

Summary
Increased blood amino acid levels (hyperaminoacidemia) stimulate pancreas expansion by unclear mechanisms. Here, by genetic and pharmacological disruption of glucagon receptor (GCGR) in mice and zebrafish, we found that the ensuing hyperaminoacidemia promotes pancreatic acinar cell proliferation and cell hypertrophy, which can be mitigated by a low protein diet in mice. In addition to mammalian target of rapamycin complex 1 (mTORC1) signaling, acinar cell proliferation required slc38a5, the most highly expressed amino acid transporter gene in both species. Transcriptomics data revealed the activation signature of yes-associated protein (YAP) in acinar cells of mice with hyperaminoacidemia, consistent with the observed increase in YAP-expressing acinar cells. Yap1 activation also occurred in acinar cells in gcgr−/− zebrafish, which was reversed by rapamycin. Knocking down yap1 in gcgr−/− zebrafish decreased mTORC1 activity and acinar cell proliferation and hypertrophy. Thus, the study discovered a previously unrecognized role of the YAP/Taz pathway in hyperaminoacidemia-induced acinar cell hypertrophy and hyperplasia.
Subject areas: Biomolecules, Molecular biology, Cell biology, Model organism
Graphical abstract
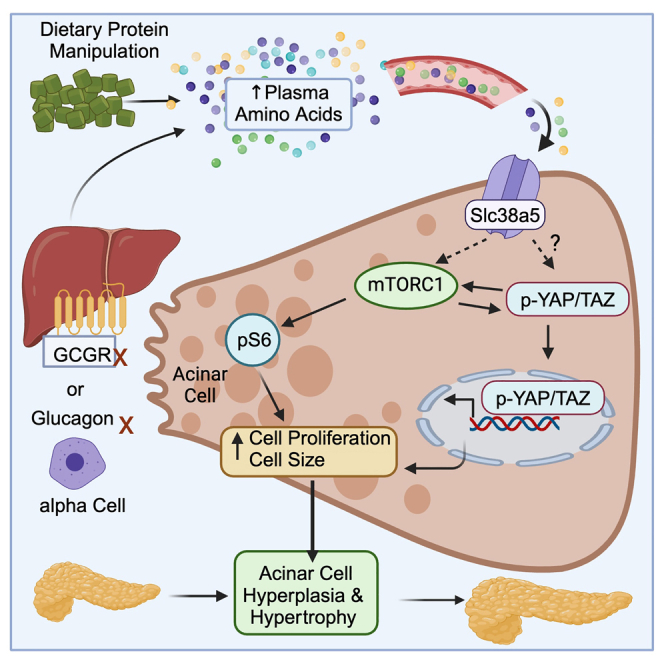
Highlights
-
•
Hyperaminoacidemia causes acinar cell hypertrophy and hyperplasia in mice and zebrafish
-
•
Zebrafish acinar cell hypertrophy and proliferation require slc38a5b
-
•
Hyperaminoacidemia activates YAP/TAZ in acinar cells
-
•
Zebrafish mTORC1 activation and acinar cell hypertrophy and proliferation entail yap1
Biomolecules; Molecular biology; Cell biology; Model organism
Introduction
Acinar cells are the most abundant cell type of the pancreas, constituting 70% of the cell number and 85% of the pancreas mass.1,2,3 They produce, store, and secrete large amounts of digestive enzymes necessary for the digestion of proteins, carbohydrates, and fats.4,5 Although pancreas mass is stable in adults, it can be changed by diet protein levels by unclear mechanisms.
Pancreas mass changes according to the levels of protein intake. Chronic protein deficiency causes pancreas atrophy both in humans and animal models.6,7 Conversely, a high-protein diet induces pancreas expansion in animal models.8,9 Both acinar cell hypertrophy and hyperplasia contribute to pancreas expansion.8 In mice, increased levels of blood amino acids (hyperaminoacidemia) and cholecystokinin (CCK) from ingestion of a high protein diet induce acinar cell proliferation and hypertrophy through activation of the mammalian target of rapamycin complex 1 (mTORC1).10,11 Whether other pathways are also necessary for hyperaminoacidemia-induced pancreas growth is unknown.
Interrupted glucagon signaling (IGS) also results in hyperaminoacidemia12,13,14,15,16 and increased pancreas mass.14,17,18,19 Hyperaminoacidemia results from decreased glucagon-stimulated amino acid (aa) catabolism through ureagenesis and gluconeogenesis in the liver.15,16,20 Unlike diet-induced hyperaminoacidemia, IGS-induced hyperaminoacidemia persists during fasting and is decoupled from enteroendocrine secretion. Nevertheless, the total pancreas mass of Gcgr−/− mice and mice with liver-specific GCGR deletion is 1.5–3.5 times larger than that of control littermates by 6 weeks of age.17,18,19 Patients with Mahvash disease, an autosomal recessive condition of biallelic GCGR loss of function, also have hyperaminoacidemia and an enlarged pancreas.18,21,22,23,24 Although IGS causes profound alpha cell hypertrophy, hyperplasia, and glucagonoma,16,17,18,19,20 the pancreas expansion is unlikely the sole result of increased endocrine cells since endocrine mass makes up less than 2% of total pancreas mass. Expansion of the exocrine pancreas is likely the major contributor.
To investigate the cellular and molecular mechanisms of hyperaminoacidemia-induced pancreas expansion, we used multiple models of IGS in mice and zebrafish. We used a low protein diet (LPD) to correct hyperaminoacidemia in an IGS mouse model. We demonstrated that IGS induces acinar cell hyperplasia and hypertrophy, which can be blunted by lowering blood aa levels with a LPD. Both acinar cell hyperplasia and hypertrophy require mTORC1. Transcriptomic analysis of acinar cells revealed Slc38a5 as the most highly expressed aa transporter and provided evidence for activation of yes-associated protein (YAP) signaling in IGS mice. Knockdown experiments in gcgr−/− zebrafish demonstrated an essential role for slc38a5b and yap1 in acinar cell hyperplasia and hypertrophy. These results uncovered multiple mechanisms of hyperaminoacidemia-induced pancreas expansion and revealed a previously unappreciated role for SLC38A5 and the YAP/TAZ pathway in mediating aa-induced acinar cell proliferation and hypertrophy.
Results
IGS causes both hyperplasia and hypertrophy of acinar cells
Pancreas expansion is a prominent phenotype in Gcgr−/− mice and patients with biallelic inactivating mutations in GCGR. To assess the mechanisms of pancreas expansion under IGS, we characterized pancreas size in two additional mouse models and one zebrafish model in which pancreas expansion has not been previously investigated. The first mouse model is Gcg−/− mice that lack the glucagon-encoding exon of the preproglucagon gene on the immunodeficient NOD scid gamma (NSG) background (Figure S1A).25 Compared to control, Gcg−/− mice exhibited lower blood glucose levels (Figure S1B), more than 2-fold increase of total serum amino acids (Figure S1C), and no change in body weight (Figure S1D). They also had increased absolute and relative pancreas weight (Figures S1E and S1F). The second mouse model is GCGR-Ab treated C57BL/6J mice (Figure S2A). Compared to IgG treatment, GCGR-Ab treatment for 8 weeks resulted in a more than 2-fold increase of serum amino acids, as reported previously (Figure S2B).15,20 At 2, 4, and 8 weeks of treatment, GCGR-Ab-treated mice had significantly decreased blood glucose levels (Figures S2C, S2G, and S2K), normal body weight (Figures S2D, S2H, and S2L), and a progressive increase absolute pancreas weight (Figures S2E, S2I, and S2M) and relative pancreas weight (Figures S2F, S2J, and S2N). We also compared individual serum amino acids in mice treated for 8 weeks. Except for tryptophan, phenylalanine, and cysteine, all other amino acids were increased by GCGR-Ab treatment (Figures S3A–S3C), confirming our previous results.20 To determine whether pancreas expansion is conserved across species, we evaluated pancreas size in gcgr−/− fish carrying Tg(ela3l:EGFP) (Figure S4A), which has hyperaminoacidemia at both larval and adult stages.26,27 Both the pancreas volume and pancreas area were significantly greater at 18 dpf compared to controls (Figures S4B–S4D). These data demonstrated that IGS in all models induces pancreas expansion.
Pancreas expansion could result from increased acinar cell size, cell proliferation, or both. Since acinar cells are the major cell type of the pancreas,1,2 we reasoned that endocrine expansion could not be responsible for the increased pancreas size. We did not observe signs of ductal expansion and did not see signs of pancreas edema on examination of the pancreas or in pancreatic sections. So, we determined acinar cell proliferation, size, and apoptosis. We found that Gcg−/− and GCGR-Ab treated mice, and gcgr−/− zebrafish significantly increased the percentage of proliferating acinar cells (Ki67+ in mice, EdU+ in zebrafish) compared to their controls (Figures 1A–1F). These data indicate that IGS stimulates acinar cell proliferation in mice and zebrafish.
Figure 1.

