

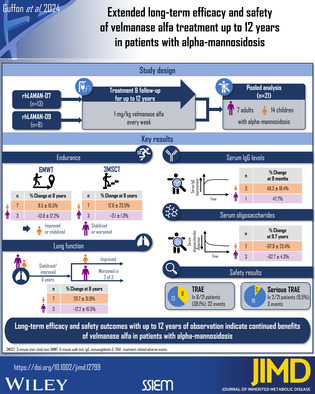
Abstract
Enzyme replacement therapy (ERT) using velmanase alfa previously showed promising efficacy and safety outcomes for up to 4 years of therapy in patients with alpha‐mannosidosis. This pooled analysis from two multicenter, open‐label phase IIIb extension trials rhLAMAN‐07 (N = 13; NCT01908712) and rhLAMAN‐09 (N = 8; NCT01908725) evaluated the long‐term effects of velmanase alfa. Sixteen patients who previously completed phase I–III rhLAMAN‐02/‐03/‐04/‐05/‐08 trials and five ERT‐naïve patients were enrolled. Patients received 1 mg/kg velmanase alfa once weekly. Endpoints included changes from treatment baseline (before initial dose of velmanase alfa in any trial) in serum oligosaccharides, 6‐minute walk test (6MWT), 3‐minute stair climb test (3MSCT), pulmonary function (forced vital capacity [FVC], % predicted), serum immunoglobulin G (IgG) levels, and adverse events. The overall cohort comprised 21 patients, divided by age at treatment baseline into pediatric (n = 14) and adult subgroups (n = 7). Distance walked according to 6MWT increased or stabilized in pediatric patients, while in adults either stabilization or slight decline was observed. Similarly, pediatric patients performed better in the 3MSCT. Changes in FVC, % predicted, were comparable in both subgroups up to ~6 years of observation, diverging thereafter. Overall, sustained serum oligosaccharide clearance and serum IgG level increase was observed upon treatment initiation and persisted until last common observation. Velmanase alfa treatment was generally well tolerated, with the majority of reported adverse events being of mild‐to‐moderate intensity. With follow‐up of up to 12 years, long‐term efficacy and safety outcomes indicate continued benefits of velmanase alfa in patients with alpha‐mannosidosis.
Keywords: alpha‐mannosidosis, enzyme replacement therapy, long‐term efficacy and safety, lysosomal storage disorder, recombinant enzymes, velmanase alfa
1. INTRODUCTION
Alpha‐mannosidosis (OMIM #248500) is a rare, autosomal recessive inherited lysosomal storage disorder with an estimated prevalence of 1:300 000–1:500 000. 1 Caused by a deficiency of alpha‐mannosidase due to pathogenic variants in the mannosidase alpha class 2B member 1 (MAN2B1) gene on chromosome 19, subsequent pathology results from the accumulation of oligosaccharides in organs including liver, kidney, spleen, and brain. 2 Characteristic clinical features include cognitive dysfunction, hearing impairment, immunodeficiency (recurrent infections), skeletal abnormalities, facial abnormalities, and motor and psychiatric symptoms. 3
Alpha‐mannosidosis features a broad, continuous spectrum of manifestations, and clinical phenotypes are not clearly distinguishable due to the high degree of heterogeneity of the disease, which makes disease progression difficult to predict. 3
To date, the genetic defect underlying alpha‐mannosidosis remains incurable, but there are therapeutic approaches available to alleviate symptoms and slow disease progression, including supportive measures, hematopoietic stem cell transplantation, and enzyme replacement therapy (ERT). 2 , 4 Hematopoietic stem cell transplantation may prevent neurocognitive decline and early death in young, severely affected patients. 5 , 6 However, the risks of serious complications must be weighed against the benefits. 5 , 7 Before the availability of ERT, disease management focused on symptomatic treatment and the prevention of complications. 4
Velmanase alfa (Lamzede®, Chiesi Farmaceutici S.p.A., Parma, Italy) 8 is the first and currently only ERT available for this disease. Approval was granted in 2018 by the European Medicines Agency 9 for the treatment of non‐neurological manifestations in patients with mild‐to‐moderate alpha‐mannosidosis, 8 and in 2023 by the U.S. Food and Drug Administration for non‐central nervous system manifestations in adult and pediatric patients with alpha‐mannosidosis. 10
In the international, multicenter, double‐blind, randomized, placebo‐controlled phase III trial rhLAMAN‐05, weekly intravenous infusions of 1 mg/kg velmanase alfa or placebo were given and the trial demonstrated a substantial decrease in serum oligosaccharides across all ages at week 52 in patients who received velmanase alfa compared with placebo. 11 Serum IgG levels significantly increased during the observation time, and motor and pulmonary function endpoints indicated a trend towards improvement with treatment, especially in pediatric patients. Velmanase alfa was well tolerated, without any significant safety or immunogenicity concerns. 11 This is in line with an integrated analysis which included data from rhLAMAN‐10 (NCT02478840), rhLAMAN‐07 (NCT01908712), and rhLAMAN‐09 (NCT01908725), based on an observation of up to 4 years. 12
The extension trials rhLAMAN‐07 and rhLAMAN‐09 offered patients from rhLAMAN‐05 and other trials to continue treatment with velmanase alfa in European countries where no compassionate use program was available. 11 , 12
Here, we present outcomes for up to 12 years of treatment with velmanase alfa using pooled data from the rhLAMAN‐07 and rhLAMAN‐09 extension trials. In view of the progressive course of alpha‐mannosidosis and the need for a lifelong ERT, this extensive follow‐up period will provide valuable information on the long‐term efficacy and safety effects of velmanase alfa. A plain language summary of this article is available in the supplementary materials S2.
