

ABSTRACT
This study aimed to explore the potential causal link between genetic predisposition to various connective tissue diseases (CTDs), namely systemic lupus erythematosus (SLE), Sjögren's syndrome (SS), polymyositis (PM), dermatomyositis (DM), systemic sclerosis (SSc), mixed connective tissue disease (MCTD), and rheumatoid arthritis (RA), and the incidence of pulmonary arterial hypertension (PAH) utilizing Mendelian randomization (MR). Employing a two‐sample MR approach, genetic variants associated with CTDs served as instrumental variables to investigate the exposure‐outcome relationship, with GWAS data sourced from the FinnGen Biobank. Comprehensive statistical analyses, including the inverse variance weighted (IVW) method, were conducted, alongside heterogeneity, pleiotropy, and sensitivity tests to ensure the robustness and validity of findings. The results revealed that in the Finnish population, no significant causal associations were identified between PAH and SLE, SS, PM, DM, MCTD, or RA. Notably, a significant association was observed between SSc and an increased risk of PAH (IVW: OR = 1.278, 95% CI = 1.061–1.540, p = 0.010). However, this finding was not replicated in other European populations. These results indicate the unique genetic and pathological pathways underlying SSc‐associated PAH, emphasizing the need for tailored screening and management protocols in this patient group.
Keywords: FinnGen, genetic association study, systemic lupus erythematosus, systemic sclerosis
1. Introduction
Pulmonary hypertension (PH) is a severe and progressive disease characterized by elevated pulmonary arterial pressure and pulmonary vascular resistance, leading to right heart failure and ultimately death if untreated. PH is defined hemodynamically as a mean pulmonary arterial pressure > 20 mmHg at rest, as assessed by right heart catheterization (RHC). The disease is classified into five groups based on similar clinical presentation, pathological findings, hemodynamic characteristics, and treatment strategy, with pulmonary arterial hypertension (PAH) categorized under Group 1 PH [1]. PAH can develop as a secondary condition to numerous underlying diseases, with connective tissue diseases (CTDs) being responsible for 15%–25% of these cases [2]. CTDs represent a heterogeneous group of systemic autoimmune disorders characterized by immune system dysfunction and the subsequent production of disease‐specific autoantibodies [3]. These conditions include systemic lupus erythematosus (SLE), Sjögren's syndrome (SS), polymyositis (PM), dermatomyositis (DM), systemic sclerosis (SSc), mixed connective tissue disease (MCTD), and rheumatoid arthritis (RA), which exhibit a wide range of systemic manifestations and their association with PAH has been a significant focus of research.
Prior observational studies indicated that the prevalence of PAH varied among different CTDs and was influenced by geographic regions and populations. PAH is particularly common in SSc and MCTD, but relatively rare in other CTDs [4]. SSc predominantly accounts for 60% to 75% of CTD‐associated PAH (CTD‐PAH) cases in Europe and North America [5, 6]. In contrast, SSc constitutes only 6%–22% of CTD‐PAH cases in Asia, where SLE is more prevalent [7]. In Japan, MCTD is the leading cause of CTD‐ PAH at 43%, followed by SLE at 29% [8]. Among patients with SS, PAH incidence is low in Western countries but notably higher in Asia, such as 10% in Japan [8] and 15% in Chinese cohorts [7, 9]. Conversely, RA shows a PAH incidence similar to that of the general population (approximately 0.35%) [10], and PAH associated with idiopathic inflammatory myopathies is exceedingly rare [11]. These studies indicated significant heterogeneity and potential confounding factors affecting the observed associations. Such inconsistencies underscore the need for rigorous methodological approaches and larger, well‐controlled studies to elucidate the true nature of these relationships.
The mechanisms underlying the observed associations between CTDs and PAH remain incompletely understood. Proposed explanations range from autoimmune‐mediated endothelial dysfunction to shared genetic susceptibilities influencing both CTD development and pulmonary vascular remodeling [4, 12, 13]. The predominant mechanism of SLE‐associated PAH (SLE‐PAH) involves the proliferation of endothelial and smooth muscle cells, fibrinoid necrosis due to vasculitis, and the deposition of immunoglobulins and complement components in the intimal and medial layers of the pulmonary vessels [14]. In contrast, endothelial dysfunction, vasculopathy, and increased vascular stiffness of small and mid‐size vessels are hallmarks of SSc‐related PAH [15]. Furthermore, genetic studies have identified potential genetic risk factors that may predispose individuals with certain CTDs to develop PAH. For example, studies have identified genetic variants associated with both SSc and PAH, suggesting overlapping genetic susceptibility loci that could influence disease susceptibility and progression [15]. These findings highlight the importance of integrating genetic data into epidemiological studies to better understand the underlying genetic architecture and pathways linking CTDs to PAH.
Clinical observational studies often grapple with the challenges of potential confounders and reverse causation, which complicates the elucidation of causal relationships between CTDs and PAH. However, a methodological approach known as Mendelian randomization (MR)—also referred to as “nature's randomized controlled trial”—offers a compelling solution [16]. As a robust statistical tool in epidemiology and genetics, MR harnesses genetic variants as instrumental variables (IVs) to estimate the causal effect of exposures on outcomes. The strength of MR lies in the random allocation of genetic variants at conception, rendering it typically more resistant to confounding and reverse causation than traditional observational designs [17]. The efficacy of MR has been demonstrated in its successful application to discern causal relationships between various diseases, such as systemic lupus erythematosus (SLE) and autoimmune liver diseases [18], RA and bronchiectasis [19], and depression and PAH [20]. Therefore, in this study, we employed a two‐sample MR approach to investigate the causal relationship between genetic predisposition to CTDs and the incidence of PAH.
2. Methods
2.1. Study Design
We carried out a two‐sample MR investigation, with CTDs as the exposure and PAH as the outcome (Figure 1). This study followed the latest STROBE‐MR guidelines for conducting MR research [16]. The validity of MR study is dependent on three essential assumptions [21]: (1) The IVs exhibit a robust correlation with the exposure; (2) Each IV does not have any association with confounding factors; (3) Each IV is linked to the outcome exclusively via the exposure, and no other routes exist for this association.
Figure 1.
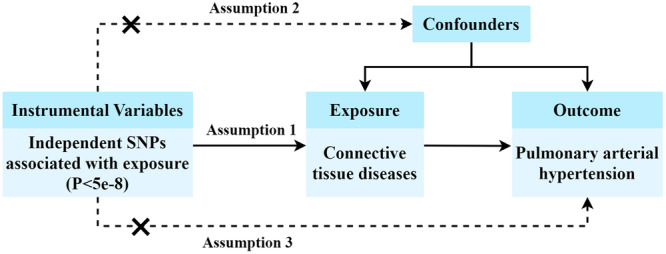
Study design of the Mendelian randomization (MR) analysis. SNPs, single nucleotide polymorphisms.
2.2. Genome‐Wide Association Studies (GWAS) Data Sources
The data sets for the primary MR analyses were obtained from the publicly accessible FinnGen Biobank. The FinnGen study is a large‐scale genomics initiative that has analyzed over 500,000 Finnish biobank samples and correlated genetic variation with health data to understand disease mechanisms and predispositions. Summary statistics for SLE, SS, PM, DM, SSc, MCTD, RA, and PAH were obtained from their respective data sets, and all patients and controls were of the Finnish population. The descriptive details of the data used in our study are presented in Table 1. Given that SSc‐associated PAH (SSc‐PAH) accounts for the majority of CTD‐PAH in European populations [5], we performed additional MR analyses using GWAS data from other European cohorts. These included two GWAS data sets for SSc and one for PAH (Supporting Information: Table S1), aimed at validating the observed associations.
