

Abstract
Patients with osteosarcoma (OS), a debilitating pediatric bone malignancy, have limited treatment options to combat aggressive disease. OS thrives on insulin growth factor (IGF)-mediated signaling that can facilitate cell proliferation. Previous efforts to target IGF-1R signaling were mostly unsuccessful, likely due to compensatory signaling through alternative splicing of the insulin receptor (IR) to the proliferative IR-A isoform. Here, we leverage splice-switching oligonucleotides (SSOs) to mitigate IR splicing toward the IR-B isoform. We show that SSOs can modulate cancer cell hallmarks and anoikis-resistant growth. Furthermore, we engineered the SSO sequence in an U7 snRNA packaged in an adeno-associated virus (AAV) to test the feasibility of viral vector-mediated gene therapy delivery. We noted modest increases in IR-B isoform levels after virus transduction, which prompted us to investigate the role of combinatorial treatments with dalotuzumab, an anti-IGF-1R monoclonal antibody. After observing additive impacts on phosphoprotein phosphorylation and anoikis-resistant growth with the dalotuzumab and SSO combination, we treated OS cells with dalotuzumab and the AAVrh74.U7 snRNA IR virus, which significantly slowed OS cell proliferation. While these viruses require further optimization, we highlight the potential for SSO therapy and viral vector delivery, as it may offer new treatment avenues for OS patients and be translated to other cancers.
Keywords: MT: Regular Issue, alternative splicing, insulin receptor, splice-switching oligonucleotides, U7 snRNA, osteosarcoma, cancer
Graphical abstract
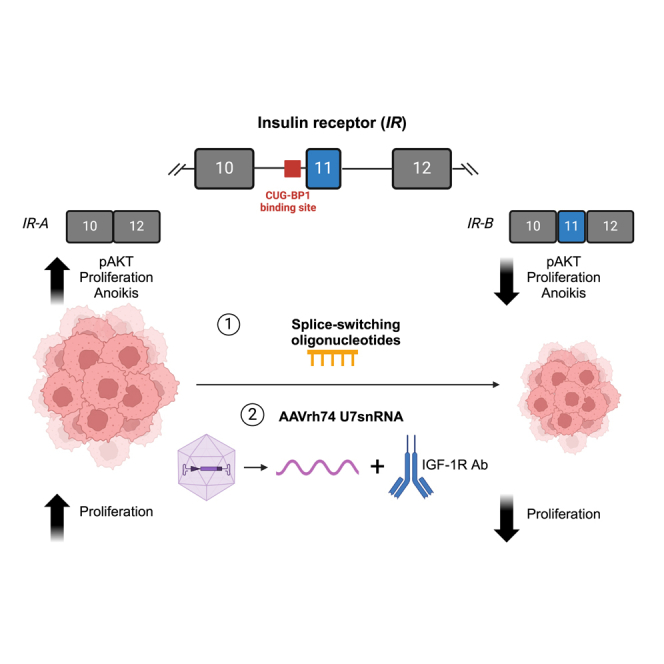
Chandler and colleagues demonstrate that therapeutic modulation of IR alternative splicing from the cancer-associated IR-A isoform to the IR-B isoform with SSOs mitigates cancer hallmarks in OS. The IR SSO can be engineered into AAVrh74.U7 snRNA viruses to decrease cell proliferation in combination with anti-IGF-1R therapies.
Introduction
Osteosarcoma (OS), the most common pediatric bone sarcoma, is characterized by a high propensity to metastasize, usually to the lungs, bones, or lymph nodes, and an overall dismal prognosis for patients with recurrent/relapsed tumors or distant spread.1,2,3,4 The 5-year survival rates for those with localized disease is as high as 70%, but this figure decreases drastically to 20% for the 15%–20% of patients who have identified metastatic disease at the time of diagnosis.1,5 With the presence of undetectable micrometastases far before clinical intervention and metastasis being the leading cause of mortality in OS and cancers in general, there is a direct need to better understand the molecular processes that promote OS tumorigenesis and also contribute to oncogenic spread.
A growing body of literature has identified various gene modules that contribute to the proliferation, migration, and cell survival in OS; among metabolic genes, the insulin receptor (IR) had the most significant impact on OS metastasis.6,7 Both IR and its closely homologous family member insulin growth factor 1R (IGF-1R) have been repeatedly implicated in the progression and malignant transformation of sarcomas, including OS.8,9,10 IGF-1R and IR are receptor tyrosine kinases that are activated by ligand binding and signal through multiple cascades, including Ras/Raf/MEK/ERK and PI3/AKT.9,11,12 The IGF-1R binds ligands IGF-1 or IGF-2 to block apoptosis and promote cell growth, but IR is a multifaceted receptor that undergoes alternative splicing to either initiate proliferative or metabolic pathways.12 A 22-exon gene, IR is differentially spliced at exon 11 to generate two distinct isoforms—namely, IR-B (includes exon 11) and IR-A (excludes exon 11). This exon, although only 36 nucleotides long and encoding 12 amino acids, is instrumental in distinguishing IR-A and IR-B’s abilities to bind the ligands IGF-2 and insulin.11 IR-B binds insulin to mediate glucose homeostasis and is the predominant isoform in adult tissues, such as the muscle, liver, and adipose tissue.9,11,12,13,14 Although IR-A can also bind insulin, it has a fivefold higher affinity to bind IGF-2, enabling it to be the primary isoform during highly proliferative stages like fetal embryogenesis and cancer.9,11,12,13,14,15
Since IGF-1R and IGF-2 upregulation is a common feature across cancer types, the IGF-1R was the target of multiple clinical trials in the 2000s.16,17 Anti-IGF-1R monoclonal antibodies (e.g., dalotuzumab, figitumumab, and ganitumab) act by inhibiting IGF-1 and IGF-2 from binding IGF-1R homodimers and IGF-1R/IR-A heterodimers, and the promise they showed in vitro was sufficient to elicit enthusiasm for phase III clinical trials across sarcomas and various other cancers.16,18 Unfortunately, monotherapies against IGF-1R were largely unsuccessful for the majority of sarcoma patients, and subsequent studies in Ewing sarcoma, another pediatric bone sarcoma, pointed to adaptive resistance through IR-A as a contributor to the failure of these single-agent trials.12,19,20 Through modulation of IR alternative splicing and upregulation of IR-A, cancer cells can use IR-A homodimers and circulating IGF-2 to achieve the same downstream proliferative pathways typically activated by IGF-1R and IGF-1.19,20 This compensatory signaling warrants dual targeting approaches that harness the potential of IGF-1R monotherapies and novel therapies against IR-A.
Because the IR-A and IR-B isoforms are produced by a single exon exclusion event, we have used splice-switching oligonucleotides (SSOs) (also referred to as antisense oligonucleotides, or ASOs) to modulate IR alternative splicing (in collaboration with Ionis Pharmaceuticals).21 SSOs are synthetic antisense RNA sequences that can bind pre-mRNA to sterically hinder splicing factors from acting at their regulatory elements, and they have been approved by the U.S. Food and Drug Administration (FDA) to correct alternative splicing events in spinal muscular atrophy (SMA) and Duchenne muscular dystrophy (DMD).22,23,24,25,26 SSO synthesis, chemistry, and physicochemical properties are critical elements in the characterization and validation of oligonucleotides as therapeutics. Specifically, we have tested IR SSOs with a phosphorothioate (PS) backbone and a 2′-O-methoxyethyl (2′MOE) modification. The feasibility and stability of multiple ASOs in this generation (approximately 30 members) have been studied both in in vitro and in vivo scenarios and are by far the most understood of RNA-targeted drugs.27,28,29 Differing only in the sequence, the members of each class of oligonucleotides have similar physicochemical characteristics and thus common pharmacokinetic and biological properties. Extensive studies in non-human primates and healthy human volunteers have assessed the stability and safety of 2′MOE ASOs. Overall, 2′MOE ASOs manufactured by Ionis Pharmaceuticals at varying doses and treatment exposures do not impact liver or kidney function, hematologic panels, or complement activation in humans.30,31,32 They also demonstrate long-term tissue half-lives while maintaining a potent splicing correction in mice and non-human primates receiving intrathecal or intracerebroventricular injections.33 Although sequence- and disease-specific effects may be observed with any candidate oligonucleotide, 2′MOE ASOs are generally stable in human tissue and safe in in vivo models.
In our previous work, we described an approach to promote IR exon 11 inclusion by targeting the negative regulatory element CUG-BP1 (CELF1).34,35 We tested a series of 2′MOE SSOs across the characterized CUG-BP1 binding site in IR intron 10 and identified a lead candidate that significantly restored IR splicing from the IR-A isoform to the IR-B isoform.21 SSO treatment reduced cell proliferation and angiogenesis in rhabdomyosarcoma cell lines, as well as vessel staining in an in vivo matrigel plug assay for angiogenesis.21
We hypothesized that toggling IR alternative splicing toward IR-B with an IR SSO candidate would be therapeutic in OS, a cancer that capitalizes on IGF/IR signaling for oncogenesis and metastatic progression. In this work, we established that OS tumors predominantly express the IR-A isoform and that SSO treatment specifically targets the CUG-BP1 binding site to shift IR splicing toward the IR-B isoform, while also decreasing AKT phosphorylation and altering cancer cell hallmarks like proliferation, growth in low adhesion, and apoptosis. To investigate the feasibility of expressing the IR SSO sequence in an AAV gene therapy model, we engineered the SSO sequence in a U7 snRNA vector system and packaged it in AAVrh74, which induced modest changes in IR-B levels but significantly reduced OS cell proliferation when combined with the anti-IGF-1R antibody dalotuzumab. Overall, our data identify the therapeutic benefits of modulating IR alternative splicing through SSOs, highlight the additive potential of splice-switching therapies in viral gene delivery models and pre-existing IGF-1R drugs, and underscore a novel strategy to mitigate cancer cell hallmarks in OS.
Results
IR is alternatively spliced to the IR-A isoform in OS patient-derived xenografts and cell lines
The IR, a 22-exon gene encoded on chromosome 19, is alternatively spliced into two isoforms depending on the inclusion or exclusion of exon 11 (IR-B or IR-A, respectively) (Figure 1A).36 The resulting two isoforms differ in the downstream signaling that they trigger, in part due to their ability to preferentially bind the ligands insulin and IGF-2. While both IR protein isoforms bind insulin, IR-A additionally binds IGF-2 at a higher affinity than insulin, a property it shares with the closely homologous IGF-1 receptor.37 Consequently, the IR-B receptor mediates differentiation and metabolic signaling, while the IR-A receptor affects growth pathways, motility, and angiogenesis during embryogenesis and tumorigenesis (Figure 1A).12,14 IR-A is upregulated in multiple cancer types, and IGF-2-mediated signaling through the IR-A receptor promotes increased proliferative signaling through activation of RAS/Raf/MEK/ERK and PI3/AKT/mTOR.12
Figure 1.

IR is alternatively spliced to the IR-A isoform in OS PDXs and cell lines
(A) Schematic depicting the exon 11 alternative splicing event in IR. The full-length isoform IR-B includes exon 11, whereas the cancer-related isoform IR-A lacks exon 11. The IR-A and IR-B proteins are closely homologous to the IGF-IR, and they exhibit differential affinity for the ligands they bind. IGF-IR and IR-A bind IGF-2 to activate growth, proliferation, and migratory signaling, whereas IR-B binds insulin to initiate metabolic and differentiation pathways. (B) Cumulative IR gene expression in normal osteoblasts (n = 2) and human OS patient tumors (n = 12) in the Sadikovic et al.38 mixed OS dataset. Analysis was performed in the R2: Genomics Analysis and Visualization platform (∗∗∗∗ p < 0.0001 by one-way ANOVA).39 (C and D) RT-PCR of IR alternative splicing in (C) PDX samples from unclassified, primary, and metastatic human OS tumors and (D) osteoblasts (OB) and human OS cell lines. RT-PCR was performed on RNA extracted from these tissues, and the isoforms were amplified using primers indicated by the black arrows in (A) and detailed in Table 1. The levels of IR-B is quantified as percent spliced in (PSI). GAPDH is shown in (C) as a loading control.
