., Kim S. Y., Cheng H. (2020). Update 2020: management of non-small cell lung cancer. Lung 198 (6), 897–907. 10.1007/s00408-020-00407-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrhein J. A., Knapp S., Hanke T. (2021). Synthetic opportunities and challenges for macrocyclic kinase inhibitors. J. Med. Chem. 64 (12), 7991–8009. 10.1021/acs.jmedchem.1c00217 [DOI] [PubMed] [Google Scholar]
- Ayoub N. M., Ibrahim D. R., Alkhalifa A. E. (2021). Overcoming resistance to targeted therapy using MET inhibitors in solid cancers: evidence from preclinical and clinical studies. Med. Oncol. 38 (12), 143. 10.1007/s12032-021-01596-6 [DOI] [PubMed] [Google Scholar]
- Baschnagel A. M., Kaushik S., Durmaz A., Goldstein S., Ong I. M., Abel L., et al. (2021). Development and characterization of patient-derived xenografts from non-small cell lung cancer brain metastases. Sci. Rep. 11 (1), 2520. 10.1038/s41598-021-81832-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauder M., Meyners C., Purder P. L., Merz S., Sugiarto W. O., Voll A. M., et al. (2021). Structure-based design of high-affinity macrocyclic FKBP51 inhibitors. J. Med. Chem. 64 (6), 3320–3349. 10.1021/acs.jmedchem.0c02195 [DOI] [PubMed] [Google Scholar]
- Bi Y., Xia C., Zhang X., Liu H. (2023). Targeted treatments after chemoradiotherapy failure in a patient with relapsed, advanced non-small cell lung cancer with on-therapy circulating tumor biomarker monitoring: a case report. Oncol. Lett. 26 (3), 407. 10.3892/ol.2023.13993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouattour M., Raymond E., Qin S., Cheng A. L., Stammberger U., Locatelli G., et al. (2018). Recent developments of c-Met as a therapeutic target in hepatocellular carcinoma. Hepatology 67 (3), 1132–1149. 10.1002/hep.29496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray F., Laversanne M., Sung H., Ferlay J., Siegel R. L., Soerjomataram I., et al. (2024). Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA a cancer J. Clin. 74 (3), 229–263. 10.3322/caac.21834 [DOI] [PubMed] [Google Scholar]
- Calles A., Kwiatkowski N., Cammarata B. K., Ercan D., Gray N. S., Jänne P. A. (2015). Tivantinib (ARQ 197) efficacy is independent of MET inhibition in non-small-cell lung cancer cell lines. Mol. Oncol. 9 (1), 260–269. 10.1016/j.molonc.2014.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Luo Y., Lin M., Chen H., Liu M., Wang Y., et al. (2022). Clinical and pathological characteristics of 11 NSCLC patients with c-MET exon 14 skipping. Transl. Cancer Res. 11 (4), 880–887. 10.21037/tcr-22-782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F., Guo D. (2019). MET in glioma: signaling pathways and targeted therapies. J. Exp. Clin. Cancer Res. 38 (1), 270. 10.1186/s13046-019-1269-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J. J., McTigue M., Nambu M., Tran-Dubé M., Pairish M., Shen H., et al. (2012). Discovery of a novel class of exquisitely selective mesenchymal-epithelial transition factor (c-MET) protein kinase inhibitors and identification of the clinical candidate 2-(4-(1-(quinolin-6-ylmethyl)-1H-[1,2,3]triazolo[4,5-b]pyrazin-6-yl)-1H-pyrazol-1-yl)ethanol (PF-04217903) for the treatment of cancer. J. Med. Chem. 55 (18), 8091–8109. 10.1021/jm300967g [DOI] [PubMed] [Google Scholar]