IGS increases acinar cell proliferation and cell size
(A and C) Representative images of acinar tissue immunofluorescence of amylase (green) and Ki67 (red). DAPI (blue) was used to label the nuclei. The pancreas sections were from Gcg+/+ or Gcg−/− mice (A) or from C57BL/J6 mice treated with IgG or GCGR-Ab (C).
(B and D) Quantification of Ki67 positive acinar cell (n = 5–7). Arrows point to Ki67+ cells.
(E) Representative immunofluorescent images of pancreas sections from 18 dpf zebrafish. Green (GFP), red (EdU), and blue (Amylase). Arrows, EdU+ acinar cells.
(F) Quantification of EdU-positive acinar cells (n = 12).
(G and I) Representative images of acinar tissue immunofluorescence of Amylase (Green), E-cadherin, and Collagen (Red). DAPI (blue) was used to label the nuclei. The pancreas sections were from Gcg+/+ or Gcg−/− mice (G) or from C57BL/J6 mice treated with IgG or GCGR-Ab (I).
(H and J) Measurements of acinar cell size (by area) in the two mouse models.
(K) Average acinar cell size of control and gcgr−/− zebrafish. Each data point is the average of more than 50 cells from one fish. Scale bar, 15 μm in (E). Scale bar, 50 μm in others. Data are represented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Student’s t test.
We next compared acinar cell size between IGS and control animals. Compared to the control, the average acinar cell size was larger in Gcg−/− mice (Figures 1G and 1H) and GCGR-Ab treated mice (Figures 1I and 1J). As peri-islet acinar cells are more hypertrophic and proliferative,28,29 we measured acinar cell size in different regions and found increased acinar cell size throughout the pancreas (Figure S5). Increased acinar cell size was also observed in gcgr−/− zebrafish (Figure 1K). These data indicate that IGS induces acinar cell hypertrophy in mice and zebrafish. TUNEL assay did not find a change in acinar cell apoptosis in GCGR-Ab treated mice (Figures S6A and S6B). Taken together, these results demonstrated that IGS induces adaptive proliferation and hypertrophy in acinar cells.
IGS-induced pancreas expansion is independent of GLP-1 and pancreatitis, but requires hyperaminoacidemia
Pharmacological activation of glucagon-like peptide-1 receptor (GLP-1R) has been shown to promote acinar cell growth and proliferation.30,31 As Gcgr−/− mice have increased α cell mass and serum GLP-1 levels,17,25,32 we evaluated whether GCGR-Ab treatment increases GLP-1/GLP-1R signaling in C57BL/J6 mice. We found that an 8-week GCGR-Ab treatment did not increase Glp1r mRNA in acinar cells compared to IgG treatment (Figure S7A). To determine whether GLP1R is necessary for IGS-induced pancreas expansion, we treated Glp1r−/− mice with GCGR-Ab or IgG for 8 weeks (Figure 2A). The treatment decreased blood glucose as expected (Figure S7B), but still induced pancreas expansion compared to IgG treatment (Figures 2B, S7C, and S7D). These results indicate that the GLP-1 pathway is unnecessary for IGS-induced pancreas expansion.
Figure 2.

Hyperaminoacidemia, but not GLP-1, contributes to the increased pancreas mass in the GCGR-Ab-treated mice
(A) Schematic of experimental design in Glp1r−/− and control mice.
(B) Pancreas mass in Glp1r−/− mice treated with IgG or GCGR-Ab (n = 9–11/treatment). ∗∗∗p < 0.001. Student’s t test.
(C) Experimental outline depicting the treatment of IGS-induced pancreas expansion by a low protein diet.
(D) Relative pancreas weight in Gcgrhep−/− and control mice on 20% or 6% protein diet.
(E) Total blood aa in Gcgrhep−/− and control mice on 20% or 6% protein diet.
(F) Experimental outline to prevent IGS-induced pancreas expansion.
(G) Relative pancreas weight in Gcgrhep−/− and control mice on 20% or 6% protein diet.
(H) Total blood aa in Gcgrhep−/− and control mice on 20% or 6% protein diet. Data are represented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. One-way ANOVA followed by Turkey’s multiple comparisons tests.
Pancreatitis has been reported to induce acinar cell proliferation.33 We, therefore, examined the presence of immune cells in the pancreas using CD45 immunofluorescence. There was no increase of CD45+ immune cells in the exocrine tissue of GCGR-Ab treated mice (Figures S7E and S7F). Furthermore, there was no change in serum CCK levels or expression of either the major CCK receptor Cckar or the minor CCK receptor Cckbr in acinar cells in GCGR-Ab treated mice compared to IgG-treated control (Figures S7G–S7I).
As hyperaminoacidemia from a protein-rich diet causes pancreas expansion, we assessed the role of hyperaminoacidemia in IGS-induced pancreas expansion. We decreased blood aa levels through diet in a fourth IGS model, hepatocyte-specific deletion of Gcgr (Gcgrhep−/− mice), with Gcgrflox mice as control.19 As dietary protein is the major contributor to blood aa,34 we changed the diet of 22-week-old Gcgrhep−/− and Gcgrflox mice from a chow diet to a regular protein diet (RPD) or a LPD for 18 weeks (Figure 2D). Gcgrhep−/− and Gcgrflox mice did not have a significant difference in body weight on the same diet, but Gcgrflox mice on the RPD weighed more than both Gcgrhep−/− and Gcgrflox mice on the LPD (Figure S8A). On the same diet, Gcgrhep−/− mice had a larger relative pancreas weight (Figures 2D and S8B) and more than 3-fold higher total plasma aa levels than Gcgrflox mice (Figure 2E). With the same genotype, mice had higher total plasma aa levels on the RPD than on the LPD (Figure 2E). However, the relative pancreas weight of Gcgrhep−/− mice on the LPD was still higher than those in Gcgrflox mice on the RPD (Figures 2D and S8B). Therefore, LPD only partially reduced pancreas expansion in Gcgrhep−/− mice.
Some of the pancreas weight increase in Gcgrhep−/− mice may have occurred before the diet switch. To test this, we started feeding Gcgrhep−/− and Gcgrflox mice the RPD or the LPD at weaning for 14 weeks (Figure 2F). Mice on LPD had a small reduction of body weight and markedly smaller absolute pancreas weight than those on the RPD (Figures S8C and S8D). As expected, on the RPD the relative pancreas weight and total blood aa levels in Gcgrhep−/− mice were higher than those of Gcgrflox mice (Figures 2G and 2H). Strikingly, there was no difference in relative pancreas weight or total blood aa levels between Gcgrhep−/− and Gcgrflox mice on the LPD, both were similar to Gcgrflox mice on the RPD (Figures 2G and 2H). Therefore, we conclude that hyperaminoacidemia is essential for IGS-induced pancreas expansion.
SLC38A5 mediates hyperaminoacidemia-induced acinar cell hyperplasia and pancreas expansion
Extracellular amino acids are transported intracellularly by aa transporters (aaTs) to exert their functions. Consistent with the expression pattern in human acinar cells,35 our acinar cell RNA-seq data indicated that Slc38a5, encoding a neutral aa transporter, was the most highly expressed aaT in mouse acinar cells (Figure 3A). Its expression was not changed by GCGR-Ab treatment (Figure 3B). To determine whether Slc38a5 is necessary for the observed acinar cell growth and proliferation, we used CRISPR/Cas9 to knock down the zebrafish ortholog slc38a5b, the second most highly expressed aaT in acinar cells (Table S1).36,37 Knockdown of slc38a5b with 2 efficient sgRNAs in zebrafish decreased the proliferation and size of acinar cells in gcgr−/− zebrafish (Figures 3C–3E) (Table S2), suggesting that Slc38a5b is crucial for IGS-induced acinar hyperplasia and hypertrophy.
Figure 3.

Slc38a5 mediates acinar cell growth
(A) The top 11 amino acid transporters expressed in mouse acinar cells (n = 5, data from RNA-seq).
(B) Slc38a5 expression in acinar cells of IgG and GCGR-Ab treated mice. CPM, count per million. Student’s t test.
(C) Representative images of zebrafish pancreas from control, gcgr−/−, and gcgr−/− with slc38a5b knockdown groups. Green, amylase; red, EdU; blue, DAPI. Scale bar, 10 μm.
(D) Quantification of EdU+ acinar cells in control, gcgr−/−, and gcgr−/− with slc38a5b knockdown group (n = 16/genotype).
(E) Acinar cell size in control, gcgr−/−, and gcgr−/− with slc38a5b knockdown groups. Each data point is the average of more than 50 cells from the same fish. Data are represented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. One-way ANOVA followed by Turkey’s Multiple Comparisons Test.
Activation of mTORC1 and YAP/TAZ contributes to hyperaminoacidemia-induced acinar cell hyperplasia and pancreas expansion
AAs are effective activators of mTORC1 pathway, an important signaling pathway for cell growth and proliferation.38,39 The levels of serine 240/244 in S6 ribosomal protein (pS6), an indicator of mTORC1 activity, were significantly increased in Gcg−/− mice and GCGR-Ab treated mice (Figures 4A–4D), indicating mTORC1 activation in acinar cells. Treatment with IgG in the presence or absence of mTORC1 inhibitor sirolimus (rapamycin) for 4 weeks significantly increased blood glucose levels as expected (Figures 4E and S9D),40,41 and did not impact body weight (Figure S9E). Importantly, sirolimus treatment abolished GCGR-Ab-induced increase in relative pancreas weight (Figures 4F and S9F). Moreover, sirolimus treatment also reduced the percentage of Ki67-positive acinar cells in GCGR-Ab-treated mice (Figures 4G and 4H). These data indicate that mTORC1 activity is required for IGS-induced pancreas expansion and acinar cell proliferation.