To understand the impact of IR in OS tumors, we first analyzed its gene expression in normal osteoblasts and pediatric OS tumor samples in the Sadikovic-14-rma_sketch_hugene10t dataset.38,39 We found that the cumulative gene expression of IR is statistically significantly higher in OS tumors than osteoblasts (p < 0.0001) (Figure 1B). We then profiled human patient-derived xenografts (PDXs) from primary and metastatic OS tumors and a panel of human OS cell lines and observed that IR is preferentially spliced to the IR-A isoform (Figures 1C and 1D). The upregulation of cumulative IR and predominance of the IR-A isoform in OS highlight a therapeutic opportunity for splice-modulating therapies that can toggle splicing toward the IR-B isoform.
SSOs targeting the negative splicing regulator CUG-BP1’s binding site robustly and specifically shift IR splicing from IR-A to IR-B
The inclusion or exclusion of IR exon 11 to produce the IR-B or IR-A isoforms is largely mediated by the positive and negative splicing regulators MBNL1 and CUG-BP1 (CELF1), respectively.34,35,40 While CUG-BP1 binding induces exon 11 skipping, MBNL1 binding promotes exon 11 inclusion. The IR pre-mRNA contains CUG-BP1 binding sites in both IR intron 10 and exon 11 and a MBNL1 site in intron 11. We previously designed SSOs spanning these regulatory elements to identify SSO candidates that could mask the binding of CUG-BP1 and increase the inclusion of exon 11.21 Initially, we performed an SSO “macro-walk” with six overlapping SSOs, each different in five-nucleotide increments, across the entire CUG-BP1 binding site (40–50 bp). Two of these six SSOs, including SSO55, significantly switched IR splicing toward IR-B. Next, we performed a “micro-walk” of 20 consecutive SSO sequences at a two-nucleotide resolution around the CUG-BP1 sequence complementary to SSO55. Out of this panel in the micro-walk, we selected SSO55, the most effective SSO to robustly switch IR alternative splicing in an IR minigene system and in endogenous settings, as the lead candidate (Figure 2A).21 To test this SSO’s ability to modulate IR splicing in OS, we transfected varying concentrations of SSO55 (denoted SSO) and a control non-specific SSO (denoted "NS SSO72" or "NS") in multiple OS cell lines and determined 100 nM to be the optimal dose that results in the IR splicing switch without ensuing toxicity (Figures 2B, S1A, and S1B). We then observed that 100 nM SSO55 transfection for 24 h in U2OS and OHS Luc-GFP(U2OS, derived from a primary OS tumor; OHS Luc-GFP, derived from a primary tumor and transduced with Luc-GFP) and 143.98.2-Luc-GFP cells (derived from the metastatic parent cell line 143B and transduced with Luc-GFP) yielded significant switching toward IR-B (p < 0.001 for all cell lines) (Figure 2C).
Figure 2.

SSOs targeting the negative splicing regulator CUG-BP1’s binding site robustly and specifically shifts splicing from IR-A to IR-B
(A) Schematic depicting the sequence of SSO55, our lead candidate, complementary to the IR intron 10 CUG-BP1 (CELF1) binding site. (B) Dose-response transfection of NS SSO72 or SSO55 for 24 h in OS cell lines. RT-PCR was performed on RNA harvested from these cells, and IR isoforms were amplified using primers in exons 10 and 12 (see Figure 1A and Table 1). (C) 100 nM NS SSO72 (“NS”) or SSO55 (“SSO”) in U2OS, 143.98.2 Luc-GFP, and OHS Luc-GFP cells for 24 h. A representative gel image shows IR isoforms and the splicing switch in the NS and SSO conditions. Statistical significance was determined using a t test (n = 3 for both U2OS, 143.98.2 Luc-GFP, and OHS Luc-GFP; ∗∗∗ p < 0.001). (D) Effect of NS/SSO55 treatment on wild-type IR minigene (IR Mg) or CUG-BP1 mutant minigene (Mutant 1) in HeLa cells. A deletion mutant was created by deleting seven nucleotides outside of the CUG-BP1 consensus sequence in the wild-type IR minigene to test the specificity of SSO55 for its target sequence.34,35,41,42 The mutagenized plasmid was verified by Sanger sequencing. Then, NS/SSO55 and wild-type IR/the Mutant 1 minigene were co-transfected in HeLa cells for 24 h. RT-PCR on RNA extracted from these cells and quantification of IR-B showed no statistically significant increase in IR splicing in the mutant minigene (n = 3 replicates, ∗ p < 0.05, ns if p > 0.05 by paired t test).
To test the specificity of the SSO candidate to IR, we initially used both NCBI-BLAST and Bowtie to confirm that SSO55 bound only the IR genomic region and no other DNA sequences with 100% complementarity.21,43 We also confirmed that SSO55 did not have predicted binding sites in global targets of CUG-BP1 regulation as identified by Xia et al. (2017), and SSO55 transfection did not change the splicing patterns of select genes from this list of targets (LMO7, PARD3, and ZDHHC16).21,44 Additionally, we mutagenized the parent IR minigene (denoted as IR Mg in Figure 2D) to create a CUG-BP1 mutant minigene. The CUG-BP1 binding site has been characterized to a 35 base pair region, and the GU-rich consensus sequence of this site (5′UGUUUGUUGU-3′) is similar to the element that the SSO55 sequence targets.34,35,41,42 We generated a deletion mutant that lacked seven nucleotides but retained greater than 50% sequence complementarity with SSO55 (denoted as Mutant 1 in Figure 2D). We transfected either the wild-type IR minigene or the deletion mutant with 100 nM NS or SSO55 for 24 h in HeLa cells, the cell line used in foundational studies that characterized the wild-type minigene.34 While we observed a statistically significant switch toward IR-B in the parent IR minigene after SSO55 treatment, there was no significant difference in IR-B levels in the deletion mutant minigene (p < 0.05 for IR Mg and p > 0.05 for the Mutant 1 Mg) (Figure 2D). We also investigated the effect of SSO55 on IR splicing at much lower concentrations (10, 30, and 50 nM). For this study, we used the wild-type minigene, the deletion mutant, and a substitution mutant minigene (denoted as Mutant 2), where we replaced three adenosines with thymidines to create discrete, single base pair alterations that maintained the thermodynamic binding properties of the wild-type sequence (Figure S2A). Here, we observed that lower concentrations of SSO55 were able to switch splicing in only the wild-type minigene but not in either mutant minigene in HeLa cells, suggesting that the integrity of the SSO55 target sequence in the CUG-BP1 binding site is essential for splicing modulation at lower doses (Figure S2B). Altogether, these results demonstrate that the splice-switching effect of SSO55 is specific to IR and occurs by targeting a key sequence element within the CUG-BP1 binding site.
The restoration of IR splicing to IR-B using SSOs targeting the IR intron 10 CUG-BP1 site consequently alters cancer hallmarks in OS cells
Upon ligand binding, both IR protein isoforms and IGF-1R execute a vast downstream signaling network mediated by the master regulator PI3/AKT.12,37,45 When insulin binds the IR-B receptor, phosphorylation of AKT activates a number of glucogenic and metabolic pathways through GSK3, AMPK, and GLUT4, among others, and simultaneously downregulates proliferative pathways via Ras/Raf/MEK/ERK, nuclear factor-κB, and JNK.12 Conversely, IGF-1 or IGF-2 binding to IGF-1R or IR-A respectively activate growth signaling and silence metabolic functions.12
Because we observed statistically significant exon 11 inclusion upon SSO55 transfection, we sought to determine whether this shift affected the IR receptor’s downstream signaling mediators, such as AKT. We transfected 100 nM NS or SSO55 in U2OS and 143.98.2 Luc-GFP cells and harvested RNA and protein at time points ranging from 4 to 72 h after transfection. We observed that pAKT levels decreased following SSO55 treatment in both U2OS and 143.98.2 Luc-GFP cells and that this decrease was time-dependent. The greatest shift toward IR-B splicing and subsequent decrease in pAKT protein levels occurred in the window of 24–48 h in U2OS and 143.98.2 Luc-GFP cells (Figure S3). Decreased pAKT levels were maintained at later time points, although the efficacy of exon 11 inclusion was reduced most likely due to the dilution effect of overall SSO among dividing cells.
OS tumors display a high plasticity and likelihood of dissemination and metastasis, a process that requires the coordination of multiple regulatory networks, many of which converge at PI3/AKT.1,6,7,46 Because our preliminary data showed that SSO55 decreases AKT phosphorylation, we sought to determine whether SSO55 treatment would also alter cancer cell hallmarks that promote tumorigenesis and metastasis. We first assessed differences in proliferation of OS cells (measured by cell confluency) following 100 nM transfection of either the control NS SSO or SSO55. We found that U2OS, 143.98.2-Luc-GFP, OHS Luc-GFP, and other OS cell lines treated with SSO55 had significantly slower proliferation compared with NS-treated cells over the course of five days (p < 0.0001 for U2OS, 143.98.2-Luc-GFP, and OHS) (Figures 3A and S1C). We also interrogated changes in anoikis, a key metastatic process when cells undergo programmed cell death following detachment from the extracellular matrix, via the growth in low attachment (GILA) assay.47 Briefly, OS cells were transfected with 100 nM NS or SSO55, seeded in low adhesion plates, and incubated for five days before luminescence was measured (Figure 3B). Cells that received SSO55 were significantly less likely to grow in low adhesion conditions, indicating their inability to overcome anoikis as compared with cells treated with NS SSO (p < 0.01 for U2OS and 143.98.2 Luc-GFP; p < 0.0001 for OHS) (Figure 3B). These changes in cell confluency and growth in low adhesion prompted us to profile changes in apoptosis via flow cytometry and real-time qPCR. Annexin 5 recognizes cells that present phosphatidylserine on their outer membrane, a property associated with early apoptosis and predictive of subsequent apoptotic processes, and it can be used to identify and quantify cell death.48,49,50 We treated 143.98.2 Luc-GFP cells with NS SSO or SSO55 for 24–72 h and stained them for annexin 5. We observed an increase in annexin 5-positive cells at 48 h after SSO55 transfection, which was sustained at 72 h (p < 0.001 at 72 h) (Figure 3C). Additional qPCR validation of a panel of apoptotic targets revealed significant upregulation of Noxa and FAS after 48 h of SSO55 transfection in U2OS and 143.98.2 Luc-GFP cells (p < 0.001 for Noxa and p < 0.01 for Fas) (Figure 3D). Noxa, a member of the BCL-2 family, is a pro-apoptotic BH3-only protein that can induce apoptosis, reactive oxygen species accumulation, and mitochondrial outer membrane permeabilization.51,52 The FAS receptor, also known as CD95 or Apo1, is a death receptor that triggers programmed cell death or apoptosis when it interacts with its ligand FasL.53 The upregulation of FAS has been documented to sensitize OS cells to chemotherapeutic drugs.54 In fact, the Fas receptor and IR signaling pathways are interconnected through their roles in apoptosis, cell survival, and metabolic regulation. Prior studies have found that a decrease in AKT signaling triggers a FAS-dependent apoptosis in cancer cells.55,56 Overall, our data indicate that the modulation of IR splicing and subsequent attenuation of the AKT pathway could be therapeutic via the induction of specific apoptotic signaling cascades.
Figure 3.

