

Abstract
Background
Eucalyptus grandis, which was first comprehensively and systematically introduced to China in the 1980s, is one of the most important fast-growing tree species in the forestry industry. However, to date, no core collection has been selected from the germplasm resources of E. grandis based on growth and genetic relationship analysis.
Results
In the present study, 545 individuals of E. grandis collected from 28 populations across 5 countries were selected for genetic diversity analysis using 16 selected SSR markers. The polymorphism information content (PIC) was employed to assess genetic diversity, yielding a mean value of 0.707. Genetic structure analysis was conducted on 492 individuals from 13 combined populations, revealing three clusters as the most suitable number. Principal coordinate analysis (PCoA) demonstrated that the populations were divided into three major clusters. Additionally, the analysis of molecular variance (AMOVA) indicated that the majority of variation occurred within populations.
Conclusions
Based on the criteria for screening the core collection, we constructed a population consisting of 158 individuals and created unique fingerprinting codes. These results provide a crucial theoretical foundation for the protection and utilization of germplasm resources of E. grandis in China, which will be helpful in the selection of genetically distant parents for future multigenerational hybridization programs.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12870-024-05970-0.
Keywords: Eucalyptus grandis, Genetic diversity, Population structure, SSR markers, Molecular fingerprint, Guangxi
Background
Multigeneration breeding, practiced to take advantage of heterosis, has always been the main strategy for the genetic improvement of forest trees [1]. This process generally begins with massive-selection in natural or unimproved populations, followed by iterations in the mating-testing-selection cycle, and elite individuals are produced [2–4]. This breeding cycle requires particular attention to the construction of a base, breeding, selection and production population [4–6]. Therefore, it is important to understand the genetic relationships of these populations, as their genetic characteristics influence the effectiveness and potential of genetic improvement and determine the collection and utilization of germplasm resources [7–9].
Eucalyptus, as a typical evergreen broad-leaved timber species, should also follow the above theory [10, 11]. As a large group of tree species native to Australia, Papua New Guinea and the Philippines [12, 13], Eucalyptus has been introduced around the world over the past century and has become one of the most important artificial commercial forest species providing a large amount of wood for industrial production and sufficient raw material for pulp and paper production [14]. At present from a global perspective, most countries, including China [10, 15, 16], Brazil [17–19], South Africa [20, 21], Australia [22, 23], etc., mainly use E. grandis × E. urophylla or its backcross hybrids for afforestation to take advantage of heterosis. It should be noted that E. grandis is a widely used hybrid parent, and the effective evaluation, protection and utilization of the germplasm resources of E. grandis are the premise and foundation for further inter- and intraspecific hybrid breeding based on the multigeneration breeding strategy for these tree species [10, 15, 24–27].
The existing Eucalyptus germplasm resources in China were all derived from early introduction experiments. Among these different tree species, E. grandis, as one of the most preferred hybrid parent species, was systematically and comprehensively introduced in the 1980s as a part of the Dongmen State Forest Farm Eucalypt Afforestation Project (1981–1989) under the Australia-China Program of Technical Cooperation [24, 28]. Subsequently, more than fifty provenances and four hundred families of E. grandis have been tested in Guangxi, Sichuan, Zhejiang and other provinces [25, 29–36]. These populations were used to evaluate suitable provenances and planting areas in China [25, 30, 35, 37], and the preliminarily elite individuals which bloomed earlier were used for random mating to obtain hybrid varieties suitable for local cultivation in the early stage [38–40]. Unfortunately, for uncontrollable reasons, much important information and germplasm resources of E. grandis have been lost in the past few years. With the exception of some sporadic distributions of E. grandis populations in Sichuan, Guangdong and Zhejiang provinces, the Dongmen improved variety base in Guangxi is the place with the most preserved germplasms in China. At present, there are 28 populations and 265 families of E. grandis with a preservation rate of approximately 7% in Dongmen, which will be used as a base population for genetic improvement under the guidance of a multigeneration breeding strategy. However, after many rounds of natural or unnatural selection, we do not know the corresponding population structure or genetic diversity, which limits further research on the genetic improvement and germplasm utilization of E. grandis in southern China [4, 41].
In recent years, the development of molecular markers and detection technologies, including RFLP (restriction fragment length polymorphism), RAPD (random amplified polymorphic DNA), AFLP (amplified fragment length polymorphism), SSR (simple sequence repeat or microsatellite sequence), and SNP (single nucleotide polymorphism) methods, has greatly promoted research on genetic diversity and genetic differentiation [42–46]. In particular, SSR markers have become increasingly popular in genetic relationship analysis due to their codominant inheritance, high polymorphism and transferability [47–50]. In previous studies, SSR markers were used to investigate the genetic relationships and origins of elite tea plant individuals (Camellia sinensis) [51], to analyze the population structure and genetic differentiation of the Populus tomentosa breeding population [52], and to provide genetic information for the conservation and breeding of black locust (Robinia pseudoacacia) [9]. In Eucalyptus, some genetic studies have also been carried out using SSR or other molecular markers, and many suitable markers have been developed for subsequent studies on population genetics [12, 13, 53–56]. On the basis of the above studies, it is appropriate and necessary to carry out genetic analysis of the existing E. grandis germplasm resources of China over time.
Core collection is a part of the entire germplasm resource selected using different methods to represent the genetic diversity of the entire germplasm resource to the greatest extent with the minimum number of resources and genetic duplication. This approach can promote the efficient evaluation, research and utilization of germplasm resources [57]. The core collection is a dynamic preservation process that is continuously updated to replenish rare and useful individuals and represents the initial population to the greatest extent ensuring that the core collection has a high level of genetic diversity [58]. Problems, such as incomplete data and technical defects, are common in early core collection research and seriously affect the core collection construction and the accuracy of evaluation [59]. Many tree species do not have a core collection: this way results in a lack of important genotypes of the tree species, and the tree species will not receive targeted protection. Therefore, by constructing a core collection, it is easier to understand the environmental adaptability and genetic characteristics of different germplasms. The core collection can represent the genetic information and genetic diversity of the entire germplasm resource.