- Cui J. J., Shen H., Tran-Dubé M., Nambu M., McTigue M., Grodsky N., et al. (2013). Lessons from (S)-6-(1-(6-(1-methyl-1H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-yl)ethyl)quinoline (PF-04254644), an inhibitor of receptor tyrosine kinase c-Met with high protein kinase selectivity but broad phosphodiesterase family inhibition leading to myocardial degeneration in rats. J. Med. Chem. 56 (17), 6651–6665. 10.1021/jm400926x [DOI] [PubMed] [Google Scholar]
- Cui J. J., Tran-Dubé M., Shen H., Nambu M., Kung P. P., Pairish M., et al. (2011). Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 54 (18), 6342–6363. 10.1021/jm2007613 [DOI] [PubMed] [Google Scholar]
- Cummings M. D., Sekharan S. (2019). Structure-based macrocycle design in small-molecule drug discovery and simple metrics to identify opportunities for macrocyclization of small-molecule ligands. J. Med. Chem. 62 (15), 6843–6853. 10.1021/acs.jmedchem.8b01985 [DOI] [PubMed] [Google Scholar]
- Dong Y., Xu J., Sun B., Wang J., Wang Z. (2022). MET-targeted therapies and clinical outcomes: a systematic literature review. Mol. Diagn Ther. 26 (2), 203–227. 10.1007/s40291-021-00568-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drusbosky L. M., Dawar R., Rodriguez E., Ikpeazu C. V. (2021). Therapeutic strategies in METex14 skipping mutated non-small cell lung cancer. J. Hematol. Oncol. 14 (1), 129. 10.1186/s13045-021-01138-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duma N., Santana-Davila R., Molina J. R. (2019). Non-small cell lung cancer: epidemiology, screening, diagnosis, and treatment. Mayo Clin. Proc. 94 (8), 1623–1640. 10.1016/j.mayocp.2019.01.013 [DOI] [PubMed] [Google Scholar]
- Eathiraj S., Palma R., Volckova E., Hirschi M., France D. S., Ashwell M. A., et al. (2011). Discovery of a novel mode of protein kinase inhibition characterized by the mechanism of inhibition of human mesenchymal-epithelial transition factor (c-Met) protein autophosphorylation by ARQ 197. J. Biol. Chem. 286 (23), 20666–20676. 10.1074/jbc.M110.213801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egile C., Kenigsberg M., Delaisi C., Bégassat F., Do-Vale V., Mestadier J., et al. (2015). The selective intravenous inhibitor of the MET tyrosine kinase SAR125844 inhibits tumor growth in MET-amplified cancer. Mol. Cancer Ther. 14 (2), 384–394. 10.1158/1535-7163.MCT-14-0428 [DOI] [PubMed] [Google Scholar]
- Ferng T. T., Terada D., Ando M., Tarver T. C., Chaudhary F., Lin K. C., et al. (2022). The irreversible FLT3 inhibitor FF-10101 is active against a diversity of FLT3 inhibitor resistance mechanisms. Mol. Cancer Ther. 21 (5), 844–854. 10.1158/1535-7163.MCT-21-0317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino T., Kobayashi Y., Suda K., Koga T., Nishino M., Ohara S., et al. (2019). Sensitivity and resistance of MET exon 14 mutations in lung cancer to eight MET tyrosine kinase inhibitors in vitro . J. Thorac. Oncol. 14 (10), 1753–1765. 10.1016/j.jtho.2019.06.023 [DOI] [PubMed] [Google Scholar]
- Fujino T., Suda K., Mitsudomi T. (2020). Emerging MET tyrosine kinase inhibitors for the treatment of non-small cell lung cancer. Expert Opin. Emerg. Drugs 25 (3), 229–249. 10.1080/14728214.2020.1791821 [DOI] [PubMed] [Google Scholar]