Figure 4.

IGS activates mTORC1 pathway in acinar cells
(A and C) Representative images of pancreas immunofluorescence from the 2 mouse models. Green, amylase; red, phosphor-S6 (240/244); blue, DAPI.
(B and D) Quantification of pS6 intensity in Gcg−/− mice (B) or antibody-treated mice (D) and their controls (n = 5/group). The intensity was normalized to control mice or the IgG treatment group. ∗p < 0.05, ∗∗p < 0.01, Student’s t test.
(E) Schematic experimental design for treating mice with sirolimus (rapamycin) treatment.
(F) Relative pancreas weight in the four groups (n = 5–6/group).
(G) Representative immunofluorescence images of acinar tissues. Amylase, green; Ki67, red; DAPI, blue. Arrows point to Ki67+ acinar cells.
(H) Quantification of Ki67+ acinar cells in the four groups (n = 5/group). Scale bar, 100 μm (A and C), 50 μm (G). Data are represented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. One-way ANOVA followed by Turkey’s Multiple Comparisons Test.
To examine other pathways that are involved in hyperaminoacidemia-induced acinar cell proliferation, we analyzed RNA-seq data from IgG- and GCGR-Ab-treated mice (Figures S10A–S10E). Ingenuity and gene ontology (GO) term analysis identified numerous upregulated pathways involved in proliferation and cell growth (Figures S10D and S10E). Of note, common YAP/TAZ target genes were upregulated in GCGR-Ab-treated acinar cells (Figure 5A). YAP and TAZ are two paralogs that regulate cell proliferation and organ size in multiple tissues via activation of the TEA domain (TEAD) transcription factors in the nucleus.38,42 They are negatively regulated by multiple mechanisms including phosphorylation-mediated degradation.42,43 In mouse pancreas, YAP expression is restricted in ductal cells as YAP suppression is necessary for normal acinar cell and endocrine cell differentiation and identity.44,45 Nevertheless, YAP has been shown to play a role in injury-induced postnatal acinar cell proliferation.46 Of genes whose expression is highly associated with YAP/TAZ activity,47 10 were upregulated by more than 1.5-fold in GCGR-Ab treated acinar cells compared to IgG-treated controls (Figure 5A). Quantitative RT-PCR analysis confirmed that expression of YAP/TAZ target genes Ctgf, Crim1, and Arhgef17, and Yap1 itself was significantly increased in the acinar cells of GCGR-Ab treated mice (Figure 5B). YAP immunofluorescence showed a 7- and 10-fold increase of YAP-high acinar cells in Gcg−/− mice and GCGR-Ab-treated mice over control, respectively (Figures 5C–5F). However, a substantial YAP signal remained cytoplasmic, indicating weak activation. Unlike in mice, yap1 is expressed in acinar cells in adult zebrafish.37 In contrast, taz expression is restricted to the ductal cells and absent in acinar cells.37 Immunofluorescence indicated that Yap1 was present in most acinar cells of control zebrafish at 18 dpf but rarely in the nucleus (<1%) (Figures 5G and 5H). Nonetheless, Yap1 was primarily nuclear in 42% of acinar cells in gcgr−/− fish, an effect that was blocked by CRISPR/Cas9-mediated knockdown of slc38a5b and rapamycin (Figures 5G, 5H, and S11). These results indicate that YAP1 is activated in acinar cells by IGS in both mice and zebrafish and its activation requires SLC38A5 and mTORC1.
Figure 5.

IGS activates the YAP/Taz pathway
(A) Upregulation of YAP target genes in acinar cells from GCGR-Ab treated mice. Data are from RNA-seq.
(B) RT-qPCR analysis of selected YAP target genes in mRNA from the pancreas of IgG and GCGR-Ab treated mice (n = 4–5/group, compared IgG vs. GCGR-Ab each gene).
(C and E) Representative immunofluorescence images of YAP (red) in acinar cells (amylase, green) in the two mouse models. Arrows point to a high expression of YAP. Scale bar, 50 μm.
(D and F) Quantifications of the percentage of acinar cells with high YAP expression (n = 5/group). p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 Student’s t test.
(G) Representative immunofluorescence images of Yap in pancreas sections of WT, gcgr−/−, and gcgr−/− with slc38a5b knockdown, and gcgr−/− with rapamycin treatment. All fish carry Tg(ela3l:EGFP) transgene that labels acinar cells with EGFP (scale bar, 10 μm). The EGFP- cells with high Yap1 signal are likely ductal cells.
(H) Quantification of the percentage of acinar cell with nuclear Yap1. n = 7, 6, and 7 for each group, respectively. Data are represented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. One-way ANOVA followed by Tukey’s multiple comparisons test.
To determine if YAP/TAZ function is necessary for IGS-induced pancreas expansion, we knocked down yap1 and taz individually and in combination in gcgr−/− zebrafish using two effective sgRNAs together for each gene (Table S2). Fish with yap1 and taz double knockdown did not survive beyond 14 dpf, consistent with the essential role of these genes in zebrafish development.48,49 Knockdown of yap1 in gcgr−/− fish decreased acinar cell proliferation (Figure 6A) and acinar cell size (Figure 6B). In contrast, the knockdown of taz in gcgr−/− fish did not affect acinar cell size (Figure S11), consistent with its lack of expression.37 As in mice, the levels of acinar cell pS6 were significantly increased in gcgr−/− fish. Interestingly, the knockdown of yap1 in gcgr−/− zebrafish reduced pS6 levels to those in WT controls (Figures 6C–6E). Taken together, these results indicate Yap signaling is necessary for mTORC1 activation and IGS-driven acinar cell hyperplasia and hypertrophy.
Figure 6.

Yap1 is required for mTORC1 activation in gcgr−/− fish
(A and B) Quantification of the percentage of EdU-labeled acinar cells and the acinar cell size in the pancreas sections WT, gcgr−/−, and gcgr−/− with yap1 knockdown.
(C) Representative immunofluorescence images of pS6(240/244) in pancreas sections. All fish carry the Tg(ela3l:EGFP) transgene that labels acinar cells with EGFP (scale bar, 10 μm).
(D) Quantification of raw pS6 signal intensity in acinar cells of these fish.
(E) Quantification of the percentage of pS6-positive acinar cells in the pancreas sections. n = 7, 6, 8 for each group). Data are represented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. One-way ANOVA followed by Tukey’s multiple comparisons test.
(F) Proposed model of IGS-induced acinar cell hyperplasia and hypertrophy.
Discussion
Pancreas mass varies several fold in humans due to genetic and nutritional differences and diseases. Dietary protein is an important determinant of pancreas size by affecting gastrointestinal hormones and aminoacidemia. High dietary protein induces proliferation and hypertrophy in acinar cells,8 which is different from the primarily hypertrophy-driven normal postnatal pancreas growth in mice.29 As IGS causes chronic hyperaminoacidemia and pancreas expansion, we investigated the underlying cellular and molecular mechanisms. We found that both proliferation and hypertrophy of acinar cells contribute to IGS-induced pancreas expansion and require mTORC1 activation. In addition, SLC38A5 and YAP1 are essential for IGS-induced acinar cell proliferation and hypertrophy (Figure 6F).
Hyperaminoacidemia is the driver of the exocrine pancreas growth in the IGS models. This is supported by abolishment of pancreas expansion in the Gcgrhep−/− mice by normalizing aminoacidemia using a LPD. It is further supported by the requirement of Slc38a5b for both hypertrophy and hyperplasia of zebrafish acinar cells. Slc38a5, the most abundant aaT in WT acinar cells, encodes a low-affinity Na+/H+ exchange coupled transporter for neutral aa, including the most abundant blood aa glutamine (Km = 3.2 mM) and alanine (Km = 2.5 mM).50 Therefore, aa uptake by SLC38A5 is low during normal aminoacidemia as the concentration of these aa is more than 5-fold lower than the Km. In IGS conditions, however, the consequent hyperaminoacidemia increases the influx of neutral aa, which in turn promotes the intake of other aa via aa harmonizers.50 Increased intracellular aa and their metabolism likely activate mTORC1 and biosynthesis necessary for acinar cell hypertrophy and proliferation expansion.15,20
Accumulating evidence indicates the presence of endocrine-exocrine crosstalk in the pancreas.48 For example, individuals with type 1 diabetes and their first-degree relatives with autoantibodies have a smaller pancreas likely due to few acinar cells.49,50,51 This may result from reduced insulin secretion by beta cells, leading to reduced insulin action on acinar cells.52,53 Also, insulin signaling in acinar cells protects them from Ca2+ overload during acute pancreatitis.53 However, our results exclude a direct role of glucagon signaling in suppressing acinar cell hyperplasia and hypertrophy since they persist in Gcgrhep−/− mice. These mice should have stronger glucagon signaling in acinar cells due to hyperglucagonemia.19 The requirement of Slc38a5 and mTORC1 for acinar cell hyperplasia and hypertrophy supports a role for glucagon signaling in determining acinar cell mass by regulating blood aa levels. However, as Slc38a5 and mTORC1 are also necessary for alpha cell hyperplasia, future studies are needed to determine whether acinar cell proliferation and hypertrophy depend on α cell hyperplasia.
