

Abstract
Cashmere grows from the secondary hair follicles (SHFs) that synchronously regenerate and degenerate in a circannual rhythm. Most studies examining factors related to cashmere growth have been performed on goat skin. However, the molecular properties and regulators preferentially expressed in SHFs are less clear. In this study, we isolated the SHFs from skin under a dissecting microscope and analysed the molecular signatures across the annual growth. It demonstrated the transcriptional profile of the isolated SHFs could accurately decipher the growth state and unequivocally distinguish three phases of the cashmere growth. We identified 1,289 molecular signatures that were differentially expressed between different phases. Real-time PCR verified the accuracy and reliability of our sequencing data for the representative genes. Taken together, the transcriptional profiles and identified molecular signatures extended previous findings, and provided a variety of promising targets for cashmere production, thereby offering valuable starting points for future work.
Subject terms: Transcriptomics, Self-renewal, RNA sequencing
Background & Summary
Cashmere is a special soft hair fibre of great resilience with high economic value because of its heat-preservation ability. Cashmere grows from the secondary hair follicles (SHFs), whereas the protective coarse hair grows from the primary hair follicles (PHFs) in cashmere goats that have these double coats1. As a type of smaller hair follicle (HF), SHFs usually surround the PHF in a follicle group2, and present with a different morphology from the PHFs3. Mature hair follicles periodically regenerate, undergoing repetitive cycles of relative quiescence (telogen), active growth (anagen), and destruction (catagen)4–7. In contrast to asynchronous growth, such as that of goat wool and horse mane, cashmere hair follicles (SHFs) shed every May for Liaoning cashmere goat, which exhibit a notably synchronized seasonal growth. We aim to unearth the dynamic features and regulatory signatures of cashmere follicles by examining the transition between quiescence, regeneration, and degeneration during annual cashmere growth.
At present, most studies related to cashmere have been performed on goat skin. However, the skin is composed of different cell types, HFs and sweat glands. Moreover, goat skin comprises two types of HFs, SHFs and PHFs. The growth and development of PHFs and SHFs are regulated by distinctive molecular mechanisms in some ways. Since the growth and apoptosis of PHFs are asynchronous, to eliminate the influence from other cell types in the skin samples, especially removing the noise signatures of the phase transition from PHFs, we performed the analysis on the isolated SHFs rather than the skin or PHFs to precisely study the molecular signatures of SHFs.
In the present study, we explored transcriptional signatures and their functional significance in cashmere follicles (SHFs) from Liaoning cashmere goats. We isolated SHF clusters under the microscope by cutting out the epidermis and removing the connective tissues. RNA sequencing experiments were performed on the collected SHFs to discover molecular signatures that were preferentially expressed in SHFs and differentially expressed in specific phases. The principal component analysis (PCA) based on the TPM (transcripts per million) data discovered three separated clusters corresponding to the telogen, anagen and catagen phases, suggesting that the expression profiles could accurately decipher the SHF states. These results uncovered the concordant expression changes of molecular signatures that function together to orchestrate the seasonal cashmere growth and shedding.
To precisely investigate the dynamic changes in gene expression patterns of SHFs, we performed RNA sequencing in 12 samples across six-time points, representing the anagen (May and September), catagen (November and December) and telogen (February and March) phases, each with two biological replicates (male and female). About 348 million reads per phase were obtained, with 1,044 million reads in total. Differentially expressed genes (DEGs) were identified. Real-time PCR verified the accuracy and reliability of our sequencing data for the representative genes. These transcriptional profiles not only revealed molecular signatures of the SHF at each state but also will provide potential avenues for identifying and characterizing regulatory pathways, networks and determinants of each state.
Methods
Secondary hair follicles tissue sampling
All experimental cashmere goats were from the Modern Agricultural Production Base Construction Engineering Centre of Liaoning Province, Liaoyang. Six cashmere goats (three male and three female) that were 1.5 years old were selected in each time points (February, March, May, September, November and December). The SHFs were isolated by cutting out the epidermis and removing the connective tissues under a dissecting microscope (SMZ150, Motic, Xiamen, China) from a piece of back skin (0.5 × 0.5 cm) of each goat. All samples were flash-frozen and stored in liquid nitrogen until processing. The detailed experiment procedure please refer to our previous work3. Total RNA was extracted from these secondary follicles using RNAiso Plus as per the manufacturer’s instructions (Takara, Dalian, China). All the experimental goats were fed with cashmere goat standard feed (NY/T 2893-2016). All the animal studies were reviewed and approved by the Ethical Committee of Shenyang Agricultural University, China (ID no. 2021121701).
RNA sequencing
To systematically investigate the dynamic changes in gene expression patterns of SHFs, we isolated SHF clusters from back skin under a dissecting microscope3 and performed RNA sequencing across six-time points, each with two biological replicates (male and female), representing the anagen (May and September), catagen (November and December) and telogen (February and March) phases. The quality of RNA was measured using the RNA Nano 6000 Assay Kit with the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). All samples had a 260/280 nm ratio between 1.91 and 1.99, RIN values exceeded 7.5 (Table 1). Then, a total of 3 µg RNA per sample pooled from three cashmere goats was used as the basis for RNA library construction by the sequencing company (Novogene, Beijing, China). RNA library construction by the sequencing company (Novogene, Beijing, China) and pair-end sequencing (150 bp) was performed on the Illumina HiSeq 4000 platform (Illumina, San Diego, CA, USA).