Table 1.
Descriptive details of the data source of CTDs and PAH.
| Phenotype | Cases | Controls | Sample size | Population | Data set |
|---|---|---|---|---|---|
| SLE | 1083 | 306,504 | 307,587 | European | finngen_R10_SLE_FG |
| SS | 2735 | 399,355 | 402,090 | European | finn‐b‐M13_SJOGREN |
| PM | 244 | 399,355 | 399,599 | European | finngen_R10_M13_POLYMYO |
| DM | 405 | 285,035 | 285,440 | European | finngen_R10_DERMATOPOLY_FG |
| SSc | 680 | 399,355 | 400,035 | European | finngen_R10_M13_SYSTSLCE |
| MCTD | 2005 | 410,176 | 412,181 | European | finngen_R10_M13_MCTD |
| RA | 13,621 | 262,844 | 276,465 | European | finngen_R10_M13_RHEUMA |
| PAH | 248 | 289,117 | 289,365 | European | R10_I9_HYPTENSPUL |
Abbreviations: CTDs, connective tissue diseases; DM, dermatomyositis; MCTD, mixed connective tissue disease; PAH, pulmonary arterial hypertension; PM, polymyositis; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; SS, Sjögren's syndrome; SSc, systemic sclerosis.
2.3. IV Selection
To meet the initial hypothesis of MR analysis, which suggests that the IVs are strongly linked to CTDs, we first performed association analysis and selected single nucleotide polymorphisms (SNPs) that demonstrated a statistically significant correlation with each CTD at the genome‐wide level (p < 5 × 10−8). To ensure the independence of the selected IVs and minimize selection bias, we proceeded with a linkage disequilibrium (LD) analysis. To obtain an optimal number of IVs, we chose a genetic distance threshold of 5000 kB and a r² range of 0.001–0.1 (Supporting Information: Table S2). To circumvent potential confounding influences from genetic variations, the secondary phenotype of each SNP was evaluated using the PhenoScanner database with a p‐value cutoff of p < 1 × 10−5. SNPs associated with confounders were eliminated to reduce pleiotropic effects. SNPs with palindromic sequences were likewise excluded. Additionally, we confirmed that the F statistic of all IVs exceeded the threshold of 10 to minimize weak instrument bias. The F statistic was calculated using the equation F = R 2(N − K − 1)/K(1 − R 2), where R 2 represents the proportion of variance in the exposure that is explained by the IVs, and N represents the sample size of the exposure data set [22].
2.4. Statistical Analysis
To ensure the reliability and validity of our findings, a variety of robust statistical methods were utilized, including the inverse variance weighted (IVW) method under a random‐effect model, the weighted median approach, the MR‐Egger regression, the simple mode and weighted mode [19]. The IVW method, used as the primary analysis for causal estimates, was most precise when all IVs are valid [23]. Additionally, we performed sensitivity analyses to assess the dependability of our findings. The MR‐Egger intercept was used to determine directional horizontal pleiotropy [24]. Subsequently, the Mendelian randomization pleiotropy residual sum and outlier (MR‐PRESSO) test was applied to detect potential horizontal pleiotropy and correct it by removing outliers [25]. The Cochrane Q test was used to evaluate heterogeneity between SNPs [26]. Furthermore, the leave‐one‐out analysis was used to investigate whether the genetic causal relationship between exposures and outcomes was influenced by a single SNP. All statistical analyses were performed using the “TwoSampleMR” and “MRPRESSO” packages in R software (version 4.3.2).
3. Results
3.1. Characteristics of the Selected SNPs for IVs
When conducting the primary MR analysis using data from the Finnish population, we extracted SNPs that were significantly related to CTDs from the GWAS (p < 5 × 10−8). To keep a balance between having enough IVs to ensure adequate statistical power and minimizing the inclusion of pleiotropic or weak instruments, we performed LD analysis with a genetic distance threshold of 5000 kB and a r² range of 0.001–0.1 varying with specific CTDs. Subsequently, SNPs related to PAH were retrieved from the PhenoScanner database and no confounder was found. Furthermore, SNPs with palindromic sequences for each CTD were excluded respectively. Finally, 13 SNPs for SLE, 17 SNPs for SS, 11 SNPs for PM, 11 SNPs for DM, 8 SNPs for SSc, 22 SNPs for MCTD, and 26 SNPs for RA were selected as IVs and included in further analyses (Supporting Information: Tables S3–S9). Similarly, we obtained IVs for the two additional European SSc data sets using the same approach (Supporting Information: Tables S10–S11). No evidence of weak‐tool bias was found in the IVs strength test (F‐statistic > 10).
3.2. Causal Estimates of Genetic Susceptibility to CTDs and PAH Risk
In the Finnish population, The MR results do not support a significant causal association between SLE, SS, PM, DM, MCTD, or RA and the risk of PAH, verified by all the five statistical methods, with IVW as the primary method in the absence of horizontal pleiotropy of IVs. The odds ratio (OR) of IVW analysis for SLE, SS, PM, DM, MCTD, and RA was 0.947 (95% confidence interval (CI) = 0.805–1.116, p = 0.517), 1.002 (95%CI = 0.836–1.203, p = 0.979), 0.902 (95%CI = 0.803–1.013, p = 0.081), 0.900 (95%CI = 0.767–1.056, p = 0.198), 1.156 (95%CI = 0.964–1.386, p = 0.118), and 0.921 (95%CI = 0.704–1.204, p = 0.545), respectively, demonstrating no statistically significant association between these CTDs and PAH. However, a significant association was found between SSc and increased risk of PAH (IVW: OR = 1.278, 95% CI = 1.061–1.540, p = 0.010) (Figure 2).
Figure 2.
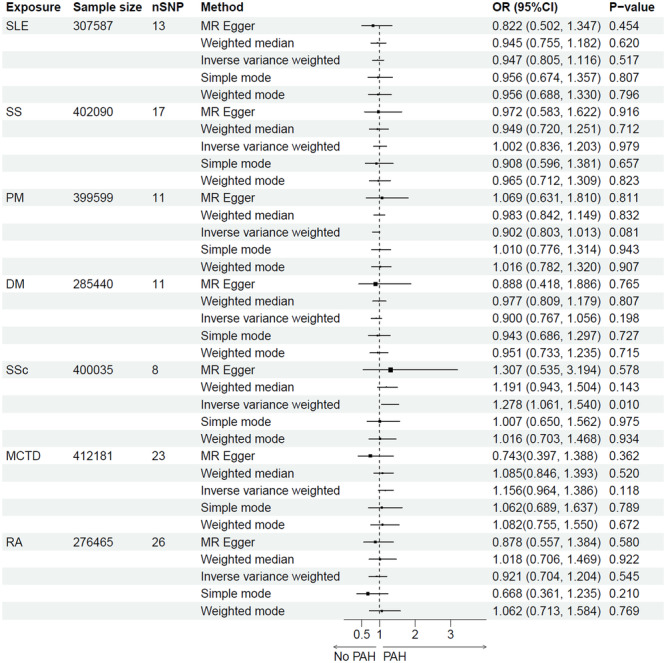
MR estimates for association of CTDs and the risk of PAH. CTDs, connective tissue diseases; DM, dermatomyositis; MCTD, mixed connective tissue disease; MR, mendelian randomization; nSNP, number of SNPs; PAH, pulmonary arterial hypertension; PM, polymyositis; OR, odds ratio; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; SS, Sjögren's syndrome; SSc, systemic sclerosis.