We show that YAP1 is necessary for IGS-induced growth of the exocrine pancreas. Suppression of YAP activity is essential for proper acinar cell differentiation.44,45 Yet, YAP activation is necessary for postnatal regeneration and homeostasis of acinar cells.46,51 For example, in adult mouse pancreas YAP is active in a rare population of Tert+ acinar cells that are capable of clonal expansion during homeostasis and after pancreatic injuries.46,51 YAP is necessary for the injury-induced proliferation of these Tert+ acinar cells.46 Since multiple pathways can activate YAP and only a small fraction of acinar cells have YAP1 activation in IGS mice, future single-cell studies are necessary to understand its activation mechanism in conditions such as hyperaminoacidemia. In zebrafish, Yap activation in acinar cells requires Slc38a5b and mTORC1. mTORC1 can regulate YAP stability through autophagy.52 Intriguingly, we found that Yap1 is necessary for mTORC1 activation in acinar cells. YAP/TAZ can stimulate mTORC1 activity via multiple mechanisms.21,53,54,55,56 We propose that the IGS-induced pancreas expansion results from the synergism between mTORC1 and YAP/TAZ signaling pathways (Figure 6F).
YAP1 activation has been reported to cause pancreatitis in mice,57,58 which was not seen in IGS mice. This may be due to the difference in the extent and intensity of YAP1 activation in these models. While previous results were derived from mice with pan-acinar cell YAP1 activation by deleting its upstream suppressors, YAP1 is only weakly activated in a small fraction of acinar cells in our models.
We show both hyperplasia and cell hypertrophy contribute to the IGS-induced expansion of the exocrine pancreas. This is similar to protein-rich diet-induced adaptive pancreas growth in rodents.8,10 However, there are several differences. First, the protein-rich diet exerts a faster response, plateauing within 7–14 days,9,10 while GCGR-Ab caused continuous growth of the exocrine pancreas for the entire 8-week treatment. Second, increased CCK is responsible for high protein diet-induced acinar cell proliferation,8 while blood CCK levels were not changed in mice with IGS. Lastly, mTORC1 is only necessary for acinar cell hypertrophy in diet-induced pancreas expansion. In contrast, mTORC1 is required for both proliferation and hypertrophy of acinar cells in IGS models. These differences are likely due, at least in part, to the difference in duration and composition of elevated blood aa in these models. While the high protein diet increases blood aa levels more than 2-fold initially, the levels gradually return to normal within 2 weeks, except for branched chain aa.59 In contrast, hyperaminoacidemia is maintained as long as glucagon signaling is disrupted and may result in higher and more persistent mTORC1 activation.
Mechanistic understanding of exocrine pancreas expansion has the potential to improve the treatment of exocrine pancreas insufficiency (EPI). Whether caused by chronic pancreatitis, cystic fibrosis, or other congenital defects, EPI requires lifelong pancreatic enzyme replacement therapy (PERT) dosed with every meal, a therapy that is both expensive and difficult to maintain. In situ expansion of acinar cells could reduce patient dependence on PERT.60 While pathways that stimulate acinar cell expansion could also increase the risk of pancreatic adenocarcinoma, an increased incidence of acinar cell cancers has not been reported in GCGR-deficient patients and animal models.17,18 Further research is needed to determine if hyperaminoacidemia-induced pancreas expansion is a viable method to improve exocrine function in EPI.
In summary, the IGS-induced pancreas expansion results from hyperaminoacidemia-induced acinar cell hyperplasia and hypertrophy. In addition to mTORC1, we identified two new components, Slc38a5 and Yap1, that are necessary for pancreas expansion in zebrafish. Although YAP/TAZ is well-known for its role in organ size control,38 its role in postnatal acinar cell growth in response to hyperaminoacidemia has not been described previously. Identification of these signaling pathways in IGS-induced pancreas expansion provides targets for therapeutic control of acinar cell proliferation and hypertrophy.
Limitations of the study
Our studies have several limitations. Although both mouse and zebrafish IGS models showed acinar cell hyperplasia and hypertrophy, diet manipulation was only done in a mouse model, and genetic manipulation of slc38a5 and yap1 was only performed in zebrafish. In addition, the knockdown of slc38a5 and yap1 was global, precluding the distinction between a cell-autonomous role and a non-cell autonomous role for these genes. Given the observed difference in Yap1 expression in zebrafish and mouse acinar cells and the stage difference between zebrafish and mice used in the study, the cross-species extrapolation, particularly to humans, should be considered preliminary and needs validation. Additional studies are also required to further clarify the mechanism of mTORC1 and YAP mutual activation in zebrafish acinar cells and whether such mutual regulation is conserved.
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Wenbiao Chen (wenbiao.chen@vanderbilt.edu).
Materials availability
Materials availability will be available upon request from the lead contact.
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon reasonable request. The RNA-seq data have been deposited in the GEO repository and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report any original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact.
Acknowledgments
This research was supported by R01DK117147 (to W.C. and A.C.P.), R01DK132669 (to E.D.D.), DK106755 (to A.C.P.), and the Department of Veterans Affairs (BX000666, A.C.P.). This research was supported by resources provided by the Vanderbilt Diabetes Research and Training Center (DK020593), including its Islet and Pancreas Analysis Core, Hormone Assay and Analytical Services Core, and Animal Metabolic Physiology Core. We thank the following colleagues for generously sharing mice: Drs. Seung Kim (Gcg−/−), Daniel Drucker (Gcgrhep−/− and Gcgrflox), and Julio Ayala (Glp1r−/−). Experiments were performed in part through the use of the Vanderbilt Cell Imaging Shared Resource, the Vanderbilt Diabetes Research and Training Center, the Islet Procurement and Analysis Core (supported by NIH grants CA68485, DK20593, DK58404, DK59637, and EY08126) and the Vanderbilt Zebrafish Aquatic Facility.
Author contributions
Conceptualization: A.C.P., C.D., E.D.D., and W.C.; investigation: C.D., Y.Z., Y.G., A.B., Z.T., K.S., E.S., B.A.C., J.S., R.J., T.M.R., and R.A.B.; writing—original draft: C.D., Y.G., and W.C.; writing—review and editing: A.C.P., E.D.D., D.C.S., J.J.W., and M.B.; visualization: C.D., Y.Z., Y.G., D.C.S., K.S., S.S., K.S., and B.A.C.; funding acquisition: A.C.P., E.D.D., and W.C.