- Garcia Jimenez D., Poongavanam V., Kihlberg J. (2023). Macrocycles in drug discovery-Learning from the past for the future. J. Med. Chem. 66 (8), 5377–5396. 10.1021/acs.jmedchem.3c00134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R., Luo J., Chang J., Rekhtman N., Arcila M., Drilon A. (2020). MET-dependent solid tumours - molecular diagnosis and targeted therapy. Nat. Rev. Clin. Oncol. 17 (9), 569–587. 10.1038/s41571-020-0377-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder P., Rai A., Talukdar V., Das P., Lakkaniga N. R. (2024). Pyrazolopyridine-based kinase inhibitors for anti-cancer targeted therapy. RSC Med. Chem. 15 (5), 1452–1470. 10.1039/d4md00003j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y., Yu Y., Miao D., Zhou M., Zhao J., Shao Z., et al. (2024). Targeting MET in NSCLC: an ever-expanding territory. JTO Clin. Res. Rep. 5 (2), 100630. 10.1016/j.jtocrr.2023.100630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J., Huang Z., Han L., Gong Y., Xie C. (2021). Mechanisms and management of 3rd-generation EGFR-TKI resistance in advanced non-small cell lung cancer (Review). Int. J. Oncol. 59 (5), 90. 10.3892/ijo.2021.5270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Cui X., Wang Z., Liu Y., Luo Y., Zhong W., et al. (2023). Safety, efficacy and pharmacokinetics of BPI-9016M in c-MET overexpression or MET exon 14 skipping mutation patients with locally advanced or metastatic non-small-cell lung cancer: a phase Ib study. BMC Cancer 23 (1), 331. 10.1186/s12885-022-10500-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Zou Q., Liu H., Qiu B., Li Q., Lin Y., et al. (2020). Management of non-small cell lung cancer patients with MET exon 14 skipping mutations. Curr. Treat. Options Oncol. 21 (4), 33. 10.1007/s11864-020-0723-5 [DOI] [PubMed] [Google Scholar]
- Huang X., Gan G., Wang X., Xu T., Xie W. (2019). The HGF-MET axis coordinates liver cancer metabolism and autophagy for chemotherapeutic resistance. Autophagy 15 (7), 1258–1279. 10.1080/15548627.2019.1580105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Guo Y., Zhou Y., Huang Q., Ru Y., Luo Y., et al. (2023). Tivantinib alleviates inflammatory diseases by directly targeting NLRP3. iScience 26 (3), 106062. 10.1016/j.isci.2023.106062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imyanitov E. N., Iyevleva A. G., Levchenko E. V. (2021). Molecular testing and targeted therapy for non-small cell lung cancer: current status and perspectives. Crit. Rev. oncology/hematology. 157, 103194. 10.1016/j.critrevonc.2020.103194 [DOI] [PubMed] [Google Scholar]
- Jin F., Lin Y., Yuan W., Wu S., Yang M., Ding S., et al. (2024). Recent advances in c-Met-based dual inhibitors in the treatment of cancers. Eur. J. Med. Chem. 272, 116477. 10.1016/j.ejmech.2024.116477 [DOI] [PubMed] [Google Scholar]
- Jing G., Yu F., Xue H. (2022). Tepotinib suppresses proliferation, invasion, migration, and promotes apoptosis of melanoma cells via inhibiting MET and PI3K/AKT signaling pathways. Oncol. Lett. 23 (6), 170. 10.3892/ol.2022.13290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen J. T., Urbanska E. M., Mollerup J. (2024). MET targeted therapy in non-small cell lung cancer patients with MET exon 14-skipping mutations. Transl. Lung Cancer Res. 13 (4), 940–946. 10.21037/tlcr-24-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiya K., Hirakawa S., Ma B., Drinnenberg I., Detmar M. (2005). Hepatocyte growth factor promotes lymphatic vessel formation and function. EMBO J. 24 (16), 2885–2895. 10.1038/sj.emboj.7600763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B. J., Kim D., Kim J. H., Kim H. S., Jang H. J. (2021). The efficacy and safety of onartuzumab in patients with solid cancers: a meta-analysis of randomized trials. Indian J. Cancer 58 (2), 232–240. 10.4103/ijc.IJC_797_18 [DOI] [PubMed] [Google Scholar]