Interestingly, when separate MR analyses were performed using two additional European SSc data sets and another European PAH data set, no significant association between SSc and PAH was observed (Supporting Information: Figures S1–S2).
3.3. Heterogeneity and Pleiotropy Tests and Sensitivity Analyses
In our primary MR analysis in the Finnish population, we conducted heterogeneity and pleiotropy tests to ensure the robustness of our causal inference. Cochran's Q test was performed to assess heterogeneity among the IVs. The test results showed no significant heterogeneity for all seven CTDs, indicating no heterogeneity between SNPs (Table 2). Additionally, MR‐Egger regression analysis was used to evaluate the presence of horizontal pleiotropy. The intercept from the MR‐Egger regression was not significantly different from zero for all CTDs, indicating no evidence of horizontal pleiotropy (Table 3). These findings support the validity of our MR analysis and suggest that the observed causal relationship is unlikely to be biased by pleiotropic effects.
Table 2.
Heterogeneity tests of MR.
| Phenotype | Method | Q‐value | df | p‐Value |
|---|---|---|---|---|
| SLE | Q MR Egger | 8.472 | 11 | 0.670 |
| Q IVW | 8.828 | 12 | 0.718 | |
| SS | Q MR Egger | 15.682 | 15 | 0.403 |
| Q IVW | 15.699 | 16 | 0.474 | |
| PM | Q MR Egger | 6.010 | 8 | 0.646 |
| Q IVW | 6.431 | 9 | 0.696 | |
| DM | Q MR Egger | 13.849 | 9 | 0.128 |
| Q IVW | 13.851 | 10 | 0.180 | |
| SSc | Q MR Egger | 7.721 | 6 | 0.259 |
| Q IVW | 7.725 | 7 | 0.358 | |
| MCTD | Q MR Egger | 19.039 | 20 | 0.519 |
| Q IVW | 21.137 | 21 | 0.451 | |
| RA | Q MR Egger | 35.712 | 24 | 0.059 |
| Q IVW | 35.809 | 25 | 0.075 |
Abbreviations: DM, dermatomyositis; IVW, inverse variance weighted; MCTD, mixed connective tissue disease; MR, Mendelian randomization; PM, polymyositis; Q, Cochran's Q test; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; SS, Sjögren's syndrome; SSc, systemic sclerosis.
Table 3.
Horizontal pleiotropy test of MR.
| Phenotype | Method | Intercept | SE | p‐Value |
|---|---|---|---|---|
| SLE | MR‐Egger regression | 0.073 | 0.122 | 0.563 |
| SS | MR‐Egger regression | 0.012 | 0.097 | 0.902 |
| PM | MR‐Egger regression | −0.117 | 0.181 | 0.535 |
| DM | MR‐Egger regression | 0.008 | 0.214 | 0.973 |
| SSc | MR‐Egger regression | −0.009 | 0.186 | 0.961 |
| MCTD | MR‐Egger regression | 0.118 | 0.082 | 0.163 |
| RA | MR‐Egger regression | 0.012 | 0.048 | 0.800 |
Abbreviations: DM, dermatomyositis; MCTD, mixed connective tissue disease; MR, Mendelian randomization; PM, polymyositis; RA, rheumatoid arthritis; SE, standard error; SLE, systemic lupus erythematosus; SS, Sjögren's syndrome; SSc, systemic sclerosis.
The sensitivity analysis employed the leave‐one‐out method, sequentially removing each SNP and comparing the causal effects of the remaining SNPs with the results of the MR analysis that included all SNPs. This approach was used to identify whether any particular SNP disproportionately influences the overall causal estimate. The analysis for CTDs showed that the causal estimates remained relatively stable and consistent across all iterations of the leave‐one‐out analysis, indicating that the MR analysis results were robust (Supporting Information: Figures S3–S9).
4. Discussion
To our knowledge, this study represents the first MR investigation into the relationship between CTDs and PAH. In the Finnish population, our results did not indicate significant causal associations between most CTDs and PAH, including SLE, SS, PM, DM, MCTD, and RA. However, the study demonstrated a significant association between SSc and an increased risk of PAH. Clinically, these findings emphasize the importance of focusing on SSc in PAH screening and management, while also advocating for further genetic research to uncover the underlying mechanisms.
SSc is a complex autoimmune condition marked by inflammation, excessive collagen accumulation, and fibrosis across multiple systems [27]. SSc‐related PAH is a severe complication that occurs in 8% to 15% of SSc patients [28]. The pathogenesis of PAH in SSc involves complex interactions between endothelial dysfunction, smooth muscle proliferation, and immune‐mediated inflammation [29]. Genetic predisposition plays a critical role in this multifactorial process. Multiple genes, including those in innate immunity (IRF5, IRF7, and TLR2), T and B cell activation (CD247, TNFAIP3, STAT4, and BLK), and the NF‐kB pathway (TNFAIP3 and TNIP1), have been implicated in the pathogenesis of SSc through GWAS and exome sequencing [30]. However, most of these studies have not included enough patients complicated with PAH, thereby hindering a definitive conclusion on whether the variants associated with the overall risk of the disease are also correlated with PAH. Despite the absence of a significant association between TGF‐β receptor polymorphisms and PAH in a cohort study of SSc‐related PAH patients [31], certain genetic dispositions may still increase PAH susceptibility, including polymorphisms in MIF, TLR2, UPAR, KCNK5, and HLA‐B35 [32]. Additionally, genes such as CSK, DDX6, DNASE1L3, and GSDMA/B may influence the vascular and fibrotic aspects of SSc [30], with some of these genes potentially contributing to the development of PAH in SSc patients. Understanding the genetic architecture of SSc‐related PAH is crucial for developing novel targeted therapies based on the underlying genetic mechanisms.
Interestingly, when we performed two additional MR analyses using SSc data from two other European cohorts and PAH data from a separate European cohort, no significant association between SSc and PAH was found. We hypothesize that this discrepancy in results may be attributed to two factors. First, the study populations differ: the primary study focused on the Finnish population, whereas the additional study involved individuals from the United Kingdom (UK) and other Western countries. There may be some degree of genetic heterogeneity between these populations. Second, in the primary study, both the exposure and outcome data were derived from the Finnish population, while in the additional study, the exposure data were obtained from the UK population, and the outcome data were derived from a mixed population comprising individuals from the United States (US), France, Germany, the Netherlands, the UK, and Italy. This difference in population composition may have contributed to the variation in results. Therefore, further studies with larger sample sizes and more diverse populations are required to validate this finding.
Genetic studies on SLE have identified numerous susceptibility loci, including alleles associated with B cell responsiveness (e.g., BANK1, BLK, and PTPN22) and innate immune response (e.g., IRF5, STAT4, and TNFAIP3) [33]. Similarly, genetic risk factors for SS included both HLA loci (e.g., HLA‐DQA1, HLA‐DQB1, and HLA‐DRA) and non‐HLA loci (e.g., IRF5, STAT4, and IL12A) [34]. However, the genetic contribution of these risk alleles to PAH in SLE and SS appears limited. Certain genetic variants have been identified as risk factors for SLE and SS‐related PAH [35, 36], while research findings on the genetic susceptibility to PAH related to MCTD, PM, DM, and RA are scarce. Despite the fact that CTDs including SLE, SS, PM, DM, MCTD, and RA may be complicated by PAH [2, 4], the current study suggests no significant genetic association between these CTDs and PAH. A possible explanation is that the genetic architecture of PAH may differ fundamentally from that of the CTDs studied. While shared genetic susceptibilities exist, their influence on PAH development might be overshadowed by other factors, such as environmental triggers, epigenetic modifications, chronic inflammation, and autoantibody presence. Besides, the polygenic nature of CTDs implies that multiple low‐effect genetic variants collectively contribute to disease susceptibility, which may not be captured adequately in MR analyses focusing on individual loci. These findings highlight the complexity of CTD‐related PAH pathogenesis and the need for multifaceted research approaches. Future studies should integrate genetic, epigenetic, and environmental data to provide a comprehensive understanding of the mechanisms driving PAH in CTD patients.
Although MR is a robust method for inferring causality that reduces confounding and reverse causation, there are several limitations to this study. Firstly, the study's focus on genetic factors may overlook the contribution of nongenetic factors, such as environmental exposures and lifestyle choices, which play significant roles in CTD and PAH pathogenesis. Integrating these factors into future analyses could provide a more holistic understanding of the disease mechanisms. Secondly, the generalizability of the findings may be limited by the population studied. The analysis using GWAS data from Finnish and other European populations yielded inconsistent results, which may be attributed to the genetic characteristics of the Finnish population, which exhibits significant distinctions from other Europeans due to unique demographic history marked by genetic bottlenecks and population isolation [37, 38]. Furthermore, this study lacks data from Asian populations, which are particularly relevant for understanding associations such as that between SLE and PAH, as these associations may differ due to genetic and epidemiological variations. This limitation highlights the need for future studies to include more diverse cohorts to ensure broader applicability and relevance of the findings. Thirdly, the complexity of CTD‐PAH pathogenesis suggests that single‐locus MR analyses may not capture the full genetic architecture. Polygenic risk scores and pathway‐based approaches could provide more comprehensive insights into the genetic underpinnings of these conditions.
In conclusion, this MR study, conducted primarily within Finnish population, revealed no significant causal association between several CTDs (SLE, SS, PM, DM, MCTD, and RA) and PAH risk, while SSc showed a significant association with an increased risk of PAH. These findings underscore the distinct genetic and pathogenic landscape of SSc‐PAH and highlight the necessity for targeted screening and management strategies in this patient population. Comprehensive studies that integrate genetic, environmental, and lifestyle factors are crucial to fully understand the complex interplay driving PAH in CTD patients. Expanding research to include diverse populations will enhance the generalizability of the findings and contribute to the development of more effective, personalized treatment approaches for PAH in patients with CTDs.
Author Contributions
Xiaoqin Wang conceptualized the study, analyzed the data, and wrote the original manuscript. Leilei Yang conceptualized the study and acquired the GWAS data. Bingjie Gu assisted in data acquisition and analysis. Dinglei Su conceptualized and supervised the study, reviewed the data analysis, wrote, and edited the manuscript. All authors read and approved the final manuscript.
Ethics Statement
This study included human subjects gathered from prior investigations to present the extensive genome‐wide association studies (GWAS). Ethical approval and informed consent of participants were obtained in all the respective original studies.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Supplementary figure 1. Forest plot of MR analysis for association of SSc and the risk of PAH in additional European populations. GWAS ID of SSc data: GCST90436638, GWAS ID of PAH data: GCST007228. MR, mendelian randomization; SSc, systemic sclerosis; PAH, pulmonary arterial hypertension; GWAS, genome‐wide association study.
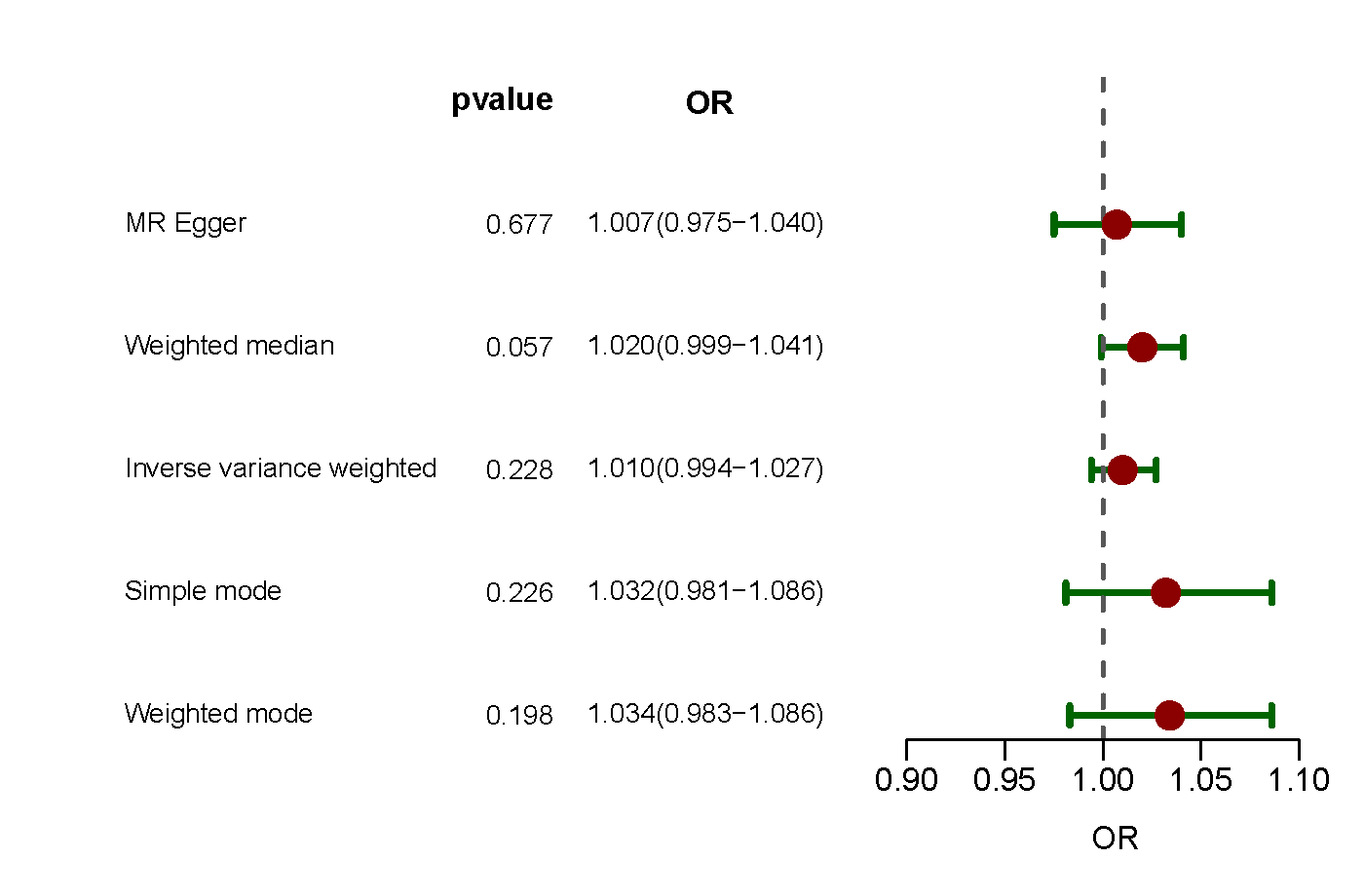{kind=link}
Supplementary figure 2. Forest plot of MR analysis for association of SSc and the risk of PAH in additional European populations. GWAS ID of SSc data: GCST90044535, GWAS ID of PAH data: GCST007228. MR, mendelian randomization; SSc, systemic sclerosis; PAH, pulmonary arterial hypertension; GWAS, genome‐wide association study.
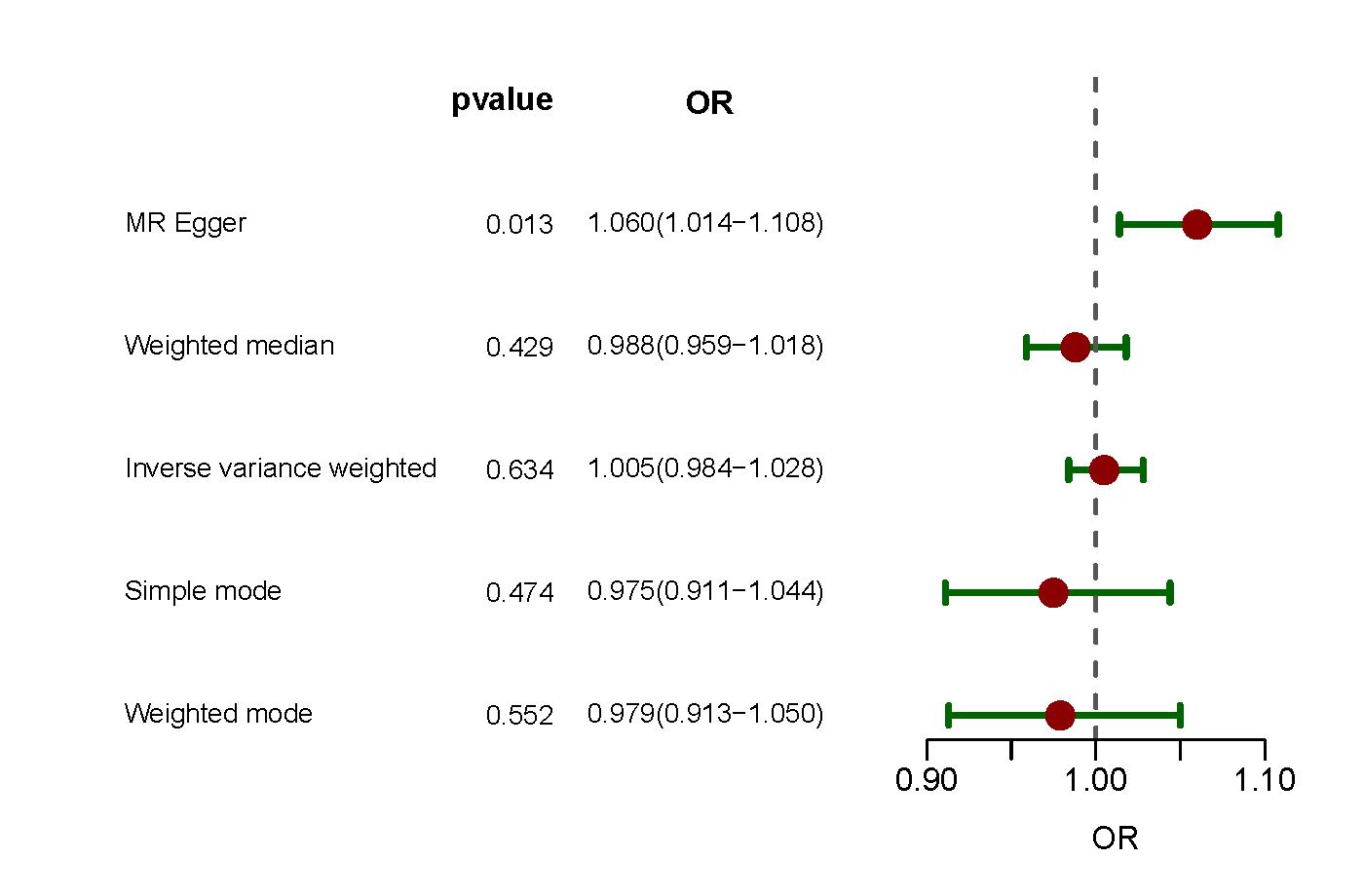{kind=link}
Supplementary figure 3. Leave‐one‐out sensitivity analysis of the MR study on the association between SLE and PAH. MR, mendelian randomization; SLE, systemic lupus erythematosus; PAH, pulmonary arterial hypertension.
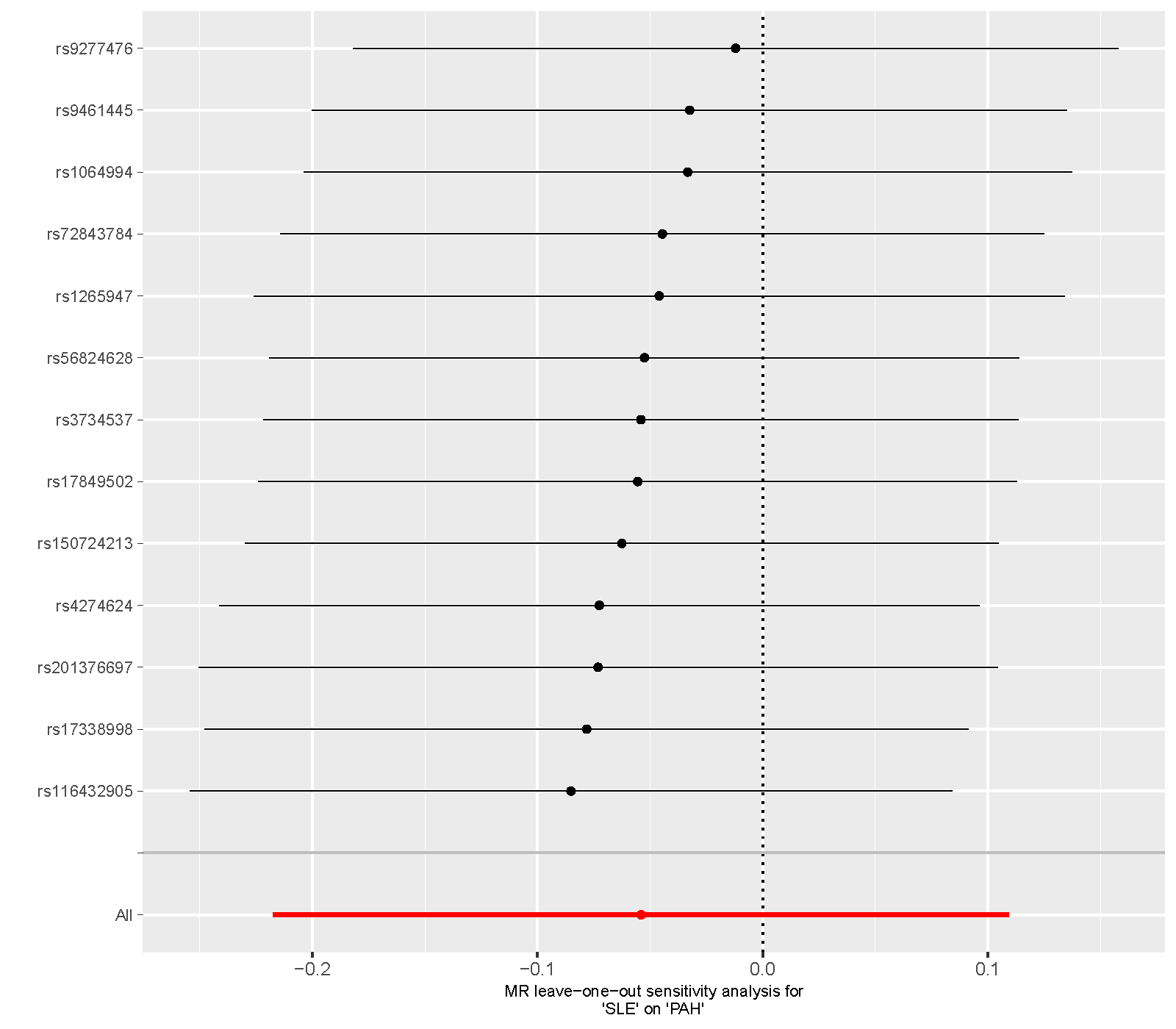{kind=link}
Supplementary figure 4. Leave‐one‐out sensitivity analysis of the MR study on the association between SS and PAH. MR, mendelian randomization; SS, Sjögren's syndrome; PAH, pulmonary arterial hypertension.
