

Abstract
Lung adenocarcinoma (LUAD) is a major contributor to cancer-related deaths, distinguished by its pronounced tumor heterogeneity and persistent challenges in overcoming drug resistance. In this study, we utilized single-cell RNA sequencing (scRNA-seq) to dissect the roles of programmed cell death (PCD) pathways, including apoptosis, necroptosis, pyroptosis, and ferroptosis, in shaping LUAD heterogeneity, immune infiltration, and prognosis. Among these, ferroptosis and pyroptosis were most significantly associated with favorable survival outcomes, highlighting their potential roles in enhancing anti-tumor immunity. Distinct PCD-related LUAD subtypes were identified, characterized by differential pathway activation and immune cell composition. Subtypes enriched with cytotoxic lymphocytes and dendritic cells demonstrated improved survival outcomes and increased potential responsiveness to immunotherapy. Drug sensitivity analysis revealed that these subtypes exhibited heightened sensitivity to targeted therapies and immune checkpoint inhibitors, suggesting opportunities for personalized treatment strategies. Our findings emphasize the interplay between PCD pathways and the tumor microenvironment, providing insights into the mechanisms underlying tumor drug resistance and immune evasion. By linking molecular and immune features to clinical outcomes, this study highlights the potential of targeting PCD pathways to enhance therapeutic efficacy and overcome resistance in LUAD. These results contribute to a growing framework for developing precise and adaptable cancer therapies tailored to specific tumor characteristics.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12672-024-01736-0.
Keywords: Lung adenocarcinoma, Programmed cell death, Single-cell RNA sequencing, Tumor heterogeneity, Drug resistance, Immune infiltration, Therapeutic targets
Introduction
Lung adenocarcinoma (LUAD) remains one of the leading causes of cancer-related mortality worldwide, accounting for a significant proportion of cancer deaths globally in recent years [1, 2]. Despite significant advancements in its diagnosis and treatment, particularly through targeted therapies and immunotherapies, the overall prognosis for LUAD patients remains poor [3–5]. The heterogeneity of LUAD, including variations in the tumor microenvironment (TME), genetic mutations, and molecular subtypes, poses challenges in diagnosis and treatment [6–8]. This complexity is especially pronounced in LUAD, where cellular diversity influences tumor progression, metastasis, and treatment outcomes [7, 8]. Thus, a deeper understanding of the molecular and cellular heterogeneity within LUAD is essential to advancing therapeutic strategies and enhancing patient survival.
A critical driver of LUAD heterogeneity is the TME, a dynamic ecosystem comprising immune cells, stromal components, and extracellular matrix [9, 10]. The TME modulates tumor growth and therapeutic response through complex interactions, with immune cells playing dual roles in either mounting anti-tumor responses or facilitating tumor evasion and immune suppression [11]. Elucidating the immune landscape of LUAD at single-cell resolution offers valuable insights into immune-tumor interactions and the identification of therapeutic targets to enhance anti-tumor immunity.
Programmed cell death (PCD) is a critical cellular process that regulates tissue homeostasis by selectively removing damaged, dysfunctional, or potentially harmful cells, thereby preserving the integrity and function of tissues [12]. Dysregulation of PCD pathways, such as apoptosis, necroptosis, pyroptosis, and autophagy, is frequently observed in cancer and contributes to tumor development, immune evasion, and resistance to therapy [13, 14]. Emerging evidence suggests that various forms of PCD may have distinct roles in the TME and that they can influence immune cell infiltration, tumor aggressiveness, and patient prognosis [15, 16]. For instance, recent studies have shown that pathways like ferroptosis, a regulated form of cell death associated with lipid peroxidation, may promote both cancer cell death and immune responses [17]. Similarly, autophagy, while primarily a cell survival mechanism, can paradoxically lead to cell death under certain conditions and impact immune responses in the tumor context [18, 19]. These findings underscore the importance of elucidating PCD-related mechanisms in LUAD to better understand how these processes interact with the immune system and influence patient outcomes.
In the context of LUAD, PCD-related pathways represent an attractive area of research due to their dual roles in both tumor suppression and potential therapeutic resistance [20]. Alterations in PCD-related gene expression and pathway activation are often linked to aggressive cancer phenotypes and poor prognosis [21, 22]. For instance, defects in apoptotic pathways can allow cancer cells to evade immune surveillance and proliferate uncontrollably. Conversely, activation of certain forms of PCD, such as pyroptosis, can enhance anti-tumor immune responses by releasing damage-associated molecular patterns (DAMPs) that recruit and activate immune cells [23, 24]. Thus, a detailed exploration of PCD-related pathways in LUAD could yield valuable insights into the mechanisms underlying tumor immune evasion and identify potential biomarkers or therapeutic targets for modulating the immune response.