2. METHODS
2.1. Study design
This pooled multicenter analysis presents data from 21 patients diagnosed with alpha‐mannosidosis from two single‐arm, open‐label phase IIIb extension trials, rhLAMAN‐07 (NCT01908712) and rhLAMAN‐09 (NCT01908725) (Figure 1A). These were designed to evaluate the long‐term efficacy and safety of velmanase alfa treatment in patients with alpha‐mannosidosis. rhLAMAN‐07 infusions were administered in France at 3 sites and in Denmark, and rhLAMAN‐09 infusions were administered in Denmark and Poland; rhLAMAN‐09 included patients based in Norway and the United Kingdom. rhLAMAN‐07 contributed 13 patients to the pooled analysis, eight of whom were previously enrolled in at least one of the following trials: rhLAMAN‐02: NCT01268358; rhLAMAN‐03: NCT01285700; rhLAMAN‐04: NCT01681940; rhLAMAN‐05: NCT01681953; rhLAMAN‐08: NCT02998879. The remaining five patients did not receive ERT prior to the start of this trial (ERT‐naïve patients) (Figure 1B). rhLAMAN‐09 contributed eight patients to the pooled analysis, who all previously participated in rhLAMAN‐02/‐03/‐04 and/or ‐05. Of the patients previously enrolled in rhLAMAN‐05, four patients had received placebo and subsequently started velmanase alfa treatment in 2013 with enrollment in rhLAMAN‐07 (n = 2) and rhLAMAN‐09 (n = 2). All patients received intravenous infusions of 1 mg/kg velmanase alfa once every week from treatment start until study end in September 2022.
FIGURE 1.
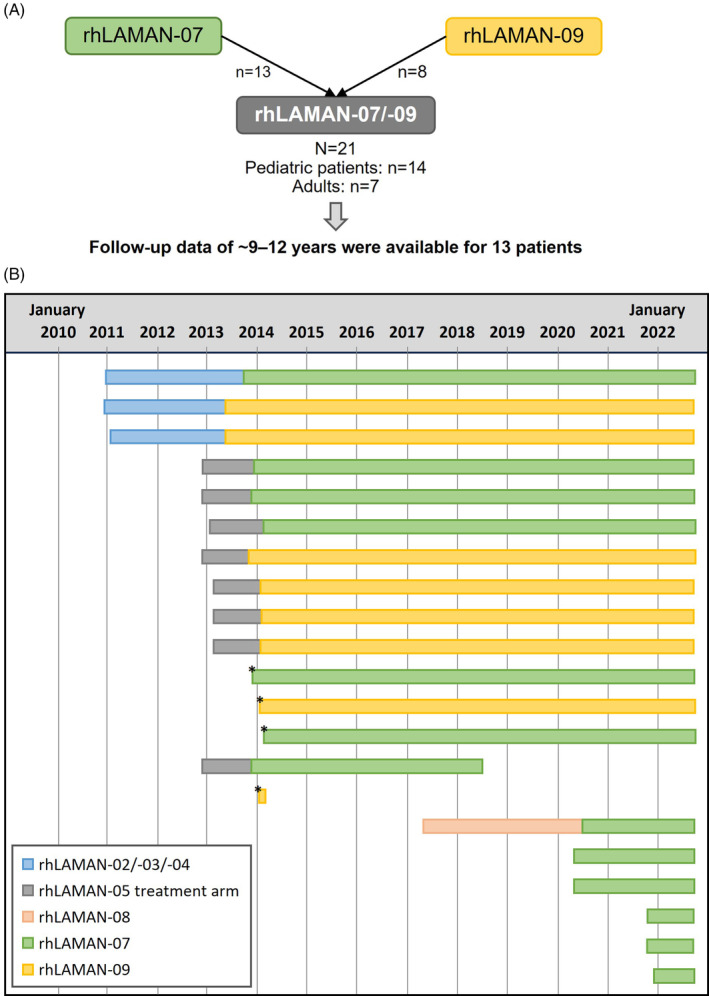
Overview of rhLAMAN‐07 and rhLAMAN‐09 treatment periods of individual patients. Overall, 21 patients were included in the pooled analysis of rhLAMAN‐07 and rhLAMAN‐09, of which 13 patients could be followed for up 12 years (Panel A). Overall exposure to velmanase alfa during participation in any rhLAMAN trial, which corresponds to the observation period for the pooled analyses from treatment baseline to study end, is shown in Panel (B) for individual patients. Thus, the observation periods included treatment periods in previous studies, except for four patients from the placebo arm of rhLAMAN‐05 (marked with an asterisk), and five patients who did not receive enzyme replacement therapy (ERT) prior to the start of rhLAMAN‐07 (ERT‐naïve patients). Two patients from the parental phase III (rhLAMAN‐05) trial withdrew consent after ~54 months of treatment in rhLAMAN‐07 and ~1 month of treatment in rhLAMAN‐09, respectively. A further six patients entered the study after 2019, five of whom were treatment‐naïve.
2.2. Patients
To be eligible for inclusion, patients had to have a confirmed diagnosis of alpha‐mannosidosis as defined by alpha‐mannosidase activity <10% of normal activity. 12 , 13 Previous participation in a prior velmanase alfa trial was required for rhLAMAN‐09, but not for rhLAMAN‐07. Further inclusion criteria were written informed consent from patients, parents, or legally authorized guardian(s) and their ability to comply with the trial protocol. Patients with known clinically significant medical conditions, pregnancy, psychosis, or a planned major surgery were excluded from participation in both trials.
2.3. Endpoints and assessments
Treatment baseline was defined as the last available value before the first dose of velmanase alfa ever infused. Treatment baseline values were therefore either from the parental study for patients previously enrolled in a trial (parental refers to the first trial each patient participated in of the velmanase alfa clinical trial program), the first dose in rhLAMAN‐07/‐09 for patients in the placebo arm of rhLAMAN‐05, or at Visit 1 for treatment‐naïve patients in rhLAMAN‐07. As the natural history of alpha‐mannosidosis typically changes during the transition phase to adulthood, the patient cohort was divided into a pediatric (<18 years) and an adult cohort (≥18 years) based on age at treatment baseline. Demographic characteristics were captured at treatment baseline.
Key efficacy endpoints, for which pooled data were analyzed, included changes from treatment baseline in: 6‐minute walk test (6MWT), 3‐minutes stair climb test (3MSCT), pulmonary function using forced vital capacity (FVC, % predicted), serum oligosaccharide (N‐acetylglucosamine‐di‐mannose, GlcNAc[Man]2) concentration, and serum immunoglobulin G (IgG) levels. Additional details on the serum oligosaccharide analyses are described in the supplementary file. A further efficacy endpoint was change from baseline in hearing ability assessed by pure tone audiometry in both ears measuring bone and air conduction (Supplementary Figures S8,S9). The test was conducted using audiometer earphones in a sound‐proof room.
Changes from treatment baseline were assessed over a period of up to 12 years of follow‐up until last common observation (LCO), reflecting the last observation interval at which data from both pediatric patients and adults were available, and last observation (LO), reflecting the very last observation interval in both subgroups independently. All efficacy assessments, except for serum oligosaccharides and immunoglobulin concentrations, were assigned to intervals ranging from 6 months (183 days) to 1 year (365 days). Multiple assessments made during a specific interval were assigned to the median visit time point after treatment baseline. Serum oligosaccharide concentrations were measured in patients from both trials at treatment baseline and assigned to intervals of 0–12 weeks after treatment start, followed by intervals every 24 weeks. Serum immunoglobulin values were assigned to 6‐month intervals and the analysis only included the subset of patients from rhLAMAN‐07 with available values at treatment baseline.
Safety endpoints included the analysis of adverse events (AEs). All AEs were coded by system organ class and preferred term using the Medical Dictionary for Regulatory Activities version 23.0. AEs emerging after the first dose of velmanase alfa in rhLAMAN‐07/‐09 were reported as treatment‐emergent AEs (TEAEs) and allocated to reporting periods consisting of 6‐month intervals based on duration since first dose. For the pooled analysis, TEAEs were summarized for the overall population.
2.4. Statistical analysis
This analysis was based on pooled data from previous trials, rhLAMAN‐07 and rhLAMAN‐09 and used descriptive statistics. For continuous variables, arithmetic mean, standard deviation (SD), standard error (SE), median, first and third quartile (Q1 and Q3), minimum and maximum values as well as absolute and percentage change from treatment baseline were calculated. In case of multiple measurements from the same patient in the same time interval, mean values were used. Key efficacy outcomes were analyzed in the overall population as well as in the pediatric and adult subgroup. Categorical data were presented by frequency counts and percentages. Missing data were not imputed. There was no formal estimation of the sample size.
3. RESULTS
3.1. Patients
A total of 21 patients with alpha‐mannosidosis were included in rhLAMAN‐07/‐09, comprising 13 (61.9%) male and eight (38.1%) female patients, all with mild‐to‐moderate phenotype in accordance with the label specified in the European Summary of Product Characteristics. 8 This overall cohort was divided by age at treatment baseline into a pediatric subgroup (n = 14) with children of a median age (range) of 9.5 (4–15) years and an adult subgroup (n = 7) of a median age of 29.0 (18–36) years. Patient characteristics of the overall cohort and the pediatric and adult subgroups are shown in Table 1 and Supplementary Table S1. The median age (range) at diagnosis was 6.5 (2–12) years in the pediatric subgroup (n = 14) and 4.0 (1–35) years in adults (n = 7).
TABLE 1.
Baseline demographics.
| Characteristic | <18 years a (n = 14) | ≥18 years a (n = 7) | Overall (N = 21) |
|---|---|---|---|
| Sex, n (%) | |||
| Female | 5 (35.7) | 3 (42.9) | 8 (38.1) |
| Male | 9 (64.3) | 4 (57.1) | 13 (61.9) |
| Age at first dose, years | |||
| Mean (SD) | 9.8 (4.0) | 25.9 (7.1) | 15.1 (9.3) |
| Median (range) | 9.5 (4; 15) | 29.0 (18; 36) | 15.0 (4; 36) |
| Q1, Q3 | 7.0; 15.0 | 18.0; 30.0 | 7.0; 18.0 |
| Initial BMI at treatment baseline b (kg/m2) | |||
| Mean (SD) | 20.0 ± 3.0 | 26.8 ± 3.1 | 22.3 ± 4.4 |
| Median (range) | 20.0 (16.0; 25.3) | 27.1 (21.8; 30.8) | 21.8 (16.0; 30.8) |
| Q1, Q3 | 18.3; 22.1 | 24.1; 28.9 | 18.3; 25.3 |
Abbreviations: Q1, quartile 1; Q3, quartile 3; SD, standard deviation.
Age categories refer to the age at treatment baseline, when the first dose of velmanase alfa ever was received.
Defined as the last available value before the first dose of velmanase alfa in the parental study, as the last available value before the first dose in rhLAMAN‐07/‐09 for patients in the placebo arm of rhLAMAN‐05, and as Visit 1 for treatment‐naïve patients in rhLAMAN‐07.
Overall, 13 patients were followed for up to ~9–12 years (Figure 1). By the end of the extension trials, six patients had reached adulthood (two patients in rhLAMAN‐07 and four in rhLAMAN‐09). One of the pediatric patients in the rhLAMAN‐07 trial, who was previously enrolled in the parental phase III (rhLAMAN‐05) trial for just over 1 year, had consent withdrawn by a parent in 2018 after a further ~4.5 years of treatment in the rhLAMAN‐07 trial, due to the patient experiencing mild‐to‐moderate infusion‐related reactions. A second patient who previously participated in the placebo arm of rhLAMAN‐05 withdrew consent in 2014, after ~1 month enrollment in rhLAMAN‐09, due to joining an aftercare program at a local hospital. One patient, who enrolled in rhLAMAN‐07 in 2020, had a follow‐up of 5 years as they started velmanase alfa treatment at a later timepoint due to the enrollment in rhLAMAN‐08, the most recent parental trial. Five patients had an even shorter follow‐up as they started treatment at enrollment in rhLAMAN‐07 in either 2020 or 2021.
Low patient numbers at specific time intervals were due to the SARS‐CoV‐2 pandemic or missed visits due to unknown reasons. During the pandemic, visits were on hold for all 13 patients. In total, one patient from rhLAMAN‐07 and four patients from rhLAMAN‐09 interrupted treatment by more than two consecutive infusions. Two of the patients from rhLAMAN‐09 missed the treatment (up to 66 infusions) for more than 1 year due to the pandemic. Further reasons for some patients lacking longer follow‐up data were more recent treatment start (n = 6) and consent withdrawals (n = 2). Serum IgG data from treatment baseline were only available for four patients (including only one adult) and limited to 2.5 years of observation.
3.2. Efficacy
3.2.1. 6MWT, 3MSCT, and FVC
Descriptive analyses of absolute and relative changes in 6MWT, 3MSCT, and FVC in the overall population and by age group are shown in Figure 2 and Supplementary Figure S1. Individual patient data stratified by the trials rhLAMAN‐07 and rhLAMAN‐09 are shown in Supplementary Figures S6,S7. Over time, the absolute distance walked on the 6MWT increased or stabilized in all pediatric patients until LO, while in adults either a stabilization or slight decline was observed, with only one adult having a large decrease. At 8 years (interval of 84–108 months) of therapy, absolute change from baseline in distance walked slightly increased in pediatric patients (n = 7, 27.8 ± 31.2 m; percentage change: 8.5 ± 10.5%), while it slightly decreased in adults (n = 3, –64.3 ± 50.2 m; percentage change: −13.8 ± 12.2%), with only one adult having a large decrease.
FIGURE 2.
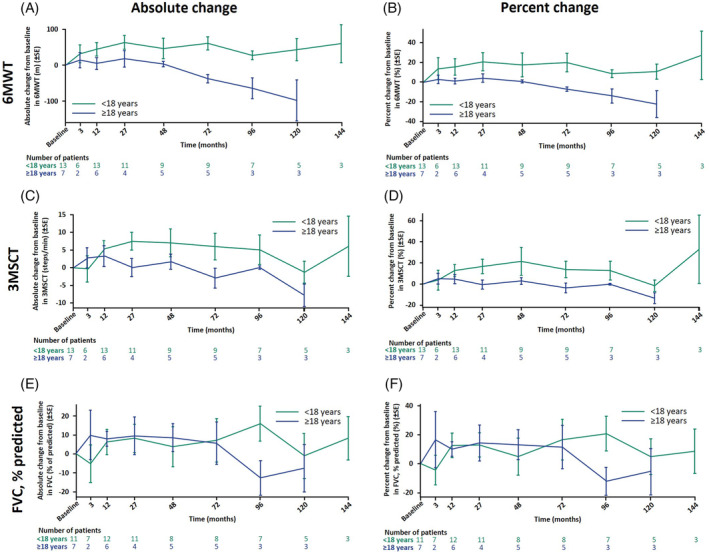
6MWT, 3MSCT, and forced vital capacity (FVC) outcomes. Mean ± SE changes are shown from treatment baseline, defined as the last available value before the first dose of velmanase alfa in the parental study, as the last available value before the first dose in rhLAMAN‐07/‐09 for patients in the placebo arm of rhLAMAN‐05, and as Visit 1 for treatment‐naïve patients in rhLAMAN‐07. X‐axes display the median of each time interval.
3MSCT, 3‐minute stair climb test, 6MWT, 6‐minute walk test; SE, standard error.
Albeit to a lesser degree, the pediatric patients also performed better on the 3MSCT than adults. At 8 years (84–108 months) of observation, pediatric patients (n = 7) showed an absolute change from baseline of 5.1 ± 11.0 steps/min versus –0.1 ± 0.8 steps/min in adults (n = 3) (percentage change: 12.6 ± 23.5% versus –0.1 ± 1.3%) (Figure 2C,D). Overall, 3MSCT results in pediatric patients initially increased and then predominantly remained stable and adult patients either stabilized or slightly declined.
Most pediatric patients improved or stabilized FVC, % predicted, throughout the observation period, while adult patients started to slowly decline in pulmonary function after a 6‐year stabilization period (Figure 2E,F). Changes from treatment baseline were similar in both subgroups for the first ~6 years of treatment, diverging thereafter: at treatment baseline, pediatric patients had a mean ± SD FVC, % predicted, of 76.2 ± 22.1% of the predicted value (n = 11), increasing by an absolute change of 7.1 ± 32.5% (n = 8; percentage change: 16.6 ± 40.0%) until 6 years (60–84 months) of treatment, while adults had a baseline of 94.4 ± 16.5% (n = 7) and an absolute change of 5.6 ± 24.9% (n = 5; percentage change: 11.4 ± 33.5%). At 8 years (84–108 months) of therapy, the change from baseline in FVC, % predicted, of pediatric patients further increased from baseline (n = 7; 15.8 ± 24.4%; percentage change: 20.7 ± 31.8%), while two of three adult patients had a drop in the FVC results (n = 3; absolute change: –12.8 ± 15.8% of predicted; percentage change: −12.2 ± 16.5%) (Supplementary Figure S1g,h). Overall, after at least 10 years of follow‐up, four pediatric patients and one adult patient had a FVC, % predicted ≥80%, while three pediatric and two adult patients had <80%.
3.2.2. Serum oligosaccharide levels
Both age groups showed similar absolute and percentage changes in serum GlcNAc(Man)2 levels from the treatment baseline over time (Figure 3, Supplementary Figures S1a,b, S4,S5). Overall mean ± SD serum oligosaccharide concentration was 8.0 ± 3.3 μmol/L in pediatric patients (n = 13) and 6.0 ± 1.3 μmol/L in adults (n = 7) at treatment baseline which consistently decreased in both subgroups until the LCO at ~9.7 years (interval of 113.2–117.8 months) of therapy by an absolute change of –5.3 ± 2.3 μmol/L in pediatric patients (n = 7; percentage change: −67.8 ± 23.4%) and −3.5 ± 0.5 μmol/L in adults (n = 3; percentage change: −62.7 ± 4.3%). Serum oligosaccharides rose again in three patients, after treatment interruption of >1 year (two patients) and antidrug antibody detection (one patient) (Supplementary Figure S1a). 14
FIGURE 3.
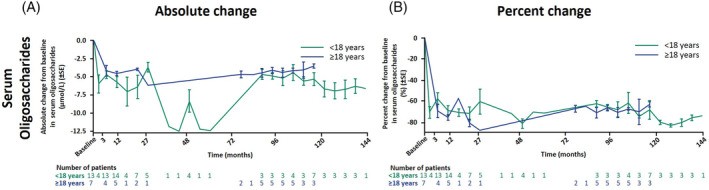
Serum oligosaccharides. Mean ± SE changes are shown from treatment baseline, defined as the last available value before the first dose of velmanase alfa in the parental study, as the last available value before the first dose in rhLAMAN‐07/‐09 for patients in the placebo arm of rhLAMAN‐05, and as Visit 1 for treatment‐naïve patients in rhLAMAN‐07. The decrease of the mean values between 38.7 and 60.8 months is due to data of only one pediatric patient being available for these time points; this patient had a higher value at treatment baseline and a stronger decrease following treatment than the other patients (Supplementary Figure S1a). X‐axes display the median of each time interval.
SE, standard error.
3.2.3. Serum IgG levels
In the analysis of serum IgG, based on four patients who started treatment in rhLAMAN‐07 and had baseline IgG levels assessed, all patients showed similar changes in IgG levels with velmanase alfa treatment (Figure 4A,B and Supplementary Figures S2,S3). Pediatric patients started with IgG levels in the lower normal range or below the normal range, depending on the age‐related range of normality that is used as a reference, 15 , 16 , 17 , 18 while the one adult patient had values just below the normal range at treatment baseline. 18 In pediatric patients the overall mean ± SD serum IgG levels markedly increased from treatment baseline level (n = 3; 6.2 ± 1.7 g/L) until the LCO at 9 (interval of 6–12) months by an absolute change of 2.8 ± 0.6 g/L (n = 3; percentage change: 49.3 ± 18.4%). At LO at 2.25 years (24–30 months), the IgG level in those patients increased by an absolute change of 4.4 ± 0.0 g/L (n = 2; percentage change: 84.3 ± 17.2%) and were within the normal range. 16 , 17 , 18 The percentage change for the adult patient (5.5 g/L at treatment baseline) was 47.7%, and the post‐treatment value of 8.1 g/L after 0.75 years (6–12 months) was within the normal range. 18 , 19 , 20 , 21 For patients who were excluded from the main analysis due to a lack of values at treatment baseline in rhLAMAN‐09 (n = 7; two adults), supplementary data show that after treatment start the IgG levels reached normal range 15 and were maintained up to 12 years in all patients except in one patient who exceeded the normal range. For two of these patients, IgG levels decreased following treatment interruption of >1 year—remaining within normal range—but then increased again after restarting treatment (Supplementary Figure S3). Supplementary data are not shown for patients from rhLAMAN‐07 who were excluded from the main analysis.
FIGURE 4.
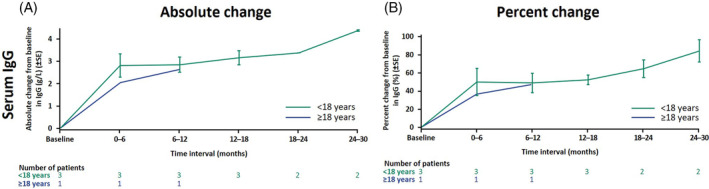
Serum IgG. All four patients (three pediatric patients, one adult) were treatment‐naïve when they were enrolled in rhLAMAN‐07/‐09. Mean ± SE changes are shown from treatment baseline at Visit 1 in rhLAMAN‐07 (treatment baseline values were not available for the remaining 17 patients of the pooled cohort).
IgG, immunoglobulin G; SE, standard error.
3.2.4. Hearing ability
Results of hearing ability tests using pure tone audiometry are summarized in Supplementary Figures S8 (rhLAMAN‐07) and S9 (rhLAMAN‐09). In general, hearing ability according to bone as well as air conduction remained mostly stable, with variable degrees of fluctuation among patients. There was considerable variability at treatment baseline, especially in pediatric patients.
3.3. Safety
The median (range) duration of exposure to velmanase alfa was 9.2 (1.1–11.8) years for pediatric patients (n = 14) and 8.8 (0.1–9.8) years for adult patients (n = 7). Table 2 provides a summary of AEs that emerged during rhLAMAN‐07/‐09 in the pooled patient cohort. Overall, 697 TEAEs were reported in 20 of 21 patients, of which 32 events in eight patients were considered treatment‐related.
TABLE 2.
Treatment‐emergent adverse events in the pooled patient cohort after enrollment in rhLAMAN‐07/‐09.
| Adverse events | Overall (N = 21) | |
|---|---|---|
| n (%) | Events | |
| Any TEAEs | 20 (95.2) | 697 |
| Treatment‐related TEAEs | 8 (38.1) | 32 |
| Serious TEAEs | 8 (38.1) | 23 |
| Serious treatment‐related TEAEs | 2 (9.5) | 3 |
| Severe TEAEs | 3 (14.3) | 4 |
| TEAEs with a fatal outcome | 0 (0.0) | 0 |
| TEAEs leading to discontinuation | 0 (0.0) | 0 |
Abbreviation: TEAEs, treatment‐emergent adverse events.
The most common MedDRA system organ class categories (>70% of patients) were infections or infestations with 198 TEAEs in 18 (85.7%) of 21 patients, followed by gastrointestinal disorders with 95 TEAEs in 17 (81.0%) patients; respiratory, thoracic, and mediastinal disorders with 41 TEAEs in 16 (76.2%) patients; general disorders and administration site conditions with 89 TEAEs in 16 (76.2%) patients; and musculoskeletal and connective tissue disorders with 57 TEAEs in 15 (71.4%) patients (Supplementary Table S2). The most frequent TEAEs included vomiting (66.7% of patients), nasopharyngitis (61.9%), pyrexia (57.1%), cough (47.6%), arthralgia (38.1%), diarrhea (38.1%), abdominal pain (33.3%), ear infection (33.3%), headache (28.6%), back pain (28.6%), and pain in extremity (28.6%).
The majority of TEAEs were mild‐to‐moderate, and only four severe events were documented in three (14.3%) patients. Eight (38.1%) patients experienced a total of 23 serious TEAEs (3.3% of all TEAEs), including three serious treatment‐related events of diarrhea, hypokalemia, and vomiting in two (9.5%) patients, which resolved on the same day or the day after. There were no TEAEs that had a fatal outcome or led to dose reduction or study discontinuation. However, one pediatric patient experienced multiple transient, mild‐to‐moderate infusion‐related reactions and withdrew consent after more than 5 years of treatment.
4. DISCUSSION
This pooled analysis of rhLAMAN‐07 and rhLAMAN‐09 provides the first long‐term assessment of efficacy and safety of weekly therapy with velmanase alfa in a population of 21 patients with alpha‐mannosidosis. Among these, 13 patients received treatment for >9 years and up to almost 12 years. Previously, efficacy and safety outcomes over 12 months were reported from the phase III trial rhLAMAN‐05, 11 as well as long‐term data based on an integrated analysis with up to 4 years of follow‐up which included patients from rhLAMAN‐10. 12 Treatment with velmanase alfa showed improvements in 6MWT, 3MSCT, and pulmonary function; a significant and persistent clearance of serum oligosaccharides; and increased serum IgG levels at last observation of up to 4 years, alongside a tolerable safety profile. 12 In rhLAMAN‐08, five children with alpha‐mannosidosis under the age of 6 years received weekly velmanase alfa therapy and were followed for 3 years. 13 The published results of the rhLAMAN‐08 trial similarly showed individual improvements in physical functioning/endurance, hearing, immunological profile, and QoL; decreased serum oligosaccharides; and acceptable safety outcomes. 13 Moreover, a multiple variable responder model used data from rhLAMAN‐05 and rhLAMAN‐10 to assess global treatment response to velmanase alfa, defined by response in 3 domains comprising pharmacodynamic, functional, and QoL outcomes. 22 This model also supported continued benefit of longer‐term treatment for up to 48 months both in pediatric and adult patients. 22
In line with these findings, this long‐term analysis of continuous, functional assessments suggested a stabilization or delay in clinical worsening. In particular, patients who started velmanase alfa treatment at a pediatric age showed stabilized or even improved outcomes with velmanase alfa, including those who became an adult during the study. Besides pediatric patients, adults also appeared to benefit from treatment, as they showed stabilized or only slightly decreased outcomes regarding endurance and FVC, % predicted. The confounding effect of childhood growth and aging is expected to differ according to age and to be especially prominent in the youngest patients and in earlier time windows. Therefore, to provide information on individual outcomes over time, individual patient data are available as supplementary materials (Supplementary Figures S1–S9).
Due to the paucity of natural history controls, it is difficult to determine to what extent any changes—especially increased endurance capabilities in 6MWT and 3MSCT—resulted from treatment versus other factors, such as clinical heterogeneity and age‐related changes. The natural history study of Beck et al. reported a broad heterogeneity of clinical phenotypes regarding 6MWT and 3MSCT results in both children and adults with alpha‐mannosidosis, with no clear trends seen in either of the patient groups. 23 Beck et al. also reported that percentage of predicted FVC remained similar in adults and decreased by up to 10% in children over 2 years, indicating that changes in pulmonary function were not clinically significant. 23 Besides clinical heterogeneity and age‐related changes, functional assessments can be influenced by factors such as motivation and training effect, as they rely on the participation of the patient. This can also make assessments challenging in young children and patients with cognitive impairment, 24 , 25 , 26 , 27 but no formal assessment of the impact of cognitive impairment on endurance was conducted in our analysis. Considering that the FVC, % predicted, as opposed to the 6MWT and 3MSCT, takes into account gender, height, and age, the observed improvements in pulmonary function likely reflect an effect of treatment rather than aging. Taken together, despite several factors potentially influencing functional outcomes, the long‐term data from this study suggest velmanase alfa improved endurance.
The 6MWT and 3MSCT assessments may capture impairments caused by balance instability and ataxia that can occur in alpha‐mannosidosis due to neurological damage, cochlea‐vestibular disorders, or muscular hypotonia. 1 As velmanase alfa does not cross the blood–brain‐barrier, the improvements seen for the 6MWT and 3MSCT may possibly be explained by velmanase alfa treatment slowing down the progression of mainly non‐central nervous system symptoms such as joint abnormalities and muscular hypotonia.
A marked reduction of serum oligosaccharide levels shortly after treatment initiation was observed, which persisted in all patients throughout the follow‐up period. It is plausible that velmanase alfa systemically targets the root 3 cause of alpha‐mannosidosis‐induced cellular dysfunctions and stabilizes or even slows down progression of the disease in some patients.
Patients with alpha‐mannosidosis are prone to recurrent infections, especially at an early age. 3 This immunodeficiency involves multiple aspects, including low IgG levels. 12 In this analysis, serum IgG levels were in the lower normal range or just below the normal range at treatment baseline and increased substantially and consistently after the start of the treatment. 15 , 16 , 17 , 18 , 28 Concentrations remained at higher, normal levels throughout the 2.5 years of observation from treatment baseline reaching up to a mean concentration of nearly 10 g/L after 24–30 months in pediatric patients, which may result in a clinically relevant improvement of immune function. 12 Interestingly, two patients who interrupted treatment for >1 year showed a re‐increase in serum IgG at first assessment after restarting treatment (Supplementary Figure S3).
The safety profile observed in our study corresponds to previously published safety findings from velmanase alfa clinical trials. 11 , 12 , 13 Three serious treatment‐related TEAEs were observed in two patients. TEAEs were most commonly infections and gastrointestinal events, and most were of mild‐to‐moderate severity. All treatment‐related TEAEs resolved and did not necessitate any dose reductions or treatment interruptions. No new safety concerns were identified with extended treatment.
Early start of ERT, preferably even before symptom onset, has been recommended for other lysosomal storage disorders to obtain better long‐term outcomes. 29 In line with our findings in this pooled rhLAMAN‐07/‐09 analysis, earlier studies have reported more pronounced efficacy benefits in pediatric patients than in adults. 11 , 12 , 22 Furthermore, a phase II study (rhLAMAN‐08) evaluated velmanase alfa in children below 6 years of age and the results suggested a clinical benefit for this young patient group. 13 Based on these observations, velmanase alfa treatment initiation at a younger age was supported as being beneficial and improving long‐term efficacy outcomes. A reduction of serum oligosaccharide accumulation at a younger age by starting velmanase alfa treatment in young children could potentially delay the development and worsening of symptoms more effectively than treatment in adults with advanced clinical manifestations and irreversible tissue damages. 29
Limitations of our integrated analysis include the small sample size, which is inherent with respect to the ultra‐rarity of the disease. Patient numbers are relatively low in the adult subgroup and at later assessment intervals due to missing data, and the lack of a control group in the context of the respective trial designs, as ethical reasons prevent the inclusion of a placebo control over extensive follow‐up periods. After conclusion of the parental trial rhLAMAN‐05, patients included in the placebo arm were switched to treatment with velmanase alfa. 11
5. CONCLUSIONS
The long‐term efficacy and safety outcomes indicate that treatment benefits of velmanase alfa are maintained over a period of up to 12 years in both pediatric and adult patients with alpha‐mannosidosis.
Treatment with velmanase alfa may successfully delay non‐central nervous system disease progression in all age groups, as many pediatric patients achieved improvements in motor and pulmonary function, and these functional outcomes in the adult cohort appeared to stabilize during long‐term treatment. Substantial decreases in serum oligosaccharide concentrations and increases in IgG levels persisted throughout the observation period regardless of age. Velmanase alfa was generally well tolerated, with mostly mild‐to‐moderate side effects. No new safety concerns were identified with extended treatment. The acceptable safety profile supports long‐term administration of velmanase alfa in patients with alpha‐mannosidosis.
AUTHOR CONTRIBUTIONS
Line Borgwardt, Nathalie Guffon, Allan Lund, and Anna Tylki‐Szymańska provided substantial contributions to the concept of this pooled analysis. All authors contributed to data acquisition, analysis, and interpretation, as well as drafting and revising the manuscript before providing their final approval.
FUNDING INFORMATION
This study was funded by Chiesi Farmaceutici S.p.A, Parma, Italy. The authors confirm independence from the sponsors; the sponsors did not influence the content of the article.
CONFLICT OF INTEREST STATEMENT
N.G. has received hospital grants as a P.I. for this study from Chiesi Farmaceutici S.p.A., and has received fees for participating as an expert on lysosomal disease management from Chiesi Farmaceutici S.p.A., Biomarin, Sanofi Genzyme, Takeda, and Ultragenyx Pharmaceutical Inc. L.B. was subinvestigator for this study from Chiesi Farmaceutici S.p.A. and received consulting fees and/or honoraria/travel support from Chiesi Farmaceutici S.p.A. A.B. was an employee at Chiesi Farmaceutici S.p.A at the time the work was conducted. F.D., A.J., H.N., and C.M. are employees at Chiesi Farmaceutici S.p.A. A.L. has received hospital grants as a P.I. for this study from Chiesi Farmaceutici S.p.A. as well as consulting fees and/or honoraria/travel support from Alexion, BioMarin, Chiesi Farmaceutici S.p.A., Sanofi Genzyme, and Shire. A.T.S. was P.I. for this study from Chiesi Farmaceutici S.p.A., and has received fees for participating as an expert on lysosomal disease management from Chiesi Farmaceutici S.p.A., Sanofi Genzyme, and Takeda.
ETHICS STATEMENT
The study protocol, patient data, and informed consent forms were approved by the Independent Ethics Committee (De Videnskabsetiske Komiteer Region Hovedstaden; Hillerød, Denmark) and the Regulatory Agency (Danish Medicines Agency; København S, Denmark) and complied with the European Federal regulations and the International Conference on Harmonization (ICH) before the patients were enrolled. This study was conducted in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki 1964, as revised in 2013), and good clinical practice guidelines as well as local guidelines and regulations.
Supporting information
Data S1. Supporting Information.
Data S2. Supporting Information.
ACKNOWLEDGMENTS
The authors thank all patients, their families, and investigators for participating in rhLAMAN‐07 and rhLAMAN‐09. Trial funding was provided by Chiesi Farmaceutici S.p.A, Parma, Italy. Medical writing support was provided by Lucie Meyer, PhD, and Tanja Kamps, PhD, of Ashfield MedComms GmbH, an Inizio company, Mannheim, Germany, and funded by Chiesi USA, Inc. Boston, MA, USA.
Guffon N, Borgwardt L, Tylki‐Szymańska A, et al. Extended long‐term efficacy and safety of velmanase alfa treatment up to 12 years in patients with alpha‐mannosidosis. J Inherit Metab Dis. 2025;48(1):e12799. doi: 10.1002/jimd.12799
Communicating Editor: Carlos R. Ferreira
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are not publicly available due to privacy or ethical restrictions. Data are available on request from the corresponding author; however, Chiesi reserves the right to deny requests for any and all legally appropriate reasons. Data requests that risk sharing participant‐level data or proprietary information will not be approved.
REFERENCES
- 1. Malm D, Riise Stensland HM, Edvardsen Ø, Nilssen Ø. The natural course and complications of alpha‐mannosidosis—a retrospective and descriptive study. J Inherit Metab Dis. 2014;37(1):79‐82. doi: 10.1007/s10545-013-9622-2 [DOI] [PubMed] [Google Scholar]
- 2. Ceccarini MR, Codini M, Conte C, et al. Alpha‐mannosidosis: therapeutic strategies. Int J Mol Sci. 2018;19(5):1500. doi: 10.3390/ijms19051500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Malm D, Nilssen Ø. Alpha‐mannosidosis. Orphanet J Rare Dis. 2008;3:21. doi: 10.1186/1750-1172-3-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Adam J, Malone R, Lloyd S, Lee J, Hendriksz CJ, Ramaswami U. Disease progression of alpha‐mannosidosis and impact on patients and carers – a UK natural history survey. Mol Genet Metab Rep. 2019;20:100480. doi: 10.1016/j.ymgmr.2019.100480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grewal SS, Shapiro EG, Krivit W, et al. Effective treatment of alpha‐mannosidosis by allogeneic hematopoietic stem cell transplantation. J Pediatr. 2004;144(5):569‐573. doi: 10.1016/j.jpeds.2004.01.025 [DOI] [PubMed] [Google Scholar]
- 6. Wall DA, Grange DK, Goulding P, Daines M, Luisiri A, Kotagal S. Bone marrow transplantation for the treatment of alpha‐mannosidosis. J Pediatr. 1998;133(2):282‐285. doi: 10.1016/s0022-3476(98)70237-9 [DOI] [PubMed] [Google Scholar]
- 7. Mynarek M, Tolar J, Albert MH, et al. Allogeneic hematopoietic SCT for alpha‐mannosidosis: an analysis of 17 patients. Bone Marrow Transplant. 2012;47(3):352‐359. doi: 10.1038/bmt.2011.99 [DOI] [PubMed] [Google Scholar]
- 8. European Medicines Agency . Lamzede EPAR summary of product characteristics. 2023.
- 9. European Medicines Agency . Lamzede (velmanase alfa) – An overview of Lamzede and why it is authorised in the EU. 2018.
- 10. U.S. Food and Drug Administration . Lamzede – Prescribing information. 2023.
- 11. Borgwardt L, Guffon N, Amraoui Y, et al. Efficacy and safety of velmanase alfa in the treatment of patients with alpha‐mannosidosis: results from the core and extension phase analysis of a phase III multicentre, double‐blind, randomised, placebo‐controlled trial. J Inherit Metab Dis. 2018;41(6):1215‐1223. doi: 10.1007/s10545-018-0185-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lund AM, Borgwardt L, Cattaneo F, et al. Comprehensive long‐term efficacy and safety of recombinant human alpha‐mannosidase (velmanase alfa) treatment in patients with alpha‐mannosidosis. J Inherit Metab Dis. 2018;41(6):1225‐1233. doi: 10.1007/s10545-018-0175-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guffon N, Konstantopoulou V, Hennermann JB, et al. Long‐term safety and efficacy of velmanase alfa treatment in children under 6 years of age with alpha‐mannosidosis: a phase 2, open label, multicenter study. J Inherit Metab Dis. 2023;46(4):705‐719. doi: 10.1002/jimd.12602 [DOI] [PubMed] [Google Scholar]
- 14. Borgwardt LG, Ceravolo F, Zardi G, Ballabeni A, Lund AM. Relationship between MAN2B1 genotype/subcellular localization subgroups, antidrug antibody detection, and long‐term velmanase alfa treatment outcomes in patients with alpha‐mannosidosis. JIMD Rep. 2023;64(2):187‐198. doi: 10.1002/jmd2.12349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dati F, Schumann G, Thomas L, et al. Consensus of a group of professional societies and diagnostic companies on guidelines for interim reference ranges for 14 proteins in serum based on the standardization against the IFCC/BCR/CAP reference material (CRM 470). International Federation of Clinical Chemistry. Community Bureau of Reference of the Commission of the European Communities. College of American Pathologists. Eur J Clin Chem Clin Biochem. 1996;34(6):517‐520. [PubMed] [Google Scholar]
- 16. Al Shamahy H, Nassar M, Salah M, Al Magrami R, Al Moyed K. Immunoglobulin levels (IgG, IgM and IgA): normal values for healthy infants and children in Sana'a City‐Yemen. Glob J Ped Neonatol Care. 2019;1(5):2019. [Google Scholar]
- 17. Bayram RO, Özdemir H, Emsen A, Türk Dağı H, Artaç H. Reference ranges for serum immunoglobulin (IgG, IgA, and IgM) and IgG subclass levels in healthy children. Turk J Med Sci. 2019;49(2):497‐505. doi: 10.3906/sag-1807-282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mayo Clinic Laboratories . Petiatric catalog: Immunoglobulins (IgG, IgA, and IgM), serum (Accessed February 23, 2024). https://pediatric.testcatalog.org/show/IMMG.
- 19. Jazayeri MH, Pourfathollah AA, Rasaee MJ, Porpak Z, Jafari ME. The concentration of total serum IgG and IgM in sera of healthy individuals varies at different age intervals. Biomed Aging Pathol. 2013;3(4):241‐245. doi: 10.1016/j.biomag.2013.09.002 [DOI] [Google Scholar]
- 20. Holma P, Pesonen P, Karjalainen MK, et al. Low and high serum IgG associates with respiratory infections in a young and working age population. EBioMedicine. 2023;94:104712. doi: 10.1016/j.ebiom.2023.104712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oyeyinka GO, Salimonu LS, Williams AI, Johnson AO, Ladipo OA, Osunkoya BO. Range of normal serum immunoglobulin (IgG, IgA and IgM) values in Nigerians. Afr J Med Med Sci. 1984;13(3–4):169‐176. [PubMed] [Google Scholar]
- 22. Harmatz P, Cattaneo F, Ardigò D, et al. Enzyme replacement therapy with velmanase alfa (human recombinant alpha‐mannosidase): novel global treatment response model and outcomes in patients with alpha‐mannosidosis. Mol Genet Metab. 2018;124(2):152‐160. doi: 10.1016/j.ymgme.2018.04.003 [DOI] [PubMed] [Google Scholar]
- 23. Beck M, Olsen KJ, Wraith JE, et al. Natural history of alpha mannosidosis a longitudinal study. Orphanet J Rare Dis. 2013;8:88. doi: 10.1186/1750-1172-8-88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vellone E, Chialà O, Boyne J, et al. Cognitive impairment in patients with heart failure: an international study. ESC Heart Fail. 2020;7(1):46‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Denissen S, De Cock A, Meurrens T, et al. The impact of cognitive dysfunction on locomotor rehabilitation potential in multiple sclerosis. J Cent Nerv Syst Dis. 2019;11:19884041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grenville J, Granell R, Dodd J. Lung function and cognitive ability in children: a UK birth cohort study. BMJ Open Respir Res. 2023;10(1):e001528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cleutjens F, Spruit MA, Ponds R, et al. Cognitive impairment and clinical characteristics in patients with chronic obstructive pulmonary disease. Chron Respir Dis. 2018;15(2):91‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Otani IM, Lehman HK, Jongco AM, et al. Practical guidance for the diagnosis and management of secondary hypogammaglobulinemia: a work group report of the AAAAI primary immunodeficiency and altered immune response committees. J Allergy Clin Immunol. 2022;149(5):1525‐1560. doi: 10.1016/j.jaci.2022.01.025 [DOI] [PubMed] [Google Scholar]
- 29. Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014;111(2):63‐72. doi: 10.1016/j.ymgme.2013.11.015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information.
Data S2. Supporting Information.
Data Availability Statement
The data that support the findings of this study are not publicly available due to privacy or ethical restrictions. Data are available on request from the corresponding author; however, Chiesi reserves the right to deny requests for any and all legally appropriate reasons. Data requests that risk sharing participant‐level data or proprietary information will not be approved.