To improve the protection and utilization of existing germplasm resources, we evaluated the genetic diversity of all preserved E. grandis individuals and populations in the Dongmen improved variety base, which is the most important eucalypt germplasm resource in China. Population structure and genetic diversity were analyzed using 16 SSR markers to establish the core collection and their molecular fingerprints. These results are very useful for constructing breeding populations and carrying out inter- and intraspecific hybridization based on multigeneration breeding strategies in the future.
Results
Phenotypic diversity of all Eucalyptus grandis populations
The mean value and coefficient of variation (CV) of the 28 populations for average tree height (HT) and average Diameter at Breast Height (DBH) were shown in Table S1. Compared with that in HT, the coefficient of variation of DBH exceeded 10% in most of the populations except for CHA, CDA and DCM. In addition, the coefficients of variation of HT and DBH were lowest in the CDA population with values of 4.50% and 1.91%, respectively. In contrast, the coefficients of variation of HT and DBH were highest for MM and PAD populations, with values of 18.64% and 32.33%, respectively.
Selection and analysis of SSR markers for all Eucalyptus grandis populations
Twelve genotypes were selected from 545 individuals of E. grandis for preliminary screening of markers. A total of 87 pairs of SSR markers with high amplification efficiency were selected. Furthermore, another twelve genotypes of E. grandis were selected for secondary screening, and 16 SSR markers with high amplification efficiency and polymorphism were ultimately obtained (Table S2).
The genetic diversity of these 16 SSR markers was analyzed (Table 1). All information on the private alleles in 545 E. grandis individuals based on 16 SSR markers was shown in Table S3. The inbreeding coefficient (FIS) was consistently positive, ranging from 0.037 (Locus EUCeSSR0620) to 0.678 (Locus EUCeSSR0957), with an average of 0.334. The Wright's fixation index (FIT) ranged from 0.137 (Locus EUCeSSR0620) to 0.728 (Locus EUCeSSR0019), with a mean of 0.412. The fixation index (FST) ranged from 0.090 (Locus EUCeSSR0615) to 0.175 (Locus EUCeSSR1125), with an average of 0.122. All polymorphism information content (PIC) values of the markers were greater than 0.55, averaging 0.707. Among the FIS, FIT, and PIC values, those of Locus EUCeSSR0620 were the lowest of all the loci.
Table 1.
Information of genetic diversity of 16 SSR markers assessed in 545 individuals
| Locus | GN | RA | FIS | FIT | FST | Fnull | PIC |
|---|---|---|---|---|---|---|---|
| EUCeSSR0701 | 34 | 181–235 | 0.654 | 0.714 | 0.174 | 0.292 | 0.739 |
| EUCeSSR0696 | 23 | 187–211 | 0.131 | 0.222 | 0.105 | 0.069 | 0.629 |
| EUCeSSR0689 | 36 | 89–135 | 0.499 | 0.575 | 0.153 | 0.210 | 0.662 |
| EUCeSSR0845 | 33 | 168–196 | 0.557 | 0.608 | 0.116 | 0.213 | 0.593 |
| EUCeSSR0056 | 31 | 214–262 | 0.168 | 0.274 | 0.127 | 0.107 | 0.784 |
| EUCeSSR0037 | 38 | 166–208 | 0.328 | 0.392 | 0.096 | 0.181 | 0.645 |
| EUCeSSR0270 | 20 | 147–187 | 0.141 | 0.221 | 0.094 | 0.071 | 0.654 |
| EUCeSSR0019 | 34 | 170–196 | 0.673 | 0.728 | 0.169 | 0.285 | 0.736 |
| EUCeSSR0957 | 69 | 220–268 | 0.678 | 0.718 | 0.123 | 0.318 | 0.884 |
| EUCeSSR0615 | 74 | 178–226 | 0.288 | 0.352 | 0.090 | 0.162 | 0.874 |
| EUCeSSR0045 | 37 | 159–181 | 0.316 | 0.379 | 0.092 | 0.142 | 0.695 |
| EUCeSSR0822 | 38 | 165–240 | 0.394 | 0.477 | 0.137 | 0.181 | 0.730 |
| EUCeSSR0592 | 48 | 223–259 | 0.288 | 0.365 | 0.108 | 0.120 | 0.695 |
| EUCeSSR1125 | 59 | 187–207 | 0.089 | 0.249 | 0.175 | 0.052 | 0.706 |
| EUCeSSR0620 | 23 | 132–153 | 0.037 | 0.137 | 0.104 | 0.052 | 0.584 |
| EUCeSSR0857 | 24 | 163–189 | 0.100 | 0.182 | 0.092 | 0.066 | 0.708 |
| Mean | 39 | 0.334 | 0.412 | 0.122 | 0.158 | 0.707 |
GN Genotype numbers, RA Range of allele sizes, FIS Inbreeding coefficient, FIT Wright’s fixation index, FST Fixation index, Fnull Estimated frequency of null alleles, PIC Polymorphism information content
Genetic diversity analysis among 13 Eucalyptus grandis populations
It is worth noting that some of the 28 populations were not suitable for further genetic diversity and genetic structure analysis because of their remote geographical distribution and small sample size. In addition, populations with relatively close geographical distributions and small genetic distance gaps could be combined, and those populations with more than 20 individuals after merging were retained and were shown in Table S4. There were 13 populations, with a total of 492 individuals, for the subsequent analysis of genetic diversity and genetic structure.