Declaration of interests
The authors declare no competing interests.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-alcam | DSHB | Cat# ZN-8 |
| anti-Chicken IgY (H+L) - Alexa Fluor® 488 | Invitrogen | Cat# A-11039; RRID: AB_2534096 |
| anti-GFP | Aves | Cat# GFP-1010; RRID: AB_2307313 |
| anti-mCherry | Novus Biologiclas | Cat# NBP2-25157; RRID: AB_2753204 |
| anti-MHC | DSHB | Cat# MF20 |
| anti-mouse IgG1 - Alexa Fluor® 488 | Invitrogen | Cat# A-21121; RRID: AB_2535764 |
| anti-mouse IgG2b - Alexa Fluor® 568 | Invitrogen | Cat# A-21144; RRID: AB_2535780 |
| anti-rabbit (H+L) Superclonal™ - Alexa Fluor® 647 | Invitrogen | Cat# A-27040; RRID: AB_2536101 |
| Bacterial and virus strains | ||
| DH5-alpha competent E.coli | New England Biolabs | Cat# C2987I |
| Chemicals, peptides, and recombinant proteins | ||
| 1-phenyl-2-thiourea (PTU) | Sigma-Aldrich | Cat# P7629-10 |
| 16% Paraformaldehyde aqueous solution | Electron Microscopy Sciences | Cat# 15710 |
| 2,3-Butanedione monoxime | Sigma-Aldrich | Cat# B0753 |
| 4-hydroxytamoxifen | Sigma-Aldrich | Cat# H7904 |
| 4’,6-Diamidino-2-phenylindole (DAPI) | Sigma-Aldrich | Cat# D9542 |
| Acetone | Sigma-Aldrich | Cat# 320110 |
| BODIPY™ FL C5-Ceramide | Invitrogen | Cat# D3521 |
| BSA | Sigma-Aldrich | Cat# A7906 |
| Chloroquine diphosphate salt | Sigma-Aldrich | Cat# C6628 |
| Chromium Nuclei Isolation Kit with RNase Inhibitor | 10x Genomics | Cat# PN-1000494 |
| CRISPR-Cas9 tracrRNA | IDT | Cat# 1072534 |
| Dimethyl sulfoxide | Sigma-Aldrich | Cat# D4540 |
| DNA Clean & Concentrator | Zymo Research | Cat# D4014 |
| Epon | Sigma-Aldrich | Cat# 45359 |
| Ethanol | Grogg Chemie | Cat# G003 |
| Fast Digerst HindIII | Thermofisher Scientific | Cat# FD0504 |
| Fast Digest BamHI | Thermofisher Scientific | Cat# FD0054 |
| Fast Digest ScaI | Thermofisher Scientific | Cat# FD0434 |
| foetal bovine serum | Sigma-Aldrich | Cat# F7524 |
| Fragment Analyzer NGS Fragment Kit | Agilent | Cat# DNF-473 |
| Gateway LR Clonase II Enzyme mix | Invitrogen | Cat# 11791020 |
| Glutaraldehyde | Agar Scientific | Cat# AGR1009 |
| Goat serum | Dominique Dutscher | Cat# S2000 |
| HiFi Cas9 Nuclease V3 | IDT | Cat# 1081060 |
| illumina NovaSeq 6000 S1 Reagent Kit v1.5 | Illumina | Cat# 20028319 |
| iScript Reverse Transcription SuperMix | Bio-Rad | Cat# 1708841 |
| KCl | Sigma-Aldrich | Cat# P9333 |
| Leibovitz's L-15 Medium | Thermo Fisher Scientific | Cat# 11415064 |
| LysoTracker™ Deep Red | Invitrogen | Cat# L12492 |
| Maxima First Strand cDNA synthesis kit | Thermo Fisher Scientific | Cat# K1671 |
| OsO4 | Electron Microscopy Sciences | Cat# 19100 |
| pENTR/D-TOPO vector | Invitrogen | Cat# K240020 |
| Phosphate buffered saline | NZYtech | Cat# MB18201 |
| PowerUp SYBR Green Master Mix | Thermo Fisher Scientific | Cat# A25742 |
| Prep User Guide | 10X Genomics | Cat# CG000505 |
| Proteinase K | Roche | Cat# 03 115 801 001 |
| Q5® High-Fidelity DNA Polymerase | New England Biolabs | Cat# M0491 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat# Q32851 |
| Sodium cacodylate trihydrate | Sigma-Aldrich | Cat# C0250 |
| T7 Endonuclease I | New England Biolabs | Cat# M0302 |
| Triton X-100 | Sigma-Aldrich | Cat# T9284 |
| TRIzol Reagent | Invitrogen | Cat# 10296010 |
| Deposited data | ||
| snRNASeq Data | NCBI GEO | GEO: GSE246850 |
| Raw data | Zenodo | Zenodo: https://doi.org/10.5281/zenodo.13982794 |
| Experimental models: Transgenic zebrafish models used in the study | ||
| Tg(actb2:mRFP-GFP-map1lc3b)udc2Tg | Allende lab | ZDB-ALT-210122-18 |
| TgKI(mRFP-map1lc3b)brn7 | This study, Mercader lab | ZDB-ALT-230926-15 |
| Tg(CMV:EGFP-map1lc3b)zf155 | Kishi lab | ZDB-ALT-091029-2 |
| TgBAC(lamp2:RFP)pd1117 | Affolter lab | ZDB-ALT-150520-1 |
| Tg(fli1a:GFP)y1 | ZIRC | ZDB-ALT-011017-8 |
| Tg(fli1a:DsRedex)um13 | Lawson lab | ZDB-ALT-100525-3 |
| Tg(kdrl:GFP)la116 | Stainier lab | ZDB-ALT-070529-1 |
| Tg(fli1a:Gal4FF)ubs3 | Affolter Lab | ZDB-ALT-120113-6 |
| Tg(kdrl:EGFP-CAAX)ubs47 | Affolter lab100 | |
| Tg(myl7:GFP)f1 | Djonov lab | ZDB-ALT-060719-2 |
| Tg(myl7:mCherry)ko08 | Kawahara lab | ZDB-ALT-090423-3 |
| Tg(EPV.Tp1-Mmu.Hbb:CreERT2,cryaa:mCherry)s959 | Singh lab | ZDB-ALT-131001-3 |
| Tg(–3.5ubi:loxP-EGFP-loxP-mCherry)cy1701 | Zon lab | ZDB-ALT-110124-1 |
| nrs (spns1hi891Tg/hi891Tg) | Kishi lab | ZDB-FISH-150901-8505 |
| Tg(UAS:spns1)brn8 | This study, Mercader lab | ZDB-ALT-230926-16 |
| Tg(UAS:myc-Notch1-intra)kca3Tg | ZIRC | ZDB-ALT-020918-8 |
| Oligonucleotides | ||
| See Table S7. | ||
| Plasmids | ||
| pKHR4 | Addgene | Cat# 74592; RRID: Addgene_74592 |
| pDestTol2pA2CrymCherry | Addgene | Cat# 64023; RRID: Addgene_64023 |
| Software and algorithms | ||
| Code and analyses | This study | https://github.com/MercaderLabAnatomy/PUB_Chavez_et_al_2023 |
| Fiji | https://fiji.sc/ | https://doi.org/10.1038/nmeth.2019 |
| Matlab | https://ch.mathworks.com/products/matlab.html | R2024a Update 3 |
| Napari | https://napari.org | https://doi.org/10.5281/zenodo.3555620 |
| R v4.0 | https://www.r-project.org/ | R version 4.0.0 |
| Seurat v4.0 | CRAN v4.0 | https://doi.org/10.1016/j.cell.2021.04.048 |
| T-MIDAS | https://github.com/MercaderLabAnatomy/T-MIDAS | https://doi.org/10.5281/zenodo.10728503 |
Experimental model and study participant details
Mice
Mice were housed under a 12-h light/12-h dark cycle in individually ventilated cages with automatic water and ad libitum access to standard rodent chow (with 24.1% protein) at Vanderbilt animal facilities. The cages had bedding with paper rolls for nesting. Some cages had refuges (huts). All experiments were conducted according to protocols and guidelines approved by the Vanderbilt University Institutional Animal Care and Use Committee. C57BL/J6 male mice and Gcg−/− (GKO) mice in NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) background were obtained from The Jackson Laboratory and bred at Vanderbilt.25,61 Glp1r−/− mice in C57BL/J6 background were provided by Dr. Julio E Ayala (Vanderbilt University).62 Gcgrflox and Gcgrhep−/− mice in C57BL/J6 background have been characterized previously (Figure S1).19 They were provided by Dr. Daniel Drucker initially was were bred at Vanderbilt. To pharmacologically interrupt glucagon signaling, C57BL/J6 mice were intraperitoneally injected with 10 mg/kg of GCGR neutralizing monoclonal antibody (GCGR-Ab) or IgG from Eli Lilly once a week for 2, 4, or 8 weeks. To be consistent with our prior study, only male mice were treated.63 Sirolimus (SIR, rapamycin, NDC 0008-1030-06, Pfizer) or PBS was given by intraperitoneal (i.p.) injection every 72 h at 1.5 mg/kg64 To correct hyperaminoacidemia, Gcgrflox and Gcgrhep−/− male and female mice (3–40 weeks old) along with age-matched control littermates were fed with isocaloric normal (20%) and low (6%) protein diets (Harlan Teklad #TD.91352 and #TD.90016, respectively). For AA measurement, serum samples were collected during the daytime from GKO and control mice at 14- to 20-week-old, from IgG or GCGR-Ab treated C57BL/J6 and Glp1r−/− mice after 8 weeks of treatment, or from Gcgrflox and Gcgrhep−/− mice at the end of experiments (Table S3).
Zebrafish
Zebrafish were raised at 27°C in an Aquatic-Habitats system and embryos were raised at 28.5°C in an incubator on a 14/10-h light/dark cycle. The age of zebrafish was expressed as days postfertilization (dpf). As larval zebrafish cannot be distinguished by sex, both males and females were used. The gcgr−/− zebrafish (gcgra−/−;gcgrb−/−) were described previously (Table S3).26 Drugs were administered in the medium.
Method details
Mice
Serum measurements
Mice were fasted for 6 h with free access to water, and blood was collected from the retro-orbital sinus. Aprotinin protease inhibitor (PentaPharm) was pre-added to collection tubes to yield a final concentration in whole blood of 1000KIU. The serum was collected and stored at −80oC until analysis. CCK and GLP-1 measurements were performed using CCK Enzyme Immunoassay kit (RayBio, #EIA-CCK) and Total GLP-1 NL-ELISA kit (Mercodia, 10-1278-01). Total serum amino acid levels were measured using an L-Amino Acid Quantification kit (Sigma MAK002). Individual amino acid levels were quantified by HPLC as described previously.20
Immunofluorescence and imaging
Pancreata were fixed, sectioned, and stained as previously described.65,66,67 Primary antibodies used in this project are listed in the key resources table. Apoptosis was assessed by TUNEL (Millipore, S7165) following the manufacturer’s instructions. Nuclei were stained with Hoechst 33342 (0.5 μg/mL, Thermo Fisher Scientific) or DAPI (300 nM, Thermo Fisher Scientific). Images were acquired with a fluorescence ScanScope (Aperio) scanner or a confocal laser-scanning microscope (LSM880, Carl Zeiss, Jena, Germany). Positive Ki67 acinar cells were quantified, and cell size was measured using HALO image analysis (Indica Labs). pS6 intensity was quantified using ImageJ.