Single-cell RNA sequencing (scRNA-seq) has emerged as a powerful tool for investigating cellular heterogeneity within tumors, enabling researchers to dissect the transcriptomic profiles of individual cells within the TME [25]. Unlike bulk RNA sequencing, which provides an averaged expression profile across all cells, scRNA-seq allows for the identification of specific cell subpopulations and their unique roles in cancer biology [26]. This approach has been instrumental in revealing rare cell types, identifying treatment-resistant clones, and characterizing immune cell infiltration patterns within tumors. In LUAD, scRNA-seq can be particularly useful for delineating the roles of specific immune cells, such as cytotoxic T cells, dendritic cells, and tumor-associated macrophages, in modulating the TME and influencing patient responses to immunotherapy [27, 28]. Furthermore, single-cell transcriptomic analysis can uncover the expression of PCD-related genes and pathways at a cellular resolution, providing a deeper understanding of how these processes vary across different cell types and tumor stages.
While previous studies have explored PCD pathways in various cancers, there is still limited knowledge regarding their roles within the TME of LUAD, especially at the single-cell level. Most research on PCD in cancer has focused on bulk tumor analyses, which overlook the heterogeneity of cell populations within the tumor [29, 30]. Consequently, the specific contributions of distinct cell subpopulations to PCD pathway activation and immune cell interactions remain poorly understood. By using scRNA-seq to analyze PCD-related pathways and immune infiltration patterns in LUAD, we can potentially uncover novel biomarkers and therapeutic targets that are specific to particular cell types or subpopulations within the tumor.
Given the pivotal role of PCD in tumor biology and immune modulation, this study aims to investigate PCD-related pathways in LUAD at the single-cell level. Specifically, we sought to determine how these pathways are differentially expressed across cell types and stages within the TME and how they correlate with immune cell infiltration. Additionally, we aimed to identify PCD-related subtypes within LUAD, examining their distinct molecular profiles, immune infiltration characteristics, and potential clinical relevance. By analyzing these subtypes, we hope to elucidate the underlying mechanisms that drive tumor heterogeneity and immune evasion, providing a foundation for the development of personalized therapies that target PCD pathways to enhance anti-tumor immunity.
Methods
Data acquisition
In this study, we included bulk RNA-seq data from 320 LUAD patients in the TCGA-LUAD cohort and retrieved gene expression datasets with complete clinical annotations from the GEO public database (GSE31210, GSE50081). The data from these cohorts were downloaded, and both gene expression and clinical information were compiled. Only samples with complete survival information, including survival time and event status, were retained to ensure the reproducibility of the study across different cohorts. After merging gene expression and clinical data from the three datasets, we obtained a final cohort of 854 samples. Additionally, four scRNA-seq datasets (GSM4506698, GSM4506699, GSM4506700, and GSM4506701) were included for further single-cell analysis.
Single-cell data analysis
To analyze the four scRNA-seq datasets, which included a total of 12,554 cells, we used the Seurat package for integrated analysis. Genes expressed in at least three cells were retained, and cells were filtered based on the following criteria: gene expression counts between 250 and 8000, mitochondrial gene content below 35%, and UMI counts above 1000. These thresholds were selected to balance the inclusion of biologically relevant cells while minimizing technical noise. Specifically, the lower bound of 250 ensures the inclusion of cells with sufficient transcriptional activity, while the upper bound of 8000 excludes doublets and potential multiplets. Similarly, a mitochondrial gene content threshold of 35% was chosen to exclude low-quality or dying cells, which often exhibit high mitochondrial gene expression. This filtering process resulted in the retention of 9254 high-quality cells for downstream analysis.
Identification of PCD-related clusters
To classify LUAD samples into clusters based on PCD-related genes, we employed the ConsensusClustersPlus R package. The optimal number of clusters was determined based on the consensus matrices and cumulative distribution function (CDF) curves of the consensus index, with the number of clusters ranging from 2 to 9. The clustering results were evaluated and visualized using principal component analysis (PCA), which illustrated the separation of clusters along the principal components. This approach helped assess the clustering structure by highlighting the distinct groupings in reduced-dimensional space. Kaplan–Meier (K–M) survival analysis with the log-rank test was then performed to compare survival differences between the two identified clusters. To assess PCD pathway activity in each LUAD patient, the single-sample gene set enrichment analysis (ssGSEA) was applied to calculate the normalized enrichment score (NES) of PCD-related pathways.
Drug sensitivity analysis
The half-maximal inhibitory concentration (IC50) of drugs was determined using the R package ‘pRRophetic.’ This analysis focused on drugs relevant to LUAD treatment, including targeted therapies and chemotherapeutic agents, obtained from the Genomics of Drug Sensitivity in Cancer (GDSC) database. IC50 values were computed based on predictive models that integrate gene expression profiles with drug sensitivity data, providing insights into the differential drug responses of identified LUAD subtypes. Statistical comparisons of drug sensitivity were conducted between clusters 1 and 2 to assess the differential drug responses across risk groups.
Immune infiltration analysis
The ESTIMATE algorithm was used to analyze differences in stromal, immune, and ESTIMATE scores between clusters, implemented through the “estimate” R package. Using the ssGSEA method in the “GSVA” R package, we compared enrichment scores to determine the relative abundance of 23 types of tumor-infiltrating immune cells. Visualization was performed with the “limma,” “ggpubr,” and “reshape2” R packages. The MCP-counter was used to estimate immune cell abundance, while CIBERSORT, EPIC, and TIMER algorithms were applied to evaluate immune cell infiltration levels in high- and low-risk groups. The results were visualized using box plots, heat maps, and violin plots.