Based on the information provided by the new populations (Table S4), out of the 13 populations (Table 2), the number of individuals (NI) within each population ranged from 21 in BRI-2 to 103 in ASS. Mean observed number of alleles (NA) ranged from 6.188 (POR) to 8.813 (ASS), averaging 7.245. Mean effective number of alleles (NE) ranged from 3.448 (POR) to 4.274 (FLO), averaging 3.845. Mean observed heterozygosity (Ho) ranged from 0.403 (POR) to 0.483 (ASS), with a mean of 0.444, and mean expected heterozygosity (He) ranged from 0.657 (POR) to 0.734 (ASS), with a mean of 0.705. Shannon's information index (I) ranged from 1.360 (POR) to 1.598 (ASS), averaging 1.497. The all I values of the populations were greater than 1.35.
Table 2.
Information of mean genetic diversity parameters of 13 Eucalyptus grandis populations
| Population | NI | NA | NE | Ho | He | Fnull | I |
|---|---|---|---|---|---|---|---|
| BRI-1 | 43 | 8.250 | 4.149 | 0.443 | 0.720 | 0.163 | 1.574 |
| BRI-2 | 21 | 6.688 | 3.628 | 0.440 | 0.681 | 0.149 | 1.444 |
| POR | 31 | 6.188 | 3.448 | 0.403 | 0.657 | 0.154 | 1.360 |
| SRA | 30 | 6.688 | 3.766 | 0.466 | 0.712 | 0.152 | 1.484 |
| WTA | 47 | 8.250 | 3.660 | 0.444 | 0.694 | 0.174 | 1.496 |
| CAI | 51 | 7.438 | 3.839 | 0.419 | 0.713 | 0.153 | 1.505 |
| SEM | 22 | 6.500 | 3.662 | 0.432 | 0.688 | 0.143 | 1.437 |
| TCR | 30 | 6.500 | 3.701 | 0.471 | 0.704 | 0.152 | 1.450 |
| TOW | 38 | 7.750 | 3.983 | 0.469 | 0.729 | 0.173 | 1.562 |
| FHG | 22 | 7.063 | 3.738 | 0.421 | 0.696 | 0.148 | 1.480 |
| ASS | 103 | 8.813 | 4.267 | 0.483 | 0.734 | 0.151 | 1.598 |
| FLO | 24 | 7.063 | 4.274 | 0.406 | 0.730 | 0.189 | 1.580 |
| SSB | 30 | 7.000 | 3.869 | 0.475 | 0.711 | 0.147 | 1.495 |
| Mean | 38 | 7.245 | 3.845 | 0.444 | 0.705 | 0.158 | 1.497 |
NI Number of individuals, NA Mean observed number of alleles, NE Mean effective number of alleles, Ho Mean observed heterozygosity, He Mean expected heterozygosity, Fnull Estimated frequency of null alleles, I Shannon’s information index
Genetic structure analysis of the Eucalyptus grandis populations
Genetic structure of 545 individuals (28 populations) and 492 individuals (13 populations) of E. grandis was analyzed by genetic distance. Among the 545 individuals, cluster I contained the smallest number of individuals (n = 40), and cluster III included the largest number (n = 282), followed by cluster II (n = 223) (Fig. S1). The genetic structure of 492 individuals was shown in Fig. S2.
Based on the above results, the genetic structure of 492 individuals from 13 populations was further analyzed. ΔK method showed that the most suitable number of clusters was K = 3 (Fig. 1A and B). Basic information of the population Q-matrix from K = 2 to K = 4 was shown in Table S5. Most of the values of population Q-matrix were not more than 0.7. The result of each run with K = 3 was shown in Fig. S3. Furthermore, the results of structural analysis with multiple K values ranged from K = 2 to K = 4 (Fig. 1C).
Fig. 1.

The suitable number of clusters and genetic structure analysis of 13 Eucalyptus grandis populations. A Values of the mean Ln P(D). B The suitable number according to the maximum K. C Genetic structure analysis of 13 populations from K = 2 to K = 4. The same color indicates the same cluster. Each population is separated by black vertical line
Principal coordinate analysis (PCoA) was performed with a matrix of pairwise FST values between 13 populations and 13 different E. grandis populations were divided into three major clusters (Fig. S4). Cluster 1 (red) was composed of two populations (WTA and FLO). Cluster 2 (green) and 3 (black) were composed of seven populations and four populations, respectively. In addition, the percentages of the first two coordinates of the principal coordinates analysis were 44.10% and 21.20%, respectively.
Population genetic differentiation analysis and Mantel test analysis
Analysis of molecular variance (AMOVA) revealed correlations revealed correlations among populations and within populations and the value of FST was 0.042. According to the results, the variance of all the E. grandis individuals was within populations rather than among populations (Table 3).
Table 3.
Basic information of analysis of molecular variance (AMOVA)
| Source of variation | d.f | Sum of squares | Variance components | Percentage of variation (%) | P-value |
|---|---|---|---|---|---|
| Among populations | 27 | 409.26 | 0.25 | 4.24 | < 0.001 |
| Within populations | 1062 | 6047.16 | 5.69 | 95.76 | < 0.001 |
| Total | 1089 | 6456.42 | 5.95 | 100 |
d.f. Degree of freedom
The Mantel test revealed no significant correlation between geographic distance and genetic distance (r = 0.013, p = 0.210), which means that the genetic difference was not due to geographic distance.
Selection and evaluation of a core collection from 545 Eucalyptus grandis individuals
Both methods were effective in screening the core collection of E. grandis. The first method analyzed Core Hunter, Power Core and Core Finder to screen out their respective core collections of E. grandis. The relationships among the core collections screened by the three screening methods were analyzed through a Venn diagram (Fig. 2). The core collection screened by Core Hunter was the most common, with 109 individuals of E. grandis, followed by 78 individuals screened by Core Finder and only 63 individuals from Power Core. Among them, the difference between the most (Core Hunter) and the least (Power Core) was 46 individuals. In addition, 27 individuals of the core collections were idebtified by the three screening methods. Their genetic diversity information was analyzed through PowerMarker 3.25 software and POPGENE32 1.32 software (Table 4). By comparing the genetic diversity of the core collections screened, it was found that the values of FIT, FST and allelic richness (AR) [60, 61] were significantly greater than the genetic diversity of the initial population. In addition, although the values of Ho in the three screening methods were roughly consistent with the value of Ho in the initial population, He significantly decreased, with a decrease of 0.105 (Core Hunter) to 0.214 (Power Core). Among the three screening methods, the value of I also decreased significantly, with the highest being 0.971 (Core Hunter) and the lowest being 0.751 (Power Core). However, compared with those of the Power Core and Core Finder populations, the genetic diversity of the core collection screened by Core Hunter was 80% or greater than the genetic diversity of the initial population.
Fig. 2.
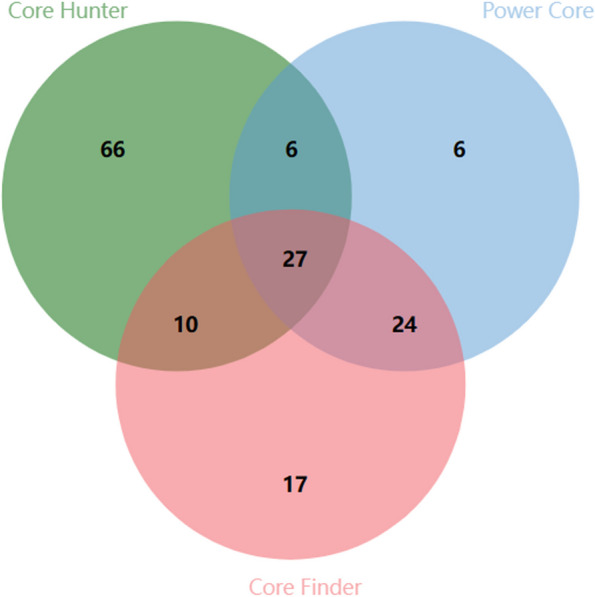
Venn diagram of core collections analyzed by three screening methods (Core Hunter, Power Core and Core Finder)
Table 4.
Genetic diversity information of three screening methods (Core Hunter, Core Finder and Power Core) for screening core collection
| Screening method | Number of individuals | Percentage of individuals | NA | NE | Ho | He | uHe | FIS | FIT | FST | AR | I |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Core Hunter | 109 | 20.00% | 3.363 | 2.746 | 0.435 | 0.542 | 0.669 | 0.200 | 0.447 | 0.318 | 5.170 | 0.971 |
| Core Finder | 78 | 14.31% | 3.182 | 2.613 | 0.439 | 0.497 | 0.620 | 0.126 | 0.433 | 0.363 | 5.190 | 0.896 |
| Power Core | 63 | 11.56% | 2.744 | 2.328 | 0.437 | 0.433 | 0.585 | 0.015 | 0.439 | 0.448 | 5.280 | 0.751 |
| Initial population | 545 | 100% | 5.453 | 3.349 | 0.429 | 0.647 | 0.685 | 0.334 | 0.412 | 0.122 | 4.470 | 1.293 |
NA Mean observed number of alleles, NE Mean effective number of alleles, Ho Observed heterozygosity, He Expected heterozygosity, uHe Unbiased expected heterozygosity, FIS Inbreeding coefficient, FIT Wright's fixation index, FST Fixation index, AR Allelic richness, I Shannon’s information index
In the second method, cluster analysis was performed using a neighbor-joining (NJ) tree. First, Nei’s genetic distance between 545 E. grandis individuals was analyzed to construct the NJ tree using PowerMarker 3.25 software (Table S6). Based on the NJ tree, branches containing only two genotypes were selected at the end of the smallest genetic distance, and one of them was deleted according to the criterion for deletion in each round of screening. A total of six rounds of screening were performed in this study, and the sample proportion was not less than 5%. The percentage of individuals preserved ranged from 100% (545 individuals) to 5.32% (29 individuals) after six rounds of screening (Table 5). After all six rounds of screening, the values of Ho and He ranged from 0.373 (6th round) to 0.454 (2nd round) and from 0.411 (6th round) to 0.617 (1st round), respectively. The Ho values of the 1st round (0.445), 2nd round (0.454) and 3rd round (0.442) were greater than that of the initial population (0.429). However, the values of He were all lower than those of the initial population. AR and I among these screenings ranged from 4.540 (1st round) to 5.180 (6th round) and 0.658 (6th round) to 1.207 (1st round), respectively. These results illustrated that the third round of screening, in which 125 individuals were preserved, was selected as the core collection of the second method.
Table 5.
Genetic diversity information of six screenings for screening core collection using cluster analysis of neighbor-joining (NJ) tree
| Round of screening | Number of individuals | Percentage of individuals | Ho | He | uHe | FIS | FIT | FST | AR | I |
|---|---|---|---|---|---|---|---|---|---|---|
| Origin | 545 | 100.00% | 0.429 | 0.647 | 0.685 | 0.334 | 0.412 | 0.122 | 4.470 | 1.293 |
| 1st round | 335 | 61.47% | 0.445 | 0.617 | 0.680 | 0.274 | 0.392 | 0.170 | 4.540 | 1.207 |
| 2nd round | 202 | 37.06% | 0.454 | 0.603 | 0.690 | 0.246 | 0.392 | 0.202 | 4.660 | 1.147 |
| 3rd round | 125 | 22.94% | 0.442 | 0.592 | 0.691 | 0.249 | 0.416 | 0.229 | 4.810 | 1.085 |
| 4th round | 77 | 14.13% | 0.424 | 0.498 | 0.634 | 0.150 | 0.444 | 0.355 | 4.910 | 0.865 |
| 5th round | 48 | 8.81% | 0.411 | 0.488 | 0.646 | 0.164 | 0.465 | 0.373 | 5.080 | 0.812 |
| 6th round | 29 | 5.32% | 0.373 | 0.411 | 0.573 | 0.112 | 0.523 | 0.479 | 5.180 | 0.658 |
Ho Observed heterozygosity, He Expected heterozygosity, uHe Unbiased expected heterozygosity, FIS Inbreeding coefficient, FIT Wright’s fixation index, FST Fixation index, AR Allelic richness, I Shannon’s information index
Discussion
Understanding the genetic relationship of the Eucalyptus grandis population and determining its genetic diversity and genetic structure are highly important for protecting and utilizing the germplasm resources of E. grandis. Although the percentage of preservation was only approximately 7%, the germplasm resources of the E. grandis population in Dongmen are comprehensive and unique and represent the provenance of E. grandis in Southwest China. At the same time, the 545 individuals selected in this study not only revealed genetic relationships but also provided help for the analysis of multigeneration breeding and the selection of hybrid combinations with relatively high genetic distances. The core collection is selected for more efficient utilization and to further screen elite breeding populations through phenotypic traits, such as the average tree height (HT) and the average diameter at breast height (DBH).
In the present study, we analyzed the genetic diversity and genetic structure of the E. grandis population. We selected twelve genotypes of E. grandis from 545 individuals, and 16 SSR markers with high amplification efficiency and high polymorphism were selected. These 16 SSR markers not only provide efficient marker information for the present study, but also for future experimental research on E. grandis. In addition, among the 16 SSR markers, some were also present in the literature. Locus EUCeSSR1125 was used to analyze the genetic diversity of Corymbia citriodora populations, and the Shannon's information index (I) and polymorphism information content (PIC) values were 2.681 and 0.911, respectively [62]. These two values were slightly greater than the values in this study (I = 1.537 and PIC = 0.706). The locus EUCeSSR0620 was used to analyze the genetic diversity of Eucalyptus cloeziana F. Muell. populations, and the value of I was 2.0216 [63]. Similarly, this value was also higher than that of this study (I = 1.078). The genetic diversity level of the population in this study was lower than that in the previous two studies, possibly because the E. grandis in this study experienced long-term high-intensity selection, and the remaining individuals fully adapted to the local growth environment.
Ho is the observed heterozygosity, and He is the expected heterozygosity. Both of these indices are critical for analyzing genetic diversity. Previously, SSR markers were used to analyze the genetic diversity of 159 E. grandis individuals from 16 provenances, and the Ho and He values were 0.743 and 0.774, respectively [64]. The reason why the values were significantly greater than those in the present study (Ho = 0.444 and He = 0.705) might be the large geographical range, high levels of population diversity and genetic differentiation among populations [64]. The mean PIC for 16 pairs of markers was 0.707 in this study. Similar results were also obtained by two previous studies. In a commercial eucalypt forest, 80 E. grandis individuals were selected for genetic diversity analysis using 17 EST-SSR markers, and the PIC was 0.648 [65]. Furthermore, 12 pairs of SSR markers were used to analyze the genetic diversity of the E. grandis population, and the PIC value was 0.54 [66]. According to the research [67], the values of the PIC in the three previous studies were all within the ideal range, but the PIC in this study was significantly greater. The reason is that this study selected markers with high amplification efficiency and high polymorphism. Furthermore, the sample size in this study was greater, the geographical range was wider, the genetic diversity of the E. grandis population was greater, and there was a certain degree of genetic differentiation between populations. At the same time, in other eucalypt species, we also found that the influence of abundant sample numbers and large geographical range coverage on genetic diversity was significant. In a previous study, the genetic diversity of 707 E. grandis individuals, including 114 open-pollinated families from 17 provenances, was analyzed [68]. These individuals were analyzed by 12 pairs of SSR markers, and the values of Ho, He and PIC were 0.432, 0.682 and 0.645, respectively. These three values of genetic diversity were very close to those in this study (Ho = 0.444, He = 0.705 and PIC = 0.707).
In this study, among the 28 populations, a few populations were removed due to containing small numbers of individuals and were not suitable for subsequent genetic structure analysis. We also combined populations with relatively close geographical provenances and small genetic distance differences. A total of 492 individuals from 13 populations were obtained. Principal coordinate analysis (PCoA) revealed three major clusters, and the clusters in the PCoA analysis were also reflected in the neighbor-joining (NJ) tree and genetic structure analysis. The genetic structure analysis provided a clear indication of the clusters and the genetic composition. The most likely number of genetic clusters was K = 3.
To determine the genetic diversity among the populations and within the populations, we carried out analysis of molecular variance (AMOVA) and the results showed that the percentage of variation within the populations was much greater than that among the populations. These results revealed that there was variation within the populations, and the following analysis was also carried out within the populations. In addition, the Mantel test showed that geographic distance was not related to genetic distance.
According to the above results, it could be found that the inbreeding coefficient (FIS) value of each population was relatively high, which indicated that the populations had a high inbreeding. This may be related to the origin source of the population in China. As an introduced tree species in China, Eucalyptus originated from the Australia. Due to the significant role of Eucalyptus in the industrial production, it had been introduced in many countries in order to improve the productivity and stability of forests, scientists have divided different climate zones according to phenological conditions and determined suitable climate zone through provenance tests [69]. With the development of the Dongmen State Forest Farm Eucalypt Afforestation Project (1981–1989) under the Australia-China Program of Technical Cooperation [24, 28], China also began to introduce targeted Eucalyptus from Australia, Brazil, South Africa and other countries. The greatest likelihood of successful introduction of woody plants lay in the similarity of climatic conditions at the origin of the tree species [70]. The provenance of introduced E. grandis was determined by comparing the climate in China and the provenance area [71]. As time passed, the number of E. grandis would gradually decrease, due to natural conditions such as climate and soil condition and artificial screening and thinning [72]. During the above introduction process, on the one hand, when E. grandis is introduced from similar climate zones in different countries, there may be close genetic relationships and repeated introductions. On the other hand, these provenances introduced into China may have originated from the same or similar original provenances earlier, and experienced the natural and unnatural selection to adapt to the local climate [73, 74]. As a result, individuals retained after intensive selection may have close genetic relationships, or even originally originated from similar provenances or families. An important characteristic of the small and isolated bottleneck populations left over from intense selection is inbreeding [75–77]. However, the adverse effects of inbreeding on plants are minimal. Compared with the reproductive mode of animal K strategy, r strategy of plant was to eliminate effects of inbreeding by increasing the seed yield and reproductive rate [78–83].