Acinar cells RNA isolation, cDNA synthesis, quantitative RT-PCR, and RNA-seq
To purify acinar cells, the mouse pancreas was digested by collagenase and acini were picked by hand.68 Total RNA was extracted from acinar cells using an RNAqueous RNA isolation kit (Ambion, Austin, TX). RNA quality control and quantity assessment (QC/QA) was performed using a Bioanalyzer instrument. The average RNA integrated Number (RIN) was 7.8 + 0.09 (IgG, 7.5–8.1) and 8.0 + 0.14 (GCGR-Ab, 7.4–8.4). cDNA was synthesized using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, 4368814) according to the manufacturer’s instructions. Quantitative PCR (qPCR) was performed using TaqMan assays (key resources table) with reagents from Applied Biosystems (Foster City, CA) as previously described.66,69,70 Actb was used for normalization. Relative changes in mRNA expression were calculated by the comparative ΔCt method.
RNA-seq was performed by MEDGENOME (Foster City, CA). About 72–137 million uniquely mapped reads were acquired per sample. Alignment was performed using STAR (v2.7.3a) aligner to the reference mouse genome (genome-build GRCm38.p6).71 The raw read counts were estimated using HTSeq (v0.11.2). Further quality control and downstream analysis were performed in the Strand NGS analysis platform v3.4 (Strand life Sciences) where read counts were normalized using TMM (Trimmed Mean of M values).72 Genes with less than 20 counts across samples were removed.
Relative pancreas weight measurement
To determine relative pancreas weight, mice were anesthetized, weighed, and the pancreas was carefully dissected and placed into a 10 cm Petri dish containing ice-cold PBS. After removing fat tissue, the pancreas was blotted with filter paper and weighed to determine the absolute pancreas weight. The weight was normalized to body weight and described as relative pancreas weight.
Zebrafish
Mutagenesis
The knockdown of slc38a5b, taz, and yap was performed according to Yin et al.73 The mutagenesis was initially determined by the heteroduplex motility assay. PCR products of the targeted region from pools of control and mutagenized embryos were subjected to Sanger sequencing and Synthego ICE or TIDE74 analysis to determine the mutagenesis rate (Table S2).
Pancreas area, volume, and acinar cell proliferation analysis
Because zebrafish pancreas is difficult to remove and weigh accurately, the pancreas size was quantified as pancreas area or pancreas volume by confocal imaging of fixed Tg(ela3l:EGFP) zebrafish.27 Proliferation by 24-h EdU labeling was done as previously described.14,26 Some fish were treated with rapamycin (200 nM) for 24 h before euthanasia. Fish were euthanized by MS-222 and fixed in 4% paraformaldehyde (PFA), equilibrated in 20% sucrose and embedded into Optimal Cutting Temperature (OCT) media to generate pancreas-containing cryosections (12 μm). EdU was detected using the Click-iT EdU Alexa Fluor 594 Imaging Kit (C10339; Invitrogen).
Acinar cells were labeled using an amylase antibody (Rabbit, Sigma, A8273). Acinar cell size was measured in Tg(ela3l:EGFP)-carrying zebrafish by dividing the GFP+ area with the number of DAPI stained nuclei. All images were collected using Zeiss LSM880 (Carl Zeiss, Jena, Germany) and analyzed by Imaris (Oxford Instruments).
Quantification and statistical analysis
Statistically significant differences were determined by two-tailed t-tests (2 groups) or one-way ANOVA (analysis of variance) followed by Tukey’s Multiple Comparisons Tests (>2 groups). A p value < 0.05 was considered statistically significant. Values reported represent mean ± SEM. In RNA-seq analysis, p-values were estimated using Z-test (Strand NGS) for differential expression. False discovery rate adjusted for multiple hypothesis testing with Benjamini-Hochberg (BH) procedure, where p-value <0.05 and fold change ≥1.5 were used to define differentially expressed genes. Differentially expressed genes were further analyzed through Ingenuity Pathway Analysis (IPA, Qiagen) and Gene Ontology (GO) analysis using DAVID v6.8.75
Published: November 22, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.111447.
Contributor Information
Alvin C. Powers, Email: al.powers@vumc.org.
Wenbiao Chen, Email: wenbiao.chen@vanderbilt.edu.
Supplemental information
References
- 1.Pin C.L., Ryan J.F., Mehmood R. Acinar cell reprogramming: a clinically important target in pancreatic disease. Epigenomics. 2015;7:267–281. doi: 10.2217/epi.14.83. [DOI] [PubMed] [Google Scholar]
- 2.Wollny D., Zhao S., Everlien I., Lun X., Brunken J., Brüne D., Ziebell F., Tabansky I., Weichert W., Marciniak-Czochra A., Martin-Villalba A. Single-Cell Analysis Uncovers Clonal Acinar Cell Heterogeneity in the Adult Pancreas. Dev. Cell. 2016;39:289–301. doi: 10.1016/j.devcel.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Tosti L., Hang Y., Debnath O., Tiesmeyer S., Trefzer T., Steiger K., Ten F.W., Lukassen S., Ballke S., Kühl A.A., et al. Single-Nucleus and In Situ RNA-Sequencing Reveal Cell Topographies in the Human Pancreas. Gastroenterology. 2021;160:1330–1344.e11. doi: 10.1053/j.gastro.2020.11.010. [DOI] [PubMed] [Google Scholar]
- 4.Case R.M. Synthesis, intracellular transport and discharge of exportable proteins in the pancreatic acinar cell and other cells. Biol. Rev. Camb. Philos. Soc. 1978;53:211–354. doi: 10.1111/j.1469-185x.1978.tb01437.x. [DOI] [PubMed] [Google Scholar]
- 5.Logsdon C.D., Ji B. The role of protein synthesis and digestive enzymes in acinar cell injury. Nat. Rev. Gastroenterol. Hepatol. 2013;10:362–370. doi: 10.1038/nrgastro.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.El-Hodhod M.A., Nassar M.F., Hetta O.A., Gomaa S.M. Pancreatic size in protein energy malnutrition: a predictor of nutritional recovery. Eur. J. Clin. Nutr. 2005;59:467–473. doi: 10.1038/sj.ejcn.1602053. [DOI] [PubMed] [Google Scholar]
- 7.Crozier S.J., D'Alecy L.G., Ernst S.A., Ginsburg L.E., Williams J.A. Molecular mechanisms of pancreatic dysfunction induced by protein malnutrition. Gastroenterology. 2009;137:1093–1101. doi: 10.1053/j.gastro.2009.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crozier S.J., Sans M.D., Wang J.Y., Lentz S.I., Ernst S.A., Williams J.A. CCK-independent mTORC1 activation during dietary protein-induced exocrine pancreas growth. Am. J. Physiol. Gastrointest. Liver Physiol. 2010;299:G1154–G1163. doi: 10.1152/ajpgi.00445.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hara H., Narakino H., Kiriyama S., Kasai T. Induction of pancreatic growth and proteases by feeding a high amino acid diet does not depend on cholecystokinin in rats. J. Nutr. 1995;125:1143–1149. doi: 10.1093/jn/125.5.1143. [DOI] [PubMed] [Google Scholar]
- 10.Green G.M., Levan V.H., Liddle R.A. Plasma cholecystokinin and pancreatic growth during adaptation to dietary protein. Am. J. Physiol. 1986;251:G70–G74. doi: 10.1152/ajpgi.1986.251.1.G70. [DOI] [PubMed] [Google Scholar]
- 11.Crozier S.J., Sans M.D., Guo L., D'Alecy L.G., Williams J.A. Activation of the mTOR signalling pathway is required for pancreatic growth in protease-inhibitor-fed mice. J. Physiol. 2006;573:775–786. doi: 10.1113/jphysiol.2006.106914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dean E.D. A Primary Role for α-Cells as Amino Acid Sensors. Diabetes. 2020;69:542–549. doi: 10.2337/dbi19-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galsgaard K.D., Winther-Sørensen M., Ørskov C., Kissow H., Poulsen S.S., Vilstrup H., Prehn C., Adamski J., Jepsen S.L., Hartmann B., et al. Disruption of glucagon receptor signaling causes hyperaminoacidemia exposing a possible liver-alpha-cell axis. Am. J. Physiol. Endocrinol. Metab. 2018;314:E93–E103. doi: 10.1152/ajpendo.00198.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gong Y., Yang B., Zhang D., Zhang Y., Tang Z., Yang L., Coate K.C., Yin L., Covington B.A., Patel R.S., et al. Hyperaminoacidemia induces pancreatic alpha cell proliferation via synergism between the mTORC1 and CaSR-Gq signaling pathways. Nat. Commun. 2023;14:235. doi: 10.1038/s41467-022-35705-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim J., Okamoto H., Huang Z., Anguiano G., Chen S., Liu Q., Cavino K., Xin Y., Na E., Hamid R., et al. Amino Acid Transporter Slc38a5 Controls Glucagon Receptor Inhibition-Induced Pancreatic α Cell Hyperplasia in Mice. Cell Metab. 2017;25:1348–1361.e8. doi: 10.1016/j.cmet.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Solloway M.J., Madjidi A., Gu C., Eastham-Anderson J., Clarke H.J., Kljavin N., Zavala-Solorio J., Kates L., Friedman B., Brauer M., et al. Glucagon Couples Hepatic Amino Acid Catabolism to mTOR-Dependent Regulation of α-Cell Mass. Cell Rep. 2015;12:495–510. doi: 10.1016/j.celrep.2015.06.034. [DOI] [PubMed] [Google Scholar]