SSO55 mitigates OS cancer cell hallmarks in vitro
(A) Proliferation curves of U2OS, 143.98.2 Luc-GFP, and OHS Luc-GFP cells transfected with 100 nM NS/SSO55. The proliferation assay measured cell confluency and was performed using Incucyte software (p < 0.0001 by the Brown-Forsythe and Welch ANOVA tests). (B) GILA assay of U2OS, 143.98.2 Luc-GFP, and OHS Luc-GFP cells. Cells were seeded and transfected with 100 nM NS/SSO55. After 24 h, all cells were reseeded at 10,000 cells/well in low attachment plates and incubated for five days. Subsequently, 100 μL Cell Titer Glo was added to each well, and luminescence was measured the Promega GlowMax Plate Reader (integration time = 1 s) (n = 4–6 for each condition, ∗∗p < 0.01, by paired t test). Cells treated with SSO55 show decreased cell growth in low adhesion conditions compared with the NS-treated controls. (C) Annexin 5 staining in 143.98.2 Luc-GFP cells treated with NS or SSO55 for 24–72 h (n = 3 for each condition, ∗∗∗p < 0.0001 by unpaired t test). Cells were seeded, transfected with NS/SSO55, and then collected and stained with Annexin 5, which was measured by flow cytometry (n = 3 for each condition, ∗∗∗p < 0.001 by unpaired t test). Splicing data corresponding to each time point is also shown. (D) qPCR analysis of apoptotic targets in U2OS and 143.98.2 Luc-GFP cells treated with NS or SSO55 for 48 h (n = 3 for U2OS, n = 6 for 143.98.2 Luc-GFP, ∗∗p < 0.01, ∗∗∗p < 0.001 by unpaired t test).
The IR SSO can be expressed as an antisense RNA in the U7 snRNA viral vector delivery system to switch IR splicing
ASO technology has revolutionized the treatment of genetic diseases with genetic diseases with concomitant aberrant splicing events, such as SMA and DMD.22,23,24,25,26 In both diseases, the use of SSOs to induce exon inclusion or exclusion has corrected the splicing defect in key genes (SMN1, DMD) and resulted in major therapeutic improvements for patients. The FDA has approved over a dozen ASO therapies for various genetic, cardiovascular, infectious, and metabolic disorders, but many of them require local, repeated administration of the therapies in the target tissues (muscle, CNS) to be effective.57 Furthermore, ASO biodistribution is largely biased to the liver, preventing the target tissues from receiving the intended dose.58,59
To overcome the barriers surrounding in vivo delivery and to promote therapeutic SSO uptake in both cancer cells and sites of OS metastasis, we turned toward delivery via viral vectors, specifically recombinant adeno-associated viruses (rAAVs) and the U7 small nuclear RNA (U7 snRNA) system. rAAVs are efficient delivery vehicles and do not integrate into the genome. The FDA has approved their use for SMA and other genetic diseases.60,61 The U7 small nuclear ribonucleoprotein (snRNP) traditionally facilitates 3′ end processing of histone RNAs through a complementary sequence to the histone downstream element (HDE) and is therefore not involved in spliceosomal processes.62 By modifying the HDE to a specific antisense sequence targeting a region of interest and substituting the U7 snRNP-specific proteins (Lsm 10 and Lsm 11) with the consensus sequence of proteins in the major spliceosome, the U7 snRNA can efficiently deliver therapeutic antisense RNAs that localize in the nucleus, where splicing occurs.62
As proof-of-principle of viral vector-mediated delivery of SSOs in cancer, we designed a modified U7 snRNA vector to express either the control NS or SSO55 sequence as an antisense RNA (Figure 4A). The modified U7 snRNA contains its own promoter that drives the expression of the NS or SSO55 sequence, Sm-binding and loop sequences (to induce the formation of a spliceosomal-like protein core that protects the snRNA), and its 3′ UTR. We validated two types of plasmids, where the U7snRNA-NS/SSO unit was encoded twice (p-N1, p-A1) or where a single U7 promoter expressed the NS/SSO sequence four times interspersed with a linker region (p-N2, p-A2) (Figure 4A). These viral plasmids were transfected with the IR minigene (see Figure 2D) in HeLa cells, and IR isoform levels were measured after 48 and 72 h post transfection to allow for sufficient expression of the plasmids (Figure 4B). Compared with the p-N1 U7 control vector, the p-A1 vector promoted a complete shift toward the IR-B isoform at both 48 and 72 h in two replicate experiments, an effect that was comparable with the synthetic SSO55 (Figure 4B). Additionally, while SSO55’s splicing switch was less pronounced at 72 h, likely in part to the oligonucleotide’s dilution during cell division, p-A1’s impact on IR-B levels was sustained. We consequently selected the p-N1 and p-A1 U7 vector pair to be packaged in rAAVs.
Figure 4.

SSO55 can be expressed as an antisense RNA in an AAV.rh74 U7 snRNA vector to modulate IR splicing in OS
(A) Schematic of the viral plasmid that expresses either the NS SSO or SSO55 sequences downstream of the U7 promoter. The histone binding elements of the native U7 snRNP have been replaced with spliceosomal components (Sm binding and loop sequences) to facilitate entry into the nucleus. The resulting NS or SSO55 sequences are expressed as antisense RNAs. Two types of viral vectors were manufactured by VectorBuilder: 1) p-N1 (encoding NS) and p-A1 (encoding SSO55) were viral plasmids with the U7-SSO-stuffer sequence unit repeated twice, and 2) p-N2 (encoding NS) and p-A2 (encoding SSO55) had a single U7-stuffer unit with four SSO repeats interspaced by a linker region. (B) Transfection of the IR minigene and viral plasmids p-N1, p-A1, p-N2, p-A2 in HeLa cells to test their efficacy in switching IR splicing. We transfected 3 μg of the IR minigene and viral plasmid in HeLa cells using Lipofectamine 3000. Cells were harvested for RNA after 48 and 72 h to allow sufficient time for plasmid expression. Minigene-specific primers were used to only measure the splicing of the minigene and not endogenous IR (see Table 1). Based on n = 2 replicates shown on the right, the p-N1 and p-A1 plasmids were selected to be packaged into AAVs. (C) Transduction of rAAV serotype 8 (AAV8) rh74 (AAVrh74) in OS cells. U2OS and 143.98.2 cells were transduced with AAV-8-eGFP or AAVrh74-eGFP empty vectors at a MOI of 1E6 and observed for up to five days post viral infection. GFP fluorescence was observed on a fluorescent microscope (scale bar, 400 μM). (D) Transduction of AAVrh74.U7snRNA-N (expressing p-N1) and AAVrh74.U7snRNA-A (expressing p-A1) viruses in U2OS and 143.98.2 Luc-GFP cells. Cells were transduced with virus at MOIs 1E6, 5E6, and 1E7, and IR splicing was measured after 48 and 72 h of transduction (n = 4 replicates, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.0001 by ordinary one-way ANOVA).
Different types of rAAVs (termed serotypes) are distinguished by their surface antigens, and these features lead to selective tissue tropism that can target rAAVs to specific tissues.60,61 However, this advantage is offset by the prevalence of rAAVs in the general environment and acquired humoral immunity against them in humans.60,61 To anticipate future challenges with in vivo delivery, we proactively tested the transduction efficacy of AAV-8 against AAVrh74, a serotype with a low seroprevalence of binding antibodies in humans, in OS cells.63,64,65 We transduced AAVrh74-eGFP and AAV-8-eGFP in U2OS and 143.98.2 cells at a multiplicity of infection (MOI) of 1E6 and observed GFP fluorescence over 5 days after transduction (Figures 4C and S4). AAVrh74 was more efficiently transduced in both cell lines compared with AAV-8 and was chosen as the packaging serotype for the U7 snRNA candidate plasmids (p-N1, p-A1).
When compared with AAVrh74.U7-N1, transduction of AAVrh74.U7-A1 at higher MOIs yielded modest but significant IR-B: IR-A splicing shifts at the 72-h time point in U2OS and 143.98.2 Luc-GFP cells (MOI 5E6 for U2OS, MOI 5E6 and 1E7 for 143.98.2 Luc-GFP) (Figure 4D). We noted that the impact of AAVrh74.U7-A1 on IR-B isoform levels was not as pronounced as SSO55. Previous applications of U7 snRNA-mediated antisense RNA expression for splicing events were in the context of genetic diseases, which usually have less dynamic microenvironments than cancer.66 Future studies to optimize the AAV.U7snRNA viral vectors will focus on observing longer transduction periods than what is shown in this work, addressing unequal cell-to-cell transduction efficiency, testing the strength of the U7 snRNA and stability of the antisense RNA, and considering the tumor microenvironment’s impact on viral transduction and antisense RNA’s expression from an U7 snRNA vector.
The IGF-1R antibody dalotuzumab acts additively with SSO55 to modulate phosphoprotein phosphorylation and with AAVrh74.U7snRNA to slow OS cell proliferation
Because we observed only a modest splicing shift toward IR-B upon AAVrh74.U7 snRNA virus transduction, we considered whether this small increase could be leveraged with an anti-IGF-1R therapy. To this end, we investigated the combination of SSO55 or later the AAVrh74.U7snRNA virus with the IGF-1R monoclonal antibody dalotuzumab (also known as MK0646), a humanized antibody that specifically inhibits IGF-1/IGF-2 binding to IGF-1R homodimers and prevents downstream activation of PI3/AKT and Ras/Raf/MEK/ERK signaling.16,67 Like other IGF-1R antagonists, dalotuzumab’s modest therapeutic benefit in patients and overall failure as a prospective treatment for IGF-driven cancers was in part attributed to IR alternative splicing and PI3-AKT signaling via IR-A.16,18,20 Therefore, we hypothesized that co-targeting of the IGF-1R and IR-A receptors with dalotuzumab and SSO55 or AAVrh74.U7snRNA, respectively, would be a therapeutic combination in OS cells.
To ascertain the individual and combined effects of dalotuzumab with SSO55 on OS cells, we performed a phosphorylation array on a broad spectrum of human phosphoproteins. We selected the metastatic cell line 143.98.2 Luc-GFP for the array and transfected cells with 100 nM NS or SSO55 in combination with dalotuzumab (Figure 5A). Because we observed the largest shift in AKT phosphorylation in this cell line at 48 h after SSO transfection (see Figure S3), we harvested these lysates at 48 h and incubated them on a multiplex antibody array of 37 human phosphoproteins (spotted in duplicate) to measure relative phosphorylation (Figures 5A, 5B, and S5A). The array contained AKT 1/2/3 (S473 and T308), ERK 1/2 (T202/Y204, T185/Y187), and GSK-3α/β (S21/9) as known targets in the IR-A signaling pathway (Table 2). Surprisingly, the addition of dalotuzumab with either NS or SSO55 abrogated phosphorylation of all but a few profiled phosphoproteins compared with SSO55 treatment alone (shown in Figure 5B and quantified in Figure S5B). AKT phosphorylation was minimally changed after SSO55 treatment alone and almost completely abolished with the addition of dalotuzumab. Since SSO55 facilitates IR-B splicing, we hypothesized that this would increase the activation of metabolic protein targets and partners. We congruently observed an increase in GSK-3α/β and GSK-3β phosphorylation after SSO55 alone and with dalotuzumab. ERK1/2 phosphorylation initially increased with SSO55 treatment, which may be due to constitutive Ras activation from Kirsten murine sarcoma virus infection in 143.98.2 Luc-GFP’s parent cell line 143B.68,69 Despite this, the addition of dalotuzumab reduced ERK1/2 phosphorylation in both the NS and SSO55 conditions. We also identified a number of additional phosphorylated targets affected by SSO55 treatment that will be candidates for future studies to assess SSO55’s impact on diverse signaling pathways and heat shock responses, among others (Figure 5B). To verify the phosphoarray findings in a cell line without constitutive Ras activation, we profiled the impact of SSO55 and dalotuzumab on pGSK-3α/β protein in U2OS cells. Indeed, we observed that protein levels of GSK3α/3β, a known substrate of AKT that participates in metabolic signaling, increase modestly in SSO55-treated U2OS cells compared with NS-treated cells, and this effect is enhanced with the addition of dalotuzumab (Figure S5C).12,70
Figure 5.