The estimated frequency of null alleles (Fnull) was calculated and analyzed. Null alleles are a common issue in the microsatellite markers, they are not caused by DNA quality or experimental technology. Instead, they primarily arise during the amplification, the in-flanking region sequences of mutations and substitutions or short alleles preferential amplification owing to inconsistent DNA template quality and quantity [84]. The influence of null alleles is very significant, which may lead to an excess of homozygotes in the population, making the Ho, He and other values lower than the normal. The genetic differentiation and genetic distance between populations can be increased. In this study, only 5 out of 16 Fnull were below 0.10, and the average Fnull was 0.158. The relatively high FIS and Fnull values might indicate that these factors influenced the estimate of genetic diversity in the E. grandis, although the natural and unnatural selection could lead to close genetic relationships [85, 86].
The genetic diversity and genetic structure analysis of 492 E. grandis individuals had been calculated by PowerMarker 3.25, STRUCTURE 2.3.3 and other software, and the cluster analysis was further carried out. However, it was worth noting that when we analyzed the genetic structure, we found that the bootstrap values were not high, only close to 50% (Fig. S2). In general, the overall bootstrap values are low, which may be caused by the number of individuals or the similarity between the individuals is too strong [87]. This phenomenon will directly cause the population genetic structure is not obvious, and have a certain impact on the final clustering result [88]. However, by Structure Harvester and NJ tree analysis (we set both the burn-in period and MCMC to 300,000 in order to increase the accuracy of calculation results), 492 individuals were able to cluster well and obtained the most suitable number of clusters, which had the similar situation in Erianthus arundinaceum and Vaccinium spp [89, 90]. It indicates that this phenomenon is relatively common in the genetic research. The inbreeding of E. grandis in each population constantly increased and genetic admixing was high because of the long-term natural and unnatural influence, resulting in the increase of FIS value and the lower Q-matrix and bootstrap values. However, due to different countries and provenances, there were differences between different populations of E. grandis in essence. But the effects of inbreeding prevented them from being visually observable in the numerical value, or the number of individuals were still insufficient. In the next few decades, it is necessary to continue comprehensive and systematic introduced in order to cluster more intuitively.
To better collect and protect the germplasm resources of E. grandis, we selected and evaluated the core collection. We compared the two methods of screening core collection with respect to genetic diversity, and ultimately, the genetic diversity values of Core Hunter met the criteria for screening the core population. All values of genetic diversity were 80% or greater of the genetic diversity values of the initial population. Therefore, the first method used the core population screened by Core Hunter as the core population of E. grandis. The first method identified 109 individuals as the core population of E. grandis. In the second method, the 3rd round of screening was selected as the core collection. In the third round of screening, 125 individuals of E. grandis were screened, the percentage of individuals was approximately 23% of the initial population, and the percentage of each genetic diversity value was approximately 80% that of the initial population. Furthermore, 76 individuals of the core collections were shared by the first method (Core Hunter) and second method (cluster analysis using an NJ tree). Excluding those 76 individuals in total, 33 individuals and 49 individuals still existed in the first and second methods, respectively. In this study, we collected all the individuals screened via the first method as the core collection of E. grandis and collected the remaining individuals via the second method as the expanded collection. Overall, 158 individuals were selected as the final core collection by the two methods (including 109 in the main core collection and 49 in the expanded collection). Genetic structure analysis of the 158 selected individuals based on a NJ tree is shown in Fig. 3. Among the 158 individuals, cluster β contained the largest number of individuals (n = 92), and cluster γ included the smallest number (n = 7), followed by cluster α (n = 59).
Fig. 3.

Genetic structure of 158 selected populations based on the neighbor-joining (NJ) tree. A total of 158 individuals were separated into three major clusters. The numbers of individuals in cluster α, cluster β and cluster γ are 59 (orange), 92 (pale blue) and 7 (pink), respectively
The experiment used as few markers as possible to completely separate the 158 individuals and built better fingerprinting codes [51, 91]. Therefore, identity analysis was performed on 158 individuals using CERVUS 3.0.7 software, and 158 individuals could ultimately be distinguished using 9 SSR markers among the 16 SSR markers. The 9 SSR markers were the EUCeSSR0270, EUCeSSR0019, EUCeSSR0957, EUCeSSR0045, EUCeSSR0822, EUCeSSR0592, EUCeSSR1125, EUCeSSR0620 and EUCeSSR0857 loci. The unique fingerprinting codes of the 158 individuals were established based on 9 SSR markers and allele sizes (Table S7). In addition, QR codes were established among the 158 individuals (Fig. S5) and three QR codes (C03, F01, I04) were shown as examples (Fig. 4). QR codes information included the ID, species, cultivation region and fingerprinting codes. Furthermore, the distributions of the HT and DBH tended to increase during core collection and raw collection/initial collection (Fig. S6). These results revealed that the core collection was highly consistent with the raw collection, and the 158 selected core collection could fully represent the entire population.
Fig. 4.

Examples of DNA Fingerprint QR codes for 158 individuals of Eucalyptus grandis. A, B and C are three examples that can be scanned to read information, which contains the ID, species, cultivation region and fingerprinting code
The 158 selected core collection of E. grandis can be used for further research, for instance, research on the selection of superior parents of E. grandis as superior hybrid parents and combinations of superior hybrid parents is lacking. Many studies have shown that genetic distance is positively correlated with heterosis and superior hybrid parents can be selected based on genetic distance [10, 25–27]. In view of phenotypic determination (screening individuals with excellent HT and DBH phenotypic traits) and analysis of genetic diversity and genetic distance, superior hybrid parents of E. grandis can be conducive to the further development of intra- and interspecific hybrid breeding of E. grandis, the construction of dominant hybrid parent combinations of E. grandis and the provision of superior parent resources for germplasm innovation research and breeding population construction. We could better select elite individuals with fast-growing, good stem type and strong stress resistance through this method to achieve better planting effects.
Conclusions
Through various analysis, 158 individuals (28.99% of the initial population) were selected as the core collection of Eucalyptus grandis. It was composed of the individuals based on the intensive natural and unnatural selection through further genetic analysis and other screenings, which could better preserve the germplasm resources of E. grandis in China with a small number of individuals and relatively rich genetic diversity. Individuals excluded in the core collection would be expanded collection, fully representing the entire population. The core germplasm can be preserved in different regions through asexual reproduction and plays an important role in genetic improvement. For example, based on the genetic distance and other analysis in this study, high-quality hybrid combinations with high genetic distance were selected from the core collection, and genetic parameters such as general combining ability and specific combining ability were calculated through the hybridization among individuals within the population and progeny test. Based on the above analysis, the excellent hybrid parents and high-quality hybrid combinations could be selected. Then, targeted breeding of excellent hybrid offspring can be carried out to obtain excellent individuals of E. grandis.
This study provides valuable genetic materials and has an important theoretical foundation for constructing breeding population of E. grandis in China, which is helpful to promote the breeding cycle in this tree species.
Materials and methods
Plant material and phenotypic data
In this study, we used all existing Eucalyptus grandis germplasms from the Dongmen improved variety base (Guangxi Dongmen Forest Farm, Chongzuo, China), which were preserved at the Dongmen State Forest Farm through the Eucalypt Afforestation Project (1981–1989) under the Australia-China Program of Technical Cooperation [24, 28], as the research material. During this project, a total of 11813 individuals of E. grandis from 33 populations and 522 families were introduced from Australia, the USA, Brazil and South Africa to Dongmen, China. After multiple rounds of natural and unnatural selection, only 805 individuals belonging to 28 populations and 265 families survived and were preserved. In the present study, 545 individuals were sampled and renumbered after individuals with serious insect pests, disease, obvious growth deficiency or redundancy were removed (Table S4). Due to differences in the number of individuals in different families, some families had only 1 or 2 individuals, and the growth of some individuals in such families was not ideal. Therefore, to ensure the integrity of the sample and the authenticity of the data, during the sampling process, we tried to ensure that all families with fewer than or equal to 3 individuals were chosen [92, 93]. The information of the above populations was shown in Fig. 5, which was made with Tableau Desktop software (https://www.tableau.com/zh-cn/products/desktop/download). To ensure the number of samples and facilitate the subsequent extraction of DNA, four to six tender leaves were harvested from each individual, and stored in a package containing silica gel until they were completely dry before moving on to the next operation.
Fig. 5.

Information of twenty-eight populations analyzed. The number of accessions analyzed is shown in parentheses. All the information of Population ID is presented in Table S4
In addition, 545 individuals of E. grandis were tested for phenotypic traits. To ensure the authenticity and validity of the phenotypic data, 545 individuals of E. grandis were measured for six years. The tested phenotypic traits included average tree height (HT) and average Diameter at Breast Height (DBH). The fixed time for measuring phenotypic traits was maintained annually. The phenotypic traits of each individual were measured three times.
DNA extraction
The dry leaves of each sample were torn into small pieces and stored in a 1.5 mL centrifuge tube. Then, 5 mm steel balls were added to each centrifuge tube, and the leaves were disrupted using a Mini-Bead beater-96 cell disrupter (Biospec Products, Bartlesville, OK, USA). Finally, approximately 0.5/g disrupted leaves were placed into a new 1.5 mL centrifuge tube. Genomic DNA was extracted using the DP360 Super Plant Genomic DNA Kit (TIANGEN Biotech, Beijing, China). After extraction, the quality of the genomic DNA was determined using 1% agarose gel, and quantitative analysis was performed using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA). All the extracted DNA was quickly stored at -20 °C freezer for further analysis.
SSR marker screening and PCR amplification
A preliminary experiment was designed to screen 240 highly polymorphic SSR markers [94]. These 240 SSR markers were screened for Eucalyptus in previous studies. However, due to the different genotypes of Eucalyptus, it is necessary to further verify which SSR markers are suitable for the materials in this study. 12 different genotypes of E. grandis were selected from the 545 individuals to maximize the selection of SSR markers with high polymorphism and high amplification efficiency. Through screening, SSR markers with poor amplification efficiency and low polymorphism were removed, and 87 SSR markers were obtained. Then, another 12 different genotypes of E. grandis were selected for rescreening. According to the two screening results, 16 pairs of SSR markers with high polymorphism and high amplification efficiency were ultimately selected. These SSR markers were synthesized by TSINGKE Biological Technology Co., Ltd. (Beijing, China).