Functional enrichment analysis of differentially expressed genes
The Seurat package’s FindAllMarkers function was used to identify differentially expressed genes (DEGs) between cluster 1 and cluster 2, with an adjusted p-value < 0.05 and absolute logFC > 0.585 as thresholds. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of DEGs were conducted using the “clusterProfiler” R package (version 4.0.5), with a false discovery rate (FDR) < 0.05 to identify significantly enriched pathways.
Construction and validation of a prognostic model
Risk scores for each patient were calculated using the following formula: Risk score = Σi Coefficient (mRNAi) × Expression (mRNAi). The cutoff value for risk stratification was determined using the “surv_cutpoint” function of the R package “survminer,” which utilizes maximally selected rank statistics to derive an optimal threshold by simulating statistically significant survival differences between groups. Patients were then divided into high- and low-risk groups based on this cutoff. Kaplan–Meier analysis was performed to compare overall survival (OS) between the high- and low-risk groups. The predictive accuracy of the prognostic model was evaluated through time-dependent receiver operating characteristic (ROC) analysis, with area under the curve (AUC) values calculated at 1-year, 3-year, and 5-year intervals to assess the model’s discriminative performance over time. Additionally, calibration curves were used to compare predicted versus observed survival probabilities, and decision curve analysis (DCA) was performed to evaluate the clinical utility and net benefit of the model across different risk thresholds. The model was further validated in the GSE31210 cohort.
Results
Single-cell sequencing analysis
In this study, we analyzed single-cell sequencing data to examine the distribution and characteristics of cell types and cell cycle phases across LUAD samples (Fig. 1). The t-SNE plot of sample type distribution across GSM4506698, GSM4506699, GSM4506700, and GSM4506701 reveals distinct clustering patterns within the dataset (Fig. 1A). We further assessed the distribution of cells in different cell cycle phases, including G1, G2M, and S, highlighting phase-specific organization within the samples (Fig. 1B). Additionally, cell types were classified as aneuploid, diploid, or undefined, with relative proportions demonstrating that diploid cells predominate in GSM4506700 and GSM4506701, whereas aneuploid cells are more prominent in GSM4506698 and GSM4506699 (Fig. 1C, D). The balance of cell cycle phase distribution is shown in Fig. 1E, with pre- and post-QC data quality comparisons available in Supplementary Fig. 1.
Fig. 1.

Single-cell RNA sequencing (scRNA-seq) analysis of lung adenocarcinoma (LUAD) samples. A t-SNE plot showing sample type distribution across four datasets: GSM4506698, GSM4506699, GSM4506700, and GSM4506701. Each color represents a different dataset, highlighting distinct clustering. B t-SNE plot illustrating cell cycle phase distribution (G1, G2M, and S) within the samples. C t-SNE plot displaying cell ploidy status, categorized as aneuploid, diploid, or undefined. D Bar plot of cell type proportions in each dataset, revealing a predominance of diploid cells in GSM4506700 and GSM4506701 and a higher proportion of aneuploid cells in GSM4506698 and GSM4506699. E Bar plot depicting cell cycle phase distribution (G1, G2M, S) across datasets, showing balanced proportions
PCD-related pathway enrichment analysis
We conducted a pathway enrichment analysis to identify variations in PCD-related pathways between malignant and normal lung cells (Fig. 2). The heatmap in Fig. 2A highlights differences in activation of key PCD pathways such as parthanatos, ferroptosis, and oxeiptosis, underscoring the distinct pathway enrichment profiles between cell types. Notably, aneuploid cells in GSM4506701 showed significant enrichment in netotic cell death pathways, while diploid cells displayed prominent activation of cuproptosis and pyroptosis pathways, indicating differential pathway engagement across cellular subtypes (Fig. 2A).
Fig. 2.

Programmed cell death (PCD) pathway enrichment in LUAD cells. A Heatmap of PCD pathway activation in malignant versus non-malignant cells across datasets, highlighting distinct activation profiles in pathways like parthanatos, ferroptosis, and oxeiptosis. B Bar plot showing activation scores for key PCD pathways in aneuploid and diploid cells within each dataset. Aneuploid cells in GSM4506701 exhibit notable enrichment in netotic cell death pathways, while diploid cells show higher activation in cuproptosis and pyroptosis pathways. C Heatmap of PCD-related gene expression across aneuploid and diploid cells, revealing differential profiles by cell type. D Bar plots showing PCD-related gene expression, with genes such as NPC2, NAPSA, and SAT1 elevated in aneuploid cells and FTL, FTL1, and FTL2 elevated in diploid cells
Identification of PCD-related subtypes
To identify PCD-related subtypes in LUAD patients, we performed consensus clustering based on PCD-related gene expression profiles. The CDF of the ConsensusClusterPlus algorithm indicated that the optimal clustering stability was achieved at k = 2 (Fig. 3A). Accordingly, 213 OS samples from the GEO and TCGA datasets were grouped into two distinct clusters: Cluster 1 (n = 451) and Cluster 2 (n = 402), as illustrated by the clear separation into two blue color blocks (Fig. 3B). Survival analysis revealed a significant prognostic difference between the clusters, with Cluster 2 demonstrating better OS compared to Cluster 1 (p < 0.001), as shown in the Kaplan–Meier curve (Fig. 3C).