- 17.Gelling R.W., Du X.Q., Dichmann D.S., Romer J., Huang H., Cui L., Obici S., Tang B., Holst J.J., Fledelius C., et al. Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc. Natl. Acad. Sci. USA. 2003;100:1438–1443. doi: 10.1073/pnas.0237106100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu R., Dhall D., Nissen N.N., Zhou C., Ren S.G. Pancreatic neuroendocrine tumors in glucagon receptor-deficient mice. PLoS One. 2011;6 doi: 10.1371/journal.pone.0023397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Longuet C., Robledo A.M., Dean E.D., Dai C., Ali S., McGuinness I., de Chavez V., Vuguin P.M., Charron M.J., Powers A.C., Drucker D.J. Liver-specific disruption of the murine glucagon receptor produces α-cell hyperplasia: evidence for a circulating α-cell growth factor. Diabetes. 2013;62:1196–1205. doi: 10.2337/db11-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dean E.D., Li M., Prasad N., Wisniewski S.N., Von Deylen A., Spaeth J., Maddison L., Botros A., Sedgeman L.R., Bozadjieva N., et al. Interrupted Glucagon Signaling Reveals Hepatic alpha Cell Axis and Role for L-Glutamine in alpha Cell Proliferation. Cell Metab. 2017;25:1362–1373.e5. doi: 10.1016/j.cmet.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu R., Nissen N.N., Dhall D., Heaney A.P. Nesidioblastosis and hyperplasia of alpha cells, microglucagonoma, and nonfunctioning islet cell tumor of the pancreas: review of the literature. Pancreas. 2008;36:428–431. doi: 10.1097/MPA.0b013e31815ceb23. [DOI] [PubMed] [Google Scholar]
- 22.Larger E., Wewer Albrechtsen N.J., Hansen L.H., Gelling R.W., Capeau J., Deacon C.F., Madsen O.D., Yakushiji F., De Meyts P., Holst J.J., Nishimura E. Pancreatic alpha-cell hyperplasia and hyperglucagonemia due to a glucagon receptor splice mutation. Endocrinol. Diabetes Metab. Case Rep. 2016;2016 doi: 10.1530/EDM-16-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robbins J., Halegoua-DeMarzio D., Basu Mallick A., Vijayvergia N., Ganetzky R., Lavu H., Giri V.N., Miller J., Maley W., Shah A.P., et al. Liver Transplantation in a Woman with Mahvash Disease. N. Engl. J. Med. 2023;389:1972–1978. doi: 10.1056/NEJMoa2303226. [DOI] [PubMed] [Google Scholar]
- 24.Li H., Zhao L., Singh R., Ham J.N., Fadoju D.O., Bean L.J.H., Zhang Y., Xu Y., Xu H.E., Gambello M.J. The first pediatric case of glucagon receptor defect due to biallelic mutations in GCGR is identified by newborn screening of elevated arginine. Mol. Genet. Metab. Rep. 2018;17:46–52. doi: 10.1016/j.ymgmr.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tellez K., Hang Y., Gu X., Chang C.A., Stein R.W., Kim S.K. In vivo studies of glucagon secretion by human islets transplanted in mice. Nat. Metab. 2020;2:547–557. doi: 10.1038/s42255-020-0213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li M., Dean E.D., Zhao L., Nicholson W.E., Powers A.C., Chen W. Glucagon receptor inactivation leads to α-cell hyperplasia in zebrafish. J. Endocrinol. 2015;227:93–103. doi: 10.1530/joe-15-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korzh S., Pan X., Garcia-Lecea M., Winata C.L., Pan X., Wohland T., Korzh V., Gong Z. Requirement of vasculogenesis and blood circulation in late stages of liver growth in zebrafish. BMC Dev. Biol. 2008;8:84. doi: 10.1186/1471-213X-8-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magami Y., Azuma T., Inokuchi H., Moriyasu F., Kawai K., Hattori T. Heterogeneous cell renewal of pancreas in mice: [(3)H]-thymidine autoradiographic investigation. Pancreas. 2002;24:153–160. doi: 10.1097/00006676-200203000-00006. [DOI] [PubMed] [Google Scholar]
- 29.Anzi S., Stolovich-Rain M., Klochendler A., Fridlich O., Helman A., Paz-Sonnenfeld A., Avni-Magen N., Kaufman E., Ginzberg M.B., Snider D., et al. Postnatal Exocrine Pancreas Growth by Cellular Hypertrophy Correlates with a Shorter Lifespan in Mammals. Dev. Cell. 2018;45:726–737.e3. doi: 10.1016/j.devcel.2018.05.024. [DOI] [PubMed] [Google Scholar]
- 30.Koehler J.A., Baggio L.L., Cao X., Abdulla T., Campbell J.E., Secher T., Jelsing J., Larsen B., Drucker D.J. Glucagon-like peptide-1 receptor agonists increase pancreatic mass by induction of protein synthesis. Diabetes. 2015;64:1046–1056. doi: 10.2337/db14-0883. [DOI] [PubMed] [Google Scholar]
- 31.Wewer Albrechtsen N.J., Albrechtsen R., Bremholm L., Svendsen B., Kuhre R.E., Poulsen S.S., Christiansen C.B., Jensen E.P., Janus C., Hilsted L., et al. Glucagon-like Peptide 1 Receptor Signaling in Acinar Cells Causes Growth-Dependent Release of Pancreatic Enzymes. Cell Rep. 2016;17:2845–2856. doi: 10.1016/j.celrep.2016.11.051. [DOI] [PubMed] [Google Scholar]
- 32.Hayashi Y., Yamamoto M., Mizoguchi H., Watanabe C., Ito R., Yamamoto S., Sun X.Y., Murata Y. Mice deficient for glucagon gene-derived peptides display normoglycemia and hyperplasia of islet alpha-cells but not of intestinal L-cells. Mol. Endocrinol. 2009;23:1990–1999. doi: 10.1210/me.2009-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams J.A. Pancreapedia: Exocrine Pancreas Knowledge Base; 2020. Regulation of Normal and Adaptive Pancreatic Growth. [DOI] [Google Scholar]
- 34.Liao S.F., Regmi N., Wu G. Homeostatic regulation of plasma amino acid concentrations. Front. Biosci. 2018;23:640–655. doi: 10.2741/4610. [DOI] [PubMed] [Google Scholar]
- 35.Rooman I., Lutz C., Pinho A.V., Huggel K., Reding T., Lahoutte T., Verrey F., Graf R., Camargo S.M.R. Amino acid transporters expression in acinar cells is changed during acute pancreatitis. Pancreatology. 2013;13:475–485. doi: 10.1016/j.pan.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 36.Jao L.E., Wente S.R., Chen W. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc. Natl. Acad. Sci. USA. 2013;110:13904–13909. doi: 10.1073/pnas.1308335110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tarifeno-Saldivia E., Lavergne A., Bernard A., Padamata K., Bergemann D., Voz M.L., Manfroid I., Peers B. Transcriptome analysis of pancreatic cells across distant species highlights novel important regulator genes. BMC Biol. 2017;15:21. doi: 10.1186/s12915-017-0362-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Csibi A., Blenis J. Hippo-YAP and mTOR pathways collaborate to regulate organ size. Nat. Cell Biol. 2012;14:1244–1245. doi: 10.1038/ncb2634. [DOI] [PubMed] [Google Scholar]
- 39.Kim J., Guan K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019;21:63–71. doi: 10.1038/s41556-018-0205-1. [DOI] [PubMed] [Google Scholar]
- 40.Fang Y., Westbrook R., Hill C., Boparai R.K., Arum O., Spong A., Wang F., Javors M.A., Chen J., Sun L.Y., Bartke A. Duration of rapamycin treatment has differential effects on metabolism in mice. Cell Metab. 2013;17:456–462. doi: 10.1016/j.cmet.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Houde V.P., Brûlé S., Festuccia W.T., Blanchard P.G., Bellmann K., Deshaies Y., Marette A. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes. 2010;59:1338–1348. doi: 10.2337/db09-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu F.X., Zhao B., Guan K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell. 2015;163:811–828. doi: 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pocaterra A., Romani P., Dupont S. YAP/TAZ functions and their regulation at a glance. J. Cell Sci. 2020;133 doi: 10.1242/jcs.230425. [DOI] [PubMed] [Google Scholar]
- 44.Gao T., Zhou D., Yang C., Singh T., Penzo-Méndez A., Maddipati R., Tzatsos A., Bardeesy N., Avruch J., Stanger B.Z. Hippo signaling regulates differentiation and maintenance in the exocrine pancreas. Gastroenterology. 2013;144:1543–1553.e1. doi: 10.1053/j.gastro.2013.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.George N.M., Day C.E., Boerner B.P., Johnson R.L., Sarvetnick N.E. Hippo signaling regulates pancreas development through inactivation of Yap. Mol. Cell Biol. 2012;32:5116–5128. doi: 10.1128/MCB.01034-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suh H.N., Kim M.J., Lee S.H., Jun S., Zhang J., Johnson R.L., Park J.-I. Yap/Taz-Activated Tert-Expressing Acinar Cells Are Required for Pancreatic Regeneration. bioRxiv. 2021 doi: 10.1101/2021.08.30.458292. Preprint at. [DOI] [Google Scholar]