{kind=link}
Supplementary figure 5. Leave‐one‐out sensitivity analysis of the MR study on the association between PM and PAH. MR, mendelian randomization; PM, polymyositis; PAH, pulmonary arterial hypertension.
{kind=link}
Supplementary figure 6. Leave‐one‐out sensitivity analysis of the MR study on the association between DM and PAH. MR, mendelian randomization; DM, dermatomyositis; PAH, pulmonary arterial hypertension.
{kind=link}
Supplementary figure 7. Leave‐one‐out sensitivity analysis of the MR study on the association between SSc and PAH. MR, mendelian randomization; SSc, systemic sclerosis; PAH, pulmonary arterial hypertension.
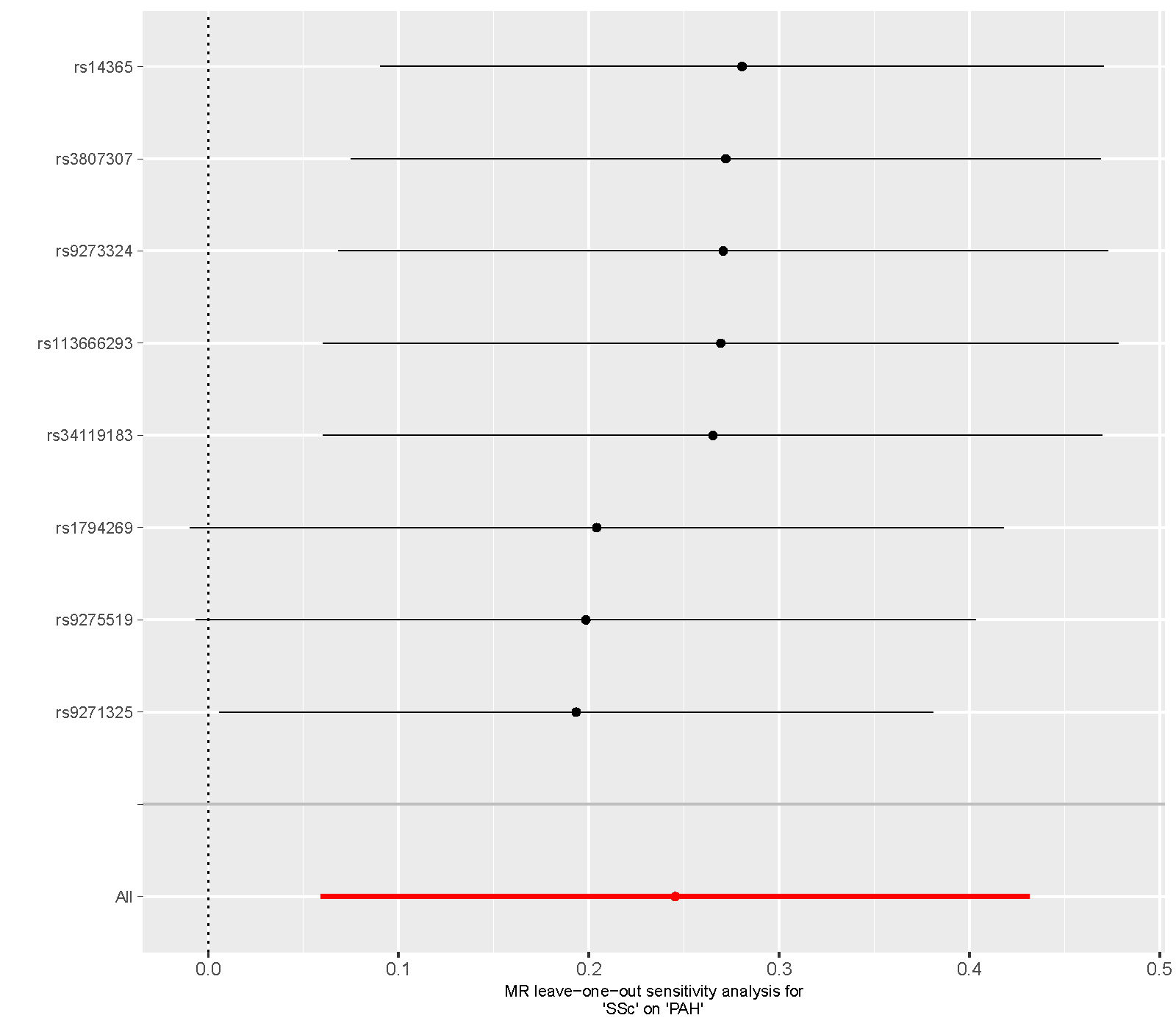{kind=link}
Supplementary figure 8. Leave‐one‐out sensitivity analysis of the MR study on the association between MCTD and PAH. MR, mendelian randomization; MCTD, mixed connective tissue disease; PAH, pulmonary arterial hypertension.
{kind=link}
Supplementary figure 9. Leave‐one‐out sensitivity analysis of the MR study on the association between RA and PAH. MR, mendelian randomization; RA, rheumatoid arthritis; PAH, pulmonary arterial hypertension.