The IGF-IR monoclonal antibody dalotuzumab alters human phosphoprotein phosphorylation in combination with SSO55 and slows OS cell proliferation with AAVrh74.U7snRNA viruses
(A) Experimental design of transfection and dalotuzumab (indicated as IGF-1R Ab throughout figure) treatment in 143.98.2 Luc-GFP cells to profile a panel of human phosphoprotein. The 48 h time point was selected due to the IR-B splicing switch and concomitant pAKT decrease previously observed in Figure S3. (B) Selected phosphoprotein spots (in duplicate) shown in the four treatment conditions: 100 nM NS only, 100 nM SSO55 only, 100 nM NS + dalotuzumab, and 100 nM SSO55 + dalotuzumab. Relative mean pixel density of each phosphoprotein duplicate is shown to the right. Quantification was performed in ImageJ by calculating relative pixel density of each spot and averaging the duplicates. The negative control on each membrane (PBS) was quantified and subtracted from the averaged values as background. The complete images and quantifications of the array membranes are shown in Figures S5A and S5B and annotated in Table 2. (C) GILA assay of U2OS and OHS cells treated with NS or SSO55 alone or in combination with dalotuzumab. Cells treated with SSO55 alone or with dalotuzumab show decreased cell transformation compared with the NS- or NS + dalotuzumab-treated controls (n = 4–6 for each condition, ∗∗p < 0.01, ∗∗∗∗p < 0.0001 by one-way ANOVA). (D) Experimental design of proliferation assay with dalotozumab and AAVrh74.U7snRNA viruses. Cells were incubated in low serum media for 24 h after transduction and then in full serum media for the duration of the assay. Dalotuzumab was replaced during media changes every 48 h. (E) Proliferation curve of U2OS transduced with AAVrh74.U7snRNA-N and rAAVrh74.U7snRNA-A viruses alone or in combination with dalotuzumab. Cells that received AAVrh74.U7snRNA-A + dalotuzumab showed significantly slower proliferation than other treatment conditions (∗∗p < 0.01, ∗∗∗p < 0.001, or ∗∗∗∗p < 0.0001 by paired t test).
Table 2.
Human proteome phosphokinase phosphoarray (R&D Systems)
| Coordinate | Phosphokinase | Phosphorylation site |
|---|---|---|
| Membrane A | ||
| A1, A2 | reference spot | – |
| B3, B4 | CREB | S133 |
| B5, B6 | EGFR | Y1086 |
| B7, B8 | eNOS | S1177 |
| B9, B10 | ERK1/2 | T202/Y204, T185/Y187 |
| C3, C4 | Fgr | Y412 |
| C5, C6 | GSK-3α/β | S21/S9 |
| C7, C8 | GSK-3β | S9 |
| C9, C10 | HSP27 | S78/S82 |
| D3, D4 | JNK 1/2/3 | T183/Y185, T221/Y223 |
| D5, D6 | Lck | Y394 |
| D7, D8 | Lyn | Y397 |
| D9, D10 | MSK1/2 | S376/S360 |
| E3, E4 | p38α | T180/Y182 |
| E5, E6 | PDGF Rβ | Y751 |
| E7, E8 | PLC-γ1 | Y783 |
| E9, E10 | Src | Y419 |
| F3, F4 | STAT2 | (pY690) |
| F5, F6 | STAT5a/b | Y694/Y699 |
| F7, F8 | WNK1 | T60 |
| F9, F10 | Yes | Y426 |
| G1, G2 | reference spot | – |
| G3, G4 | Β-catenin | – |
| G9, G10 | PBS (negative control) | – |
| Membrane B | ||
| A11, A12 | Akt 1/2/3 | T308 |
| A13, A14 | Akt 1/2/3 | S473 |
| A17, A18 | reference spot | – |
| B11, B12 | Chk-2 | T68 |
| B13, B14 | c-Jun | S63 |
| C11, C12 | p53 | S15 |
| C13, C14 | p53 | S46 |
| C15, C16 | p53 | S392 |
| D11, D12 | p70 S6 kinase | T389 |
| D13, D14 | p70 S6 kinase | T421/S424 |
| D15, D16 | PRAS40 | T246 |
Protocol, phosphoarray coordinates, and template: https://resources.rndsystems.com/pdfs/datasheets/ary003c.pdf?_ga=2.111712964.1319397147.1706157690-1571267669.1695235735.
Based on the distinct phosphorylation changes in a variety of phosphoproteins that was induced by the combination of SSO55 and dalotuzumab rather than SSO55 alone, we examined whether these two agents would also change properties of anoikis in OS cells. We repeated the GILA assay in OS cells and observed that simultaneous treatment with SSO55 and dalotuzumab statistically significantly decreased growth in low adhesion compared with control cells (p ≤ 0.05 for U2OS and 143.98.2) (Figures 5C and S6A), highlighting that dalotuzumab could enhance SSO55’s favorable effects on cancer cell hallmarks.
Our phosphoarray and GILA results suggested that dalotuzumab’s widespread impact on downstream signaling in both NS- and SSO55-treated cells may be a favorable mechanism to augment the modest increase in IR-B levels observed with the AAVrh74.U7 snRNA virus. We treated both U2OS and 143.98.2 Luc-GFP cells with dalotozumab and the AAVrh74.U7 snRNA control or IR viruses, with the caveat that Rh74-eGFP more efficiently transduced U2OS cells than 143.98.2 Luc-GFP cells and the former would be the more reliable cell line to measure cell proliferation (Figures 4C, 5D, and S6B). To maximize the number of U2OS cells that would be transduced, we used an MOI of 1E7 and observed that AAVrh74.U7-A and dalotuzumab (indicated as IGF-1R Ab in the figure) were more effective in significantly slowing OS cell proliferation (measured by cell confluency) than the virus alone or the control virus with dalotuzumab (p < 0.0001 in all conditions) (Figure 5E).
In comparison with AAVrh74.U7snRNA transduction, SSO transfection was highly efficient in significantly switching IR splicing toward the IR-B isoform, which was also sufficient to reduce proliferation and anoikis-resistant growth. However, in the absence of a major splicing correction, combinatorial therapy wherein we can target both the IGF-1R and IR-A, as illustrated through the dual use of dalotuzumab and AAVrh74.U7snRNA IR virus, might be additive and elicit anti-tumorigenic molecular and functional changes in cancer cells.
Discussion
Alternative splicing is a pivotal biological process that enables genes to generate multiple protein isoforms, contributing to transcript variation and proteome diversity. However, recent advancements in high-throughput sequencing have revealed that splicing deregulation, which impacts a wide array of genes, plays a substantial role in various hallmarks of the tumorigenic process, including angiogenesis and metastasis.71,72 It is imperative that to understand the mechanistic details of cancers as well as to treat them effectively, we tap into this level of regulation and identify splicing targets that contribute to tumorigenicity.
While the introduction of chemotherapy has greatly improved OS patient survival since the disease was first described in the 1950s, the overall treatment regimen and the dismal outlook for patients with metastases has remained stagnant.73 The IGF-2/AKT pathway is highly upregulated in all OS patients, and copy number gains of the IGF-1R gene have been found in OS patients with corresponding IGF-1R overexpression.74 Additionally, IGF-2 is also known to protect OS cells against chemotherapy.75 However, unfortunately the inhibition of IGF-1R using monoclonal antibodies like cixutumumab or dalotuzumab or even with tyrosine kinase inhibitors has not resulted in successful tumor remission.20 Enzymatic quantification has shown that the alternatively spliced IR-A receptor demonstrates increased binding affinity for IGF-2 as compared with the full-length IR-B receptor.15 As such, in the setting of anti-IGF-1R monotherapy, the IGF-2:IR-A signaling pathway could be contributing to lack of clinical success as it circumvents IGF-1R inhibition and promotes oncogenic growth responses.19,20
In this work, we have shown that IR-A is the dominant isoform in OS tumors, and induction of the IR-B isoform with our IR SSO candidate across multiple OS cell lines resulted in changes in cell proliferation, GILA, and apoptosis. To advance the use of SSO technology in cancer, which currently has limitations in delivery to target tissues and biodistribution, we engineered the IR SSO as an antisense RNA in a rAAV vector with a U7 promoter.57,58,59 This approach has been used to deliver ASOs in genetic diseases but has not yet been reported in a cancer model to our knowledge.62,76,77,78 The transduction of OS cells with AAVrh74.U7snRNA virus resulted in a modest but significant splicing shift from IR-A to IR-B, which prompted us to investigate the potential for combinatorial treatment regimens with the anti-IGF-1R monoclonal antibody dalotuzumab. We found that dalotuzumab acted in an additive manner with our SSO to alter phosphokinase phosphorylation and anoikis-resistant growth. Additionally, dalotuzumab enhanced the effect of the AAVrh74.U7snRNA virus to slow OS cell proliferation.
Multiple classes of RNA therapeutics have been shown to impact the expression of disease-causing genes—ASOs, siRNAs, microRNAs, and CRISPR-Cas9, to name a few.79 Within these classifications, ASOs (also known as SSOs) have received the most support for clinical applications from the FDA, with 10 therapeutics receiving approval as of 2020.80,81,82 We have characterized the use of a second-generation SSO with a PS backbone and 2′MOE modification on the ribose sugars to modulate IR splicing and induce functional changes in OS cells. The PS backbone stabilizes the ASOs to nuclease degradation, reduces hydrophobicity, and enhances protein binding, properties that helps increase cell uptake and intracellular distribution.83,84 The 2′MOE modification improves pharmacokinetics, increases the half-life of these ASOs, and enhances binding affinity to target RNA. These synthesized oligonucleotides thus exhibit increased binding to serum proteins such as albumin, which maintains them in circulation long enough for tissue distribution. Following intravenous or subcutaneous administration, the phase of distribution from plasma to tissues ranges from minutes to a few hours, followed by a prolonged elimination phase that can last for several weeks. Additionally, oligonucleotides containing both PS and sugar modifications such as 2′MOE have tissue half-lives measured in weeks.85 PS and MOE-modified single-stranded ASOs can be administered by all routes, and their oral bioavailability and absorption profiles have been demonstrated in animals and humans.85,86
Of note, SSOs, specifically those with unmodified chemistries, have been previously described to impact Toll-like receptors (TLRs) 3, 7, 8, and 9 and mounting of innate immune responses.87 However, the addition of sugar moieties and other modifications has been shown to abrogate an innate immune response that may be triggered through TLR and interferon signaling.81,88 To further develop SSOs as a viable therapeutic option in OS and other cancers, our future studies will focus on SSO55’s impact on gene expression, including immune genes, to elucidate potential off-target effects after IR splicing modulation.