Fig. 3.

Identification of PCD-related clusters in LUAD patients. A Cumulative distribution function (CDF) plot from consensus clustering, identifying k = 2 as the optimal cluster number. B Consensus matrix for k = 2, illustrating clear separation of LUAD samples into Cluster 1 and Cluster 2. C Kaplan–Meier survival curve comparing overall survival between clusters, with Cluster 2 showing significantly improved survival (p < 0.0001). D Violin plot of PCD scores by cluster, indicating a significantly higher PCD score in Cluster 1. E Heatmap of PCD-related gene expression across clusters, with genes like PCBP2, HSBP1, BAG3, JUN, and DAP1 highly expressed in Cluster 2. F Heatmap of pathway enrichment scores for PCD pathways (alkaliptosis, cuproptosis, autophagy, oxeiptosis, pyroptosis), showing notable differences between clusters
Using the ssGSEA algorithm, we calculated PCD scores for each cluster, finding that Cluster 1 exhibited significantly higher PCD pathway activity than Cluster 2 (p < 0.05), highlighting distinct biological characteristics (Fig. 3D). A heatmap of PCD-related gene expression (Fig. 3E) showed that Cluster 2 had higher expression of genes such as PCBP2, HSBP1, BAG3, JUN, and DAP1, suggesting a PCD gene profile associated with a more favorable prognosis. Pathway enrichment analysis further revealed significant differences in activation levels of key pathways, including alkaliptosis, cuproptosis, autophagy, oxeiptosis, and pyroptosis (Fig. 3F). These findings indicate that each cluster engages distinct PCD mechanisms, contributing to their unique TME and clinical behavior. Furthermore, Fig. 4 explores the differential drug sensitivity across these subtypes, providing insights into potential therapeutic vulnerabilities and treatment strategies for each cluster.
Fig. 4.
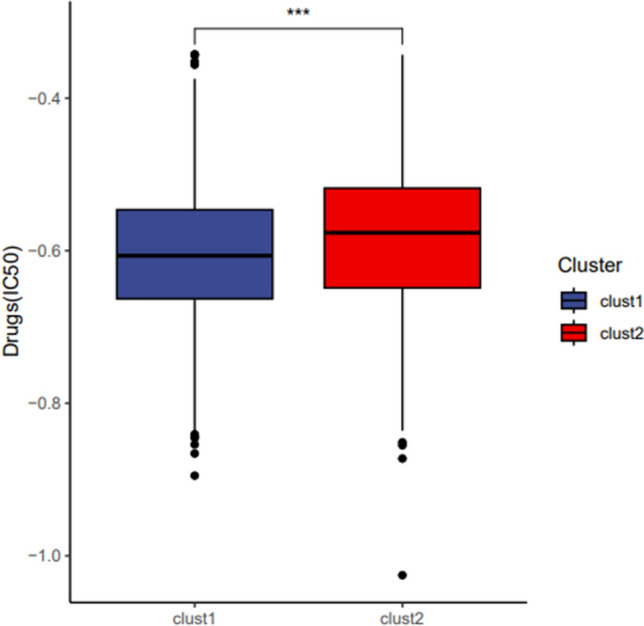
Drug sensitivity analysis in PCD-related LUAD clusters. Box plot of IC50 values for selected drugs between Cluster 1 and Cluster 2, highlighting significant drug sensitivity differences, with Cluster 2 showing higher sensitivity to certain drugs, suggesting cluster-specific therapeutic vulnerabilities
Analysis of immune infiltration differences between PCD-related subtypes
We further investigated immune cell infiltration differences within the tumor immune microenvironment across the two subtypes. Using the ESTIMATE algorithm, we calculated immune infiltration levels for each cluster subtype. The results (Fig. 5A) indicate that Cluster 2 exhibits significantly higher immune, stromal, and ESTIMATE scores compared to Cluster 1, whereas Cluster 1 shows a notably higher tumor purity. Following this, we examined the distribution characteristics of immune cell infiltration across the clusters. As displayed in the heatmap (Fig. 5B), Cluster 2 demonstrates significantly elevated immune cell infiltration levels, particularly for cell types such as B lineage cells, cytotoxic lymphocytes, monocytic lineage cells, and dendritic cells. This heightened immune response in Cluster 2 aligns with its observed survival advantage over Cluster 1.
Fig. 5.

Immune infiltration in PCD-related LUAD clusters. A Violin plots comparing immune score, stromal score, ESTIMATE score, and tumor purity between clusters (using ESTIMATE algorithm). Cluster 2 shows significantly higher immune and stromal scores and a higher ESTIMATE score, while Cluster 1 has higher tumor purity. B Heatmap of immune cell infiltration levels by cluster, with elevated infiltration of immune cell types (e.g., B cells, cytotoxic lymphocytes, monocytes, dendritic cells) in Cluster 2. C Box plot of immune checkpoint expression between clusters, showing significantly higher expression of checkpoints (e.g., YTHDF1, NRP1, B2M, TNFSF15) in Cluster 2, indicating potential responsiveness to immunotherapy
Additionally, expression differences in immune checkpoints between clusters were assessed using box plots (Fig. 5C). Results reveal that immune checkpoint markers, including YTHDF1, NRP1, B2M, and TNFSF15, are expressed at significantly higher levels in Cluster 2, suggesting that patients in this cluster may be more responsive to immunotherapy.