Table 1.
RNA quantification and quality control variables including 260/280 ratio and RNA integrity number (RIN).
| Month | Phase | Sex | RNA Concentration (ng/μL) | 260/280 Ratio | RIN |
|---|---|---|---|---|---|
| February | Telogen | Female | 264.7 | 1.92 | 8.1 |
| February | Telogen | Male | 485.2 | 1.93 | 8.5 |
| March | Telogen | Female | 314.8 | 1.94 | 8.6 |
| March | Telogen | Male | 724.6 | 1.94 | 8.6 |
| May | Anagen | Female | 657.2 | 1.99 | 8.4 |
| May | Anagen | Male | 472.0 | 1.94 | 8.1 |
| September | Anagen | Female | 408.3 | 1.93 | 8.2 |
| September | Anagen | Male | 505.7 | 1.99 | 8.5 |
| November | Catagen | Female | 279.5 | 1.91 | 7.9 |
| November | Catagen | Male | 211.2 | 1.99 | 8.5 |
| December | Catagen | Female | 409.2 | 1.91 | 7.5 |
| December | Catagen | Male | 361.9 | 1.94 | 7.9 |
Data analysis for transcriptome data
All raw RNA-seq sequence data (FASTQ files) have been deposited on the NCBI Gene Expression Omnibus (for details, see Data Records). Clean data were obtained after trimming adapter sequences from the raw sequence reads and removing low-quality reads. About 348 million reads per phase were obtained, with 1,044 million reads in total. Detailed information on each sample and sequencing data are listed in Table 2. The gene expression level was quantified by using Kallisto (v.0.46.1)8 to map to Capra hircus (GCF_001704415.1_ARS1) primary transcript sequences, and by using the Sleuth (v.0.30.0)9 program.
Table 2.
Information of samples and sequencing data.
| Month | Phase | Sex | Sequencing Data (G) |
|---|---|---|---|
| February | Telogen | Female | 13.91 |
| February | Telogen | Male | 14.08 |
| March | Telogen | Female | 12.97 |
| March | Telogen | Male | 15.53 |
| May | Anagen | Female | 13.08 |
| May | Anagen | Male | 13.70 |
| September | Anagen | Female | 12.04 |
| September | Anagen | Male | 12.49 |
| November | Catagen | Female | 11.88 |
| November | Catagen | Male | 12.32 |
| December | Catagen | Female | 12.33 |
| December | Catagen | Male | 12.36 |
Differential expression analysis
To identify factors in the transition between consecutive phases, we assayed genes that were differentially expressed with potential roles in the phase transformation during the hair cycle. Given the appreciable difference in gene expression profiles between May and September, we compared those of May and September to the other phases. The gene abundances were obtained by Kallisto-Sleuth pipeline in TPM8. Differentially expressed genes between the different phases were identified using the Sleuth package in R (v.4.0.2)9. The filtered criteria used were: (1) TPM ≥ 20 in at least two samples to remove low-abundance transcripts, (2) an adjusted p-value < 0.05, and (3) the threshold of expression change is log2(Fold Change) ≥ 1.
Data Records
Raw RNA-seq data files and TPM values are available on the Gene Expression Omnibus (GEO) database with accession no. GSE22147210. In addition, the identified DEGs are available on the figshare repository (10.6084/m9.figshare.27126594)11.
Technical Validation
Principal component analysis (PCA)
A principal component analysis based on TPM data discovered three separated clusters corresponding to telogen, anagen and catagen phase, respectively (Fig. 1). It revealed that SHFs could be divided into three main clusters that represent three phases of the hair growth cycle. Anagen was clustered closer to catagen than telogen, which was markedly distinct compared to the others. Early anagen (May) showed a profile similar to that of mid-anagen (September), although there was one sample of November clustered with September. Together, the results strongly support the validity and reliability of our data.
Fig. 1.
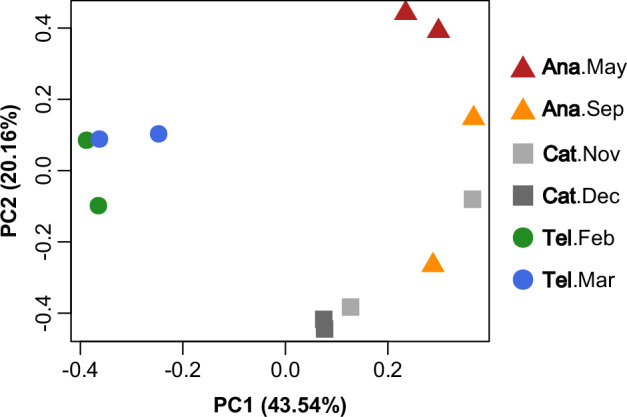
Principal component analysis for all samples. Ana—anagen; Cat—catagen; Tel—telogen.
Experimental validation of transcriptomic data
Real-time PCR verified the accuracy and reliability of our sequencing data for the representative genes in anagen (or catagen) compared to telogen in SHFs. Consistent with and extending on our sequencing data, these regulators were either upregulated in the telogen phase (Pparg, Igfbp3 and Zfp36l2) or in the anagen phase (Col1a2) or in both anagen and catagen (Lgals1) (Fig. 2a, b). The correlation analysis between RNA-seq and RT-qPCR results was conducted using the corrplot package (v.0.84) in R for the representative genes. The values were transformed into fold change (the ratio of mean value in anagen or catagen to that in telogen) for comparison (Fig. 2c). Both the RT-qPCR results and sequencing data showed that the expressions of these genes were upregulated in the phases as the expected regulatory functions indicated (Fig. 2). Taken together, the PCR results were in agreement with those from mRNA-seq data, thereby providing compelling evidence of the accuracy and reliability of our sequencing data.
Fig. 2.

Expression profiles of the representative genes. (a) Gene expression (mean TPM between two biological replicates from RNA-Seq) profiles of the representative genes in anagen, catagen and telogen. (b) Relative expression of representative genes in three phases detected with RT-qPCR. n = 3 and data are means ± SDs in all bar graphs. *p < 0.05, **p < 0.01, ***p < 0.001. Significant analysis was performed via one-sided Student’s t-test. (c) The correlation analysis was performed between RNA-seq and RT-qPCR. Values are expressed in log2 base. Ana—anagen; Cat—catagen; Tel—telogen. Circle or triangle represents anagen or catagen compared to telogen, respectively. Genes are annotated in different colour.
Acknowledgements
We are grateful to Dan Guo of the Modern Agricultural Production Base Construction Engineering Centre of Liaoning Province and Hong Zhang of Shenyang Agricultural University-Wellhope Veterinary Teaching Hospital for their kind help and cooperation during the animal experiments.
Author contributions
H.X. conceived and designed the study; Y.L., M.W., X.L., H.D., J.Z. and X.Z. collected the samples; H.X., Y.L., M.W., Z.L. and S.S. performed bioinformatics analyses; Y.L. and M.W. prepared figures and tables; H.X. and Y.L. drafted the manuscript; H.X. and S.Z. revised the manuscript; H.X. contributed to funding acquisition and all authors approved the final manuscript.
Code availability
Software and their versions used for RNA-seq analysis were described in Methods. No custom code was used to generate or process the data described in the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Yanlei Liu, Minglin Wang.
References
- 1.Dong, Y. et al. Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (Capra hircus). Nat Biotechnol31, 135–141, 10.1038/nbt.2478 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Yang, C. H. et al. Melatonin promotes secondary hair follicle development of early postnatal cashmere goat and improves cashmere quantity and quality by enhancing antioxidant capacity and suppressing apoptosis. J Pineal Res67, e12569, 10.1111/jpi.12569 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Wang, M. et al. Discovery and Functional Analysis of Secondary Hair Follicle miRNAs during Annual Cashmere Growth. Int J Mol Sci24, 10.3390/ijms24021063 (2023). [DOI] [PMC free article] [PubMed]
- 4.Lavker, R. M. et al. Hair follicle stem cells. J Investig Dermatol Symp Proc8, 28–38, 10.1046/j.1523-1747.2003.12169.x (2003). [DOI] [PubMed] [Google Scholar]
- 5.Paus, R. & Cotsarelis, G. The biology of hair follicles. N Engl J Med341, 491–497, 10.1056/nejm199908123410706 (1999). [DOI] [PubMed] [Google Scholar]
- 6.Woo, J., Suh, W. & Sung, J. H. Hair Growth Regulation by Fibroblast Growth Factor 12 (FGF12). Int J Mol Sci23, 10.3390/ijms23169467 (2022). [DOI] [PMC free article] [PubMed]
- 7.Liang, A. et al. Signaling pathways in hair aging. Front Cell Dev Biol11, 1278278, 10.3389/fcell.2023.1278278 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nature biotechnology34, 525–527, 10.1038/nbt.3519 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Pimentel, H., Bray, N. L., Puente, S., Melsted, P. & Pachter, L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods14, 687–690, 10.1038/nmeth.4324 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Xue, H., Wang, M., Liu, Z., Liu, Y. & Sheng, S. Molecular Signatures and Functional Analysis of Secondary Hair Follicles in Cashmere Goat During Annual Cashmere Growth. NCBI Gene Expression Omnibus (GEO)https://identifiers.org/geo/GSE221472 (2023).
- 11.Xue, H., Liu, Y., Wang, M., Liu, Z. & Sheng, S. Differentially expressed genes of secondary hair follicles during annual cashmere growth. Figshare10.6084/m9.figshare.27126594 (2024).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Xue, H., Liu, Y., Wang, M., Liu, Z. & Sheng, S. Differentially expressed genes of secondary hair follicles during annual cashmere growth. Figshare10.6084/m9.figshare.27126594 (2024).
Data Availability Statement
Software and their versions used for RNA-seq analysis were described in Methods. No custom code was used to generate or process the data described in the manuscript.