Our in vitro success of mitigating some cancer cell hallmarks with SSO treatment, compounded with our desire to address barriers of SSO therapy (such as targeted delivery to tumors) in cancer, prompted us to engineer the IR SSO55 sequence as a transgene expressed via the U7 snRNA AAV gene therapy tool. While AAVrh74.U7snRNA did not switch IR splicing as efficiently as the synthetic SSO, the chemistry of the oligonucleotide versus the antisense RNA produced from the AAV or the strength of the U7 promoter could impact its efficacy. It is unclear how effective the U7 promoter is in driving downstream antisense sequence expression in cancer. Tumor cells are comparatively more hypoxic and nutrient starved than healthy cells, and these stresses can be used as leverage in gene therapy to selectively activate transgene expression.89 In the case of spliceosomal snRNPs, modified U7, U6, and U1 snRNAs are all capable of delivering antisense sequences to the nucleus.90,91,92 However, unlike U7, U1 and U6 are abundant core components of the spliceosome, which may interfere with the efficacy of modified U1 and U6 snRNAs used as gene therapy.66
In addition to our choice of promoter, our AAVrh74.U7 snRNA viruses may have demonstrated fewer splice-switching capabilities due to choice of serotype and AAV capsid design. While we selected AAVrh74 due to the low seroprevalence of binding antibodies in humans and prior successful use in DMD gene therapy development, the U7 snRNA vectors could theoretically be packaged into one of several other studied AAV serotypes that may be more efficiently transduced in OS cells or transforming osteoblasts.61,63,65,93,94,95,96 Further optimization of the viral capsid may also be key in increasing delivery efficacy.60,89 The viral capsid can be enhanced through the use of adaptors or bispecific antibodies that interact with cell surface receptors or inserting target cell-specific moieties in the viral capsid.89,97 Prior studies have shown that OS cells express epidermal growth factor receptor (EGFR), fibroblast growth factor receptor, vascular endothelial growth factor receptor, αvB3 and αvB5 integrins, and transferrin, and any of these proteins could improve AAV attachment and entry in OS.98
Taken in the context of emerging therapies for OS, the approach of using SSOs and AAV-mediated delivery has the same goals as immunotherapies (monoclonal antibodies and chimeric antigen receptor [CAR]-T cell therapy, to name a few), and small molecule inhibitors—to modulate OS cell expression and sensitize it to other chemotherapeutic agents or a host-mediated immune response.99,100 OS tumors demonstrate intra- and inter-tumor heterogeneity and numerous molecular alterations that induce metastatic transformation, making regimens that target OS-specific surface markers and disease-causing pathways a therapeutic necessity.101,102 Multiple studies and clinical trials have focused on OS surface proteins by using immune checkpoint inhibitors and T cell therapy targeting PD-1/PD-L1 and CTLA-4, bispecific T cell engagers targeting GD2, and CAR-T cell therapy with HER2 and dextrotecan.99,100,103,104,105 While these immunomodulatory therapies have shown initial preclinical and clinical promise for OS, issues with tumor specificity and immunosuppressive microenvironment remain.99,100,103 Additional described approaches include direct targeting of the DNA damage repair, angiogenesis, and other molecular pathways enriched in OS (reviewed in106,107,108). Excitingly, monoclonal antibodies aimed at the mTOR pathway (sirolimus and related drugs) and multi-level inhibition of receptor tyrosine kinases (EGFR, platelet-derived growth factor, IGF-1R, and vascular endothelial growth factor receptors, to name a few) have been especially helpful for patients with unresectable or relapsed disease.99,108 To this end, mitigating IGF-1R/IR signaling via SSOs/AAV.U7 snRNA viruses and anti-IGF-1R antibodies is a new approach to deregulating key growth pathways at the level of pre-mRNA and protein in OS. Nevertheless, further in vitro and in vivo characterization of the IR SSO and AAVrh74.U7 snRNA viruses presented in our work are paramount to characterizing their efficacy and clinical utility as therapeutics, both alone and in combination with IGF-1R inhibitors, chemotherapies, and/or immunotherapies.
Altogether, we offer SSOs as a modality to switch IR alternative splicing toward the non-proliferative IR-B isoform and to be combined with anti-IGF-1R therapies. Our results demonstrate that SSOs can mediate cancer cell hallmarks, suggesting that modulating alternative splicing has the power to shift cancer cells away from oncogenic, proliferative signaling and sensitize them to chemotherapeutics or other agents. Importantly, our perspective on advancing SSO therapeutics through viral vector delivery is an innovative approach that still requires further design optimization and consideration of the dynamic tumor microenvironment, heterogeneity of tumor cells, or complexity of the coordinated gene networks that promote tumorigenesis. Our results shed light on the role of IR alternative splicing in cancer and the testing of novel therapeutics against disease-inducing splicing events, an approach can be translated to other cancers to improve the clinical outcomes in pediatric populations.
Materials and methods
Cell lines and PDXs
Cells and cell culture
The human cell lines Saos-2 (HTB-85, OS), U2OS (HTB-96, OS), SJSA-1 (CRL-2098, OS), HeLa (CRM-CCL-2, cervical adenocarcinoma) were obtained from American Type Culture Collection (Manassas, VA). OS-17, OS-25, and OHS were kind gifts from Ryan Roberts (Nationwide Children’s Hospital, Columbus, OH). The OS cell line 143.98.2 was obtained from ATCC (CRL-11226). The 143.98.2 and OHS cells were transduced with firefly luciferase-T2A-GFP expressing lentiviral vector (PLV-10172, Cellomics Technology, Halethorpe, MD) and selected with 0.75 μg/mL puromycin for 2 weeks to become stable reporter cell lines named 143.98.2-Luc-GFP and OHS-Luc-GFP. OS-17, OS-26, OHS, Saos-2, U2OS, and SJSA-1 were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. HeLa and 143.98.2 Luc-GFP were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. All cell lines were verified by short tandem repeat genotyping and confirmed to be free of mycoplasma by IDEXX Bioanalytics or in-house mycoplasma testing.
PDXs
PDXs 1–6 (OS) were obtained as snap-frozen vials of tissue chunks from the Tumor Core at Nationwide Children’s Hospital (Columbus, OH). Tissues were designated as derived from primary or metastatic tumors by the Tumor Core at Nationwide Children’s Hospital. The tissue specimens were ground using a mortar and pestle in liquid nitrogen. Approximately 1000 ng RNA was then extracted from the specimens using the RNeasy (Qiagen, Cat# 74106, Hilden, Germany). Reverse transcription (RT) was performed using approximately 1 μg RNA as described by Jacob et al. (2013).109
Plasmids and minigenes
The IR 10-11-12 minigene was a kind gift from Nick Webster.110 The IR mutant and substitution minigenes were generated using the Quikchange Lightning mutagenesis kit (Agilent, Cat# 210519, Santa Clara, CA) and primers listed in Table 1.
Table 1.
Primer sequences
| Primer name | Sequence (5′-3′) |
|---|---|
| Endogenous IR exon 10, forward | GGCTGAAGCTGCCCTCGAG |
| Endogenous IR exon 12, reverse | GCGACTCCTTGTTCACCACC |
| IR minigene exon 10, forward | TAATACGACTCACTATAGGGC |
| IR minigene exon 12, reverse | GCTGCAATAAACAAGTTCTGC |
| IR deletion minigene (mutagenesis, forward) | GGCCTCCAAGTGTCAGAGTCTGTCTAATGAAGTTCC |
| IR deletion minigene (mutagenesis, reverse) | GGAACTTCATTAGACAGACTCTGACACTTGGAGGCC |
| IR substitution minigene (mutagenesis, forward) | TCATTAGACAGACCACAGGGCACAGACACTTGGAGGCCAC |
| IR substitution minigene (mutagenesis, reverse) | GTGGCCTCCAAGTGTCTGTGCCCTGTGGTCTGTCTAATGA |
| Puma (forward) | CCCTGGAGGGTCCTGTACAA |
| Puma (reverse) | CTCTGTGGCCCCTGGGTAA |
| Bax (forward) | CCCCGAGAGGTCTTTTTCCG |
| Bax (reverse) | GGCGTCCCAAAGTAGGAGA |
| Noxa (forward) | GCTGGAAGTCGAGTGTGCTA |
| Noxa (reverse) | CCTGAGCAGAAGAGTTTGGA |
| Fas (forward) | GGGGTGGCTTTGTCTTCTTCTTTTG |
| Fas (reverse) | ACCTTGGTTTTCCTTTCTGTGCTTTCT |
RNA isolation, RTs, and PCRs and quantitative real-time PCRs
RNA was isolated from cells using the RNeasy Plus Mini Kit (Qiagen, Cat#74134). RT reactions were performed with 500–1000 ng of RNA using Transcriptor RT enzyme (Sigma Aldrich, Cat# 03531287001, Burlington, MA) or SuperScript IV Reverse Transcriptase (Invitrogen, Cat# 18090010, Carlsbad, CA). PCRs for endogenous IR were performed using primers in Exons 10 and 12 (see Table 1 for sequences) that distinguished the IR-A (135 bp) and IR-B (160 bp) isoforms. PCRs for the IR minigene were performed using minigene-specific primers in Exons 10 and 12 that would not pick up the endogenous IR (see Table 1 for sequences). PCRs were performed with the following program unless specified otherwise: 94°C for 5 min, 35 cycles of 94°C for 30 s, 65°C for 30 s, 72°C for 1 min, and 72°C for 7 min. PCR products were then run on a 2% agarose gel at 150 V for 105 min and visualized on a Gel Doc XR + Gel Documentation System (Biorad, Hercules, CA).
All real-time qPCR reactions were performed with standard PCR conditions using a CFX96 Touch Real-Time PCR Detection system (Biorad). Real-time PCR reactions were carried out using the SYBR Green PCR Master Mix (Applied Biosystems, Cat# 4309155, Waltham, MA). The primers used to amplify the apoptosis target transcripts have been listed in Table 1. All PCR reactions were carried out with at least three technical replicates, and the amplification of single PCR products in each reaction was confirmed using a dissociation curve.
Dalotuzumab, SSOs, and transfections
Dalotuzumab (also known as MK-0646) was kindly provided by Peter Houghton and later purchased from Med Chem Express (Cat# HY-P99284, Monmouth Junction, NJ). For experiments involving dalotuzumab, 0.04 mg was added to cells with 1 mL of fresh media. Cells were then incubated for 30 min before harvest or received fresh media + dalotuzumab every 48 h.
The SSO targeting CUG-BP1 in intron 10 of the IR gene and a non-specific control SSO have been previously described by Khurshid et al.21 (2022) and were kindly manufactured by Ionis Pharmaceuticals. The SSO55 sequence is 5′-CACAGTCTCGGGTCACCA-3′, and the NS SSO72 sequence is 5′-TTAGTTTAATCACGCTCG-3'.
For transfections, cells were seeded in the appropriate media in six-well plates and transfected with 10 μL Lipofectamine 2000 (Invitrogen, Cat# 11668500) in 1 mL OptiMEM media unless otherwise stated. After a 4 h incubation, OptiMEM was removed and replaced with the appropriate media for each cell line.
Functional assays
Proliferation
U2OS and 143.98.2 cells were treated with 100 nM NS and SSO55 (using Lipofectamine 2000) and placed in IncuCyte for 60–72 h. The IncuCyte (Sartorius, Göttingen, Germany) allows for live cell imaging and images cells after every 2–4 h. The data was collected at the end of 72 h and plotted in Prism.
GILA assay
Cells were seeded in six-well plates (Corning, Cat# 7007, Corning, NY) and transfected the following day with 100 nM NS or SSO55 as described above. After 24 h, cells were stained with Trypan blue (Gibco, Cat# 1520061, Grand Island, NY) and counted using a hemocytometer. Cells were then reseeded in an ultra-low attachment 96-well plate (Corning, Cat# 3474) at a concentration of 10,000 cells/well and incubated at 37°C for 5 days. On day 5, 100 μL Cell Titer Glo (Promega, Cat# G7570, Madison, WI) was added to each well, and the plate was incubated on a shaker at room temperature for 25 min. Then, 150 μL from each well was transferred to a white bottom 96-well plate, and luminescence was quantified on the Promega GlowMax Plate Reader (integration time = 1 s).
Annexin 5 staining
NS- and SSO-treated 143.98.2 Luc-GFP cells were harvested using Accutase (Gibco, Cat# A1110501) for detachment at indicated time points for Annexin V/7ADD staining (Annexin A5 Apoptosis Detection Kit, Cat # 640930, Biolegend, San Diego, CA). Cells were then washed twice with PBS and spun down to a pellet. Cell pellets were resuspended in 100 μL Annexin V Binding Buffer containing 5 μL of APC Annexin V and 5 μL of 7-AAD and incubated at room temperature in the dark for 15 min. After incubation, an additional 400uL of Annexin V Binding Buffer were added to the cell suspension. Data were acquired on NovoCyte (Agilent) and were analyzed with FlowJo software 10.10.0 (BD Bioscience, Ashland, OR).
Proteome profiler array
The Proteome Profiler Human Phospho-Kinase Array was purchased from R&D Systems (Cat# ARY003C, Minneapolis, MN). The prepared blots came with specific phospho-kinase antibodies blotted on nitrocellulose membranes in duplicate. 143.98.2 Luc-GFP cells were seeded at 200,000 cells/well in a 6-well plate and transfected with 100 nM NS or SSO55 the following day. After 48 h following transfection, cells were harvested, and whole-cell lysates were collected for the array and processed according to the R&D Systems protocol. The membranes were visualized using the array’s chemilumiscent agent and developed on X-ray film. The signal produced on the film for each antibody correlated with the phosphorylation present in either the NS- or SSO55-treated cells. Phosphorylation was quantified by capturing the relative pixel density of each spot in ImageJ and subtracting a background signal from the average of each duplicate set.