We then analyzed pathway enrichment differences between the two subtypes. GO enrichment analysis showed that DEGs between the clusters are primarily enriched in pathways such as small GTPase-mediated signal transduction, positive regulation of cell development, neuronal cell body, and DNA-binding transcription factor binding (Fig. 6A). Meanwhile, KEGG pathway enrichment analysis highlighted that DEGs are significantly enriched in pathways like PI3K-Akt signaling, human papillomavirus infection, and Kaposi sarcoma-associated herpesvirus infection (Fig. 6B).
Fig. 6.

Pathway enrichment analysis of differentially expressed genes (DEGs) between clusters. A Circular plot of Gene Ontology (GO) enrichment for DEGs, showing significant pathways, including small GTPase signaling, positive regulation of cell development, and transcription factor binding. B Circular plot of KEGG pathway enrichment for DEGs, revealing significant pathways such as PI3K-Akt signaling, human papillomavirus infection, and Kaposi sarcoma-associated herpesvirus infection
In terms of prognosis, Fig. 7A presents the distinction between high-risk and low-risk subtypes within the TCGA dataset. A scatter plot of survival status against risk scores shows a tendency for high-risk patients to be deceased, while low-risk patients are more likely to be alive (Fig. 7B). We then assessed the differential expression of genes such as BAMBI, TMCC2, NOX4, DKK1, and CBS between the two subtypes (Fig. 7C).
Fig. 7.
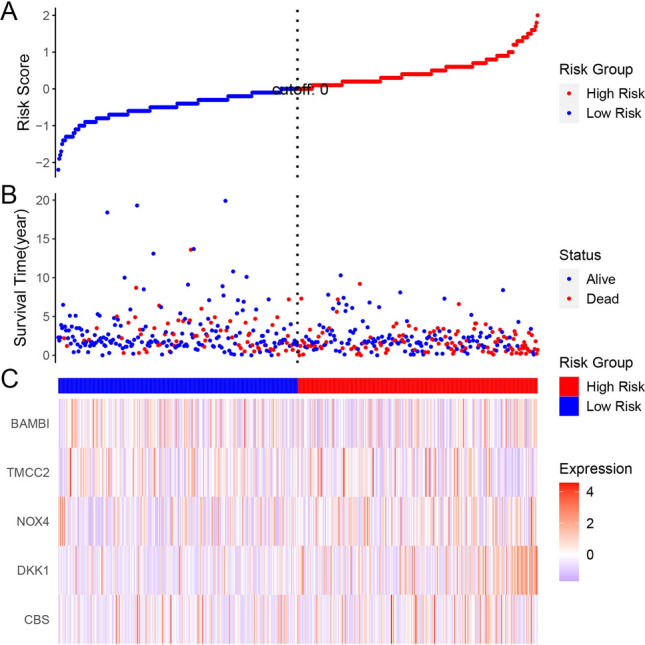
Risk stratification based on PCD-related genes in TCGA dataset. A Risk score distribution plot with a cutoff separating high-risk and low-risk groups. B Scatter plot of survival status versus risk score, showing higher mortality among high-risk patients. C Heatmap of gene expression (BAMBI, TMCC2, NOX4, DKK1, CBS) in high-risk versus low-risk groups, illustrating distinct expression patterns associated with risk
Kaplan–Meier survival curves from the TCGA dataset (Fig. 8A) confirm a significant survival difference, with the low-risk group showing markedly better prognosis than the high-risk group. The ROC curves (Fig. 8B) further support the model’s predictive power, with AUC values of 0.85, 0.88, and 0.84 at 1, 3, and 5 years, respectively, indicating high specificity and sensitivity in prognosis prediction. Similarly, in the GSE31210 dataset, Kaplan–Meier survival curves reveal significant survival differences between the high- and low-risk groups, with ROC analysis further validating the model’s prognostic accuracy (Fig. 8C, D).
Fig. 8.

Prognostic analysis of PCD-related subtypes in TCGA and GSE31210 datasets. A Kaplan–Meier survival curves for high- and low-risk groups in TCGA dataset, with the low-risk group showing better prognosis (p < 0.0001). B ROC curve for TCGA dataset, with AUC values of 0.85, 0.88, and 0.84 at 1, 3, and 5 years, demonstrating strong predictive performance. C Kaplan–Meier survival curves for GSE31210 dataset, reaffirming significant survival differences between risk groups. D ROC curve for GSE31210, supporting the model’s prognostic accuracy with high AUC values
Discussion
This study investigates the role of PCD pathways in the heterogeneity and immune microenvironment of LUAD at the single-cell level, revealing their critical impact on patient prognosis. By analyzing PCD-related gene expression patterns, we identified two distinct LUAD subtypes characterized by differences in immune cell infiltration, PCD pathway activation, and clinical outcomes. These findings highlight the intricate interplay between PCD mechanisms and the TME, offering new insights into LUAD heterogeneity and potential therapeutic targets.