- Kim J., Park K. E., Jeong Y. S., Kim Y., Park H., Nam J. H., et al. (2020). Therapeutic efficacy of ABN401, a highly potent and selective MET inhibitor, based on diagnostic biomarker test in MET-addicted cancer. Cancers (Basel) 12 (6), 1575. 10.3390/cancers12061575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. H., Kim H. (2017). Progress of antibody-based inhibitors of the HGF-cMET axis in cancer therapy. Exp. Mol. Med. 49 (3), e307. 10.1038/emm.2017.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauf J. A., Luckett K. A., Chen K. Y., Voza F., Socci N. D., Ghossein R., et al. (2018). Hgf/Met activation mediates resistance to BRAF inhibition in murine anaplastic thyroid cancers. J. Clin. Invest 128 (9), 4086–4097. 10.1172/JCI120966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch J. P., Aebersold D. M., Zimmer Y., Medová M. (2020). MET targeting: time for a rematch. Oncogene 39 (14), 2845–2862. 10.1038/s41388-020-1193-8 [DOI] [PubMed] [Google Scholar]
- Lei T., Xu T., Zhang N., Zou X., Kong Z., Wei C., et al. (2023). Anlotinib combined with osimertinib reverses acquired osimertinib resistance in NSCLC by targeting the c-MET/MYC/AXL axis. Pharmacol. Res. 188, 106668. 10.1016/j.phrs.2023.106668 [DOI] [PubMed] [Google Scholar]
- Leonetti A., Sharma S., Minari R., Perego P., Giovannetti E., Tiseo M. (2019). Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 121 (9), 725–737. 10.1038/s41416-019-0573-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A., Yang J. J., Zhang X. C., Zhang Z., Su J., Gou L. Y., et al. (2017). Acquired MET Y1248H and D1246N mutations mediate resistance to MET inhibitors in non-small cell lung cancer. Clin. Cancer Res. 23 (16), 4929–4937. 10.1158/1078-0432.CCR-16-3273 [DOI] [PubMed] [Google Scholar]
- Liu Y., Li Y., Wang Y., Lin C., Zhang D., Chen J., et al. (2022). Recent progress on vascular endothelial growth factor receptor inhibitors with dual targeting capabilities for tumor therapy. J. Hematol. Oncol. 15 (1), 89. 10.1186/s13045-022-01310-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi A. M., Sangiolo D., Vigna E. (2024). MET oncogene targeting for cancer immunotherapy. Int. J. Mol. Sci. 25 (11), 6109. 10.3390/ijms25116109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S., Fang J., Li X., Cao L., Zhou J., Guo Q., et al. (2021). Once-daily savolitinib in Chinese patients with pulmonary sarcomatoid carcinomas and other non-small-cell lung cancers harbouring MET exon 14 skipping alterations: a multicentre, single-arm, open-label, phase 2 study. Lancet Respir. Med. 9 (10), 1154–1164. 10.1016/S2213-2600(21)00084-9 [DOI] [PubMed] [Google Scholar]
- Lu X. Y., Smaill J. B., Ding K. (2020). Medicinal chemistry strategies for the development of kinase inhibitors targeting point mutations. J. Med. Chem. 63 (19), 10726–10741. 10.1021/acs.jmedchem.0c00507 [DOI] [PubMed] [Google Scholar]
- Ma C., Cui S., Xu R. (2024). Developments of Fms-like tyrosine kinase 3 inhibitors as anticancer agents for AML treatment. Curr. Med. Chem. 31 (29), 4657–4686. 10.2174/0109298673277543231205072556 [DOI] [PubMed] [Google Scholar]
- Marona P., Górka J., Kotlinowski J., Majka M., Jura J., Miekus K. (2019). C-Met as a key factor responsible for sustaining undifferentiated phenotype and therapy resistance in renal carcinomas. Cells 8 (3), 272. 10.3390/cells8030272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazieres J., Paik P. K., Garassino M. C., Le X., Sakai H., Veillon R., et al. (2023). Tepotinib treatment in patients with MET exon 14-skipping non-small cell lung cancer: long-term follow-up of the VISION Phase 2 nonrandomized clinical trial. JAMA Oncol. 9 (9), 1260–1266. 10.1001/jamaoncol.2023.1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazieres J., Vioix H., Pfeiffer B. M., Campden R. I., Chen Z., Heeg B., et al. (2023). MET exon 14 skipping in NSCLC: a systematic literature review of epidemiology, clinical characteristics, and outcomes. Clin. Lung Cancer 24 (6), 483–497. 10.1016/j.cllc.2023.06.008 [DOI] [PubMed] [Google Scholar]
- Michaelides I. N., Collie G. W., Börjesson U., Vasalou C., Alkhatib O., Barlind L., et al. (2023). Discovery and optimization of the first ATP competitive type-III c-MET inhibitor. J. Med. Chem. 66 (13), 8782–8807. 10.1021/acs.jmedchem.3c00401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min W., Yang H., Wang D., Chen C., Wang Y., Hou Y., et al. (2024). Discovery of potent and selective c-Met degraders for hepatocellular carcinoma treatment. J. Med. Chem. 67 (14), 12314–12330. 10.1021/acs.jmedchem.4c01004 [DOI] [PubMed] [Google Scholar]
- Mulcahy E. Q. X., Colόn R. R., Abounader R. (2020). HGF/MET signaling in malignant brain tumors. Int. J. Mol. Sci. 21 (20), 7546. 10.3390/ijms21207546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X., Li H. J., Fang S. B., Li Q. Y., Wu Y. C. (2020). Structure-based discovery of novel 4-(2-fluorophenoxy)quinoline derivatives as c-Met inhibitors using isocyanide-involved multicomponent reactions. Eur. J. Med. Chem. 193, 112241. 10.1016/j.ejmech.2020.112241 [DOI] [PubMed] [Google Scholar]
- Nan X., Wang Q. X., Xing S. J., Liang Z. G. (2023). Design, synthesis, and biological evaluation of thiazole/thiadiazole carboxamide scaffold-based derivatives as potential c-Met kinase inhibitors for cancer treatment. J. Enzyme Inhib. Med. Chem. 38 (1), 2247183. 10.1080/14756366.2023.2247183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S. K., Tangen C., Thompson I. M., Jr, Balzer-Haas N., George D. J., Heng D. Y. C., et al. (2021). A comparison of sunitinib with cabozantinib, crizotinib, and savolitinib for treatment of advanced papillary renal cell carcinoma: a randomised, open-label, phase 2 trial. Lancet 397 (10275), 695–703. 10.1016/S0140-6736(21)00152-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh P. K., Ghate M. D. (2018). Recent advances in the discovery of small molecule c-Met Kinase inhibitors. Eur. J. Med. Chem. 143, 1103–1138. 10.1016/j.ejmech.2017.08.044 [DOI] [PubMed] [Google Scholar]
- Parsons B. M., Meier D. R., Richmond C. S., Gurda G. T., Lofgren K. A., Burkard M. E., et al. (2020). Acquisition of cabozantinib-sensitive MET D1228N mutation during progression on crizotinib in MET-amplified triple-negative breast cancer. Clin. Breast Cancer 20 (4), e433–e438. 10.1016/j.clbc.2020.02.003 [DOI] [PubMed] [Google Scholar]
- Pievsky D., Pyrsopoulos N. (2016). Profile of tivantinib and its potential in the treatment of hepatocellular carcinoma: the evidence to date. J. Hepatocell. Carcinoma 3, 69–76. 10.2147/JHC.S106072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pothula S. P., Xu Z., Goldstein D., Pirola R. C., Wilson J. S., Apte M. V. (2020). Targeting HGF/c-MET Axis in pancreatic cancer. Int. J. Mol. Sci. 21 (23), 9170. 10.3390/ijms21239170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu L., Lin H., Dai S., Guo M., Chen X., Jiang L., et al. (2023). Structural insight into the macrocyclic inhibitor TPX-0022 of c-Met and c-Src. Comput. Struct. Biotechnol. J. 21, 5712–5718. 10.1016/j.csbj.2023.11.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recondo G., Bahcall M., Spurr L. F., Che J., Ricciuti B., Leonardi G. C., et al. (2020). Molecular mechanisms of acquired resistance to MET tyrosine kinase inhibitors in patients with MET exon 14-mutant NSCLC. Clin. Cancer Res. 26 (11), 2615–2625. 10.1158/1078-0432.CCR-19-3608 [DOI] [PubMed] [Google Scholar]
- Redmer T., Schumann E., Peters K., Weidemeier M. E., Nowak S., Schroeder H. W. S., et al. (2024). MET receptor serves as a promising target in melanoma brain metastases. Acta Neuropathol. 147 (1), 44. 10.1007/s00401-024-02694-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remon J., Hendriks L. E. L., Mountzios G., García-Campelo R., Saw S. P. L., Uprety D., et al. (2023). MET alterations in NSCLC-current perspectives and future challenges. J. Thorac. Oncol. 18 (4), 419–435. 10.1016/j.jtho.2022.10.015 [DOI] [PubMed] [Google Scholar]
- Rocco D., Gravara L. D., Palazzolo G., Gridelli C. (2024). The treatment of a new entity in advanced non-small cell lung cancer: MET exon 14 skipping mutation. Curr. Med. Chem. 31 (21), 3043–3056. 10.2174/0929867331666230803094432 [DOI] [PubMed] [Google Scholar]
- Salgia R., Pharaon R., Mambetsariev I., Nam A., Sattler M. (2021). The improbable targeted therapy: KRAS as an emerging target in non-small cell lung cancer (NSCLC). Cell Rep. Med. 2 (1), 100186. 10.1016/j.xcrm.2020.100186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi P., Oh Y. T., Zhang G., Yao W., Yue P., Li Y., et al. (2016). Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett. 380 (2), 494–504. 10.1016/j.canlet.2016.07.021 [DOI] [PubMed] [Google Scholar]
- Spagnolo C. C., Ciappina G., Giovannetti E., Squeri A., Granata B., Lazzari C., et al. (2023). Targeting MET in non-small cell lung cancer (NSCLC): a new old story? Int. J. Mol. Sci. 24 (12), 10119. 10.3390/ijms241210119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R., Hou Z., Zhang Y., Jiang B. (2022). Drug resistance mechanisms and progress in the treatment of EGFR‑mutated lung adenocarcinoma (Review). Oncol. Lett. 24 (5), 408. 10.3892/ol.2022.13528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z. G., Yang Y. A., Zhang Z. G., Zhu H. L. (2019). Optimization techniques for novel c-Met kinase inhibitors. Expert Opin. Drug Discov. 14 (1), 59–69. 10.1080/17460441.2019.1551355 [DOI] [PubMed] [Google Scholar]
- Sweeney P. L., Suri Y., Basu A., Koshkin V. S., Desai A. (2023). Mechanisms of tyrosine kinase inhibitor resistance in renal cell carcinoma. Cancer Drug Resist 6 (4), 858–873. 10.20517/cdr.2023.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler L. C., Le A. T., Chen N., Nijmeh H., Bao L., Wilson T. R., et al. (2022). MET gene amplification is a mechanism of resistance to entrectinib in ROS1+ NSCLC. Thorac. Cancer 13 (21), 3032–3041. 10.1111/1759-7714.14656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Li J., Qu L., Tang X., Song X., Yang F., et al. (2022). Discovery of D6808, a highly selective and potent macrocyclic c-Met inhibitor for gastric cancer harboring MET gene alteration treatment. J. Med. Chem. 65 (22), 15140–15164. acs.jmedchem.2c00981. 10.1021/acs.jmedchem.2c00981 [DOI] [PubMed] [Google Scholar]
- Wang L., Jia Q., Chu Q., Zhu B. (2023). Targeting tumor microenvironment for non-small cell lung cancer immunotherapy. Chin. Med. J. Pulm. Crit. Care Med. 1 (1), 18–29. 10.1016/j.pccm.2022.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N., Zhang Y., Wu J., Zhu Y., Wu Y., Huang B., et al. (2024). MET overexpression correlated with prognosis of EGFR-mutant treatment-naïve advanced lung adenocarcinoma: a real-world retrospective study. Clin. Transl. Oncol. 26 (7), 1696–1707. 10.1007/s12094-024-03391-x [DOI] [PubMed] [Google Scholar]
- Wang N., Zhu Y., Wu Y., Huang B., Wu J., Zhang R., et al. (2023). MET overexpression in EGFR L858R mutant treatment-naïve advanced lung adenocarcinoma correlated with poor prognosis: a real-world retrospective study. J. Cancer Res. Clin. Oncol. 149 (7), 3219–3228. 10.1007/s00432-022-04225-5 [DOI] [PubMed] [Google Scholar]
- Wang Q., Yang S., Wang K., Sun S. Y. (2019). MET inhibitors for targeted therapy of EGFR TKI-resistant lung cancer. J. Hematol. Oncol. 12 (1), 63. 10.1186/s13045-019-0759-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf J., Seto T., Han J. Y., Reguart N., Garon E. B., Groen H. J., et al. (2020). Capmatinib in MET exon 14-mutated or MET-amplified non-small-cell lung cancer. N. Engl. J. Med. 383 (10), 944–957. 10.1056/NEJMoa2002787 [DOI] [PubMed] [Google Scholar]
- Wu J., Lin Z. (2022). Non-Small cell lung cancer targeted therapy: drugs and mechanisms of drug resistance. Int. J. Mol. Sci. 23 (23), 15056. 10.3390/ijms232315056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. L., Cheng Y., Zhou J., Lu S., Zhang Y., Zhao J., et al. (2020). Tepotinib plus gefitinib in patients with EGFR-mutant non-small-cell lung cancer with MET overexpression or MET amplification and acquired resistance to previous EGFR inhibitor (INSIGHT study): an open-label, phase 1b/2, multicentre, randomised trial. Lancet Respir. Med. 8 (11), 1132–1143. 10.1016/S2213-2600(20)30154-5 [DOI] [PubMed] [Google Scholar]
- Wu Y. L., Smit E. F., Bauer T. M. (2021). Capmatinib for patients with non-small cell lung cancer with MET exon 14 skipping mutations: a review of preclinical and clinical studies. Cancer Treat. Rev. 95, 102173. 10.1016/j.ctrv.2021.102173 [DOI] [PubMed] [Google Scholar]
- Wu Z. X., Li J., Dong S., Lin L., Zou C., Chen Z. S. (2021). Tepotinib hydrochloride for the treatment of non-small cell lung cancer. Drugs Today (Barc). 57 (4), 265–275. 10.1358/dot.2021.57.4.3238323 [DOI] [PubMed] [Google Scholar]
- Xu Y., Gu L., Li Y., Zhao R., Jian H., Xie W., et al. (2022). Integrative genomic analysis of drug resistance in MET exon 14 skipping lung cancer using patient-derived xenograft models. Front. Oncol. 12, 1024818. 10.3389/fonc.2022.1024818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav A. K., Wang S., Shin Y. M., Jang B. C. (2024). PHA-665752’s antigrowth and proapoptotic effects on HSC-3 human oral cancer cells. Int. J. Mol. Sci. 25 (5), 2871. 10.3390/ijms25052871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M. C., Wang H. Y., Wang Q. B., Zhang Z., Zhang C. Y. (2016). Allosteric inhibition of c-Met kinase in sub-microsecond molecular dynamics simulations induced by its inhibitor, tivantinib. Phys. Chem. Chem. Phys. 18 (15), 10367–10374. 10.1039/c5cp07001e [DOI] [PubMed] [Google Scholar]
- Yang J. J., Maloney N. J., Nguyen K. A., Worswick S., Smogorzewski J., Bach D. Q. (2021). Sweet syndrome as an adverse reaction to tyrosine kinase inhibitors: a review. Dermatol Ther. 34 (1), e14461. 10.1111/dth.14461 [DOI] [PubMed] [Google Scholar]
- Yang M., Mandal E., Liu F. X., O'Hara R. M., Jr, Lesher B., Sanborn R. E. (2024). Non-small cell lung cancer with MET amplification: review of epidemiology, associated disease characteristics, testing procedures, burden, and treatments. Front. Oncol. 13, 1241402. 10.3389/fonc.2023.1241402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L., Chen X., Zhou F. (2021). EGFR-mutant NSCLC: emerging novel drugs. Curr. Opin. Oncol. 33 (1), 87–94. 10.1097/CCO.0000000000000701 [DOI] [PubMed] [Google Scholar]
- Yin J., Hu W., Fu W., Dai L., Jiang Z., Zhong S., et al. (2019). HGF/MET regulated epithelial-mesenchymal transitions and metastasis by FOSL2 in non-small cell lung cancer. Onco Targets Ther. 12, 9227–9237. 10.2147/OTT.S217595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoh K., Hirashima T., Saka H., Kurata T., Ohe Y., Hida T., et al. (2021). Savolitinib ± osimertinib in Japanese patients with advanced solid malignancies or EGFRm NSCLC: ph1b TATTON Part C. Target Oncol. 16 (3), 339–355. 10.1007/s11523-021-00806-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka H., Azuma K., Yamamoto N., Takahashi T., Nishio M., Katakami N., et al. (2015). A randomized, double-blind, placebo-controlled, phase III trial of erlotinib with or without a c-Met inhibitor tivantinib (ARQ 197) in Asian patients with previously treated stage IIIB/IV nonsquamous nonsmall-cell lung cancer harboring wild-type epidermal growth factor receptor (ATTENTION study). Ann. Oncol. 26 (10), 2066–2072. 10.1093/annonc/mdv288 [DOI] [PubMed] [Google Scholar]
- Zhang Q. W., Ye Z. D., Shi L. (2019). c-Met kinase inhibitors: an update patent review (2014-2017). Expert Opin. Ther. Pat. 29 (1), 25–41. 10.1080/13543776.2019.1552261 [DOI] [PubMed] [Google Scholar]
- Zhang Y., Yang Q., Zeng X., Wang M., Dong S., Yang B., et al. (2021). MET amplification attenuates lung tumor response to immunotherapy by inhibiting STING. Cancer Discov. 11 (11), 2726–2737. 10.1158/2159-8290.CD-20-1500 [DOI] [PubMed] [Google Scholar]
- Zhao S., Wu W., Jiang H., Ma L., Pan C., Jin C., et al. (2021). Selective inhibitor of the c-Met receptor tyrosine kinase in advanced hepatocellular carcinoma: No beneficial effect with the use of tivantinib? Front. Immunol. 12, 731527. 10.3389/fimmu.2021.731527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Ji Q., Li Q. (2021). Resistance to anti-EGFR therapies in metastatic colorectal cancer: underlying mechanisms and reversal strategies. J. Exp. Clin. Cancer Res. 40 (1), 328. 10.1186/s13046-021-02130-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Yao Z., Bai H., Duan J., Wang Z., Wang X., et al. (2021). Treatment-related adverse events of PD-1 and PD-L1 inhibitor-based combination therapies in clinical trials: a systematic review and meta-analysis. Lancet Oncol. 22 (9), 1265–1274. 10.1016/S1470-2045(21)00333-8 [DOI] [PubMed] [Google Scholar]
- Zou H. Y., Li Q., Lee J. H., Arango M. E., Burgess K., Qiu M., et al. (2012). Sensitivity of selected human tumor models to PF-04217903, a novel selective c-Met kinase inhibitor. Mol. Cancer Ther. 11 (4), 1036–1047. 10.1158/1535-7163.MCT-11-0839 [DOI] [PubMed] [Google Scholar]