Western blot and antibodies
U2OS and 143.98.2 Luc-GFP cells were seeded at 300,000 cells/well or 200,000 cells/well respectively in a 6-well plate. After transfection with 100 nM NS or SSO55 via Lipofectamine 2000 for specified time points, cells were harvested, and whole-cell lysates were isolated using cell lysis buffer (Cell Signaling, Cat# 9803, Danvers, MA) supplemented with a protease/phosphatase inhibitor cocktail (Thermo Scientific, Cat# 78440, Waltham, MA). Protein concentrations were calculated using the Pierce BCA Protein Assay Kit (Thermo Scientific, Cat# 23225), and approximately 30 μg protein was loaded onto a denaturing NuPAGE 4 to 12% Bis-Tris gel (Invitrogen, Cat# NP0336BOX) with a Precision Plus protein standard (Bio-Rad, Cat# 1610376, Hercules, CA). Gels were run at 120 V for 105 min, and the separated proteins were transferred to polyvinylidene fluoride membranes on ice at 35 V for 60 min. Membranes were blocked for 30 min in casein buffer (Thermo Scientific, Cat# 37582) and incubated overnight at 4°C in the following antibodies: pAKT (Ser473) (1:1,000, Cell Signaling, Cat# 4060S, clone D9E), panAKT (1:1,000, Cell Signaling, Cat# 4691S, clone C67E7), pGSK3α/β (1:1,000, Cell Signaling, Cat#9331S), and GAPDH (1:2,000, Cell Signaling, Cat#2118S). For GAPDH, the membranes were incubated in the primary antibody for 60 min at room temperature. Membranes were washed in Tris-buffered saline with 0.1% Tween 20 (TBST) thrice and then incubated with a secondary antibody conjugated to horseradish peroxidase (Cell Signaling, Cat# 7074S) for 60 min at room temperature. After additional TBST washes, the membranes were covered in ECL Western blotting substrate (Thermo Scientific, Cat# 32106) and developed on X-ray film.
U7-snRNA viral vectors
Plasmid construction and AAV viral vector production
The U7 snRNA viral vectors used to express the IR NS and SSO55 sequences were constructed and packaged by Vector Builder into AAVrh74. The Vector Builder IDs for the viral vectors shown in Figure 4 are VB230608-1954gzn (p-N1), VB230608-1856jdp (p-A1), VB230621-1559www (p-N2), VB230405-1136ksw (p-A2), and these IDs can be used to obtain the vector details on www.vectorbuilder.com.
AAV-eGFP and AAVrh74.U7snRNA virus transduction
U2OS and 143.98.2 Luc-GFP were seeded at appropriate densities in full serum media (10% FBS RPMI for U2OS, 10% DMEM for 143.98.2) 1 day before virus transduction. During the transduction period, media were replaced with 2% FBS RPMI or DMEM, and virus was added to the cells in a dropwise fashion. Full serum media was replaced after 24 h of virus transduction and every 48 h during the experimental timeline. Cells transduced with fluorescent eGFP vectors were imaged on a fluorescent microscope under the GFP lamp at 10× magnification.
Data and code availability
All data presented in this manuscript are included in the main and the supplemental figures. Additional data, materials, or reagents can be requested from the authors via email at dawn.chandler@nationwidechildrens.org and timothy.cripe@nationwidechildrens.org.
Acknowledgments
We thank Melissa Sammons and Lindsey Ryan (Tumor Core, Abigail Wexner Research Institute, Nationwide Children’s Hospital) for their assistance in providing OS PDX samples; Callie Barber (BSGP, The Ohio State University) for technical assistance and generating the mutant minigene shown in Figure 2; and Chandler and Cripe lab members for their input on experimental techniques and critical review of this manuscript. Figures were created in Biorender.com.
This work was funded in part by the OSU CMBP RNA Center, Cancer Free Kids Research Alliance, Elsa U. Pardee Foundation, Sunbeam Foundation, and NIH grants U54CA232561, T32CA269052, UL1TR002733, RO1CA262873, R03CA259856, and R21CA195324.
Author contributions
These studies were conceptualized by D.S.C., T.P.C., S.K. A.S.V., M.M., P.Y.W., and N.W. Methodology was designed by D.S.C., T.P.C., S.K. A.S.V., M.M., P.Y.W., and N.W. Experiments were performed by S.K. A.S.V., M.M., J.K., and P.Y.W. Resources for the experiments were graciously provided by R.D.R. and F.R. The data presented here was curated by S.K., A.S.V., and M.M., and visualized by A.S.V. and S.K. The original manuscript draft was written by A.S.V and S.K., and additional versions were reviewed and edited by D.S.C., T.P.C., S.K., A.S.V., M.M., P.Y.W., N.W., J.K., and R.D.R. Project supervision and administration were overseen by D.S.C. and T.P.C, and funding was acquired by D.S.C., T.P.C., and A.S.V.
Declaration of interests
The AAVrh74.U7 snRNA viruses shown in this work are the subject of an international patent application (USSN 63/572,178) with A.S.V., P.Y.W., N.W., D.S.C., and T.P.C. as inventors. T.P.C. is a member of the Molecular Therapy Oncology board and Editor-in-Chief. D.S.C. is a member of the Molecular Therapy Oncology editorial board. F.R. is affiliated with Ionis Pharmaceuticals.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omton.2024.200908.
Supplemental information
References
- 1.Sheng G., Gao Y., Yang Y., Wu H. Osteosarcoma and metastasis. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.780264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mirabello L., Troisi R.J., Savage S.A. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. Int. J. Cancer. 2009;125:229–234. doi: 10.1002/ijc.24320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kager L., Zoubek A., Pötschger U., Kastner U., Flege S., Kempf-Bielack B., Branscheid D., Kotz R., Salzer-Kuntschik M., Winkelmann W., et al. Primary metastatic osteosarcoma: presentation and outcome of patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J. Clin. Oncol. 2003;21:2011–2018. doi: 10.1200/JCO.2003.08.132. [DOI] [PubMed] [Google Scholar]
- 4.Beird H.C., Bielack S.S., Flanagan A.M., Gill J., Heymann D., Janeway K.A., Livingston J.A., Roberts R.D., Strauss S.J., Gorlick R. Osteosarcoma. Nat. Rev. Dis. Prim. 2022;8:77. doi: 10.1038/s41572-022-00409-y. [DOI] [PubMed] [Google Scholar]
- 5.Society A.C. 2023. Survival Rates for Osteosarcoma. [Google Scholar]
- 6.Huang W., Xiao Y., Wang H., Chen G., Li K. Identification of risk model based on glycolysis-related genes in the metastasis of osteosarcoma. Front. Endocrinol. 2022;13 doi: 10.3389/fendo.2022.1047433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guan X., Guan Z., Song C. Expression profile analysis identifies key genes as prognostic markers for metastasis of osteosarcoma. Cancer Cell Int. 2020;20:104. doi: 10.1186/s12935-020-01179-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rikhof B., de Jong S., Suurmeijer A.J.H., Meijer C., van der Graaf W.T.A. The insulin-like growth factor system and sarcomas. J. Pathol. 2009;217:469–482. doi: 10.1002/path.2499. [DOI] [PubMed] [Google Scholar]
- 9.Vella V., Milluzzo A., Scalisi N.M., Vigneri P., Sciacca L. Insulin Receptor Isoforms in Cancer. Int. J. Mol. Sci. 2018;19:3615. doi: 10.3390/ijms19113615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuijjer M.L., Peterse E.F.P., van den Akker B.E.W.M., Briaire-de Bruijn I.H., Serra M., Meza-Zepeda L.A., Myklebost O., Hassan A.B., Hogendoorn P.C.W., Cleton-Jansen A.-M. IR/IGF1R signaling as potential target for treatment of high-grade osteosarcoma. BMC Cancer. 2013;13:245. doi: 10.1186/1471-2407-13-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Meyts P. Endotext; 2016. The Insulin Receptor and its Signal Transduction Network. [Google Scholar]
- 12.Belfiore A., Malaguarnera R. Insulin receptor and cancer. Endocr. Relat. Cancer. 2011;18:R125–R147. doi: 10.1530/ERC-11-0074. [DOI] [PubMed] [Google Scholar]
- 13.Malaguarnera R., Belfiore A. The insulin receptor: a new target for cancer therapy. Front. Endocrinol. 2011;2:93. doi: 10.3389/fendo.2011.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belfiore A., Malaguarnera R., Vella V., Lawrence M.C., Sciacca L., Frasca F., Morrione A., Vigneri R. Insulin receptor isoforms in physiology and disease: an updated view. Endocr. Rev. 2017;38:379–431. doi: 10.1210/er.2017-00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denley A., Bonython E.R., Booker G.W., Cosgrove L.J., Forbes B.E., Ward C.W., Wallace J.C. Structural determinants for high-affinity binding of insulin-like growth factor II to insulin receptor (IR)-A, the exon 11 minus isoform of the IR. Mol. Endocrinol. 2004;18:2502–2512. doi: 10.1210/me.2004-0183. [DOI] [PubMed] [Google Scholar]
- 16.Wang P., Mak V.C., Cheung L.W. Drugging IGF-1R in cancer: New insights and emerging opportunities. Genes Dis. 2023;10:199–211. doi: 10.1016/j.gendis.2022.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh P., Alex J.M., Bast F. Insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-1R) signaling systems: novel treatment strategies for cancer. Med. Oncol. 2014;31:805. doi: 10.1007/s12032-013-0805-3. [DOI] [PubMed] [Google Scholar]
- 18.de Groot S., Röttgering B., Gelderblom H., Pijl H., Szuhai K., Kroep J.R. Unraveling the Resistance of IGF-Pathway Inhibition in Ewing Sarcoma. Cancers. 2020;12:3568. doi: 10.3390/cancers12123568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buck E., Gokhale P.C., Koujak S., Brown E., Eyzaguirre A., Tao N., Rosenfeld-Franklin M., Lerner L., Chiu M.I., Wild R., et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Mol. Cancer Therapeut. 2010;9:2652–2664. doi: 10.1158/1535-7163.MCT-10-0318. [DOI] [PubMed] [Google Scholar]
- 20.Garofalo C., Manara M.C., Nicoletti G., Marino M.T., Lollini P.L., Astolfi A., Pandini G., López-Guerrero J.A., Schaefer K.-L., Belfiore A., et al. Efficacy of and resistance to anti-IGF-1R therapies in Ewing's sarcoma is dependent on insulin receptor signaling. Oncogene. 2011;30:2730–2740. doi: 10.1038/onc.2010.640. [DOI] [PubMed] [Google Scholar]
- 21.Khurshid S., Montes M., Comiskey D.F., Shane B., Matsa E., Jung F., Brown C., Bid H.K., Wang R., Houghton P.J., et al. Splice-switching of the insulin receptor pre-mRNA alleviates tumorigenic hallmarks in rhabdomyosarcoma. npj Precis. Oncol. 2022;6:1–11. doi: 10.1038/s41698-021-00245-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burghes A.H.M., McGovern V.L. Antisense oligonucleotides and spinal muscular atrophy: skipping along. Genes Dev. 2010;24:1574–1579. doi: 10.1101/gad.1961710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sivanesan S., Howell M., DiDonato C., Singh R. Antisense oligonucleotide mediated therapy of spinal muscular atrophy. Transl. Neurosci. 2013;4:1–7. doi: 10.2478/s13380-013-0109-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wood M.J.A., Talbot K., Bowerman M. Spinal muscular atrophy: antisense oligonucleotide therapy opens the door to an integrated therapeutic landscape. Hum. Mol. Genet. 2017;26:R151–R159. doi: 10.1093/hmg/ddx215. [DOI] [PubMed] [Google Scholar]
- 25.Aartsma-Rus A., Krieg A.M. FDA approves eteplirsen for Duchenne muscular dystrophy: the next chapter in the eteplirsen saga. Nucleic Acid Therapeut. 2017;27:1–3. doi: 10.1089/nat.2016.0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mendell J.R., Rodino-Klapac L.R., Sahenk Z., Roush K., Bird L., Lowes L.P., Alfano L., Gomez A.M., Lewis S., Kota J., et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013;74:637–647. doi: 10.1002/ana.23982. [DOI] [PubMed] [Google Scholar]
- 27.Altmann K.-H., Dean N.M., Fabbro D., Freier S.M., Geiger T., Häner R., Hüsken D., Martin P., Monia B.P., Müller M., et al. Second generation of antisense oligonucleotides: from nuclease resistance to biological efficacy in animals. Chimia. 1996;50:168. [Google Scholar]
- 28.Bennett C.F., Baker B.F., Pham N., Swayze E., Geary R.S. Pharmacology of antisense drugs. Annu. Rev. Pharmacol. Toxicol. 2017;57:81–105. doi: 10.1146/annurev-pharmtox-010716-104846. [DOI] [PubMed] [Google Scholar]
- 29.Crooke S.T. Molecular mechanisms of antisense oligonucleotides. Nucleic Acid Therapeut. 2017;27:70–77. doi: 10.1089/nat.2016.0656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crooke S.T., Baker B.F., Pham N.C., Hughes S.G., Kwoh T.J., Cai D., Tsimikas S., Geary R.S., Bhanot S. The effects of 2′-O-methoxyethyl oligonucleotides on renal function in humans. Nucleic Acid Therapeut. 2018;28:10–22. doi: 10.1089/nat.2017.0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crooke S.T., Baker B.F., Kwoh T.J., Cheng W., Schulz D.J., Xia S., Salgado N., Bui H.-H., Hart C.E., Burel S.A., et al. Integrated safety assessment of 2′-O-methoxyethyl chimeric antisense oligonucleotides in nonhuman primates and healthy human volunteers. Mol. Ther. 2016;24:1771–1782. doi: 10.1038/mt.2016.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swayze E.E., Siwkowski A.M., Wancewicz E.V., Migawa M.T., Wyrzykiewicz T.K., Hung G., Monia B.P., Bennett C.F. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res. 2007;35:687–700. doi: 10.1093/nar/gkl1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rigo F., Chun S.J., Norris D.A., Hung G., Lee S., Matson J., Fey R.A., Gaus H., Hua Y., Grundy J.S., et al. Pharmacology of a central nervous system delivered 2′-O-methoxyethyl–modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J. Pharmacol. Exp. Therapeut. 2014;350:46–55. doi: 10.1124/jpet.113.212407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sen S., Talukdar I., Webster N.J.G. SRp20 and CUG-BP1 modulate insulin receptor exon 11 alternative splicing. Mol. Cell Biol. 2009;29:871–880. doi: 10.1128/MCB.01709-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kosaki A., Nelson J., Webster N.J. Identification of intron and exon sequences involved in alternative splicing of insulin receptor pre-mRNA. J. Biol. Chem. 1998;273:10331–10337. doi: 10.1074/jbc.273.17.10331. [DOI] [PubMed] [Google Scholar]
- 36.Kahn C.R., White M.F. The insulin receptor and the molecular mechanism of insulin action. J. Clin. Invest. 1988;82:1151–1156. doi: 10.1172/JCI113711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kido Y., Nakae J., Accili D. Clinical review 125: The insulin receptor and its cellular targets. J. Clin. Endocrinol. Metab. 2001;86:972–979. doi: 10.1210/jcem.86.3.7306. [DOI] [PubMed] [Google Scholar]
- 38.Sadikovic B., Yoshimoto M., Chilton-MacNeill S., Thorner P., Squire J.A., Zielenska M. Identification of interactive networks of gene expression associated with osteosarcoma oncogenesis by integrated molecular profiling. Hum. Mol. Genet. 2009;18:1962–1975. doi: 10.1093/hmg/ddp117. [DOI] [PubMed] [Google Scholar]
- 39.R2: Genomics Analysis and Visualization Platform. http://r2.amc.nl
- 40.Sen S., Talukdar I., Liu Y., Tam J., Reddy S., Webster N.J.G. Muscleblind-like 1 (Mbnl1) promotes insulin receptor exon 11 inclusion via binding to a downstream evolutionarily conserved intronic enhancer. J. Biol. Chem. 2010;285:25426–25437. doi: 10.1074/jbc.M109.095224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beisang D., Rattenbacher B., Vlasova-St Louis I.A., Bohjanen P.R. Regulation of CUG-binding protein 1 (CUGBP1) binding to target transcripts upon T cell activation. J. Biol. Chem. 2012;287:950–960. doi: 10.1074/jbc.M111.291658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Masuda A., Andersen H.S., Doktor T.K., Okamoto T., Ito M., Andresen B.S., Ohno K. CUGBP1 and MBNL1 preferentially bind to 3′ UTRs and facilitate mRNA decay. Sci. Rep. 2012;2:209. doi: 10.1038/srep00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xia H., Chen D., Wu Q., Wu G., Zhou Y., Zhang Y., Zhang L. CELF1 preferentially binds to exon-intron boundary and regulates alternative splicing in HeLa cells. Biochim. Biophys. Acta. Gene Regul. Mech. 2017;1860:911–921. doi: 10.1016/j.bbagrm.2017.07.004. [DOI] [PubMed] [Google Scholar]
- 45.Haeusler R.A., McGraw T.E., Accili D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018;19:31–44. doi: 10.1038/nrm.2017.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y.H., Han X.D., Qiu Y., Xiong J., Yu Y., Wang B., Zhu Z.Z., Qian B.P., Chen Y.X., Wang S.F., et al. Increased expression of insulin-like growth factor-1 receptor is correlated with tumor metastasis and prognosis in patients with osteosarcoma. J. Surg. Oncol. 2012;105:235–243. doi: 10.1002/jso.22077. [DOI] [PubMed] [Google Scholar]
- 47.Rotem A., Janzer A., Izar B., Ji Z., Doench J.G., Garraway L.A., Struhl K. Alternative to the soft-agar assay that permits high-throughput drug and genetic screens for cellular transformation. Proc. Natl. Acad. Sci. USA. 2015;112:5708–5713. doi: 10.1073/pnas.1505979112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lakshmanan I., Batra S.K. Protocol for apoptosis assay by flow cytometry using annexin V staining method. Bio. Protoc. 2013;3:e374. doi: 10.21769/bioprotoc.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin S.J., Reutelingsperger C.P., McGahon A.J., Rader J.A., van Schie R.C., LaFace D.M., Green D.R. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J. Exp. Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Logue S.E., Elgendy M., Martin S.J. Expression, purification and use of recombinant annexin V for the detection of apoptotic cells. Nat. Protoc. 2009;4:1383–1395. doi: 10.1038/nprot.2009.143. [DOI] [PubMed] [Google Scholar]
- 51.Roufayel R., Younes K., Al-Sabi A., Murshid N. BH3-only proteins Noxa and Puma are key regulators of induced apoptosis. Life. 2022;12:256. doi: 10.3390/life12020256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oda E., Ohki R., Murasawa H., Nemoto J., Shibue T., Yamashita T., Tokino T., Taniguchi T., Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 53.Nagata S. 1996. Fas-mediated Apoptosis. Mechanisms of Lymphocyte Activation and Immune Regulation VI: Cell Cycle and Programmed Cell Death in the Immune System; pp. 119–124. [Google Scholar]
- 54.Rao-Bindal K., Zhou Z., Kleinerman E.S. MS-275 sensitizes osteosarcoma cells to Fas ligand-induced cell death by increasing the localization of Fas in membrane lipid rafts. Cell Death Dis. 2012;3:e369. doi: 10.1038/cddis.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suhara T., Kim H.-S., Kirshenbaum L.A., Walsh K. Suppression of Akt signaling induces Fas ligand expression: involvement of caspase and Jun kinase activation in Akt-mediated Fas ligand regulation. Mol. Cell Biol. 2002;22:680–691. doi: 10.1128/MCB.22.2.680-691.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Osaki M., Kase S., Adachi K., Takeda A., Hashimoto K., Ito H. Inhibition of the PI3K-Akt signaling pathway enhances the sensitivity of Fas-mediated apoptosis in human gastric carcinoma cell line, MKN-45. J. Cancer Res. Clin. Oncol. 2004;130:8–14. doi: 10.1007/s00432-003-0505-z. [DOI] [PubMed] [Google Scholar]
- 57.Godfrey C., Desviat L.R., Smedsrød B., Piétri-Rouxel F., Denti M.A., Disterer P., Lorain S., Nogales-Gadea G., Sardone V., Anwar R., et al. Delivery is key: lessons learnt from developing splice-switching antisense therapies. EMBO Mol. Med. 2017;9:545–557. doi: 10.15252/emmm.201607199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gökirmak T., Nikan M., Wiechmann S., Prakash T.P., Tanowitz M., Seth P.P. Overcoming the challenges of tissue delivery for oligonucleotide therapeutics. Trends Pharmacol. Sci. 2021;42:588–604. doi: 10.1016/j.tips.2021.04.010. [DOI] [PubMed] [Google Scholar]
- 59.Hammond S.M., Aartsma-Rus A., Alves S., Borgos S.E., Buijsen R.A.M., Collin R.W.J., Covello G., Denti M.A., Desviat L.R., Echevarría L., et al. Delivery of oligonucleotide-based therapeutics: challenges and opportunities. EMBO Mol. Med. 2021;13 doi: 10.15252/emmm.202013243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Santiago-Ortiz J.L., Schaffer D.V. Adeno-associated virus (AAV) vectors in cancer gene therapy. J. Contr. Release. 2016;240:287–301. doi: 10.1016/j.jconrel.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Issa S.S., Shaimardanova A.A., Solovyeva V.V., Rizvanov A.A. Various AAV serotypes and their applications in gene therapy: an overview. Cells. 2023;12:785. doi: 10.3390/cells12050785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schümperli D., Pillai R.S. The special Sm core structure of the U7 snRNP: far-reaching significance of a small nuclear ribonucleoprotein. Cell. Mol. Life Sci. 2004;61:2560–2570. doi: 10.1007/s00018-004-4190-0. [DOI] [PubMed] [Google Scholar]
- 63.Mendell J.R., Connolly A.M., Lehman K.J., Griffin D.A., Khan S.Z., Dharia S.D., Quintana-Gallardo L., Rodino-Klapac L.R. Testing preexisting antibodies prior to AAV gene transfer therapy: rationale, lessons and future considerations. Mol. Ther. Methods Clin. Dev. 2022;25:74–83. doi: 10.1016/j.omtm.2022.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Louis Jeune V., Joergensen J.A., Hajjar R.J., Weber T. Pre-existing anti–adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum. Gene Ther. Methods. 2013;24:59–67. doi: 10.1089/hgtb.2012.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goedeker N.L., Dharia S.D., Griffin D.A., Coy J., Truesdale T., Parikh R., Whitehouse K., Santra S., Asher D.R., Zaidman C.M. Evaluation of rAAVrh74 gene therapy vector seroprevalence by measurement of total binding antibodies in patients with Duchenne muscular dystrophy. Ther. Adv. Neurol. Disord. 2023;16 doi: 10.1177/17562864221149781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gadgil A., Raczyńska K.D. U7 snRNA: A tool for gene therapy. J. Gene Med. 2021;23 doi: 10.1002/jgm.3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Scartozzi M., Bianconi M., Maccaroni E., Giampieri R., Berardi R., Cascinu S. Dalotuzumab, a recombinant humanized mAb targeted against IGFR1 for the treatment of cancer. Curr. Opin. Mol. Therapeut. 2010;12:361–371. [PubMed] [Google Scholar]
- 68.Botter S.M., Arlt M.J., Born W., Fuchs B. Bone Cancer. Elsevier; 2015. Mammalian models of bone sarcomas; pp. 349–363. [Google Scholar]
- 69.Boyle D.B., Coupar B.E. Identification and cloning of the fowlpox virus thymidine kinase gene using vaccinia virus. J. Gen. Virol. 1986;67:1591–1600. doi: 10.1099/0022-1317-67-8-1591. [DOI] [PubMed] [Google Scholar]
- 70.Hermida M.A., Dinesh Kumar J., Leslie N.R. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv. Biol. Regul. 2017;65:5–15. doi: 10.1016/j.jbior.2017.06.003. [DOI] [PubMed] [Google Scholar]
- 71.Bradley R.K., Anczuków O. RNA splicing dysregulation and the hallmarks of cancer. Nat. Rev. Cancer. 2023;23:135–155. doi: 10.1038/s41568-022-00541-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peng Q., Zhou Y., Oyang L., Wu N., Tang Y., Su M., Luo X., Wang Y., Sheng X., Ma J., Liao Q. Impacts and mechanisms of alternative mRNA splicing in cancer metabolism, immune response, and therapeutics. Mol. Ther. 2022;30:1018–1035. doi: 10.1016/j.ymthe.2021.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harris M.A., Hawkins C.J. Recent and ongoing research into metastatic osteosarcoma treatments. Int. J. Mol. Sci. 2022;23:3817. doi: 10.3390/ijms23073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ameline B., Kovac M., Nathrath M., Barenboim M., Witt O., Krieg A.H., Baumhoer D. Overactivation of the IGF signalling pathway in osteosarcoma: a potential therapeutic target? J. Pathol. Clin. Res. 2021;7:165–172. doi: 10.1002/cjp2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shimizu T., Sugihara E., Yamaguchi-Iwai S., Tamaki S., Koyama Y., Kamel W., Ueki A., Ishikawa T., Chiyoda T., Osuka S., et al. IGF2 preserves osteosarcoma cell survival by creating an autophagic state of dormancy that protects cells against chemotherapeutic stress. Cancer Res. 2014;74:6531–6541. doi: 10.1158/0008-5472.CAN-14-0914. [DOI] [PubMed] [Google Scholar]
- 76.Aupy P., Zarrouki F., Sandro Q., Gastaldi C., Buclez P.-O., Mamchaoui K., Garcia L., Vaillend C., Goyenvalle A. Long-term efficacy of AAV9-U7snRNA-mediated exon 51 skipping in mdx52 Mice. Mol. Ther. Methods Clin. Dev. 2020;17:1037–1047. doi: 10.1016/j.omtm.2020.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Biferi M.G., Cohen-Tannoudji M., Cappelletto A., Giroux B., Roda M., Astord S., Marais T., Bos C., Voit T., Ferry A., Barkats M. A new AAV10-U7-mediated gene therapy prolongs survival and restores function in an ALS mouse model. Mol. Ther. 2017;25:2038–2052. doi: 10.1016/j.ymthe.2017.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Goyenvalle A., Vulin A., Fougerousse F., Leturcq F., Kaplan J.-C., Garcia L., Danos O. Rescue of dystrophic muscle through U7 snRNA-mediated exon skipping. Science. 2004;306:1796–1799. doi: 10.1126/science.1104297. [DOI] [PubMed] [Google Scholar]
- 79.Takakura K., Kawamura A., Torisu Y., Koido S., Yahagi N., Saruta M. The clinical potential of oligonucleotide therapeutics against pancreatic cancer. Int. J. Mol. Sci. 2019;20:3331. doi: 10.3390/ijms20133331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dhuri K., Bechtold C., Quijano E., Pham H., Gupta A., Vikram A., Bahal R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. 2020;9:2004. doi: 10.3390/jcm9062004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Roberts T.C., Langer R., Wood M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020;19:673–694. doi: 10.1038/s41573-020-0075-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Egli M., Manoharan M. Chemistry, structure and function of approved oligonucleotide therapeutics. Nucleic Acids Res. 2023;51:2529–2573. doi: 10.1093/nar/gkad067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Geary R.S., Yu R., Leeds J.M., Watanabe T.A., Henry S.P., Levin A.A., Templin M.V. Pharmacokinetic properties in animals. Antisense Drug Technol. 2001:119–154. Marcel Dekker Inc. [Google Scholar]
- 84.Levin A.A., Henry S.P. Toxicology of Oligonucleotide Therapeutics and Understanding the Relevance of the Toxicities. Pharmaceut. Sci. Encycl. Drug Discov. Dev. Manuf. 2010:1–38. [Google Scholar]
- 85.Geary R.S., Baker B.F., Crooke S.T. Clinical and preclinical pharmacokinetics and pharmacodynamics of mipomersen (Kynamro®): a second-generation antisense oligonucleotide inhibitor of apolipoprotein B. Clin. Pharmacokinet. 2015;54:133–146. doi: 10.1007/s40262-014-0224-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tillman L.G., Geary R.S., Hardee G.E. Oral delivery of antisense oligonucleotides in man. J. Pharmacol. Sci. (Tokyo, Jpn.) 2008;97:225–236. doi: 10.1002/jps.21084. [DOI] [PubMed] [Google Scholar]
- 87.Ohto U., Shibata T., Tanji H., Ishida H., Krayukhina E., Uchiyama S., Miyake K., Shimizu T. Structural basis of CpG and inhibitory DNA recognition by Toll-like receptor 9. Nature. 2015;520:702–705. doi: 10.1038/nature14138. [DOI] [PubMed] [Google Scholar]
- 88.Yoshida T., Hagihara T., Uchida Y., Horiuchi Y., Sasaki K., Yamamoto T., Yamashita T., Goda Y., Saito Y., Yamaguchi T., et al. Introduction of sugar-modified nucleotides into CpG-containing antisense oligonucleotides inhibits TLR9 activation. Sci. Rep. 2024;14 doi: 10.1038/s41598-024-61666-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hacker U.T., Bentler M., Kaniowska D., Morgan M., Büning H. Towards clinical implementation of adeno-associated virus (AAV) vectors for cancer gene therapy: current status and future perspectives. Cancers. 2020;12:1889. doi: 10.3390/cancers12071889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gorman L., Mercatante D.R., Kole R. Restoration of correct splicing of thalassemic β-globin pre-mRNA by modified U1 snRNAs. J. Biol. Chem. 2000;275:35914–35919. doi: 10.1074/jbc.M006259200. [DOI] [PubMed] [Google Scholar]
- 91.Noonberg S.B., Scott G.K., Garovoy M.R., Benz C.C., Hunt C.A. In vivo generation of highly abundant sequence-specific oligonucleotides for antisense and triplex gene regulation. Nucleic Acids Res. 1994;22:2830–2836. doi: 10.1093/nar/22.14.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Good P.D., Krikos A.J., Li S.X., Bertrand E., Lee N.S., Giver L., Ellington A., Zaia J.A., Rossi J.J., Engelke D.R. Expression of small, therapeutic RNAs in human cell nuclei. Gene Ther. 1997;4:45–54. doi: 10.1038/sj.gt.3300354. [DOI] [PubMed] [Google Scholar]
- 93.Shoti J., Qing K., Keeler G.D., Duan D., Byrne B.J., Srivastava A. Development of capsid-and genome-modified optimized AAVrh74 vectors for muscle gene therapy. Mol. Ther. Methods Clin. Dev. 2023;31 doi: 10.1016/j.omtm.2023.101147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.ELEVIDYS (delandistrogene moxeparvovec) FDA Highlights of prescribing information. https://www.fda.gov/media/169679/download?attachment
- 95.John A.A., Xie J., Yang Y.-S., Kim J.-M., Lin C., Ma H., Gao G., Shim J.-H. AAV-mediated delivery of osteoblast/osteoclast-regulating miRNAs for osteoporosis therapy. Mol. Ther. Nucleic Acids. 2022;29:296–311. doi: 10.1016/j.omtn.2022.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee L.R., Peacock L., Lisowski L., Little D.G., Munns C.F., Schindeler A. Targeting adeno-associated virus vectors for local delivery to fractures and systemic delivery to the skeleton. Mol. Ther. Methods Clin. Dev. 2019;15:101–111. doi: 10.1016/j.omtm.2019.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lv Y.-F., Zhang H., Cui Z., Ma C.-J., Li Y.-L., Lu H., Wu H.-Y., Yang J.-L., Cao C.-Y., Sun W.-Z., Huang X.F. Gene delivery to breast cancer by incorporated EpCAM targeted DARPins into AAV2. BMC Cancer. 2023;23:1220. doi: 10.1186/s12885-023-11705-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Witlox M.A., Lamfers M.L., Wuisman P.I.J.M., Curiel D.T., Siegal G.P. Evolving gene therapy approaches for osteosarcoma using viral vectors: review. Bone. 2007;40:797–812. doi: 10.1016/j.bone.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Moukengue B., Lallier M., Marchandet L., Baud’huin M., Verrecchia F., Ory B., Lamoureux F. Origin and therapies of osteosarcoma. Cancers. 2022;14:3503. doi: 10.3390/cancers14143503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gill J., Gorlick R. Advancing therapy for osteosarcoma. Nat. Rev. Clin. Oncol. 2021;18:609–624. doi: 10.1038/s41571-021-00519-8. [DOI] [PubMed] [Google Scholar]
- 101.Wang Y., Tian X., Zhang W., Zhang Z., Lazcano R., Hingorani P., Roth M.E., Gill J.D., Harrison D.J., Xu Z., et al. Comprehensive surfaceome profiling to identify and validate novel cell-surface targets in osteosarcoma. Mol. Cancer Therapeut. 2022;21:903–913. doi: 10.1158/1535-7163.MCT-21-0836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.PosthumaDeBoer J., Witlox M.A., Kaspers G.J.L., van Royen B.J. Molecular alterations as target for therapy in metastatic osteosarcoma: a review of literature. Clin. Exp. Metastasis. 2011;28:493–503. doi: 10.1007/s10585-011-9384-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lu Y., Zhang J., Chen Y., Kang Y., Liao Z., He Y., Zhang C. Novel immunotherapies for osteosarcoma. Front. Oncol. 2022;12 doi: 10.3389/fonc.2022.830546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wedekind M.F., Wagner L.M., Cripe T.P. Immunotherapy for osteosarcoma: where do we go from here? Pediatr. Blood Cancer. 2018;65 doi: 10.1002/pbc.27227. [DOI] [PubMed] [Google Scholar]
- 105.Ahmed N., Brawley V.S., Hegde M., Robertson C., Ghazi A., Gerken C., Liu E., Dakhova O., Ashoori A., Corder A., et al. Human epidermal growth factor receptor 2 (HER2)–specific chimeric antigen receptor–modified T cells for the immunotherapy of HER2-positive sarcoma. J. Clin. Oncol. 2015;33:1688–1696. doi: 10.1200/JCO.2014.58.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hu Z., Wen S., Huo Z., Wang Q., Zhao J., Wang Z., Chen Y., Zhang L., Zhou F., Guo Z., et al. Current status and prospects of targeted therapy for osteosarcoma. Cells. 2022;11:3507. doi: 10.3390/cells11213507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li S., Zhang H., Liu J., Shang G. Targeted therapy for osteosarcoma: a review. J. Cancer Res. Clin. Oncol. 2023;149:6785–6797. doi: 10.1007/s00432-023-04614-4. [DOI] [PubMed] [Google Scholar]
- 108.Lilienthal I., Herold N. Targeting molecular mechanisms underlying treatment efficacy and resistance in osteosarcoma: a review of current and future strategies. Int. J. Mol. Sci. 2020;21:6885. doi: 10.3390/ijms21186885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jacob A.G., O'Brien D., Singh R.K., Comiskey D.F., Jr., Littleton R.M., Mohammad F., Gladman J.T., Widmann M.C., Jeyaraj S.C., Bolinger C. Stress-induced isoforms of MDM2 and MDM4 correlate with high-grade disease and an altered splicing network in pediatric rhabdomyosarcoma. Neoplasia. 2013;15:1049–1063. doi: 10.1593/neo.13286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sen S., Jumaa H., Webster N.J.G. Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat. Commun. 2013;4:1336. doi: 10.1038/ncomms2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data presented in this manuscript are included in the main and the supplemental figures. Additional data, materials, or reagents can be requested from the authors via email at dawn.chandler@nationwidechildrens.org and timothy.cripe@nationwidechildrens.org.