

Abstract
Serangium japonicum (Coleoptera; Coccinellidae) plays a crucial role as a predatory coccinellid in ecosystems, exhibiting adept predation on diverse whitefly species and effectively regulating their population dynamics. Nonetheless, the absence of high-quality genomic data has hindered our comprehension of the molecular mechanisms underlying this predatory beetle. This study performed genome sequencing of S. japonicum using the PacBio HiFi long reads and Hi-C data. The genome spans 433.74 Mb, which includes 104 contigs and 17 scaffolds, with a contig N50 size of 11.44 Mb and a scaffold N50 size of 42.67 Mb. A substantial portion of the genome, totaling 433.04 Mb (99.84%), was anchored to 10 chromosomes. BUSCO analysis demonstrates a high genomic completeness of 97.8% (n = 1,376), comprising 97.3% single-copy genes and 0.5% duplicated genes. The genome includes 54.66% (237.06 Mb) repetitive elements and 12,299 predicted protein-coding genes. The chromosome-level genome of S. japonicum offers important genomic insights that enhance our understanding of the evolution and ecology of the Coccinellidae family.
Subject terms: Genome, Genomics
Background & Summary
Ladybird beetles, members of the Coccinellidae family within the order Coleoptera, encompass over 6,000 species globally1. Predominantly predatory, ladybird beetles are natural enemies of agricultural pests such as aphids, mealybugs, scale insects, and mites2,3. However, there are also phytophagous and mycophagous species, with the phytophagous ones having the potential to inflict substantial damage on economically significant crops4. Ladybirds are also a focal point of chemical ecology research, given that many species exhibit aposematic coloration and secrete toxic alkaloid compounds when disturbed5. Due to their diverse forms, behaviors, and ecological roles in agriculture, ladybird beetles are extensively studied as model organisms in ecology and evolutionary biology6,7.
Serangium japonicum exhibits high prey intake, a brief generation cycle, an extended adult lifespan, and substantial reproductive capacity as a predatory species8. It effectively targets several whitefly species, including Bemisia tabaci9–11, Dialeurodes citri12, and Aleurocanthus camelliae13. This predatory behavior renders S. japonicum a beneficial insect in agriculture and underscores its crucial role in integrated pest managemen14. The genome assembly of twelve species from the family Coccinellidae has reached the chromosome level in the NCBI database (accessed June 2024). However, neither genome assemblies for S. japonicum nor chromosomal-level genomes for Serangium species have been reported.
To better understand the genetic basis of S. japonicum’s adaptability and predatory behavior. We successfully assembled the chromosome-level genome of S. japonicum by integrating data from PacBio HiFi, Illumina, and Hi-C data. We comprehensively annotated repeats, non-coding RNAs (ncRNAs), and protein-coding genes. The high-quality genome of S. japonicum marks a substantial progression in the study of Coccinellidae, offering important insights into its evolutionary trajectory and ecological roles.
Methods
Sample collection and sequencing
The S. japonicum samples utilized in this study were collected in May 2022 from Baiyun Mountain in Guangzhou, Guangdong Province, and were reared under controlled conditions for 10 generations (26 ± 1 °C, 70%–75% relative humidity, 14 L:10D). Adult individuals were thoroughly rinsed with phosphate-buffered saline and then rapidly flash-frozen in liquid nitrogen.
Genomic DNA was extracted using the FastPure® Blood/Cell/Tissue/Bacteria DNA Isolation Mini Kit (Vazyme Biotech Co., Ltd, Nanjing, China), while RNA was extracted with TRIzol reagent (YiFeiXue Tech, Nanjing, China). A total of 12 adult individuals of mixed gender were used for transcriptome sequencing. For Hi-C sequencing, 6 adult individuals of mixed gender were employed, and the library construction involved formaldehyde cross-linking, chromatin digestion with the restriction enzyme MboI, end repair, DNA cyclization, and DNA purification15. All short-read libraries were sequenced on the Illumina NovaSeq6000 platform. Additionally, for PacBio sequencing, DNA was extracted from 10 adult individuals of mixed gender, and a library with an insert size of 20 kb was prepared using the SMRTbellTM Express Template Prep Kit 2.0. This library was subsequently sequenced on the PacBio Sequel II platform in HiFi mode. All procedures related to library preparation and sequencing were performed by Berry Genomics (Beijing, China). Finally, the PacBio HiFi reads accounted for 39.97 Gb (59.81×), Hi-C reads totaled 110.14 Gb (164.80×), RNA-seq data amounted to 17.41 Gb, and RNA-ONT data totaled 11.56 Gb (Table 1). The PacBio HiFi long reads achieved a scaffold N50 of 15.08 kb, with an average length of 15.17 kb.