Our results align with recent studies that highlight the substantial heterogeneity within LUAD. Previous work has shown that variations in cell types, immune cell infiltration, and gene expression profiles contribute to different responses to therapy and clinical outcomes among LUAD patients [31, 32]. However, while many studies have focused on bulk RNA sequencing data, our single-cell approach allowed us to dissect this heterogeneity at a more granular level, identifying specific PCD-related subtypes with unique immune landscapes and gene expression patterns.
A critical finding in our study is the differentiation of LUAD subtypes based on PCD pathway activation, with one subtype showing increased immune infiltration and the other exhibiting higher tumor purity. This result is consistent with studies indicating that high immune infiltration within tumors, particularly of cytotoxic T cells and natural killer cells, often correlates with better survival outcomes [33, 34]. Our identification of a subtype with elevated levels of immune cell infiltration and PCD-related gene expression suggests a possible mechanism by which certain LUAD patients achieve a favorable prognosis through endogenous immune responses. This finding echoes research by Lavin et al. [35] and Lambrechts et al. [31], who similarly identified immune-rich LUAD subtypes with superior survival outcomes.
Our analysis revealed differential activation of various PCD pathways, including autophagy, ferroptosis, and pyroptosis, across the LUAD subtypes. This finding aligns with research demonstrating that these pathways can play dual roles in cancer progression and immune modulation, depending on the context. For instance, autophagy has been implicated in both promoting tumor cell survival under stress conditions and enhancing anti-tumor immune responses by stimulating the presentation of tumor antigens [36]. The high expression of autophagy-related genes in one of our LUAD subtypes suggests that these tumors may utilize autophagy to evade immune responses and survive in the TME. This is consistent with findings from Amaravadi et al. [37], who reported that autophagy inhibitors could sensitize tumors to immune-mediated killing, thereby enhancing the efficacy of immunotherapy. Similarly, ferroptosis, a form of cell death driven by iron-dependent lipid peroxidation, has been shown to influence the TME by releasing DAMPs that recruit and activate immune cells [38]. Our study’s observation of elevated ferroptosis-related gene expression in certain cell subpopulations supports the idea that ferroptosis may enhance anti-tumor immunity. This is consistent with studies by Wang et al. [39], who found that ferroptosis induction in LUAD could lead to increased infiltration of immune cells, including dendritic cells and macrophages, thereby promoting an immune-supportive TME.
Our immune infiltration analysis showed that certain immune cell types, such as cytotoxic lymphocytes and dendritic cells, were more abundant in the PCD-high subtype, which correlated with better survival outcomes. This is consistent with research indicating that a “hot” TME, rich in immune cells, is generally associated with more favorable responses to immunotherapy [40]. Importantly, our finding that immune checkpoint molecules, such as B2M, NRP1, and TNFSF15, were more highly expressed in the immune-rich subtype suggests that these patients may benefit more from immune checkpoint inhibitors (ICIs). This aligns with studies demonstrating that high expression of checkpoint markers is often correlated with increased immune infiltration and improved responsiveness to immunotherapies in LUAD [41]. Previous research has also suggested that the expression of immune checkpoints can vary widely across cancer subtypes, impacting the efficacy of immunotherapy [42]. Our study contributes to this body of evidence by identifying distinct immune checkpoint expression profiles across PCD-related subtypes, underscoring the need for subtype-specific immunotherapy strategies. In particular, the high checkpoint expression in our immune-rich subtype may make these patients prime candidates for combination therapies that include ICIs along with agents targeting specific PCD pathways.
The functional enrichment analysis in our study identified several pathways that were differentially activated between the LUAD subtypes, including the PI3K-Akt signaling pathway and small GTPase-mediated signal transduction. The PI3K-Akt pathway has long been recognized as a key regulator of cell proliferation, survival, and metabolism in cancer [43]. Its enrichment in one of our subtypes suggests that targeted inhibition of PI3K-Akt signaling could be beneficial for patients with this subtype, particularly if combined with treatments that modulate PCD pathways. Similarly, the enrichment of small GTPase signaling pathways in the PCD-high subtype aligns with findings from other studies that suggest GTPases play important roles in modulating immune cell migration and activation within the TME [44]. Targeting small GTPase signaling in combination with immunotherapy may therefore enhance immune infiltration and improve therapeutic efficacy for this subtype.
Our study’s prognostic analysis of the PCD-related subtypes showed significant differences in survival outcomes, with the immune-rich subtype demonstrating better OS. This finding is in line with studies suggesting that higher immune infiltration generally correlates with improved prognosis in LUAD [34]. Furthermore, our survival analysis using TCGA and GSE31210 datasets confirmed the robustness of our subtype classification and its potential utility as a prognostic tool. The ability to stratify LUAD patients into distinct prognostic subtypes based on PCD-related gene expression and immune infiltration profiles could have significant implications for personalized treatment. Patients identified as part of the immune-rich, PCD-high subtype may benefit more from immunotherapy, while those in the lower PCD or immune-suppressed subtype might require alternative approaches, such as PCD pathway-targeted therapies combined with immune modulators. This approach aligns with the current trend in oncology toward personalized medicine and precision immunotherapy [45].
Limitations
Despite the insights provided by our study, there are several limitations that should be acknowledged. First, our study primarily relied on retrospective data from public databases, which may introduce biases related to patient demographic variability and treatment heterogeneity. To mitigate these potential biases, we ensured that datasets were harmonized by applying consistent preprocessing steps, including normalization of gene expression data and exclusion of samples with incomplete clinical annotations. Additionally, we performed stratified analyses to evaluate the potential impact of demographic factors such as age, sex, and smoking status, and included these variables as covariates in survival analyses to account for their influence. While these steps aimed to reduce bias, the inherent heterogeneity in treatment regimens and clinical follow-up across cohorts may still limit the generalizability of our findings. Future studies should incorporate more detailed clinical metadata and consider prospective cohort designs to validate these results. Integrating data from diverse populations and accounting for treatment heterogeneity in statistical models will be essential to improve the robustness and applicability of our conclusions.
In conclusion, our study provides valuable insights into the role of PCD pathways in LUAD heterogeneity, immune infiltration, and prognosis. By using scRNA-seq to dissect PCD-related subtypes, we highlight the potential of these pathways as biomarkers and therapeutic targets. The distinct immune infiltration patterns and checkpoint profiles observed across subtypes emphasize the need for tailored therapeutic approaches that account for the unique PCD and immune characteristics of each patient’s tumor. Ultimately, our findings contribute to a growing understanding of the complex interplay between cell death mechanisms and the immune system in LUAD, with implications for improving personalized treatment strategies and patient outcomes.
Supplementary Information
{kind=link}
Supplementary Figure 1. Quality Control of scRNA-seq Data for LUAD Samples.Scatter plots of QC metrics, including nFeature_RNA and nCount_RNA, with correlation coefficients indicating data quality.Violin plots of nFeature_RNA, nCount_RNA, percent.mt, and percent.Ribo, showing sequencing variability across datasets.Violin plots showing RNA feature and mitochondrial content distributions post-QC.PCA plot of scRNA-seq data for dimensional reduction, colored by dataset, illustrating clustering patterns across samples
Acknowledgements
None.
Author contributions
SH designed the study. LL performed data analysis and drafted the manuscript. SH revised the manuscript. All authors read and approved the final manuscript.
Funding
None.
Data availability
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Myers DJ, Wallen JM. Lung adenocarcinoma. In: StatPearls. 2024, StatPearls Publishing Copyright © 2024, StatPearls Publishing LLC.: Treasure Island (FL) ineligible companies. Disclosure: Jason Wallen declares no relevant financial relationships with ineligible companies.
- 2.Yu Y, et al. Predicting potential therapeutic targets and small molecule drugs for early-stage lung adenocarcinoma. Biomed Pharmacother. 2024;174: 116528. [DOI] [PubMed] [Google Scholar]
- 3.Butnor KJ. Controversies and challenges in the histologic subtyping of lung adenocarcinoma. Transl Lung Cancer Res. 2020;9(3):839–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalinke L, Janes SM. Two phenotypes that predict prognosis in lung adenocarcinoma. Eur Respir J. 2022;60(1):2200569. [DOI] [PubMed] [Google Scholar]
- 5.Song Y, Kelava L, Kiss I. MiRNAs in lung adenocarcinoma: role, diagnosis, prognosis, and therapy. Int J Mol Sci. 2023;24(17):13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bischoff P, et al. Single-cell RNA sequencing reveals distinct tumor microenvironmental patterns in lung adenocarcinoma. Oncogene. 2021;40(50):6748–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diao X, Guo C, Li S. Identification of a novel anoikis-related gene signature to predict prognosis and tumor microenvironment in lung adenocarcinoma. Thorac Cancer. 2023;14(3):320–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen Y, et al. Cross-talk between cuproptosis and ferroptosis regulators defines the tumor microenvironment for the prediction of prognosis and therapies in lung adenocarcinoma. Front Immunol. 2022;13:1029092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu J, et al. Prediction of prognosis, immunogenicity and efficacy of immunotherapy based on glutamine metabolism in lung adenocarcinoma. Front Immunol. 2022;13: 960738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo W, et al. Distinct immune microenvironment of lung adenocarcinoma in never-smokers from smokers. Cell Rep Med. 2023;4(6): 101078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun X, et al. Single-cell dissection reveals the role of aggrephagy patterns in tumor microenvironment components aiding predicting prognosis and immunotherapy on lung adenocarcinoma. Aging. 2023;15(23):14333–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kopeina GS, Zhivotovsky B. Programmed cell death: past, present and future. Biochem Biophys Res Commun. 2022;633:55–8. [DOI] [PubMed] [Google Scholar]
- 13.D’Arcy MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int. 2019;43(6):582–92. [DOI] [PubMed] [Google Scholar]
- 14.Obeng E. Apoptosis (programmed cell death) and its signals—a review. Braz J Biol. 2021;81(4):1133–43. [DOI] [PubMed] [Google Scholar]
- 15.Hsu SK, et al. Inflammation-related pyroptosis, a novel programmed cell death pathway, and its crosstalk with immune therapy in cancer treatment. Theranostics. 2021;11(18):8813–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu J, et al. Programmed cell death tunes tumor immunity. Front Immunol. 2022;13: 847345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang M, et al. Programmed cell death in tumor immunity: mechanistic insights and clinical implications. Front Immunol. 2023;14:1309635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fang C, et al. IFN-γ-induced ER stress impairs autophagy and triggers apoptosis in lung cancer cells. Oncoimmunology. 2021;10(1):1962591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang W, et al. Identification of cuproptosis and immune-related gene prognostic signature in lung adenocarcinoma. Front Immunol. 2023;14:1179742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Devarakonda S, Morgensztern D, Govindan R. Genomic alterations in lung adenocarcinoma. Lancet Oncol. 2015;16(7):e342–51. [DOI] [PubMed] [Google Scholar]
- 21.Sun HF, et al. Single-cell RNA sequencing reveals cellular and molecular reprograming landscape of gliomas and lung cancer brain metastases. Clin Transl Med. 2022;12(11): e1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei Q, et al. Molecular subtypes of lung adenocarcinoma patients for prognosis and therapeutic response prediction with machine learning on 13 programmed cell death patterns. J Cancer Res Clin Oncol. 2023;149(13):11351–68. [DOI] [PubMed] [Google Scholar]
- 23.Cui Y, et al. B4GALT1 promotes immune escape by regulating the expression of PD-L1 at multiple levels in lung adenocarcinoma. J Exp Clin Cancer Res. 2023;42(1):146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu C, et al. KRAS-G12D mutation drives immune suppression and the primary resistance of anti-PD-1/PD-L1 immunotherapy in non-small cell lung cancer. Cancer Commun. 2022;42(9):828–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng C, et al. A review of single-cell RNA-Seq annotation, integration, and cell–cell communication. Cells. 2023;12(15):1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang A, et al. Integration of single-cell RNA sequencing and bulk RNA sequencing data to establish and validate a prognostic model for patients with lung adenocarcinoma. Front Genet. 2022;13: 833797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding D, et al. Machine learning developed a programmed cell death signature for predicting prognosis and immunotherapy benefits in lung adenocarcinoma. Transl Oncol. 2023;38: 101784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang N, et al. Development and validation of polyamines metabolism-associated gene signatures to predict prognosis and immunotherapy response in lung adenocarcinoma. Front Immunol. 2023;14:1070953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin W, et al. GLIPR2: a potential biomarker and therapeutic target unveiled—insights from extensive pan-cancer analyses, with a spotlight on lung adenocarcinoma. Front Immunol. 2024;15:1280525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song P, et al. Identification and validation of a novel signature based on NK cell marker genes to predict prognosis and immunotherapy response in lung adenocarcinoma by integrated analysis of single-cell and bulk RNA-sequencing. Front Immunol. 2022;13: 850745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lambrechts D, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. 2018;24(8):1277–89. [DOI] [PubMed] [Google Scholar]
- 32.Reuben A, et al. TCR repertoire intratumor heterogeneity in localized lung adenocarcinomas: an association with predicted neoantigen heterogeneity and postsurgical recurrence. Cancer Discov. 2017;7(10):1088–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Newman DR, et al. Expression of WNT5A in idiopathic pulmonary fibrosis and its control by TGF-β and WNT7B in human lung fibroblasts. J Histochem Cytochem. 2016;64(2):99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thorsson V, et al. The immune landscape of cancer. Immunity. 2018;48(4):812-830.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lavin Y, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell. 2017;169(4):750-765.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Q, et al. HIF-1α-induced expression of m6A reader YTHDF1 drives hypoxia-induced autophagy and malignancy of hepatocellular carcinoma by promoting ATG2A and ATG14 translation. Signal Transduct Target Ther. 2021;6(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Amaravadi RK, Kimmelman AC, Debnath J. Targeting autophagy in cancer: recent advances and future directions. Cancer Discov. 2019;9(9):1167–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stockwell BR, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, et al. The function and mechanism of ferroptosis in cancer. Apoptosis. 2020;25(11–12):786–98. [DOI] [PubMed] [Google Scholar]
- 40.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–30. [DOI] [PubMed] [Google Scholar]
- 41.Hellmann MD, et al. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell. 2018;33(5):843-852.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rizvi NA, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13(2):140–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fritz G, Henninger C, Huelsenbeck J. Potential use of HMG-CoA reductase inhibitors (statins) as radioprotective agents. Br Med Bull. 2011;97:17–26. [DOI] [PubMed] [Google Scholar]
- 45.Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity. 2020;52(1):17–35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Quality Control of scRNA-seq Data for LUAD Samples.Scatter plots of QC metrics, including nFeature_RNA and nCount_RNA, with correlation coefficients indicating data quality.Violin plots of nFeature_RNA, nCount_RNA, percent.mt, and percent.Ribo, showing sequencing variability across datasets.Violin plots showing RNA feature and mitochondrial content distributions post-QC.PCA plot of scRNA-seq data for dimensional reduction, colored by dataset, illustrating clustering patterns across samples
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.