- 47.Wang Y., Xu X., Maglic D., Dill M.T., Mojumdar K., Ng P.K.S., Jeong K.J., Tsang Y.H., Moreno D., Bhavana V.H., et al. Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep. 2018;25:1304–1317.e5. doi: 10.1016/j.celrep.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kimelman D., Smith N.L., Lai J.K.H., Stainier D.Y. Regulation of posterior body and epidermal morphogenesis in zebrafish by localized Yap1 and Wwtr1. Elife. 2017;6 doi: 10.7554/eLife.31065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miesfeld J.B., Gestri G., Clark B.S., Flinn M.A., Poole R.J., Bader J.R., Besharse J.C., Wilson S.W., Link B.A. Yap and Taz regulate retinal pigment epithelial cell fate. Development. 2015;142:3021–3032. doi: 10.1242/dev.119008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gauthier-Coles G., Vennitti J., Zhang Z., Comb W.C., Xing S., Javed K., Bröer A., Bröer S. Quantitative modelling of amino acid transport and homeostasis in mammalian cells. Nat. Commun. 2021;12:5282. doi: 10.1038/s41467-021-25563-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neuhöfer P., Roake C.M., Kim S.J. Acinar cell clonal expansion in pancreas homeostasis and carcinogenesis. Nature. 2021;597:715–719. doi: 10.1038/s41586-021-03916-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liang N., Zhang C., Dill P., Panasyuk G., Pion D., Koka V., Gallazzini M., Olson E.N., Lam H., Henske E.P., et al. Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. J. Exp. Med. 2014;211:2249–2263. doi: 10.1084/jem.20140341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hansen C.G., Ng Y.L.D., Lam W.L.M., Plouffe S.W., Guan K.L. The Hippo pathway effectors YAP and TAZ promote cell growth by modulating amino acid signaling to mTORC1. Cell Res. 2015;25:1299–1313. doi: 10.1038/cr.2015.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu J.K.H., Du W., Shelton S.J., Oldham M.C., DiPersio C.M., Klein O.D. An FAK-YAP-mTOR Signaling Axis Regulates Stem Cell-Based Tissue Renewal in Mice. Cell Stem Cell. 2017;21:91–106.e6. doi: 10.1016/j.stem.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Park Y.Y., Sohn B.H., Johnson R.L., Kang M.H., Kim S.B., Shim J.J., Mangala L.S., Kim J.H., Yoo J.E., Rodriguez-Aguayo C., et al. Yes-associated protein 1 and transcriptional coactivator with PDZ-binding motif activate the mammalian target of rapamycin complex 1 pathway by regulating amino acid transporters in hepatocellular carcinoma. Hepatology. 2016;63:159–172. doi: 10.1002/hep.28223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vaidyanathan S., Salmi T.M., Sathiqu R.M., McConville M.J., Cox A.G., Brown K.K. YAP regulates an SGK1/mTORC1/SREBP-dependent lipogenic program to support proliferation and tissue growth. Dev. Cell. 2022;57:719–731.e8. doi: 10.1016/j.devcel.2022.02.004. [DOI] [PubMed] [Google Scholar]
- 57.Tamura T., Kodama T., Sato K., Murai K., Yoshioka T., Shigekawa M., Yamada R., Hikita H., Sakamori R., Akita H., et al. Dysregulation of PI3K and Hippo signaling pathways synergistically induces chronic pancreatitis via CTGF upregulation. J. Clin. Invest. 2021;131 doi: 10.1172/JCI143414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu J., Gao M., Nipper M., Deng J., Sharkey F.E., Johnson R.L., Crawford H.C., Chen Y., Wang P. Activation of the intrinsic fibroinflammatory program in adult pancreatic acinar cells triggered by Hippo signaling disruption. PLoS Biol. 2019;17 doi: 10.1371/journal.pbio.3000418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anderson H.L., Benevenga N.J., Harper A.E. Associations among food and protein intake, serine dehydratase, and plasma amino acids. Am. J. Physiol. 1968;214:1008–1013. doi: 10.1152/ajplegacy.1968.214.5.1008. [DOI] [PubMed] [Google Scholar]
- 60.Murtaugh L.C., Keefe M.D. Regeneration and repair of the exocrine pancreas. Annu. Rev. Physiol. 2015;77:229–249. doi: 10.1146/annurev-physiol-021014-071727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nicholson B., Manner C.K., Kleeman J., MacLeod C.L. Sustained nitric oxide production in macrophages requires the arginine transporter CAT2. J. Biol. Chem. 2001;276:15881–15885. doi: 10.1074/jbc.M010030200. [DOI] [PubMed] [Google Scholar]
- 62.Scrocchi L.A., Brown T.J., MaClusky N., Brubaker P.L., Auerbach A.B., Joyner A.L., Drucker D.J. Glucose intolerance but normal satiety in mice with a null mutation in the glucagon-like peptide 1 receptor gene. Nat. Med. 1996;2:1254–1258. doi: 10.1038/nm1196-1254. [DOI] [PubMed] [Google Scholar]
- 63.Wang M.Y., Dean E.D., Quittner-Strom E., Zhu Y., Chowdhury K.H., Zhang Z., Zhao S., Li N., Ye R., Lee Y., et al. Glucagon blockade restores functional beta-cell mass in type 1 diabetic mice and enhances function of human islets. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2022142118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dai C., Walker J.T., Shostak A., Padgett A., Spears E., Wisniewski S., Poffenberger G., Aramandla R., Dean E.D., Prasad N., et al. Tacrolimus- and sirolimus-induced human beta cell dysfunction is reversible and preventable. JCI Insight. 2020;5 doi: 10.1172/jci.insight.130770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cai Q., Brissova M., Reinert R.B., Pan F.C., Brahmachary P., Jeansson M., Shostak A., Radhika A., Poffenberger G., Quaggin S.E., et al. Enhanced expression of VEGF-A in β cells increases endothelial cell number but impairs islet morphogenesis and β cell proliferation. Dev. Biol. 2012;367:40–54. doi: 10.1016/j.ydbio.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dai C., Kayton N.S., Shostak A., Poffenberger G., Cyphert H.A., Aramandla R., Thompson C., Papagiannis I.G., Emfinger C., Shiota M., et al. Stress-impaired transcription factor expression and insulin secretion in transplanted human islets. J. Clin. Invest. 2016;126:1857–1870. doi: 10.1172/jci83657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reinert R.B., Brissova M., Shostak A., Pan F.C., Poffenberger G., Cai Q., Hundemer G.L., Kantz J., Thompson C.S., Dai C., et al. Vascular endothelial growth factor-a and islet vascularization are necessary in developing, but not adult, pancreatic islets. Diabetes. 2013;62:4154–4164. doi: 10.2337/db13-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brissova M., Fowler M., Wiebe P., Shostak A., Shiota M., Radhika A., Lin P.C., Gannon M., Powers A.C. Intraislet endothelial cells contribute to revascularization of transplanted pancreatic islets. Diabetes. 2004;53:1318–1325. doi: 10.2337/diabetes.53.5.1318. [DOI] [PubMed] [Google Scholar]
- 69.Dai C., Brissova M., Hang Y., Thompson C., Poffenberger G., Shostak A., Chen Z., Stein R., Powers A.C. Islet-enriched gene expression and glucose-induced insulin secretion in human and mouse islets. Diabetologia. 2012;55:707–718. doi: 10.1007/s00125-011-2369-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dai C., Hang Y., Shostak A., Poffenberger G., Hart N., Prasad N., Phillips N., Levy S.E., Greiner D.L., Shultz L.D., et al. Age-dependent human beta cell proliferation induced by glucagon-like peptide 1 and calcineurin signaling. J. Clin. Invest. 2017;127:3835–3844. doi: 10.1172/JCI91761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Robinson M.D., Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yin L., Jao L.E., Chen W. Generation of Targeted Mutations in Zebrafish Using the CRISPR/Cas System. Methods Mol. Biol. 2015;1332:205–217. doi: 10.1007/978-1-4939-2917-7_16. [DOI] [PubMed] [Google Scholar]
- 74.Brinkman E.K., Chen T., Amendola M., van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42 doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon reasonable request. The RNA-seq data have been deposited in the GEO repository and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report any original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact.