

Abstract
INTRODUCTION
Factors responsible for the deposition of pathological tau in the brain are incompletely understood. This study links macroscale tau deposition in the human brain to cerebrospinal fluid (CSF) flow dynamics using resting‐state functional magnetic resonance imaging (rsfMRI).
METHODS
Low‐frequency (< 0.1 Hz) resting‐state global brain activity is coupled with CSF flow and potentially reflects CSF dynamics‐related clearance. We examined the correlation between rsfMRI measures of CSF inflow and global activity (gBOLD–CSF coupling) as a predictor, interacting with amyloid beta (Aβ), of tau and cortical thickness (dependent variables) across Alzheimer's Disease Neuroimaging Initiative (ADNI) participants from cognitively unimpaired through mild cognitive impairment (MCI) and Alzheimer's disease (AD).
RESULTS
Tau deposition in Aβ+ participants, accompanied by cortical thinning and cognitive decline, is associated with decreased gBOLD–CSF coupling. Tau mediates the relationship between coupling and thickness.
DISCUSSION
Findings suggest that resting‐state global brain activity and CSF movements comodulate Alzheimer's tau deposition, presumably related to CSF clearance.
Highlights
A non‐invasive functional magnetic resonance imaging (fMRI) assessment of a CSF clearance‐related process is carried out.
Global brain activity is coupled with CSF inflow in human fMRI during resting state.
Global fMRI–CSF coupling is correlated with tau in Alzheimer's disease (AD).
This coupling measure is also associated with cortical thickness, mediated by tau.
Keywords: Alzheimer's disease pathology, brain atrophy, cerebrospinal fluid (CSF) flow, CSF dynamics‐related clearance, global resting‐state fMRI signal, tau deposition
1. BACKGROUND
Alzheimer's disease (AD) is pathologically characterized by the extracellular accumulation of amyloid beta (Aβ) plaques and the intracellular accumulation of hyperphosphorylated tau in the form of neurofibrillary tangles. 1 Converging evidence has shown that Aβ accelerates tau phosphorylation and promotes tau aggregation and oligomerization. 2 Tau plays an especially critical role in cognitive decline, 3 , 4 brain atrophy, particularly cortical thinning, 5 , 6 and neuronal and synaptic loss. 3 , 7 The deposition of tau aggregates in AD follows a stereotypical pattern, beginning in the entorhinal cortex and hippocampus and then propagating to areas that have been characterized in post mortem and imaging studies as Braak stages. 8 , 9 The neural mechanism underlying such a stereotyped pattern of tau accumulation remains elusive. Current hypotheses have attributed tau spreading to altered neural activity 10 , 11 and a “prion‐like” mechanism manifested as abnormal tau seeds transferring through anatomically connected brain regions demonstrated in both cell and animal models 12 and via functionally connected cortical regions in humans. 13 , 14 , 15
Beyond its aggregation patterns, tau clearance has received increasing attention. 16 For example, recent animal studies have identified the critical role of glymphatic function in clearing brain wastes, including Aβ and tau, via a pathway involving cerebrospinal fluid (CSF) flow and the exchange between CSF and interstitial fluid (ISF). 16 , 17 , 18 In mice, the sleep–wake cycle regulates ISF tau, and sleep deprivation can significantly increase ISF and CSF tau as well as tau spreading, 19 presumably due to inadequate sleep‐dependent CSF dynamics‐related clearance. 17 While progress has been made in assessing CSF movement‐related clearance, 18 , 20 , 21 , 22 , 23 , 24 a real‐time non‐invasive imaging method is still needed to evaluate relationships between CSF dynamics and clearance in humans. However, CSF clearance is linked to spontaneous low‐frequency (<0.1 Hz) resting‐state global brain activity assessed with the global blood‐oxygen‐level‐dependent (gBOLD) signal in functional magnetic resonance imaging (fMRI) 25 , 26 , 27 ; observation of this phenomenon during light sleep or low arousal states supports the importance of this process in clearing brain waste 28 , 29 , 30 due to the strong sleep‐dependent effect of CSF clearance. 17 CSF movement, a key determinant of CSF clearance, 16 , 17 , 18 is coupled with gBOLD during both sleep 31 and wakefulness. 25 This global brain activity and CSF movement (gBOLD–CSF) coupling has recently been proposed to reflect CSF dynamics potentially related to clearance and is also correlated with cortical Aβ in AD, 25 , 27 cognitive decline in AD and Parkinson's disease, 25 , 26 and age. 32
Although early resting‐state fMRI (rsfMRI) studies assumed the gBOLD signal was noise, 30 an increasing number of studies have suggested that gBOLD reflects slow global neural activity. 28 , 33 , 34 , 35 Specifically, concurrent fMRI‐electrophysiology studies in primates have shown correlations between the gamma‐band power of local field potentials in the visual cortex and rsfMRI across widespread brain regions, suggesting a significant portion of gBOLD is directly linked to neural activity. 28 Moreover, converging evidence also has demonstrated that gBOLD fluctuation indicates a brain state related to vigilance, and its amplitude increase during light sleep or low arousal states 28 , 29 , 30 , 34 implies a potential link with sleep‐dependent CSF clearance. 17 , 19 In addition, cortical co‐activations at prominent gBOLD peaks display larger signal increases in the sensory‐motor network, accompanied by deactivations in the nucleus basalis, a critical component of the cholinergic system. 34 Furthermore, gBOLD correlates with strong sympathetic changes, including pupil size, 33 , 36 respiratory and cardiac pulsation, 37 , 38 , 39 , 40 , 41 and heart rate variability. 42 These sympathetic activities may constrict pial arteries and facilitate CSF movements or indirectly affect the slow (<0.1 Hz) modulation of cardiac and respiratory pulsations, accepted as the driving forces of CSF flow. 43 , 44 Thus, despite the lack of direct evidence, gBOLD–CSF coupling is a plausible surrogate measure of the global brain and CSF dynamics related to clearance.
Therefore, a key question arises: Does coupling between global brain activity and CSF movement (gBOLD–CSF coupling) as a measure of CSF clearance explain the amount of cortical tau deposition across participants? To address the hypothetical question, we examined multimodal data from the Alzheimer's Disease Neuroimaging Initiative‐3 (ADNI3; see the full ADNI investigators list at the Appendix 1) to investigate the relationship between gBOLD–CSF coupling, tau deposition measured with PET, cortical thickness, and cognitive function.
2. METHODS
2.1. Participants and data
We included 95 participants from ADNI3 who had available tau positron emission tomography (tau‐PET) (18F‐Flortaucipir [FTP]), Aβ‐PET (either [18F]florbetaben [FBB] or [18F]florbetapir [FBP]), rsfMRI (TR = 0.607 s only), and structural MRI (cortical thickness) data. Participants included four different clinical diagnosis (http://adni.loni.usc.edu/study‐design/): six AD patients, 22 with mild cognitive impairment (MCI), five participants with subjective memory concern (SMC), and 62 healthy controls. We further categorized the participants into cognitively impaired (AD and MCI) and unimpaired (SMC and control) groups. Cognitive performance was measured with the Montreal Cognitive Assessment (MoCA). Our cohort thus includes 46 Aβ− and 49 Aβ+ participants, including 21 unimpaired Aβ+ and 28 impaired Aβ+ individuals. All participants provided written informed consent. Ethical approval from the individual Institutional Review Board (IRB; http://adni.loni.usc.edu/wp‐content/uploads/2013/09/DOD‐ADNI‐IRB‐Approved‐Final‐protocol‐08072012.pdf) was granted to the investigators at each ADNI participating site. All the ADNI data were collected per the principles of the Declaration of Helsinki.
Aβ‐PET status, tau‐PET regional standardized uptake value ratios (SUVRs), rsfMRI scans, cortical thickness, and MoCA were obtained from the same study visit (the time interval between pairwise modalities was no more than 183 days 45 ). The file “UC Berkeley—AV1451 PVC 8 mm Res Analysis [ADNI2,3] (version: 2023‐02‐17)” was used to provide the tau‐PET SUVR. 46 , 47 Both “UC Berkeley—AV45 8 mm Res Analysis [ADNIGO,2,3] (version: 2023‐02‐17)” and “UC Berkeley—FBB 8 mm Res Analysis [ADNI3] (version: 2023‐02‐17)” were used to provide the Aβ‐PET status (thresholds for Aβ+ were AV45–Aβ > 1.11 SUVR or FBB–Aβ > 1.08 SUVR using the whole cerebellum as reference region). 48 Cortical thickness was downloaded from ADNI as “UCSF—Cross‐Sectional FreeSurfer (6.0) [ADNI3] (Version: 2022‐08‐17).” MoCA score was also directly acquired from ADNI as “Montreal Cognitive Assessment (MoCA) [ADNIGO,2,3].” All these data, as well as the rsfMRI and cortical thickness, are publicly accessible on the ADNI website (http://adni.loni.usc.edu/). It is worth noting that the cognitively impaired group included only individuals who were Aβ+, while the cognitively unimpaired group included Aβ+ and Aβ− individuals.
2.2. Image acquisition and preprocessing
All rsfMRI scans were acquired using 3 Tesla MR scanners from multiple ADNI participating sites following a unified protocol (http://adni.loni.usc.edu/methods/documents/mri‐protocols/). The MRI data used in the current study were collected on Siemens MRI scanners (Siemens Medical Solutions, Siemens, Erlangen, Germany). Each MRI session began with a T1‐weighted (T1w) MPRAGE sequence (flip angle = 9°, spatial resolution = 1 × 1 × 1 mm3, echo time [TE] = 3.0 ms, repetition time [TR] = 2300 ms), which was used for cortical thickness, anatomical segmentation, and registration. 49 During rsfMRI acquisition, 976 fMRI volumes were collected with an echo‐planar image (EPI) sequence with TR/TE = 607/32 ms (flip angle = 50°, spatial resolution = 2.5 × 2.5 × 2.5 mm3, slice thickness = 2.5 mm; see details at: http://adni.loni.usc.edu/methods/documents/).
RESEARCH IN CONTEXT
Systematic review: A systematic literature search was carried out using traditional (eg, PubMed) sources to identify articles studying tau deposition and clearance in Alzheimer's disease (AD). Despite the different approaches that have been used to investigate cerebrospinal fluid (CSF) flow clearance of tau, human studies on tau clearance with non‐invasive fMRI have never been formally conducted.
Interpretation: Our findings suggest the decreased coupling between CSF flow and global brain activity could play a critical role in promoting tau pathology, presumably through reduced CSF clearance. Tau mediates the link between CSF clearance and brain atrophy.
Future directions: Future studies may investigate a causal association between impaired CSF flow and tau accumulation through a longitudinal study. Moreover, direct evidence is needed to link CSF inflow fMRI signal and CSF dynamics‐related clearance. Prospective studies should also examine whether global brain activity and associated physiological processes, including cardiac and respiratory functions, co‐modulate CSF dynamics‐related clearance.
PET imaging was acquired according to standardized protocols at each ADNI site. FTP‐PET data were acquired from 75 to 105 min after injection of 10 mCi tracer. FBP‐PET and FBB‐PET data were acquired from the 50 to 70 min after injection of 10 mCi tracer and the 90 to 110 min after injection of 8.1 mCi tracer, 48 , 50 respectively (https://adni.loni.usc.edu/wp‐content/uploads/2012/10/ADNI3‐Procedures‐Manual_v3.0_20170627.pdf).
We preprocessed structural MRI using FreeSurfer version 7.1 (https://surfer.nmr.mgh.harvard.edu/fswiki/DownloadAndInstall5.3) 51 to derive FreeSurfer regions of interest (ROIs), that is, DKT‐68 parcel, 52 in participants’ native space and extract the parcel‐based cortical thickness. Following a previous study, 25 we preprocessed the rsfMRI data using FSL (https://fsl.fmrib.ox.ac.uk/fsl/fslwiki) 53 and AFNI (https://afni.nimh.nih.gov/) 54 with a modification of excluding rsfMRI sessions with excessive head motion (session‐mean frame‐wise displacement [FD] larger than 0.6 mm or the maximal FD larger than 3 mm). 55 The general procedures for rsfMRI preprocessing include motion correction, skull stripping, spatial smoothing (full width at half maximum [FWHM] = 4 mm), and temporal filtering (bandpass filter, 0.01 to 0.1 Hz). As in previous studies, 25 , 40 we also removed the first 20 and last 20 volumes for each rsfMRI session to reduce the edge effect from the temporal filtering and to ensure a steady magnetization.
All participants (N = 95) had Aβ‐PET (N = 32 FBP; N = 63 FBB) and tau‐PET data. To generate the PET data in DKT‐68 parcels, several preprocessing steps were performed, including image averaging, spatial smoothing, and registration to the (structural) MRI space to extract the tau or Aβ intensity in gray matter and each DKT‐68 parcel. 52 We then normalized parcel‐wise tau with the inferior cerebellar reference region to derive the tau SUVR and further applied partial volume correction (PVC) to reduce the influence of low image resolution and limited tissue sampling. 47 Regarding the Aβ SUVR, we normalized the FBP or FBB intensity in target gray matter using the whole cerebellum reference region.
2.3. Extraction of gBOLD and CSF inflow signals
We derived the gBOLD signal by averaging the rsfMRI (Z‐normalized) time series across all voxels in the gray matter region (see a representative example in Figure S1B, upper, green, corresponding to the signal in Figure S1A, green). 25 We used the Harvard‐Oxford cortical and subcortical structural atlases (https://neurovault.org/collections/262/) to define the gray matter mask, which was then transformed into the native fMRI space of each participant referring to previous studies. 25 , 27 Thus, the subject‐specific gray matter mask was generated for each subject. The rsfMRI in individual original space went through the above preprocessing procedures (not including nuisance regression; to avoid the CSF signal regression attenuating the CSF inflow signal and to avoid the motion parameter regression weakening the gBOLD signal; refer to the detailed explanation and preprocessing steps in previous studies 25 , 26 , 40 ). The preprocessed rsfMRI signal was averaged across each individual's gray matter mask to derive the gBOLD signal for each participant.
To derive the CSF inflow signal, the preprocessed fMRI in the original individual space was averaged across the CSF voxels at the bottom slice of fMRI acquisition to maximize sensitivity to the CSF inflow effect following previous studies. 25 , 27 Because fresh spins in the bottom slice that did not experience any radiofrequency pulse flow into the imaging volume, their signals are higher than those of local spins that have reached the steady state. All participants have individual CSF masks (Figure S1B, lower, corresponding to the signal in Figure S1A, purple) with similar voxel numbers (∼14).
2.4. Coupling between gBOLD and CSF inflow signals
We also calculated the cross‐correlation functions between the gBOLD signal and the CSF inflow signal (by assessing Pearson's correlation) at the negative peak of the mean cross‐correlation, a lag of +4.856 s (arrow in Figure S1C), to evaluate the gBOLD–CSF coupling for each participant, as was done previously. 25
2.5. Correlating gBOLD–CSF coupling with cortical Aβ, tau, thickness, and MoCA
We first compared gBOLD–CSF coupling (adjusted for age and sex) between Aβ− and Aβ+ groups (two‐sample t test). For each of these subgroups (ie, the Aβ−, Aβ+, unimpaired Aβ+, and impaired Aβ+) and the entire cohort, we correlated the gBOLD–CSF coupling (age‐ and sex‐adjusted) with cortical tau SUVR in each DKT‐68 parcel and then mapped this to the brain surface (using WorkBench software [version 1.5.0; https://www.humanconnectome.org/software/workbench‐command]) to see the spatial distribution of correlation coefficients. We further tested the coupling‐tau correlation in four ROIs, including Braak V–VI ROI, Braak III–IV ROI, 8 , 9 temporal meta‐ROI, 56 and entorhinal cortex across participants within each subgroup. We quantified the regional tau burden by (volume‐weighted) averaging FTP SUVR (PVC‐ed; normalized to the inferior cerebellar gray matter region) in these DKT‐68 parcels belonging to Braak V–VI and III–IV ROIs, as well as the temporal meta‐ROI (referring to: https://adni.bitbucket.io/reference/docs/UCBERKELEYAV1451/UCBERKELEY_AV1451_Methods_2021‐01‐14.pdf).
Similarly, we calculated the regional thickness by averaging the thickness in these DKT‐68 parcels belonging to Braak V–VI and III–IV ROIs, as well as the temporal meta‐ROI.
2.6. Determining the mediating role of tau in the coupling–thickness association
After examining the aforementioned relationships, we investigated whether tau mediated the effect of gBOLD–CSF coupling on cortical thickness. We first averaged tau SUVR and cortical thickness across all participants and compared their distribution patterns by correlating them across DKT‐68 parcels. Similarly, the tau difference between Aβ+ and Aβ− groups (reflecting the tau spreading with AD progression) was spatially compared with that difference in thickness. We further evaluated the inter‐participant similarity between tau and thickness in each of the above four ROIs across participants within each of these different subgroups, including the Aβ−, Aβ+, unimpaired Aβ+, and impaired Aβ+ ones. We then examined the hypothesis that tau mediated the association between gBOLD–CSF coupling and cortical thickness using a mediation analysis 57 in the Braak V–VI ROI, Braak III–IV ROI, or temporal meta‐ROI among the groups of the whole cohort, Aβ+, and impaired Aβ+, among which the coupling–tau and coupling–thickness associations were significant in Figures 1 and 2. The possibility that coupling mediated the relationship between tau and thickness was also tested in these ROIs.
FIGURE 1.
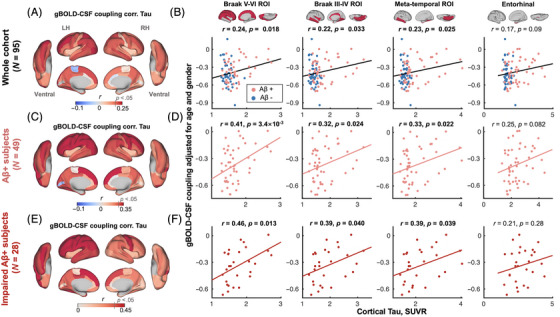
gBOLD–CSF coupling is correlated with cortical tau across whole cohort and Aβ+ participants. (A) gBOLD–CSF coupling was positively correlated with tau in most cortical regions across all participants, that is, participants with weaker (less negative) coupling had more tau deposition. (B) gBOLD–CSF coupling (strength) significantly decreased with more tau deposition in Braak V–VI, Braak III–IV, and temporal meta‐ROI across the whole cohort (all r > 0.22, all p < 0.033; N = 95). This association was marginally significant (r = 0.17, p = 0.09) for tau in entorhinal cortex. The FDR was used for the multiple comparison correction (p corrected values were 0.044, 0.044, 0.044, and 0.09 for the first column to the fourth column). (C–F) These associations between gBOLD–CSF coupling and regional tau were also evident among Aβ+ and/or specifically impaired Aβ+ participants (all r > 0.32, all p < 0.040; in Braak V–VI, Braak III–IV, and temporal meta‐ROI), but not for the Aβ− and unimpaired Aβ+ participants (Figure S2). In Figure 1D, the p corrected values were 0.014, 0.032, 0.032, and 0.082. In Figure 1F, the p corrected values were .052, .053, .053, and .28. Aβ+: cortical AV45–Aβ > 1.11 SUVR or cortical FBB–Aβ > 1.08 SUVR. Impaired group: AD and MCI participants. Each point represents one participant. The linear regression line was estimated based on the linear least‐squares fitting (the same hereinafter unless noted otherwise). Aβ, amyloid beta; AD, Alzheimer's disease; FBB, [18F]florbetaben; FDR, false discovery ratio; gBOLD–CSF, global brain activity and CSF movement; MCI, mild cognitive impairment; ROI, region of interest; SUVR, standardized uptake value ratio.