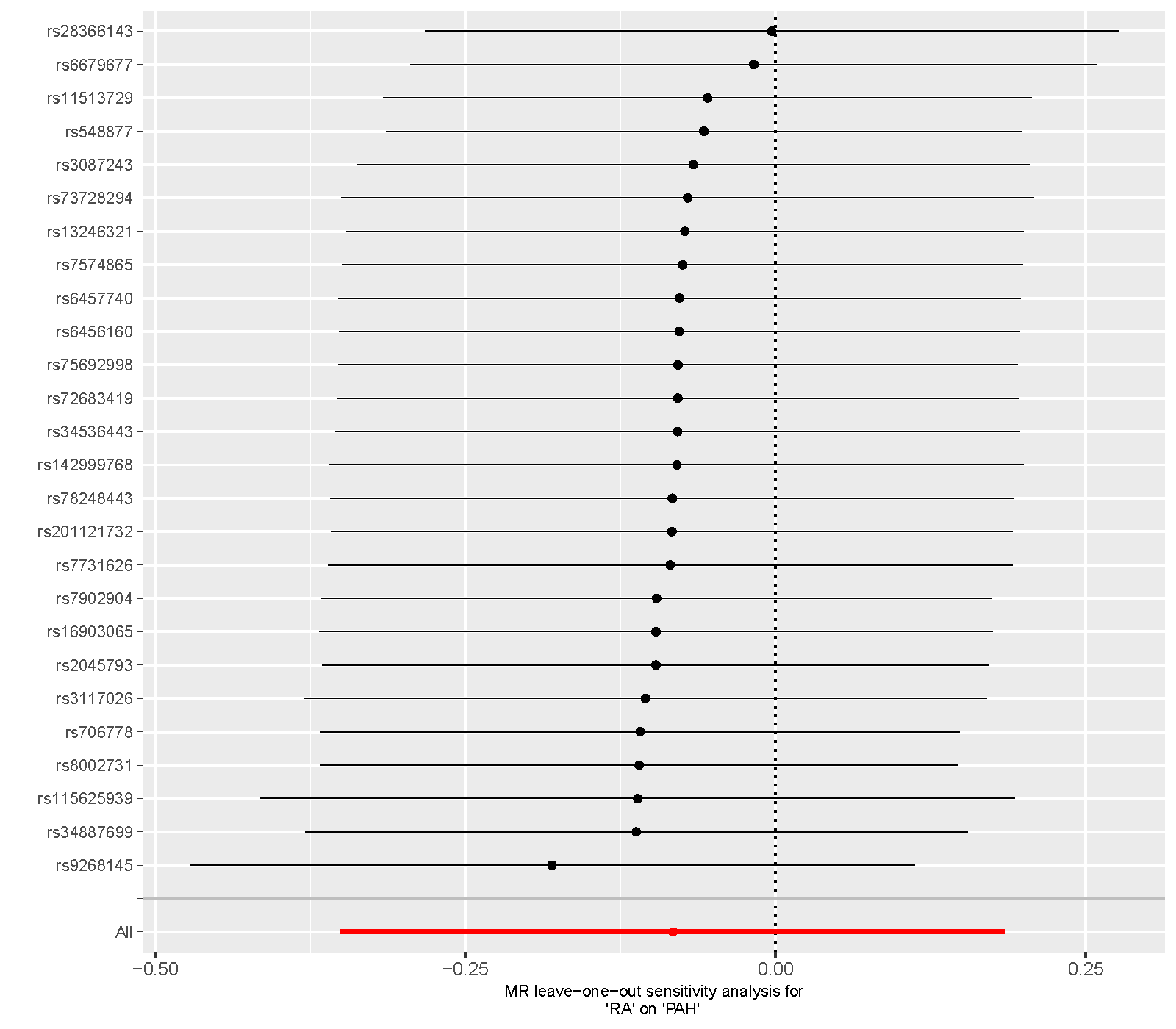{kind=link}
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Acknowledgments
We want to acknowledge the participants and investigators of the FinnGen study.
References
- 1. Hassoun P. M., “Pulmonary Arterial Hypertension,” New England Journal of Medicine 385 (2021): 2361–2376. [DOI] [PubMed] [Google Scholar]
- 2. Lewis C., Sanderson R., Vasilottos N., Zheutlin A., and Visovatti S., “Pulmonary Arterial Hypertension in Connective Tissue Diseases Beyond Systemic Sclerosis,” Heart Failure Clinics 19 (2023): 45–54. [DOI] [PubMed] [Google Scholar]
- 3. Mulhearn B., Tansley S. L., and McHugh N. J., “Autoantibodies in Connective Tissue Disease,” Best Practice & Research Clinical Rheumatology 34 (2020): 101462. [DOI] [PubMed] [Google Scholar]
- 4. Thoreau B. and Mouthon L., “Pulmonary Arterial Hypertension Associated With Connective Tissue Diseases (CTD‐PAH): Recent and Advanced Data,” Autoimmunity Reviews 23 (2024): 103506. [DOI] [PubMed] [Google Scholar]
- 5. Peacock A. J., Murphy N. F., McMurray J. J. V., Caballero L., and Stewart S., “An Epidemiological Study of Pulmonary Arterial Hypertension,” European Respiratory Journal 30 (2007): 104–109. [DOI] [PubMed] [Google Scholar]
- 6. McGoon M. D. and Miller D. P., “Reveal: A Contemporary Us Pulmonary Arterial Hypertension Registry,” European Respiratory Review 21 (2012): 8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hao Y. J., Jiang X., Zhou W., et al., “Connective Tissue Disease‐Associated Pulmonary Arterial Hypertension in Chinese Patients,” European Respiratory Journal 44 (2014): 963–972. [DOI] [PubMed] [Google Scholar]
- 8. Shirai Y., Yasuoka H., Okano Y., Takeuchi T., Satoh T., and Kuwana M., “Clinical Characteristics and Survival of Japanese Patients With Connective Tissue Disease and Pulmonary Arterial Hypertension: a Single‐Centre Cohort,” Rheumatology 51 (2012): 1846–1854. [DOI] [PubMed] [Google Scholar]
- 9. Zhao J., Wang Q., Liu Y., et al., “Clinical Characteristics and Survival of Pulmonary Arterial Hypertension Associated With Three Major Connective Tissue Diseases: A Cohort Study in China,” International Journal of Cardiology 236 (2017): 432–437. [DOI] [PubMed] [Google Scholar]
- 10. Montani D., Henry J., O'Connell C., et al., “Association Between Rheumatoid Arthritis and Pulmonary Hypertension: Data From the French Pulmonary Hypertension Registry,” Respiration 95 (2018): 244–250. [DOI] [PubMed] [Google Scholar]
- 11. Sanges S., Yelnik C. M., Sitbon O., et al., “Pulmonary Arterial Hypertension in Idiopathic Inflammatory Myopathies: Data From the French Pulmonary Hypertension Registry and Review of the Literature,” Medicine 95 (2016): e4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Budhram B., Weatherald J., and Humbert M., “Pulmonary Hypertension in Connective Tissue Diseases Other Than Systemic Sclerosis,” Seminars in Respiratory and Critical Care Medicine 45 (2024): 419–434. [DOI] [PubMed] [Google Scholar]
- 13. Khangoora V., Bernstein E. J., King C. S., and Shlobin O. A., “Connective Tissue Disease‐Associated Pulmonary Hypertension: A Comprehensive Review,” Pulmonary Circulation 13 (2023): e12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Parperis K., Velidakis N., Khattab E., Gkougkoudi E., and Kadoglou N. P. E., “Systemic Lupus Erythematosus and Pulmonary Hypertension,” International Journal of Molecular Sciences 24 (2023): 5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bruni C., Guignabert C., Manetti M., Cerinic M. M., and Humbert M., “The Multifaceted Problem of Pulmonary Arterial Hypertension in Systemic Sclerosis,” The Lancet Rheumatology 3 (2021): e149–e159. [DOI] [PubMed] [Google Scholar]
- 16. Skrivankova V. W., Richmond R. C., Woolf B. A. R., et al., “Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization: The STROBE‐MR Statement,” Journal of the American Medical Association 326 (2021): 1614–1621. [DOI] [PubMed] [Google Scholar]
- 17. Zheng J., Baird D., Borges M. C., et al., “Recent Developments in Mendelian Randomization Studies,” Current Epidemiology Reports 4 (2017): 330–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang W., Jin T., Zheng W., et al., “Identifying the Genetic Association Between Systemic Lupus Erythematosus and the Risk of Autoimmune Liver Diseases,” Journal of Autoimmunity 145 (2024): 103188. [DOI] [PubMed] [Google Scholar]
- 19. Chen Z., Li X., Shi H., Huang Y., and Liu J., “Causal Relationship Between Rheumatoid Arthritis and Bronchiectasis: A Bidirectional Mendelian Randomization Study,” Arthritis Research & Therapy 26 (2024): 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Z., Kutty S., Peng W., et al., “Causal Association of Depression, Anxiety, Cognitive Performance, the Brain Cortical Structure With Pulmonary Arterial Hypertension: A Mendelian Randomization Study,” Journal of Affective Disorders 356 (2024): 356–362. [DOI] [PubMed] [Google Scholar]
- 21. Burgess S., Scott R. A., Timpson N. J., Davey Smith G., and Thompson S. G., “Using Published Data in Mendelian Randomization: A Blueprint for Efficient Identification of Causal Risk Factors,” European Journal of Epidemiology 30 (2015): 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin T., Zhou F., Mao H., Xie Z., and Jin Y., “Vitamin D and Idiopathic Pulmonary Fibrosis: A Two‐Sample Mendelian Randomization Study,” BMC Pulmonary Medicine 23 (2023): 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burgess S., Dudbridge F., and Thompson S. G., “Combining Information on Multiple Instrumental Variables in Mendelian Randomization: Comparison of Allele Score and Summarized Data Methods,” Statistics in Medicine 35 (2016): 1880–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bowden J., Davey Smith G., and Burgess S., “Mendelian Randomization With Invalid Instruments: Effect Estimation and Bias Detection through Egger Regression,” International Journal of Epidemiology 44 (2015): 512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Verbanck M., Chen C. Y., Neale B., and Do R., “Detection of Widespread Horizontal Pleiotropy in Causal Relationships Inferred From Mendelian Randomization Between Complex Traits and Diseases,” Nature Genetics 50 (2018): 693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hemani G., Zheng J., Elsworth B., et al., “The MR‐Base Platform Supports Systematic Causal Inference Across the Human Phenome,” eLife 7 (2018): e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Denton C. P. and Khanna D., “Systemic Sclerosis,” The Lancet 390 (2017): 1685–1699. [DOI] [PubMed] [Google Scholar]