Table 1.
Statistics of the genome sequencing data for Serangium japonicum.
| Libraries | Insert sizes (bp) | Clean data (Gb) | Sequencing coverage (x) |
|---|---|---|---|
| PacBio HiFi | 15Kb | 39.97 | 59.81 |
| Hi-C | 350 | 110.14 | 164.80 |
| RNA-sr | 350 | 17.41 | — |
| RNA-ONT | — | 11.56 | — |
Estimation of genomic characteristics
We analyzed the genomic features of S. japonicum using a K-mer approach, with K-mer counts generated by BBTools v38.8216 (K = 21). This analysis estimated a genome size of approximately 422.19 Mb and revealed substantial repeat content (39.6%) and heterozygosity (1.94%), as characterized by GenomeScope v2.017 (Fig. 1).
Fig. 1.
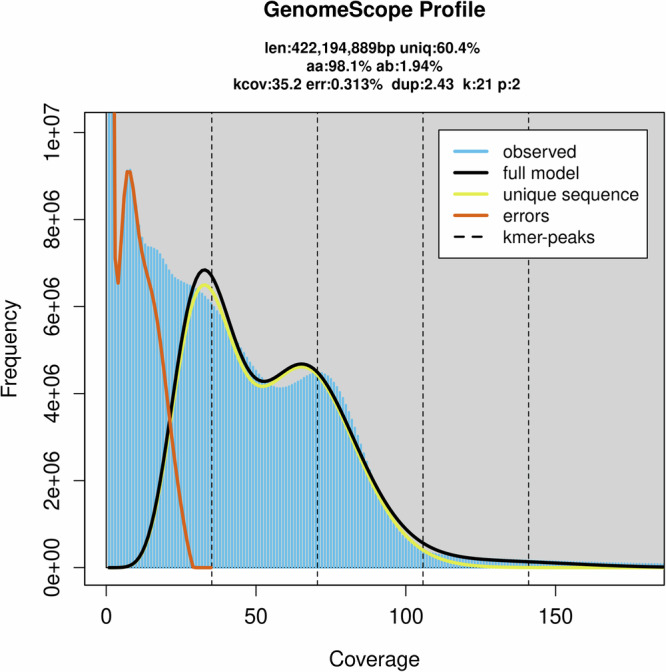
Estimated characteristics of the Serangium japonicum genome based on a 21-mer count histogram from Illumina short-read data.
Genome assembly
We initially employed Hifiasm v0.16.118 with default parameters for the preliminary assembly of PacBio HiFi long reads. Purge_dups v1.2.519 was applied to remove heterozygous regions in the assembly based on contig similarity. A haploid cutoff of 70 was set to identify contigs as haplotigs. Quality control of the Hi-C data and read alignment was performed using Juicer v1.6.220. Next, we used the 3D-DNA v18092221 to anchor contigs into chromosomes. The contig assembly results were carefully examined, and any errors in the assembly were manually corrected using Juicebox v.1.11.0821. To detect possible contaminants, we conducted BLASTN-like searches with MMseqs2 v1322 against the NCBI nucleotide and UniVec databases. Additionally, we used blastn (BLAST + v2.11.0)23 specifically with the UniVec database to detect vector contaminants. Sequences with over 90% similarity to entries in these databases were marked as possible contaminants. For sequences with similarity exceeding 80%, further verification was performed using online BLASTN against the NCBI nucleotide database. To ensure the purity of the assembled scaffolds, sequences potentially originating from bacterial and human sources were systematically removed.
The final assembly of the S. japonicum genome achieved a chromosome-level resolution, encompassing a total size of 433.74 Mb, comprising 17 scaffolds and 104 contigs. The longest scaffold and contig length is 82.69 and 28.54 Mb, with scaffold N50 length of 42.67 Mb and contig N50 length of 11.44 Mb, demonstrating exceptionally high assembly continuity. The genome maintains a GC content of 27.90% (Table 2). The majority of contigs, accounting for 99.84% and totaling 433.04 Mb, were anchored into ten chromosomes. These chromosomes exhibited lengths varying from 21.42 Mb to 82.69 Mb. (Figs 2; 3).
Table 2.
Genome assembly statistics for Serangium japonicum.
| Content | Values |
|---|---|
| Assembly size (bp) | 433,734,680 |
| Number of chromosomes (sizes) | 10 (433,038,680 bp) |
| Number of scaffolds/contigs | 17/104 |
| Longest scaffold/contig (Mb) | 82.692/28.539 |
| N50 scaffold/contig length (Mb) | 42.67/11.44 |
| GC content (%) | 27.90 |
| BUSCO | C:97.8% [S:97.3%, D:0.5%], F:0.1%, M: 2.1%, n = 1,367 |
C: complete BUSCOs; D: complete and duplicated BUSCOs; F: fragmented BUSCOs; M: missing BUSCOs.
Fig. 2.
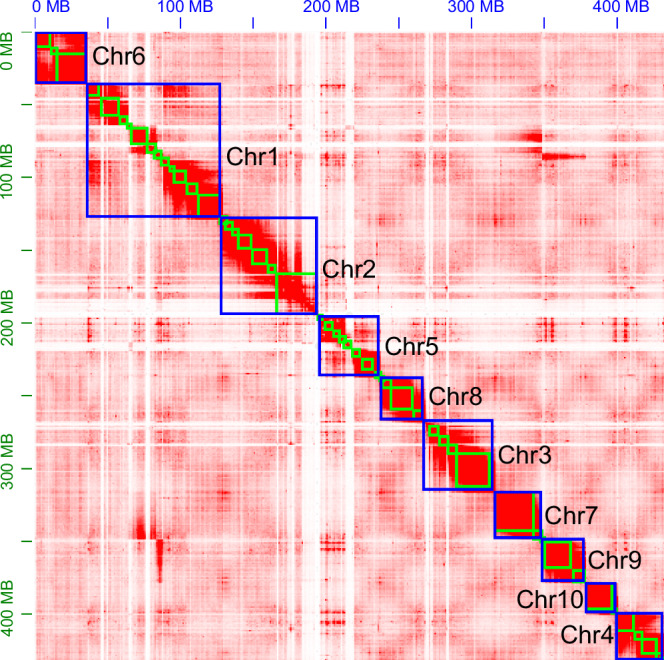
The heatmap of the Serangium japonicum genome showing chromosome-level scaffolding with ten anchored chromosomes. Red indicates high intra-chromosomal contact frequencies.
Fig. 3.

Circular genome map of Serangium japonicum, showing the distribution of genomic features across ten chromosomes. Rings represent chromosome length, GC content, gene density, and repeat elements (DNA transposons, SINEs, LINEs, LTRs, and simple repeats). The central image depicts an adult S. japonicum.
Genome annotation
We established a de novo repeat library of S. japonicum focusing on the distinctive structure and de novo prediction of repeat sequences. Employing RepeatModeler v2.0.424 with the ‘-LTRStruct’. This newly constructed database was subsequently integrated with Dfam 3.525 and RepBase-2018102626 databases to form a comprehensive reference dataset for repeat sequences. Subsequently, RepeatMasker v4.1.427 was employed to conduct S. japonicum genome identification of repetitive elements using a custom-built library. Our analysis identified a total of 731,474 repeat sequences, encompassing 54.66% of the genome (237.06 Mb). The predominant repeat sequence categories include unclassified elements (34.19%), LTR transposons (2.28%), LINE transposons (6.93%), DNA transposons (6.90%), and Simple repeat (1.42%) (Table 3).
Table 3.
Genome assembly and annotation statistics of Serangium japonicum.
| Characteristics | S. japonicum |
|---|---|
| Genome assembly | |
| Genome Size (Mb) | 433.74 |
| Number of scaffolds | 17 |
| Number of chromosomes | 10 |
| Scaffold N50 length (Mb) | 42.67 |
| GC (%) | 27.90 |
| BUSCO completeness (%) | 97.8 |
| Protein-coding genes | |
| Number | 12,229 |
| Mean gene length (bp) | 12,065.7 |
| BUSCO completeness (%) | 97.0 |
| Repetitive elements | |
| Size (Mb) | 237.06 (54.66%) |
| DNA transposons (Mb) | 29.93 (6.90%) |
| Simple repeat (Mb) | 6.16 (1.42%) |
| LINEs (Mb) | 30.16 (6.93%) |
| LTRs (Mb) | 9.52 (2.28%) |
| Unclassified (Mb) | 148.28 (34.19%) |
| ncRNA | |
| Number of ncRNA | 1,270 |
| rRNA | 861 |
| miRNA | 55 |
| snRNA | 78 |
We employed Infernal v1.1.428 along with the Rfam v14.10 database29 for the annotation of ncRNAs within the S. japonicum genome. Additionally, tRNAscan-SE v2.0.930 was utilized for the prediction of tRNA sequences, with low-confidence tRNAs subsequently filtered using the ‘EukHigh Confidence Filter’ script. The analysis revealed 1,270 ncRNAs in the S. japonicum genome, mainly including 3 lncRNAs, 2 ribozymes, 78 snRNAs, 55 miRNAs, 247 tRNAs, and 861 rRNAs (Table 3).
The protein-coding gene annotation for S. japonicum was performed using MAKER v3.01.0331, which integrates transcribed RNA, ab initio gene predictions, and homologous proteins. Transcribed RNA was aligned using HISAT2 v2.2.132, and the resulting RNA-seq alignments facilitated genome-guided assembly through StringTie v2.1.133. Ab initio gene predictions were performed using BRAKER v2.1.634, which integrates GeneMark-ES/ET/EP 4.68_lic34, GeneMark-ETP35, and Augustus v3.5.036. These tools were automatically trained on RNA sequence alignments and reference proteins sourced from the OrthoDB v11 database37. Gene predictions were performed using GeMoMa v1.938, incorporating protein sequences from six different species (Drosophila melanogaster (GCA_029775095.1)39, Apis mellifera (GCA_019321825.1)40, Chrysoperla carnea (GCA_905475395.1)41, Tribolium castaneum (GCA_000002335.3)42, Coccinella septempunctata (GCA_907165205.1)43, and Harmonia axyridis (GCA_011033045.2)44). Finally, the MAKER pipeline predicted sum of 12,299 protein-coding genes, with an average gene length of 12,065.7 bp. Each gene had an average of 6.1 exons, with an average exon length of 305.6 bp. On average, each gene had 5.1 introns, with an intron length averaging 2,116.1 bp. Furthermore, genes contained about 5.9 coding sequence regions (CDS), which averaged 266.8 bp in length (Table 3). The completeness of the protein sequences was evaluated with BUSCO v5.0.445, yielding an impressive score of 97.0% (n = 1,367). This encompassed 84.9% (1,160) single-copy, 12.1% (166) duplicated, 0.2% (3) fragmented, and 2.8% (38) missing BUSCOs, indicating high-quality predictions.
We employed Diamond v2.0.11.146 in highly sensitive mode (‘–very-sensitive -e 1e-5’) to conduct gene function searches against the UniProtKB database. Subsequently, InterProScan 5.58-91.047 and eggNOG-mapper v2.1.548 were utilized to simultaneously query five databases: Pfam49, SMART50, Superfamily51, CDD52, and Gene3D53. These analyses aimed to predict conserved protein sequences and domains within the gene set, as well as provide insights into Gene Ontology (GO) terms and pathways (KEGG, Reactome). InterPro identified protein domains for 10,178 protein-coding genes, while InterPro and eggNOG-mapper jointly annotated GO terms for 9,074 genes and assigned KEGG pathway entries to 4,356 genes.
Data Records
The genomic project of Serangium japonicum has been uploaded to NCBI. The datasets for Hi-C, transcriptome, RNA-ONT, and PacBio HiFi are accessible using the identifiers SRR2925231954, SRR2925232055, SRR2925231856 and SRR2925232257. The assembled genome has been submitted to the NCBI database under the accession number GCA._040543525.258. The annotation results have been uploaded in figshare59.
Technical Validation
The completeness of the assembly was evaluated using BUSCO v5.0.445, referencing the Insecta database (n = 1,367). The results indicated a BUSCO completeness of 97.8%, comprising 97.3% single-copy gene, 0.5% duplicated gene, 0.1% fragmented gene, and 2.1% missing gene. The analysis involved using Minimap2 and SAMtools software to align the reads from PacBio, Illumina, and RNA sequencing to the final assembly. Furthermore, the alignment rates of the RNA-sr, RNA-ONT, and HiFi data were observed to be 93.76%, 99.39%, and 99.85%, respectively. These findings substantiate the exceptional quality of the S. japonicum genome assembly.
Acknowledgements
This study was supported by grants from the·Key Science and Technology Project of CNTC (110202101053: LS-13), Key Science and Technology Projects·of YNTC (2023530000241004), the Science & Technology Fundamental Resources Investigation Program (2022FY100504), the Science and Technology Program of Guangzhou (202206010113), and National Natural Science Foundation of China (31970441).
Author contributions
X.W. contributed to the research design. K.N., Z.L. and H.L. collected the samples. J.J., M.Y. and Y.X. analyzed the data. M.Y. and C.H. wrote the draft manuscript and revised the manuscript. All co-authors contributed to this manuscript and approved it.
Code availability
No specific script was used in this work. All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding bioinformatic software.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Maolin Ye, Yonghui Xie.
References
- 1.Magro, A., Lecompte, E., Magné, F., Hemptinne, J. L. & Crouau-Roy, B. Phylogeny of ladybirds (Coleoptera: Coccinellidae): are the subfamilies monophyletic? Mol Phylogenet Evol.54, 833–848 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Bianchi, F. J. & Van der Werf, W. The effect of the area and configuration of hibernation sites on the control of aphids by Coccinella septempunctata (Coleoptera: Coccinellidae) in agricultural landscapes: a simulation study. Environ Entomol.32, 1290–1304 (2003). [Google Scholar]
- 3.Koch, R. L., Venette, R. C. & Hutchison, W. D. Influence of alternate prey on predation of monarch butterfly (Lepidoptera: Nymphalidae) larvae by the multicolored Asian lady beetle (Coleoptera: Coccinellidae). Environ Ecol.34, 410–416 (2005). [Google Scholar]
- 4.Giorgi, J. A. et al. The evolution of food preferences in Coccinellidae. Biol Control.51, 215–231 (2009). [Google Scholar]
- 5.Seago, A. E., Giorgi, J. A., Li, J. H. & Ślipiński, A. Phylogeny, classification and evolution of ladybird beetles (Coleoptera: Coccinellidae) based on simultaneous analysis of molecular and morphological data. Mol Phylogenet Evol.60, 137–151 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Maredia, K. M., Gage, S. H., Landis, D. A. & Scriber, J. M. Habitat use patterns by the seven-spotted lady beetle (Coleoptera: Coccinellidae) in a diverse agricultural landscape. Biol Control.2, 159–165 (1992). [Google Scholar]
- 7.Robertson, J. A., Giorgi, J. A., Li, J. & Ślipiński, S. A. Searching for natural lineages within the Cerylonid series (Coleoptera: Cucujoidea). Mol Phylogenet Evol.46, 193–205 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Sun, Y. X., Hao, Y. N., Riddick, E. W. & Liu, T. X. Factitious prey and artificial diets for predatory lady beetles: current situation, obstacles, and approaches for improvement: a review. Biocontrol Sci Technol.27, 601–619 (2017). [Google Scholar]
- 9.Ren, S. X., Wang, Z. Z., Qiu, B. L. & Yuan, X. The pest status of Bemisia tabaci in China and non-chemical control strategies. Entomol Sin.18, 279–288 (2001). [Google Scholar]
- 10.Sahar, F. & Ren, S. X. Interaction of Serangium japonicum (Coleoptera: Coccinellidae), an obligate predator of whitefly with immature stages of Eretmocerus sp. (Hymenoptera:Aphelinidae) with whitefly host (Homoptera: Aleyrodidae). Asian J. Plant Sci.3, 243–246 (2004). [Google Scholar]
- 11.Li, P., Chen, Q. Z. & Liu, T. X. Effects of a juvenile hormone analog, pyriproxyfen, on Serangium japonicum (Coleoptera: Coccinellidae), a predator of Bemisia tabaci (Hemiptera: Aleyrodidae). Biol Control.86, 7–13 (2015). [Google Scholar]
- 12.Kaneko, S. Seasonal and yearly change in adult abundance of a predacious ladybird Serangium japonicum (Coleoptera: Coccinellidae) and the citrus whitefly Dialeurodes citri (Hemiptera: Aleyrodidae) in citrus groves. Appl Entomol Zool.52, 481–489 (2017). [Google Scholar]
- 13.Ozawa, A. & Uchiyama, T. Effects of pesticides on adult ladybird beetle Serangium japonicum (Coleoptera: Coccinellidae), a potential predator of the tea spiny whitefly Aleurocanthus camelliae (Hemiptera: Aleyrodidae). Jap J Appl Entomol Zool.60, 45–49 (2016). [Google Scholar]
- 14.Tian, M., Zhang, S. Z. & Liu, T. X. Advances in research on the biology and ecology of Serangium japonicum Chapin, a predator of Bemisia tabaci (Gennadius). Chinese Journal of Applied Entomology.57, 800–805 (2020). [Google Scholar]
- 15.Belton, J. M. et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods.58, 268–276 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bushnell, B. BBtools. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 1 October 2022) (2014).
- 17.Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics33, 2202–2204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods.18, 170–175 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics.36, 2896–2898 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst.3, 95–98 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science.356, 92–95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steinegger, M. & Soding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol.35, 1026–1028 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol.215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 24.Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA117, 9451–9457 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hubley, R. et al. The Dfam database of repetitive DNA families. Nucleic Acids Res.44, D81–D89 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bao, W. et al. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. Dna.6, 11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org (accessed on 1 October 2022) (2013–2015).
- 28.Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics.29, 2933–2935 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res.33, D121–124 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan, P. P. & Lowe, T. M. TRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol Biol.1962, 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holt, C. & Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. Bmc Bioinformatics.12, 491 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim, D., Langmead, B. & Salzberg, S. L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods.12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol.20, 278 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bruna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. Nar Genom. Bioinform.3, lqaa108 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bruna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP: Eukaryotic gene prediction with self-training in the space of genes and proteins. Nar Genom. Bioinform.2, lqaa26 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res.32, W309–W312 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuznetsov, D. et al. OrthoDB V11: Annotation of orthologs in the widest sampling of organismal diversity Nucleic Acids Res.51, D445–D451, 10.1093/nar/gkac998 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O. & Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. Bmc Bioinformatics.19, 189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoskins, R. A. et al. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome research.25, 445–458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gibbs, R. A. et al. Insights into social insects from the genome of the honeybee Apis mellifera. Nature.443, 931–949 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crowley, L. M. The genome sequence of the common green lacewing, Chrysoperla carnea (Stephens, 1836). Wellcome open research.6, 334 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tribolium Genome Sequencing Consortium The genome of the model beetle and pest Tribolium castaneum. Nature.452, 949–955 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Crowley, L. The genome sequence of the seven-spotted ladybird, Coccinella septempunctata Linnaeus, 1758. Wellcome open research.6, 319 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen, M. Y. et al. A chromosome-level assembly of the harlequin ladybird Harmonia axyridis as a genomic resource to study beetle and invasion biology. Mol Ecol Resour.21, 1318–1332 (2021). [DOI] [PubMed] [Google Scholar]
- 45.Waterhouse, R. M. et al. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol.35, 543–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND Nat Methods.18, 366–368, 10.1038/s41592-021-01101-x (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Finn, R. D. et al. InterPro in 2017—Beyond protein family and domain annotations. Nucleic Acids Res.45, D190–D199 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huerta-Cepas, J. et al. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol.34, 2115–2122 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res.47, D427–D432 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Letunic, I. & Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res.46, D493–D496 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilson, D. et al. SUPERFAMILY—Sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res.37, D380–D386 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marchler-Bauer, A. et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res.45, D200–D203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lewis, T. E. et al. Gene3D: Extensive Prediction of Globular Domains in Proteins. Nucleic Acids Res.46, D1282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.NCBI Sequence Read Archivehttps://identifiers.org/ncbi/insdc.sra:SRR29252319 (2024).
- 55.NCBI Sequence Read Archivehttps://identifiers.org/ncbi/insdc.sra:SRR29252320 (2024).
- 56.NCBI Sequence Read Archivehttps://identifiers.org/ncbi/insdc.sra:SRR29252318 (2024).
- 57.NCBI Sequence Read Archivehttps://identifiers.org/ncbi/insdc.sra:SRR29252322 (2024).
- 58.NCBI Assemblyhttps://identifiers.org/ncbi/insdc.gca:GCA_040543525.2 (2024).
- 59.Jin, J. Genome annotation of Serangium japonicum. figshare. Dataset. 10.6084/m9.figshare.25904044 (2024).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- NCBI Sequence Read Archivehttps://identifiers.org/ncbi/insdc.sra:SRR29252319 (2024).
- NCBI Sequence Read Archivehttps://identifiers.org/ncbi/insdc.sra:SRR29252320 (2024).
- NCBI Sequence Read Archivehttps://identifiers.org/ncbi/insdc.sra:SRR29252318 (2024).
- NCBI Sequence Read Archivehttps://identifiers.org/ncbi/insdc.sra:SRR29252322 (2024).
- NCBI Assemblyhttps://identifiers.org/ncbi/insdc.gca:GCA_040543525.2 (2024).
- Jin, J. Genome annotation of Serangium japonicum. figshare. Dataset. 10.6084/m9.figshare.25904044 (2024).
Data Availability Statement
No specific script was used in this work. All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding bioinformatic software.