FIGURE 2.
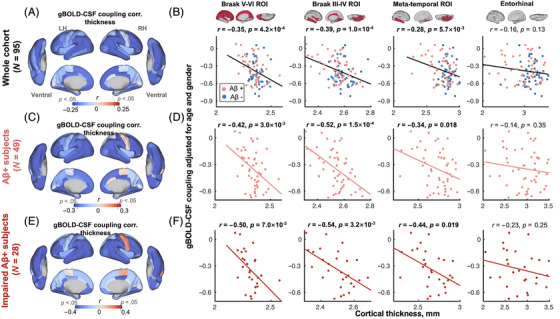
gBOLD–CSF coupling is correlated with cortical thickness across whole cohort and Aβ+ participants. (A) Participants with weaker (less negative) gBOLD–CSF coupling had thinner cortices in the majority of brain regions, especially in the frontal, parietal, and temporal lobes, including DMN and FPN. (B) The gBOLD–CSF coupling strength significantly decreased with thinner cortices in Braak V–VI, Braak III–IV, and temporal meta‐ROIs across the whole cohort (all r < −0.28, all p < 5.7×10−3; N = 95). Although not significant, the coupling–thickness association in the entorhinal region showed a trend similar to those in other ROIs noted above (r = −0.16, p = 0.13). The FDR was used for the multiple comparison correction (p corrected values were 8.0 × 10−4, 4.0 × 10−4, 7.6 × 10−3, and 0.13 for the first to fourth columns). (C–F) Among Aβ+ participants, particularly impaired Aβ+ ones, the coupling–thickness association remained striking (all r < −0.34, all p < 0.019 in Braak V–VI, Braak III–IV, and temporal meta‐ROI in D and F), while this was not the case in the Aβ− and unimpaired Aβ+ participants (Figure S5). In Figure 2D, the p corrected values were 6.0 × 10−3, 6.0 × 10−4, .024, and .35. In Figure 2F, the p corrected values were .014, .013, .025, and .25. Each point represents one participant. Aβ, amyloid beta; DMN, default mode network; FPN, frontoparietal network; gBOLD–CSF, global brain activity and CSF movement; ROI, region of interest.
2.7. Statistical analysis
A two‐sample t test was performed for group comparisons on continuous measures. We used Fisher's exact test 58 to compare categorical measures (ie, sex) between subgroups characterized for Aβ pathology progression. Pearson's correlation was used to quantify the linear correlation between different variables. Single‐level mediation analysis 57 was performed to test the hypothesis of tau mediating the coupling–thickness link. We also applied this mediation analysis to test the other possibility that coupling could mediate the tau–thickness relationship. An interaction effect test was also applied for the Aβ (or the diagnostic information) and coupling measure on the tau deposition. A p‐value of no more than 0.05 was considered statistically significant. We applied a multiple comparison correction with the false discovery rate (FDR) method for our major results.
To test the effect of head motion on our coupling metrics and tau, we evaluated the participant‐wise head motion with mean FD 59 and correlated this with tau in the four aforementioned ROIs and gBOLD–CSF coupling across participants from both the entire cohort and each subgroup. The coupling–tau association was retested after regressing out the participant‐wise head motion measure, the mean FD, from gBOLD–CSF coupling, and then correlating the coupling with tau in the Braak V–VI ROI, Braak III–IV ROI, and temporal meta‐ROI among the whole cohort, Aβ+, or impaired Aβ+ participants.
Similar to the foregoing analyses on the coupling–tau association, we also correlated the gBOLD–CSF coupling with cortical thickness in the same sets of ROIs or MoCA scores across the same groups of participants.
3. RESULTS
3.1. Cohort demographics
Participant characteristics are shown in Table 1. Aβ+ and Aβ− participants were different (p < 0.001) in age and MoCA but had a similar sex ratio. The Aβ− participants were also relatively younger than both the unimpaired and impaired Aβ+ individuals (both p < 3.7×10−3). The impaired Aβ+ participants had a higher proportion of males than Aβ− and unimpaired Aβ+ participants (p = 0.042).
TABLE 1.
Participant characteristics.
| N = 95 | Aβ− (N = 46) | Aβ+ (N = 49) | p‐value |
|---|---|---|---|
| Age | 69.4 (7.8) | 75.8 (6.9) | 6.5×10−5 |
| Sex (M/F) | 17/29 | 23/26 | 0.4 |
| Diagnosis (AD:MCI:SMC:Control) | 0:0:1:45 | 6:22:4:17 | – |
| MoCA | 25.1 (2.8) (2 N/A) | 22.3 (4.8) (1 N/A) | 1.0×10−3 |
Note: Data represent the mean (SD) unless otherwise indicated. A two‐sample t test was applied to compare the continuous measures, while a Fisher's exact test between groups was used for the sex ratio. Aβ+: cortical AV45–Aβ > 1.11 SUVR or cortical FBB–Aβ > 1.08 SUVR.
Abbreviations: Aβ, amyloid beta; AD, Alzheimer's disease group; M/F, male/female; MCI, mild cognitive impairment; MoCA, Montreal Cognitive Assessment; SMC, subjective memory concern; SUVR, standardized uptake value ratio, referring to whole cerebellum reference region.
3.2. fMRI‐based CSF clearance measure is related to tau across Aβ+ participants
CSF inflow rsfMRI signal was negatively correlated with gBOLD signal with a +4.856‐s time lag (Figure S1C; with a representative example in Figure S1, A and B), similar to the previous finding. 25 The gBOLD–CSF coupling appeared to be stronger in the Aβ− group than in the Aβ+ group, showing a trend similar to that in the previous study, 25 although not significant (Figure S1D). Importantly, the coupling was positively correlated with tau in most cortical regions across all participants, that is, participants with more cortical tau deposition had weaker (less negative) coupling (Figure 1A). This association was significant for regional tau deposition in the Braak V–VI ROI (isocortical regions), Braak III–IV ROI (limbic area; see Ref. 9 for detailed cortical regions), and temporal meta‐ROI 56 across the whole cohort (all r > 0.22, all p < 0.033; N = 95; p corrected were 0.044, 0.044, 0.044, and 0.09 for the first column to the fourth column; Figure 1B). A marginally significant correlation was found between the coupling measure and tau in the entorhinal cortex (r = 0.17, p = 0.09). The positive coupling–tau relationship was evident for all Aβ+ and also just for the impaired Aβ+ participants (Figure 1, C–F; p corrected for Figure 1D: 0.014, 0.032, 0.032, 0.082; p corrected for Figure 1F: 0.052, 0.053, 0.053, 0.28), but not for the unimpaired Aβ+ participants alone (Figure S2B). The Aβ and gBOLD–CSF coupling showed a significant interaction effect on tau in the Braak V–VI ROI (p = 4.3 × 10−3), Braak III–IV ROI (p = 0.020), and temporal meta‐ROI (p = 0.021), while the diagnostic group and coupling showed no significant interaction effect on tau in the ROIs (all p > 0.099), which was related to the limited sample size of the cognitively impaired group. Among the Aβ− participants, individuals with stronger coupling showed higher tau deposition in the Braak V–VI ROI, Braak III–IV ROI, and temporal meta‐ROI, but this was marginally significant (all p > 0.052; Figure S2A). Of note, head motion during fMRI was not associated with either tau or gBOLD–CSF coupling (Figure S3), and tau remained strongly correlated with the gBOLD–CSF coupling after adjusting for head motion (Figure S4).
Among the entire cohort, participants with stronger gBOLD–CSF coupling (more negative) had higher MoCA scores (r = −0.23, p = 0.027; N = 92). This trend was also found across Aβ+ individuals and the impaired Aβ+ ones, although not significant (both r < −0.25, both p < 0.11).
3.3. Tau partially mediates the strong association between gBOLD–CSF coupling and cortical thickness
We next examined the coupling–thickness association, as tau is closely associated with cortical atrophy and ultimately leads to cognitive decline. 3 , 4 , 60 Similar to the coupling–tau links discussed earlier, gBOLD–CSF coupling was also significantly related to cortical thickness in widespread cortical regions, including the Braak V–VI ROI, Braak III–IV ROI, and temporal meta‐ROI, across the entire group of participants, and more specifically among the impaired Aβ+ participants (Figure 2; see detailed p uncorrected and p corrected in the figure caption), but not in the Aβ− and unimpaired Aβ+ groups alone (Figure S5).
Cortical tau and thickness showed a similar spatial distribution pattern averaged across the entire cohort and when contrasting Aβ+ and Aβ− participants (both p < 1.9×10−4; Figure 3A and B). Cortical tau and thickness were also closely associated across participants (all p < 0.05; Figure S6). Given the corresponding spatial pattern and the predictive role of tau in brain atrophy, 60 we used mediation analysis to examine whether tau in Braak V–VI ROI, Braak III–IV ROI, and the temporal meta‐ROI mediated the observed relationship between gBOLD–CSF coupling and cortical thickness in the same regions. We found that tau in the three ROIs played a significant role in mediating the coupling–thickness link across the whole cohort (p < 0.021; Figure 3C; see detailed p corrected in the figure caption). Among Aβ+ participants, the mediation effect was marginally significant (p < 0.063) for the Braak III–IV ROI and the temporal meta‐ROI and significant for the Braak V–VI (p = 0.039; Figure 3D). The mediation was less robust in impaired Aβ+ participants (Figure 3E; only significant in the meta‐temporal region, p = 0.035), which may be attributed to the limited sample size of this group (N = 28). Together, these results suggest that tau is a mediator of the association between CSF dynamics of clearance relevance, reflected by gBOLD–CSF coupling and cortical thickness.
FIGURE 3.
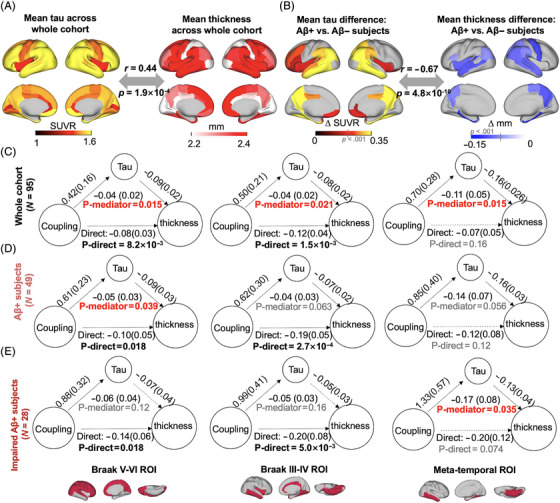
The association between coupling metrics and thickness is mediated by tau. (A and B) Averaged map of cortical tau and thickness across all participants (r = 0.44, p = 1.9×10−4), as well as the difference between Aβ− and Aβ+ participants (r = −0.67, p = 4.8×10−10), indicative of tau and atrophy distributions were similar. This close association between tau pathology and atrophy is also consistent with their significant correlation across participants (Figure S6). (C–E) Across entire cohort and Aβ+ participants, tau mediated the significant association in Figure 2 between gBOLD–CSF coupling and thickness throughout cortex, including Braak V–VI, Braak III–IV, and meta‐temporal area (p < 0.039, except for the marginally significant relationship [p < 0.063] in Braak III–IV and meta‐temporal among Aβ+ participants), whereas the mediation effect was weaker for impaired Aβ+ ones, which might be attributed to the limited sample size. In Figure 3C, the p corrected values were 0.021, 0.021, and 0.021. In Figure 3D, the p corrected values were .063, .063, and .063. In Figure 3E, the p corrected values were 0.16, 0.16, and 0.11. Significant direct and mediation effects were highlighted with black and red bold formatting, respectively. gBOLD–CSF, global brain activity and CSF movement.
We also examined whether coupling mediated the tau–thickness relationship (Figure S7) and found the mediation was significant for the Braak V–VI and Braak III–IV ROIs (p < 0.042), although it was marginally significant in the Braak III–IV among Aβ+ participants (p = 0.055).
4. DISCUSSION
We show an association between resting‐state global brain activity and tau pathology, including the pattern of tau deposition and relevant brain atrophy and cognitive decline. The coupling between global brain activity, quantified by gBOLD, and CSF movement was associated with tau deposition in widespread neocortical regions that approximate the later stages of tau deposition. This finding was evident in the whole cohort but more striking for the Aβ+ and impaired Aβ+ participants, as would be expected since the tau burden is likely to be greater for these groups. The Aβ and gBOLD–CSF coupling showed a significant interaction effect on tau. Weaker gBOLD–CSF coupling, which may indicate declining CSF clearance, was also associated with reduced cortical thickness in the same regions, likely related to the mediating role of tau. Together, these results suggest that resting‐state global brain activity helps to determine the stereotyped pattern of tau deposition in the neocortex among individuals with elevated Aβ, presumably via its effect on CSF dynamics‐related clearance.
Pathological tau aggregation has received increasing attention because of its strong relation to brain atrophy and cognitive impairment. 3 , 7 While a majority of recent studies have attributed the stereotyped pattern of tau accumulation over cortical regions to neural activity and anatomical and functional connectivity, 10 , 11 , 12 , 13 , 14 , 15 , 61 , 62 other data have repeatedly demonstrated that the brain's clearance system affects tau pathology 16 through CSF flow. 18 , 19 This pathway clears brain waste through CSF movement pushing the exchange between CSF and ISF and its solutes, including Aβ and tau. 18 , 19
Our study investigated the association between tau deposition in several Braak stages, indicative of different stages of tau pathology 8 , 9 and the coupling between global brain BOLD and CSF inflow signals, as a unique evaluation of CSF dynamics‐related clearance. Previous studies suggested that Aβ facilitated the spread and pathogenicity of tau. 63 Consistently, particularly among the participants with elevated Aβ, our study emphasizes that gBOLD–CSF coupling could also affect tau deposition and further affect cortical thickness, particularly in the neocortex, including Braak III–IV, V–VI, and meta‐ROI regions, but not in the Braak I region, that is, entorhinal cortex, where tau deposits in normal aging without Aβ elevation. 64 This would imply the different mechanisms of tau deposition in the entorhinal cortex and other Braak staging regions and that tau deposition in the latter appears to be more complicated and related to the comodulation of Aβ and CSF clearance assessed by the gBOLD–CSF coupling. In addition, our observations suggest that the coupling‐related CSF clearance of tau is more sensitive to AD stages with fast tau accumulation (ie, Aβ+ stage). In this stage, tau deposition in entorhinal cortex is elevated but could have slower accumulation that is more difficult to capture by the coupling measure. Furthermore, the spatial location of atrophy is closely associated with tau deposition, especially when Aβ is elevated. 63 This may also explain why the coupling was correlated with thickness in the regions where tau was significantly associated with the coupling metrics.
Our results showed a close association between the global BOLD and CSF inflow fMRI signal at rest. Several lines of evidence support a link between resting‐state global brain activity and CSF dynamics‐related clearance. First, global brain activity, measured by gBOLD fMRI signal and whole‐brain electrophysiology signals, 30 , 34 is coupled with CSF movement, 25 , 26 , 27 , 31 , 32 a key determinant of CSF clearance. 16 , 17 , 18 This coupling is particularly striking during sleep, 31 when glymphatic CSF clearance can be 20‐fold stronger than wakefulness. 17 More recently, the coupling between gBOLD and CSF inflow rsfMRI signals is correlated with various AD risk factors, cortical Aβ deposition, 25 , 27 older age, 32 and cognitive decline in AD and Parkinson's disease (PD), 25 , 26 further supporting the relationship between global brain activity and CSF dynamics‐related clearance. Second, neuronal firing cascades that underlie global spontaneous brain events 33 and gBOLD signal are often accompanied by the modulation of sympathetic outflow, including cardiac and respiratory pulsations, 37 , 38 , 39 , 40 , 41 heart rate variability, 42 and pupil size. 33 , 36 The sympathetic activity could not only facilitate peri‐arterial CSF movements via arterial constriction 41 , 65 but also arouse slow (<0.1 Hz) modulations of cardiac and respiratory pulsations, considered as the major driving forces of glymphatic CSF movement. 43 , 44 Third, the intrinsic subcortical vasoactive pathways, 65 particularly the basal‐cortical projections 66 relevant to the cholinergic system and astrocytes, are involved with the modulation of global brain activity on vascular tone. 41 Importantly, while a small proportion of perivascular neurons have direct contact with the vessel wall, most abut astrocytic endfeet 65 constituting the astroglial aquaporin‐4 (AQP4) channels that facilitate CSF flow. 18 A recent study suggested that intrinsic large astrocytic Ca2+ spikes were coupled with the negative gBOLD peaks. 67 In short, global brain activity and specific neural and physiological factors play a critical role in supporting CSF flow and thus could affect AD progression by moderating the “accumulation‐removal balance” of toxic proteins, such as Aβ and tau.
In our study, we observed the strength of gBOLD–CSF coupling reflecting CSF dynamics‐related clearance decreased with tau deposition. Recent studies in animal models have repeatedly demonstrated the role of CSF clearance or glymphatic function in tau aggregation. 19 , 68 , 69 , 70 Using intracortical injection of human tau into mice, a previous study tracked the tau clearance pathway and found that tau could be removed by CSF flow, especially in traumatic brain injury, a risk factor for tau aggregation. 68 Further investigations on tau and CSF clearance suggested that tau was cleared from the brain by an AQP4‐dependent mechanism. 69 , 70 For example, a recent mouse study suggested that impaired CSF–ISF exchange and AQP4 polarization, especially using an AQP4 inhibitor, in the glymphatic system could exacerbate or even induce pathogenic accumulation of tau. 69 The same study further showed an inverse association between CSF clearance and tau deposition in the healthy mouse cortex. 69 In addition, the glymphatic CSF clearance was hypothesized to affect the cell‐to‐cell propagation of tau in brain, 71 since tau can be secreted and taken up by both neurons and glia, 72 and tau secretion to the extracellular space plays an important role in intracellular tau spreading. 68 , 73 Beyond these mouse studies, a recent human study identified the relationship between reduced whole‐brain CSF clearance activity, assessed with diffusion tensor image analysis along the perivascular space (DTI‐ALPS), and the deposition of tau using PET along with cognitive decline. 74 All these studies suggest the role of CSF movement clearing tau, which supports the idea that the CSF clearance‐related gBOLD–CSF coupling is related to tau deposition.
It is not surprising that we observed that tau mediated the association between CSF clearance and cortical thickness. Our results showing an association between coupling and atrophy were consistent with a recent study showing that the impaired CSF clearance in TAR DNA‐binding protein 43 transgenic mice, mimicking the pathology of amyotrophic lateral sclerosis, was accompanied by neocortical atrophy. 75 More importantly, middle‐aged AQP4 knock‐out mice showed elevated tau in both CSF and hippocampus, as well as severe brain atrophy with thinner cortices and hippocampus. 70 This brain atrophy was attributed to neuronal loss, including the reduction of dentate granule cells and pyramidal cell layer neurons in the piriform cortex, presumably modulated by tau aggregation induced by the AQP4 deficiency. 70 Of note, our results may also pose the possibility that the coupling mediates the tau–thickness association. This could result from the very strong association between tau and thickness and their significant correlations with the coupling metrics. However, it is hard to determine the causal relationship between variables, and a longitudinal study is needed.
There are a few limitations of the present study. First, we used a cross‐sectional analysis that did not have information about rates of tau accumulation. In addition, it is possible that elevated tau accumulation in the cortical brain influences CSF dynamics and clearance or even global brain activity, which further leads to a decreased gBOLD–CSF coupling. A longitudinal design should be used to test the hypothesis that the tau accumulation rate is linked to fMRI‐based gBOLD–CSF coupling or an alternative hypothesis that the tau deposition leads to a decrease of CSF inflow or global brain activity and the coupling measure over years. Second, direct evidence that gBOLD–CSF coupling reflects CSF dynamics‐related clearance is needed to validate and extend our findings that global brain activity affects CSF flow and may further modulate CSF clearance. We also note that some of the main results were marginally significant or not significant after multiple comparison corrections, which could come from imaging noise and the imperfect methodology to quantify the coupling strength between CSF inflow and gBOLD. Third, the lack of electroencephalogram (EEG) monitoring of sleep or arousal state was also a limitation of this study. Although it is challenging to acquire EEG‐fMRI data for both healthy aging and AD patients, at rest or during sleep, it would be interesting for future studies to investigate the role of sleep and arousal state in the association between tau pathology and the coupling between global brain activity and CSF inflow.
In summary, this study provides initial evidence that CSF dynamics, reflected by the coupling between global brain activity and the CSF movement signals, is closely associated with tau pathology in people with elevated Aβ, presumably because poorer CSF clearance promotes abnormal protein aggregation.
AUTHOR CONTRIBUTIONS
Feng Han and William J Jagust conceptualized and designed the study; Feng Han, JiaQie Lee, Jacob Ziontz, Tyler Ward, Susan M Landau, and William J Jagust curated the data and performed analyses; Feng Han, Xi Chen, Suzanne L Baker, and Theresa M Harrison and William J Jagust designed and performed assays; Feng Han. and William J Jagust wrote the original draft, and JiaQie Lee; Feng Han, Xi Chen, Jacob Ziontz, Tyler Ward, Susan M Landau, Suzanne L Baker, Theresa M Harrison and William J Jagust reviewed and edited the manuscript; William J Jagust supervised the project; and all authors read and approved the manuscript.
CONFLICT OF INTEREST STATEMENT
W.J.J. has served as a consultant for Biogen, Eisai, Clario, and Lilly. The remaining authors declare no competing financial interests. Author disclosures are available in the Supporting Information.
CONSENT STATEMENT
All human subjects provided written informed consent.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
This work is supported by funding from the following: NIH (U19AG024904 to W.J.J. and S.M.L., K01AG078443 to T.M.H., F31AG079595 to J.Z.) and BrightFocus Foundation (A2021004F to X.C.). Data collection and sharing for this project was funded by the ADNI (National Institutes of Health Grant U01 AG024904; Principal Investigator: Michael Weiner) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012; Principal Investigator: Michael Weiner). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering (NIBIB), and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
APPENDIX I. COLLABORATORS
ADNI investigators list
Infrastructure investigators
Michael Weiner, MD (University of California [UC], San Francisco, Leadership and Infrastructure Principal Investigator [PI]); Paul Aisen, MD (UC San Diego [UCSD], Alzheimer's Disease Cooperative Study [ADCS] PI and Director of Coordinating Center Clinical Core); Gerald Novak, PhD (Janssen Alzheimer Immunotherapy, ADNI 2 Private Partner Scientific Board [PPSB] Chair); Robert C. Green, MD, MPH (Boston University, Data and Publication Committee [DPC]); Tom Montine, MD, PhD (University of Oregon, Resource Allocation Review Committee); Ronald Petersen, MD, PhD (Mayo Clinic, Clinical Core Leader, Core PI); Paul Aisen, MD (UCSD, Clinical Core Leader); Anthony Gamst, PhD (UCSD, Clinical Informatics and Operations); Ronald G. Thomas, PhD (UCSD, Clinical Informatics and Operations); Michael Donohue, PhD (UCSD, Clinical Informatics and Operations); Sarah Walter, MSc (UCSD, Clinical Informatics and Operations); Devon Gessert, BS (UCSD, Clinical Informatics and Operations); Tamie Sather, BS (UCSD, Clinical Informatics and Operations); Laurel Beckett, PhD (UC Davis, Biostatistics Core Leader); Danielle Harvey, PhD (UC Davis, Biostatistics Co‐investigator); Anthony Gamst, PhD (UCSD, Biostatistics Co‐investigator); Michael Donohue, PhD (UCSD, Biostatistics Co‐investigator); John Kornak, PhD (UC Davis, Biostatistics Co‐investigator); Clifford R. Jack, Jr., MD (Mayo, MRI Core Leader); Anders Dale, PhD (UCSD, MRI Co‐investigator); Matthew Bernstein, PhD (Mayo, MRI Co‐investigator); Joel Felmlee, PhD (Mayo, MRI Co‐investigator); Nick Fox, MD (Univ College London, MRI Co‐investigator); Paul Thompson, PhD (UCLA, MRI Co‐investigator); Norbert Schuff, PhD (UCSF, MRI Co‐investigator); Gene Alexander, PhD (Banner Alzheimer's Institute, MRI Co‐investigator); Charles DeCarli, MD (UC Davis, MRI Co‐investigator); William Jagust, MD (UC Berkeley, PET Core Leader); Dan Bandy, MS, CNMT (Banner Alzheimer's Institute, PET Co‐investigator); Robert A. Koeppe, PhD (Univ Michigan, PET Co‐investigator); Norm Foster, MD (Univ Utah, PET Co‐investigator); Eric M. Reiman, MD (Banner Alzheimer's Institute, PET Co‐investigator); Kewei Chen, PhD (Banner Alzheimer's Institute, PET Co‐investigator); Chet Mathis, MD (Univ Pittsburgh, PET Co‐investigator); John Morris, MD (Washington University, Neuropathology Core Leader); Nigel J. Cairns, PhD, MRCPath (Washington University, Neuropathology Co‐investigator); Lisa Taylor‐Reinwald, BA, HTL (ASCP); (Washington University, Neuropathology Coinvestigator); J.Q. Trojanowki, MD, PhD (Univ Pennsylvania, Biomarkers Core Leader); Les Shaw, PhD (Univ Pennsylvania, Biomarkers Co‐investigator); Virginia M.Y. Lee, PhD, MBA (Univ Pennsylvania, Biomarkers Co‐investigator); Magdalena Korecka, PhD (Univ Pennsylvania, Biomarkers Co‐investigator); Arthur W. Toga, PhD (UCLA, Informatics Core Leader); Karen Crawford (UCLA, Informatics Co‐investigator); Scott Neu, PhD (UCLA, Informatics Co‐investigator); Andrew J. Saykin, PsyD (Indiana University, Genetics Core Leader); Tatiana M. Foroud, PhD (Indiana University, Genetics Co‐Investigator); Steven Potkin, MD UC (UC Irvine, Genetics Co‐Investigator); Li Shen, PhD (Indiana University, Genetics Co‐Investigator); Zaven Kachaturian, PhD (Khachaturian, Radebaugh & Associates (KRA), Inc Alzheimer's Association's Ronald and Nancy Reagan's Research Institute, Early project development Co‐investigator); Richard Frank, MD, PhD (General Electric, Early project development Co‐investigator); Peter J. Snyder, PhD (University of Connecticut, Early project development Co‐investigator); Susan Molchan, PhD (National Institute on Aging/National Institutes of Health, Co‐investigator).
Site investigators
Oregon Health and Science University: Jeffrey Kaye, MD (Oregon Health Science University, Co‐investigator); Sara Dolen, BS (Oregon Health Science University, Co‐investigator); Joseph Quinn, MD (Oregon Health Science University, Co‐investigator); University of Southern California: Lon S. Schneider, MD (University of Southern California, Co‐investigator); Sonia Pawluczyk, MD (University of Southern California, Co‐investigator); Bryan M. Spann, DO, PhD (University of Southern California, Co‐investigator); University of California–San Diego: James Brewer, MD, PhD (University of California–San Diego, Co‐investigator); Helen Vanderswag, RN (University of California–San Diego, Co‐investigator); University of Michigan: Judith L. Heidebrink, MD, MS (University of Michigan, Co‐investigator); Joanne L. Lord, LPN, BA, CCRC (University of Michigan, Co‐investigator); Mayo Clinic, Rochester: Ronald Petersen, MD, PhD (Mayo Clinic, Rochester, Co‐investigator); Kris Johnson, RN (Mayo Clinic, Rochester, Co‐investigator); Baylor College of Medicine: Rachelle S. Doody, MD, PhD (Baylor College of Medicine, Co‐investigator); Javier Villanueva‐Meyer, MD (Baylor College of Medicine, Co‐investigator); Munir Chowdhury, MS (Baylor College of Medicine, Co‐investigator); Columbia University Medical Center: Yaakov Stern, PhD (Columbia University Medical Center, Co‐investigator); Lawrence S. Honig, MD, PhD (Columbia University Medical Center, Co‐investigator); Karen L. Bell, MD (Columbia University Medical Center, Co‐investigator); Washington University, St. Louis: John C. Morris, MD, PhD (Washington University, Co‐investigator); Mark A. Mintun, MD (Washington University, Co‐investigator); Stacy Schneider APRN, BC, GNP (Washington University, Co‐investigator); University of Alabama at Birmingham: Daniel Marson, JD, PhD (University of Alabama at Birmingham, Co‐investigator); Randall Griffith, PhD, ABPP (University of Alabama at Birmingham, Co‐investigator); David Clark, MD (University of Alabama at Birmingham, Co‐investigator); Mount Sinai School of Medicine: Hillel Grossman MD (Mount Sinai School of Medicine, Co‐investigator); Effie Mitsis, PhD (Mount Sinai School of Medicine, Co‐investigator); Aliza Romirowsky, BA (Mount Sinai School of Medicine, Co‐investigator); Rush University Medical Center: Leyla deToledo‐Morrell, PhD (Rush University Medical Center, Co‐investigator); Raj C. Shah, MD (Rush University Medical Center, Co‐investigator); Wein Center: Ranjan Duara, MD (Wein Center, Co‐investigator); Daniel Varon, MD (Wein Center, Co‐investigator); Peggy Roberts CNA (Wein Center, Co‐investigator); Johns Hopkins University: Marilyn Albert, PhD (Johns Hopkins University); Chiadi Onyike, MD (Johns Hopkins University); Stephanie Kielb, RN (Johns Hopkins University); New York University: Henry Rusinek, PhD (New York University, Co‐investigator); Mony J de Leon, EdD (New York University, Co‐investigator); Lidia Glodzik, MD PhD (New York University, Co‐investigator); Duke University Medical Center: P. Murali Doraiswamy, MD (Duke University Medical Center, Co‐investigator); Jeffrey R. Petrella, MD (Duke University Medical Center, Co‐investigator); University of Pennsylvania: Steven E. Arnold, M.D. (University of Pennsylvania, Co‐investigator); Jason H. Karlawish, MD (University of Pennsylvania, Co‐investigator); David Wolk, MD (University of Pennsylvania, Co‐investigator); University of Kentucky: Charles D. Smith, MD (University of Kentucky, Co‐investigator); Greg Jicha, MD (University of Kentucky, Co‐investigator); Peter Hardy, PhD (University of Kentucky, Co‐investigator); University of Pittsburgh: Oscar L. Lopez, MD (University of Pittsburgh, Co‐investigator); MaryAnn Oakley, MA (University of Pittsburgh, Co‐investigator); Donna M. Simpson, CRNP, MPH (University of Pittsburgh, Co‐investigator); University of Rochester Medical Center: M. Saleem Ismail MD (University of Rochester Medical Center, Co‐investigator); Connie Brand RN (University of Rochester Medical Center, Co‐investigator); University of California, Irvine: Ruth A. Mulnard, DNSc, RN, FAAN (University of California, Irvine, Co‐investigator); Gaby Thai, MD (University of California, Irvine, Co‐investigator); Catherine Mc‐Adams‐Ortiz, MSN, RN, A/GNP (University of California, Irvine, Co‐investigator); University of Texas Southwestern Medical School: Ramon Diaz‐Arrastia, MD, PhD (University of Texas Southwestern Medical School, Co‐investigator); Kristen Martin‐Cook, MA (University of Texas Southwestern Medical School, Co‐investigator); Michael DeVous, PhD (University of Texas Southwestern Medical School, Co‐investigator); Emory University: Allan I. Levey, MD, PhD (Emory University, Co‐investigator); James J. Lah, MD, PhD (Emory University, Co‐investigator); Janet S. Cellar, RN, MSN (Emory University, Co‐investigator); University of Kansas, Medical Center: Jeffrey M. Burns, MD (University of Kansas, Medical Center, Co‐investigator); Heather S. Anderson, MD (University of Kansas, Medical Center, Co‐investigator); Russell H. Swerdlow, MD (University of Kansas, Medical Center, Co‐investigator); University of California, Los Angeles: George Bartzokis, MD (University of California, Los Angeles, Co‐investigator); Daniel H.S. Silverman, MD, PhD (University of California, Los Angeles, Co‐investigator); Po H. Lu, PsyD (University of California, Los Angeles, Co‐investigator); Liana Apostolova, MD (University of California, Los Angeles, Co‐investigator): Mayo Clinic, Jacksonville: Neill R. Graff‐Radford MBBCH, FRCP (Mayo Clinic, Jacksonville, Co‐investigator); Francine Parfitt, MSH, CCRC (Mayo Clinic, Jacksonville, Co‐investigator); Heather Johnson, MLS, CCRP (Mayo Clinic, Jacksonville, Co‐investigator); Indiana University: Martin Farlow, MD (Indiana University, Co‐investigator); Scott Herring, RN (Indiana University, Co‐investigator); Ann M. Hake, MD (Indiana University, Co‐investigator); Yale University School of Medicine: Christopher H. van Dyck, MD (Yale University School of Medicine, Co‐investigator); Richard E. Carson, PhD (Yale University School of Medicine, Co‐investigator); Martha G. MacAvoy, PhD (Yale University School of Medicine, Co‐investigator); McGill Univ., Montreal‐Jewish General Hospital: Howard Chertkow, MD (McGill Univ., Montreal‐Jewish General Hospital, Co‐investigator); Howard Bergman, MD (McGill Univ., Montreal‐Jewish General Hospital, Co‐investigator); Chris Hosein, M.Ed (McGill Univ., Montreal‐Jewish General Hospital, Co‐investigator); Sunnybrook Health Sciences, Ontario: Sandra Black, MD FRCP(C) (Sunnybrook Health Sciences, Ontario, Co‐investigator); Dr Bojana Stefanovic (Sunnybrook Health Sciences, Ontario, Co‐investigator); Curtis Caldwell, PhD (Sunnybrook Health Sciences, Ontario, Co‐investigator); U.B.C. Clinic for AD & Related Disorders Ging‐Yuek Robin Hsiung, MD, MHSc, FRCPC (U.B.C. Clinic for AD & Related Disorders, Co‐investigator); Howard Feldman, MD FRCP(C) (U.B.C. Clinic for AD & Related Disorders, Co‐investigator); Michele Assaly, MA (U.B.C. Clinic for AD & Related Disorders, Co‐investigator); Cognitive Neurology—St. Joseph's, Ontario: Andrew Kertesz, MD (Cognitive Neurology—St. Joseph's, Ontario, Co‐investigator); John Rogers, MD (Cognitive Neurology—St. Joseph's, Ontario, Co‐investigator); Dick Trost, PhD (Cognitive Neurology—St. Joseph's, Ontario, Co‐investigator); Cleveland Clinic Lou Ruvo Center for Brain Health Charles Bernick, MD (Cleveland Clinic Lou Ruvo Center for Brain Health, Co‐investigator); Donna Munic, PhD (Cleveland Clinic Lou Ruvo Center for Brain Health, Co‐investigator); Northwestern University: Chuang‐Kuo Wu MD PhD (Northwestern University, Co‐investigator); Nancy Johnson, PhD (Northwestern University, Co‐investigator); Marsel Mesulam, MD (Northwestern University, Co‐investigator); Premiere Research Institute (Palm Beach Neurology): Carl Sadowsky MD (Premiere Research Institute [Palm Beach Neurology], Co‐investigator); Walter Martinez MD (Premiere Research Institute [Palm Beach Neurology], Co‐investigator); Teresa Villena MD (Premiere Research Institute [Palm Beach Neurology], Co‐investigator); Georgetown University Medical Center: Raymond Scott Turner, MD, PhD (Georgetown University Medical Center, Co‐investigator); Kathleen Johnson, NP (Georgetown University Medical Center, Co‐investigator); Brigid Reynolds, NP (Georgetown University Medical Center, Co‐investigator); Brigham and Women's Hospital: Reisa A. Sperling, MD (Brigham and Women's Hospital, Co‐investigator); Meghan Frey (Brigham and Women's Hospital, Co‐investigator); Keith A. Johnson, MD (Brigham and Women's Hospital, Co‐investigator); Stanford University: Allyson Rosen, PhD (Stanford University, Co‐investigator); Jared Tinklenberg, MD (Stanford University, Co‐investigator); Wes Ashford, MD, PhD (Stanford University, Co‐investigator); Banner Sun Health Research Institute Marwan Sabbagh, MD, FAAN, CCRI (Banner Sun Health Research Institute, Co‐investigator); Christine Belden PsyD. (Banner Sun Health Research Institute, Co‐investigator); Sandra Jacobson MD (Banner Sun Health Research Institute, Co‐investigator); Boston University: Ronald Killiany, PhD (Boston University, Co‐investigator); Alexander Norbash, MD (Boston University, Co‐investigator); Anil Nair, MD (Boston University, Co‐investigator); Howard University: Thomas O. Obisesan, MD, MPH (Howard University, Co‐investigator); Saba Wolday, M.Sc. (Howard University, Co‐investigator); Salome K. Bwayo, Pharm.D. (Howard University, Co‐investigator); Case Western Reserve University: Alan Lerner, MD (Case Western Reserve University, Co‐investigator); Leon Hudson, MPH (Case Western Reserve University, Co‐investigator); Paula Ogrocki, PhD (Case Western Reserve University, Co‐investigator); University of California, Davis—Sacramento: Charles DeCarli, MD (University of California, Davis—Sacramento, Co‐investigator); Evan Fletcher, PhD (University of California, Davis—Sacramento, Co‐investigator); Owen Carmichael, PhD (University of California, Davis—Sacramento, Co‐investigator); Neurological Care of CNY: Smita Kittur, MD (Neurological Care of CNY, Co‐investigator); Parkwood Hospital: Michael Borrie, MD (Parkwood Hospital, Co‐investigator); T‐Y Lee, PhD (Parkwood Hospital, Co‐investigator); Dr Rob Bartha, PhD (Parkwood Hospital, Co‐investigator); University of Wisconsin: Sterling Johnson, PhD (University of Wisconsin, Co‐investigator); Sanjay Asthana, MD (University of Wisconsin, Co‐investigator); Cynthia M. Carlsson, MD (University of Wisconsin, Co‐investigator); University of California, Irvine—BIC: Steven G. Potkin, MD (University of California, Irvine—BIC, Co‐investigator); Adrian Preda, MD (University of California, Irvine—BIC, Co‐investigator); Dana Nguyen, PhD (University of California, Irvine—BIC, Co‐investigator); Banner Alzheimer's Institute: Pierre Tariot, MD (Banner Alzheimer's Institute, Co‐investigator); Adam Fleisher, MD (Banner Alzheimer's Institute, Co‐investigator); Stephanie Reeder, BA (Banner Alzheimer's Institute, Co‐investigator); Dent Neurologic Institute Vernice Bates, MD (Dent Neurologic Institute, Co‐investigator); Horacio Capote, MD (Dent Neurologic Institute, Co‐investigator); Michelle Rainka, PhD (Dent Neurologic Institute, Co‐investigator); Barry A. Hendin, MD (Dent Neurologic Institute, Co‐investigator); The Ohio State University: Douglas W. Scharre, MD (The Ohio State University, Co‐investigator); Maria Kataki, MD, PhD (The Ohio State University, Co‐investigator); Albany Medical College: Earl A. Zimmerman, MD (Albany Medical College, Co‐investigator); Dzintra Celmins, MD (Albany Medical College, Co‐investigator); Alice D. Brown, FNP (Albany Medical College, Co‐investigator); Hartford Hospital, Olin Neuropsychiatry Research Center: Godfrey D. Pearlson MD (Hartford Hospital, Olin Neuropsychiatry Research Center, Coinvestigator); Karen Blank, MD (Hartford Hospital, Olin Neuropsychiatry Research Center, Co‐investigator); Karen Anderson, RN (Hartford Hospital, Olin Neuropsychiatry Research Center, Co‐investigator); Dartmouth‐Hitchcock Medical Center: Andrew J. Saykin, PsyD (Dartmouth‐Hitchcock Medical Center, Co‐investigator); Robert B. Santulli, MD (Dartmouth‐Hitchcock Medical Center, Co‐investigator); Eben S. Schwartz, PhD (Dartmouth‐Hitchcock Medical Center, Co‐investigator); Wake Forest University Health Sciences: Jeff D. Williamson, MD, MHS (Wake Forest University Health Sciences, Co‐investigator); Kaycee M. Sink, MD, MS (Wake Forest University Health Sciences, Co‐investigator); Franklin Watkins, MD (Wake Forest University Health Sciences, Co‐investigator); Rhode Island Hospital: Brian R. Ott, MD (Rhode Island Hospital, Co‐investigator); Henry Querfurth, MD (Rhode Island Hospital, Co‐investigator); Geoffrey Tremont, PhD (Rhode Island Hospital, Co‐investigator); Butler Hospital Stephen Salloway, MD, MS (Butler Hospital, Co‐investigator); Paul Malloy, PhD (Butler Hospital, Co‐investigator); Stephen Correia, PhD (Butler Hospital, Co‐investigator); UC San Francisco Howard J. Rosen, MD (UC San Francisco, Co‐investigator); Bruce L. Miller, MD (UC San Francisco, Co‐investigator); Medical University South Carolina Jacobo Mintzer, MD, MBA (Medical University South Carolina, Co‐investigator); Crystal Flynn Longmire, PhD (Medical University South Carolina, Co‐investigator); Kenneth Spicer, MD, PhD (Medical University South Carolina, Co‐investigator).
Han F, Lee JQ, Chen X, et al. Global brain activity and its coupling with cerebrospinal fluid flow is related to tau pathology. Alzheimer's Dement. 2024;20:8541–8555. 10.1002/alz.14296
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in the analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp‐content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
DATA AVAILABILITY STATEMENT
The multimodal data, including subject characteristics, rsfMRI, structural MRI, Aβ‐PET, tau‐PET, and MoCA measures are all publicly available at the ADNI website upon the approval of the data use application (http://adni.loni.usc.edu/). The ADNI was launched in 2003 as a public‐private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early AD. For up‐to‐date information, see www.adni‐info.org. All the codes used in the present study are available from the corresponding author upon request.
REFERENCES
- 1. Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. doi: 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bloom GS. Amyloid‐β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014;71:505‐508. doi: 10.1001/jamaneurol.2013.5847 [DOI] [PubMed] [Google Scholar]
- 3. Bejanin A, Schonhaut DR, La Joie R, et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer's disease. Brain. 2017;140:3286‐3300. doi: 10.1093/brain/awx243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sperling RA, Mormino EC, Schultz AP, et al. The impact of amyloid‐beta and tau on prospective cognitive decline in older individuals. Ann Neurol. 2019;85:181‐193. doi: 10.1002/ana.25395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harrison TM, Du R, Klencklen G, Baker SL, Jagust WJ. Distinct effects of beta‐amyloid and tau on cortical thickness in cognitively healthy older adults. Alzheimers Dement. 2021;17:1085‐1096. doi: 10.1002/alz.12249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ossenkoppele R, Smith R, Ohlsson T, et al. Associations between tau, Aβ, and cortical thickness with cognition in Alzheimer disease. Neurology. 2019;92:e601‐e612. doi: 10.1212/WNL.0000000000006875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. DeVos SL, Miller RL, Schoch KM, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9:eaag0481. doi: 10.1126/scitranslmed.aag0481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389‐404. doi: 10.1007/s00401-006-0127-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schöll M, Lockhart SN, Schonhaut DR, et al. PET imaging of tau deposition in the aging human brain. Neuron. 2016;89:971‐982. doi: 10.1016/j.neuron.2016.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pooler AM, Phillips EC, Lau DHW, Noble W, Hanger DP. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013;14:389‐394. doi: 10.1038/embor.2013.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu JW, Hussaini SA, Bastille IM, et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci. 2016;19:1085‐1092. doi: 10.1038/nn.4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mudher A, Colin M, Dujardin S, et al. What is the evidence that tau pathology spreads through prion‐like propagation?. Acta Neuropathol Commun. 2017;5:99. doi: 10.1186/s40478-017-0488-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ossenkoppele R, Iaccarino L, Schonhaut DR, et al. Tau covariance patterns in Alzheimer's disease patients match intrinsic connectivity networks in the healthy brain. NeuroImage Clin. 2019;23:101848. doi: 10.1016/j.nicl.2019.101848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Franzmeier N, Rubinski A, Neitzel J, et al. Functional connectivity associated with tau levels in ageing, Alzheimer's, and small vessel disease. Brain. 2019;142:1093‐1107. doi: 10.1093/brain/awz026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cope TE, Rittman T, Borchert RJ, et al. Tau burden and the functional connectome in Alzheimer's disease and progressive supranuclear palsy. Brain. 2018;141:550‐567. doi: 10.1093/brain/awx347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tarasoff‐Conway JM, Carare RO, Osorio RS, et al. Clearance systems in the brain—implications for Alzheimer disease. Nat Rev Neurol. 2015;11:457‐470. doi: 10.1038/nrneurol.2015.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373‐377. doi: 10.1126/science.1241224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Iliff JJ, Wang M, Liao Y, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. 2012;4:147ra111. doi: 10.1126/scitranslmed.3003748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Holth JK, Fritschi SK, Wang C, et al. The sleep‐wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science. 2019;363:880‐884. doi: 10.1126/science.aav2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smets NG, Strijkers GJ, Vinje V, Bakker ENTP. Cerebrospinal fluid turnover as a driver of brain clearance. NMR Biomed. 2024;37:e5029. doi: 10.1002/nbm.5029 [DOI] [PubMed] [Google Scholar]
- 21. Iliff JJ, Lee H, Yu M, et al. Brain‐wide pathway for waste clearance captured by contrast‐enhanced MRI. J Clin Invest. 2013;123:1299‐1309. doi: 10.1172/JCI67677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ota M, Sato N, Nakaya M, et al. Relationships between the deposition of amyloid‐β and tau protein and glymphatic system activity in Alzheimer's disease: diffusion tensor image study. J Alzheimers Dis. 2022;90:295‐303. doi: 10.3233/JAD-220534 [DOI] [PubMed] [Google Scholar]
- 23. Taoka T, Masutani Y, Kawai H, et al. Evaluation of glymphatic system activity with the diffusion MR technique: diffusion tensor image analysis along the perivascular space (DTI‐ALPS) in Alzheimer's disease cases. Jpn J Radiol. 2017;35:172‐178. doi: 10.1007/s11604-017-0617-z [DOI] [PubMed] [Google Scholar]
- 24. Sepehrband F, Barisano G, Sheikh‐Bahaei N, et al. Volumetric distribution of perivascular space in relation to mild cognitive impairment. Neurobiol Aging. 2021;99:28‐43. doi: 10.1016/j.neurobiolaging.2020.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Han F, Chen J, Belkin‐Rosen A, et al. Reduced coupling between cerebrospinal fluid flow and global brain activity is linked to Alzheimer disease‐related pathology. PLoS Biol. 2021;19:e3001233. doi: 10.1371/journal.pbio.3001233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Han F, Brown GL, Zhu Y, et al. Decoupling of global brain activity and cerebrospinal fluid flow in Parkinson's disease cognitive decline. Mov Disord. 2021;36:2066‐2076. doi: 10.1002/mds.28643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Han F, Liu X, Mailman RB, Huang X, Liu X. Resting‐state global brain activity affects early β‐amyloid accumulation in default mode network. Nat Commun. 2023;14:7788. doi: 10.1038/s41467-023-43627-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schölvinck ML, Maier A, Ye FQ, Duyn JH, Leopold DA. Neural basis of global resting‐state fMRI activity. Proc Natl Acad Sci U S A. 2010;107:10238‐10243. doi: 10.1073/pnas.0913110107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fukunaga M, Horovitz SG, van Gelderen P, et al. Large‐amplitude, spatially correlated fluctuations in BOLD fMRI signals during extended rest and early sleep stages. Magn Reson Imaging. 2006;24:979‐992. doi: 10.1016/j.mri.2006.04.018 [DOI] [PubMed] [Google Scholar]
- 30. Liu TT, Nalci A, Falahpour M. The global signal in fMRI: nuisance or information?. Neuroimage. 2017;150:213‐229. doi: 10.1016/j.neuroimage.2017.02.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fultz NE, Bonmassar G, Setsompop K, et al. Coupled electrophysiological, hemodynamic, and cerebrospinal fluid oscillations in human sleep. Science. 2019;366:628‐631. doi: 10.1126/science.aax5440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Han F, Liu X, Yang Y, Liu X. Sex‐specific age‐related changes in glymphatic function assessed by resting‐state functional magnetic resonance imaging. BioRxiv. 2023. doi: 10.1101/2023.04.02.535258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu X, Leopold DA, Yang Y. Single‐neuron firing cascades underlie global spontaneous brain events. Proc Natl Acad Sci U S A. 2021;118:e2105395118. doi: 10.1073/pnas.2105395118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu X, De Zwart JA, Schölvinck ML, et al. Subcortical evidence for a contribution of arousal to fMRI studies of brain activity. Nat Commun. 2018;9:395. doi: 10.1038/s41467-017-02815-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu X. Decoupling between brain activity and cerebrospinal fluid movement in neurological disorders. J Magn Reson Imaging. 2024;60(5):1743‐1752. doi: 10.1002/jmri.29148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pais‐Roldán P, Takahashi K, Sobczak F, et al. Indexing brain state‐dependent pupil dynamics with simultaneous fMRI and optical fiber calcium recording. Proc Natl Acad Sci U S A. 2020;117:6875‐6882. doi: 10.1073/pnas.1909937117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Özbay PS, Chang C, Picchioni D, et al. Sympathetic activity contributes to the fMRI signal. Commun Biol. 2019;2:421. doi: 10.1038/s42003-019-0659-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chang C, Cunningham JP, Glover GH. Influence of heart rate on the BOLD signal: the cardiac response function. Neuroimage. 2009;44:857‐869. doi: 10.1016/j.neuroimage.2008.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Birn RM, Diamond JB, Smith MA, Bandettini PA. Separating respiratory‐variation‐related fluctuations from neuronal‐activity‐related fluctuations in fMRI. Neuroimage. 2006;31:1536‐1548. doi: 10.1016/j.neuroimage.2006.02.048 [DOI] [PubMed] [Google Scholar]
- 40. Gu Y, Han F, Sainburg LE, Liu X. Transient arousal modulations contribute to resting‐state functional connectivity changes associated with head motion parameters. Cereb Cortex. 2020;30:5242‐5256. doi: 10.1093/cercor/bhaa096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Özbay PS, Chang C, Picchioni D, et al. Contribution of systemic vascular effects to fMRI activity in white matter. Neuroimage. 2018;176:541‐549. doi: 10.1016/j.neuroimage.2018.04.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chang C, Metzger CD, Glover GH, Duyn JH, Heinze HJ, Walter M. Association between heart rate variability and fluctuations in resting‐state functional connectivity. Neuroimage. 2013;68:93‐104. doi: 10.1016/j.neuroimage.2012.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Iliff JJ, Wang M, Zeppenfeld DM, et al. Cerebral arterial pulsation drives paravascular CSF‐interstitial fluid exchange in the murine brain. J Neurosci. 2013;33:18190‐18199. doi: 10.1523/JNEUROSCI.1592-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yamada S, Miyazaki M, Yamashita Y, et al. Influence of respiration on cerebrospinal fluid movement using magnetic resonance spin labeling. Fluids Barriers CNS. 2013;10:36. doi: 10.1186/2045-8118-10-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Harrison TM, Maass A, Adams JN, Du R, Baker SL, Jagust WJ. Tau deposition is associated with functional isolation of the hippocampus in aging. Nat Commun. 2019;10:4900. doi: 10.1038/s41467-019-12921-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Veitch DP, Weiner MW, Aisen PS, et al. Using the Alzheimer's Disease Neuroimaging Initiative to improve early detection, diagnosis, and treatment of Alzheimer's disease. Alzheimers Dement. 2022;18:824‐857. doi: 10.1002/alz.12422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Baker SL, Maass A, Jagust WJ. Considerations and code for partial volume correcting [18F]‐AV‐1451 tau PET data. Data Brief. 2017;15:648‐657. doi: 10.1016/j.dib.2017.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Royse SK, Minhas DS, Lopresti BJ, et al. Validation of amyloid PET positivity thresholds in centiloids: a multisite PET study approach. Alzheimers Res Ther. 2021;13:99. doi: 10.1186/s13195-021-00836-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jack CR Jr, Bernstein MA, Fox NC, et al. The Alzheimer's Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging. 2008;27:685‐691. doi: 10.1002/jmri.21049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Giorgio J, Jagust WJ, Baker S, et al. A robust and interpretable machine learning approach using multimodal biological data to predict future pathological tau accumulation. Nat Commun. 2022;13:1887. doi: 10.1038/s41467-022-28795-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fischl B. FreeSurfer. Neuroimage. 2012;62:774‐781. doi: 10.1016/j.neuroimage.2012.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Desikan RS, Ségonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968‐980. doi: 10.1016/j.neuroimage.2006.01.021 [DOI] [PubMed] [Google Scholar]
- 53. Smith SM, Jenkinson M, Woolrich MW, et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage. 2004;23:S208‐S219. doi: 10.1016/j.neuroimage.2004.07.051 [DOI] [PubMed] [Google Scholar]
- 54. Cox RW. AFNI: software for analysis and visualization of functional magnetic resonance neuroimages. Comput Biomed Res. 1996;29:162‐173. doi: 10.1006/cbmr.1996.0014 [DOI] [PubMed] [Google Scholar]
- 55. Palmqvist S, Schöll M, Strandberg O, et al. Earliest accumulation of β‐amyloid occurs within the default‐mode network and concurrently affects brain connectivity. Nat Commun. 2017;8:1214. doi: 10.1038/s41467-017-01150-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jack CR Jr, Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimers Dement. 2017;13:205‐216. doi: 10.1016/j.jalz.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shrout PE, Bolger N. Mediation in experimental and nonexperimental studies: new procedures and recommendations. Psychol Methods. 2002;7:422‐445. doi: 10.1037/1082-989X.7.4.422 [DOI] [PubMed] [Google Scholar]
- 58. Fisher RA. On the interpretation of χ2 from contingency tables, and the calculation of P. J R Stat Soc. 1922;85:87‐94. [Google Scholar]
- 59. Power JD, Barnes KA, Snyder AZ, Schlaggar BL, Petersen SE. Spurious but systematic correlations in functional connectivity MRI networks arise from subject motion. Neuroimage. 2012;59:2142‐2154. doi: 10.1016/j.neuroimage.2011.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. La Joie R, Visani AV, Baker SL, et al. Prospective longitudinal atrophy in Alzheimer's disease correlates with the intensity and topography of baseline tau‐PET. Sci Transl Med. 2020;12:eaau5732. doi: 10.1126/scitranslmed.aau5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Clavaguera F, Bolmont T, Crowther RA, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909‐913. doi: 10.1038/ncb1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. de Calignon A, Polydoro M, Suárez‐Calvet M, et al. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73:685‐697. doi: 10.1016/j.neuron.2011.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jagust W. Imaging the evolution and pathophysiology of Alzheimer disease. Nat Rev Neurosci. 2018;19:687‐700. doi: 10.1038/s41583-018-0067-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Maass A, Landau S, Baker SL, et al. Comparison of multiple tau‐PET measures as biomarkers in aging and Alzheimer's disease. Neuroimage. 2017;157:448‐463. doi: 10.1016/j.neuroimage.2017.05.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hamel E. Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol. 2006;100:1059‐1064. doi: 10.1152/japplphysiol.00954.2005 [DOI] [PubMed] [Google Scholar]
- 66. Hamel E. Cholinergic modulation of the cortical microvascular bed. Prog Brain Res. 2004;145:171‐178. doi: 10.1016/S0079-6123(03)45012-7 [DOI] [PubMed] [Google Scholar]
- 67. Wang M, He Y, Sejnowski TJ, Yu X. Brain‐state dependent astrocytic Ca2+ signals are coupled to both positive and negative BOLD‐fMRI signals. Proc Natl Acad Sci USA. 2018;115:E1647‐E1656. doi: 10.1073/pnas.1711692115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Iliff JJ, Chen MJ, Plog BA, et al. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci. 2014;34:16180‐16193. doi: 10.1523/JNEUROSCI.3020-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Harrison IF, Ismail O, Machhada A, et al. Impaired glymphatic function and clearance of tau in an Alzheimer's disease model. Brain. 2020;143:2576‐2593. doi: 10.1093/brain/awaa179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ishida K, Yamada K, Nishiyama R, et al. Glymphatic system clears extracellular tau and protects from tau aggregation and neurodegeneration. J Exp Med. 2022;219:e20211275. doi: 10.1084/jem.20211275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lopes DM, Llewellyn SK, Harrison IF. Propagation of tau and α‐synuclein in the brain: therapeutic potential of the glymphatic system. Transl Neurodegener. 2022;11:19. doi: 10.1186/s40035-022-00293-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Brunello CA, Merezhko M, Uronen R‐L, Huttunen HJ. Mechanisms of secretion and spreading of pathological tau protein. Cell Mol Life Sci. 2020;77:1721‐1744. doi: 10.1007/s00018-019-03349-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jucker M, Walker LC. Self‐propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45‐51. doi: 10.1038/nature12481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hsu J‐L, Wei Y‐C, Toh CH, et al. Magnetic resonance images implicate that glymphatic alterations mediate cognitive dysfunction in Alzheimer disease. Ann Neurol. 2023;93:164‐174. doi: 10.1002/ana.26516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zamani A, Walker AK, Rollo B, et al. Impaired glymphatic function in the early stages of disease in a TDP‐43 mouse model of amyotrophic lateral sclerosis. Transl Neurodegener. 2022;11:17. doi: 10.1186/s40035-022-00291-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Data Availability Statement
The multimodal data, including subject characteristics, rsfMRI, structural MRI, Aβ‐PET, tau‐PET, and MoCA measures are all publicly available at the ADNI website upon the approval of the data use application (http://adni.loni.usc.edu/). The ADNI was launched in 2003 as a public‐private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early AD. For up‐to‐date information, see www.adni‐info.org. All the codes used in the present study are available from the corresponding author upon request.