- 28. Atsumi T., Bae S. C., Gu H., et al., “Risk Factors for Pulmonary Arterial Hypertension in Patients With Systemic Lupus Erythematosus: A Systematic Review and Expert Consensus,” ACR Open Rheumatology 5 (2023): 663–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bahi M., Li C., Wang G., and Korman B. D., “Systemic Sclerosis‐Associated Pulmonary Arterial Hypertension: From Bedside to Bench and Back Again,” International Journal of Molecular Sciences 25 (2024): 4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Orvain C., Assassi S., Avouac J., and Allanore Y., “Systemic Sclerosis Pathogenesis: Contribution of Recent Advances in Genetics,” Current Opinion in Rheumatology 32 (2020): 505–514. [DOI] [PubMed] [Google Scholar]
- 31. Koumakis E., Wipff J., Dieudé P., et al., “TGFβ Receptor Gene Variants in Systemic Sclerosis‐Related Pulmonary Arterial Hypertension: Results From a Multicentre EUSTAR Study of European Caucasian Patients,” Annals of the Rheumatic Diseases 71 (2012): 1900–1903. [DOI] [PubMed] [Google Scholar]
- 32. Jiang Y., Turk M. A., and Pope J. E., “Factors Associated With Pulmonary Arterial Hypertension (PAH) in Systemic Sclerosis (SSc),” Autoimmunity Reviews 19 (2020): 102602. [DOI] [PubMed] [Google Scholar]
- 33. Catalina M. D., Owen K. A., Labonte A. C., Grammer A. C., and Lipsky P. E., “The Pathogenesis of Systemic Lupus Erythematosus: Harnessing Big Data to Understand the Molecular Basis of Lupus,” Journal of Autoimmunity 110 (2020): 102359. [DOI] [PubMed] [Google Scholar]
- 34. Thorlacius G. E., Björk A., and Wahren‐Herlenius M., “Genetics and Epigenetics of Primary Sjögren Syndrome: Implications for Future Therapies,” Nature Reviews Rheumatology 19 (2023): 288–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qian J., Chen Y., Yang X., et al., “Association Study Identified HLA‐DQA1 as a Novel Genetic Risk of Systemic Lupus Erythematosus‐Associated Pulmonary Arterial Hypertension,” Arthritis & Rheumatology 75 (2023): 2207–2215. [DOI] [PubMed] [Google Scholar]
- 36. Li M., Shi Y., Zhao J., Wang Q., Li M., and Zhao X., “Identification of Potential Susceptibility Genes in Patients With Primary Sjögren's Syndrome‐Associated Pulmonary Arterial Hypertension Through Whole Exome Sequencing,” Arthritis Research & Therapy 25 (2023): 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Locke A. E., Steinberg K. M., Chiang C. W. K., et al., “Exome Sequencing of Finnish Isolates Enhances Rare‐Variant Association Power,” Nature 572 (2019): 323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kerminen S., Cerioli N., Pacauskas D., et al., “Changes in the Fine‐Scale Genetic Structure of Finland Through the 20th Century,” PLoS Genetics 17 (2021): e1009347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1. Forest plot of MR analysis for association of SSc and the risk of PAH in additional European populations. GWAS ID of SSc data: GCST90436638, GWAS ID of PAH data: GCST007228. MR, mendelian randomization; SSc, systemic sclerosis; PAH, pulmonary arterial hypertension; GWAS, genome‐wide association study.
Supplementary figure 2. Forest plot of MR analysis for association of SSc and the risk of PAH in additional European populations. GWAS ID of SSc data: GCST90044535, GWAS ID of PAH data: GCST007228. MR, mendelian randomization; SSc, systemic sclerosis; PAH, pulmonary arterial hypertension; GWAS, genome‐wide association study.
Supplementary figure 3. Leave‐one‐out sensitivity analysis of the MR study on the association between SLE and PAH. MR, mendelian randomization; SLE, systemic lupus erythematosus; PAH, pulmonary arterial hypertension.
Supplementary figure 4. Leave‐one‐out sensitivity analysis of the MR study on the association between SS and PAH. MR, mendelian randomization; SS, Sjögren's syndrome; PAH, pulmonary arterial hypertension.
Supplementary figure 5. Leave‐one‐out sensitivity analysis of the MR study on the association between PM and PAH. MR, mendelian randomization; PM, polymyositis; PAH, pulmonary arterial hypertension.
Supplementary figure 6. Leave‐one‐out sensitivity analysis of the MR study on the association between DM and PAH. MR, mendelian randomization; DM, dermatomyositis; PAH, pulmonary arterial hypertension.
Supplementary figure 7. Leave‐one‐out sensitivity analysis of the MR study on the association between SSc and PAH. MR, mendelian randomization; SSc, systemic sclerosis; PAH, pulmonary arterial hypertension.
Supplementary figure 8. Leave‐one‐out sensitivity analysis of the MR study on the association between MCTD and PAH. MR, mendelian randomization; MCTD, mixed connective tissue disease; PAH, pulmonary arterial hypertension.
Supplementary figure 9. Leave‐one‐out sensitivity analysis of the MR study on the association between RA and PAH. MR, mendelian randomization; RA, rheumatoid arthritis; PAH, pulmonary arterial hypertension.
Supporting information.
Supporting information.
Supporting information.
Supporting information.