The TP-M13-SSR polymerase chain reaction (PCR) system was performed in a 20 μL reaction system including 2 μL (30 ng/μL) of genomic DNA, 10 μL 2 × Accurate Taq Master Mix (TSINGKE, Beijing, China), 0.08 μL forward marker, 0.32 μL reverse marker, 0.4 μL M13 labeled marker and 7.2 μM ddH2O [95, 96]. PCR amplification was performed using a Bio-Rad T100 Thermo Cycler (Bio-Rad Laboratories Inc. Hercules, CA, USA) with the following conditions: initial denaturation for 4 min at 94 °C; 8 cycles of 30 s at 94 °C, 30 s at 60 °C, and 30 s at 72 °C; 10 cycles of 30 s at 94 °C, 30 s at 52 °C and 30 s at 72 °C; 10 cycles of 30 s at 94 °C, 30 s at 56 °C and 30 s at 72 °C; 10 cycles of 30 s at 94 °C, 30 s at 60 °C and 30 s at 72 °C; and a final extension at 72 °C for 10 min and holding at 4 °C [97]. The PCR products were subjected to capillary electrophoresis using an ABI 3730XL DNA sequencer (Applied Biosystems, CA, USA). The results were analyzed using GeneMarker 2.2.0 software (SoftGenetics LLC., PA, USA), which can determine allele sizes and peak intensities [98].
Genetic diversity analysis
We sorted the range of allele sizes (RA) and calculated the genotype numbers (GN) and polymorphism information content (PIC) for the SSR markers using PowerMarker 3.25 software [99]. According to previous research [67], when the PIC value is greater than 0.5, it is very informative, when the PIC value is between 0.25 and 0.50, it is somewhat informative, and when it is less than 0.25, the PIC value is not very informative. The genotype data were converted into different formats for further statistical analysis using Convert 1.3.1 software [100]. The inbreeding coefficient (FIS), Wright's fixation index (FIT) and fixation index (FST) for each SSR marker were estimated using GenAlEx 6.51 software [101]. Estimated frequency of null alleles (Fnull) was analyzed by FreeNA software [102]. The number of individuals (NI), mean observed number of alleles (NA), mean effective number of alleles (NE), mean observed heterozygosity (Ho), mean expected heterozygosity (He) and Shannon’s information index (I) for the populations were calculated using POPGENE32 1.32 software [103]. Allelic richness (AR) was analyzed by HP-Rare Desktop Software [104].
Population structure and principal coordinates analysis (PCoA)
The population structure was analyzed using the Bayesian clustering algorithm in STRUCTURE 2.3.3 software [105]. In this analysis, we evaluated 1 to 15 K values, and each K value was calculated in 10 runs. Each run was performed with a burn-in period of 300,000 iterations and a Markov chain Monte Carlo (MCMC) of 300,000 replications. The online program Structure Harvester [106] was used to determine the most likely number of clusters (K) based on the ΔK method [107]. Bar plots of the Q-matrix results were generated with CLUMPP 1.1.2 software [108] and Distruct 1.1 software [109]. PowerMarker 3.25 software [99] was used to analyze the genetic relationships based on Nei’s distance analysis to construct a neighbor-joining (NJ) tree, which was further visualized using FIGTREE 1.3.1 software (http://tree.bio.ed.ac.uk/software/figtree/) [110]. The different genotypes of E. grandis were grouped via principal coordinate analysis (PCoA) in the GenAlEx 6.51 software [101]. PCoA was based on the data type of the tri-distance matrix and the distance-standardized method to determine the differences among the populations and within the populations.
Differentiation analysis and Mantel analysis
Analysis of molecular variance (AMOVA) was performed using Arlequin 3.5 software [111, 112], and statistical significance was determined from 1,000 permutations. According to the genetic distance and geographic distance data, the Mantel test was used to analyze the correlations between them via the GenAlEx 6.51 software [101].
Core collection analysis
The core collection of E. grandis was constructed by different methods. In the first method, Core Hunter [113], Power Core [114] and Core Finder [115] could be used to screen the core collection of 545 individuals of E. grandis, analyze their genetic diversity, and compare it with the genetic diversity of the initial population. For the most suitable core collection, each value of genetic diversity should not be less than 80% of the initial population. In the second method, one of the only two genotypes at the end of the branches in the NJ tree was deleted. The criteria for deletion were as follows: first, the proportion of the core collection was approximately 5%-30% of the initial population; second, the percentage of each value of the core collection should be approximately 80% of the initial population and the phenotype of the core collection could not vary by approximately 20% [116]. CERVUS 3.0.7 software (Field Genetics Ltd., London, UK) was used to select and construct the fingerprinting codes [117, 118]. In addition, QR codes were created using an Ostools online program (OSCHINA) (https://tool.oschina.net/qr/) to distinguish all selected individuals. Furthermore, a Venn diagram was created through a Jvenn online program (http://jvenn.toulouse.inra.fr/app/example.html) to analyze and compare the relationships between various methods of screening the core population.
Supplementary Information
Acknowledgements
The authors are grateful to Jianzhong Wang, Tao Xiong and Yingwei Zhao from Guangxi Dongmen Forest Farm for field data collection and additional help.
Authors’ contributions
JY and XK conceived and designed the research. CL and LZ conducted the experiments. LZ, ZP, XL and ZL collected the materials. CL, LZ and TL analyzed the data. CL and JY wrote the manuscript. XK provided valuable suggestions and reviewed the manuscript. CL and LZ contributed equally to this work. All authors read and approved the final manuscript.
Funding
This research was supported by the National Key R&D Program of China during the 14th Five-year Plan Period (2021YFD2200105) and the 5·5 Engineering Research & Innovation Team Project of Beijing Forestry University (BLRC2023C06).
Data availability
The datasets generated and analyzed during the current study are available in the Supplementary Materials.
Declarations
Ethics approval and consent to participate
The plant material of Eucalyptus grandis used in this research was collected from Guangxi Dongmen Forest Farm at Dongmen Town and its surrounding areas in Chongzuo City, China (22°20′20″N, 107°50′52″E), which was done with the permission of the local management department. Eucalyptus grandis has been widely cultivated over the past few decades, which is neither from the wild nor at risk of extinction. All samples collected are stored in the National Engineering Research Center of Tree Breeding and Ecological Restoration, Beijing Forestry University (Beijing, China). The experiments were performed in accordance with all relevant Chinese laws.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Chenhe Li and Lei Zhang contributed equally to this work.
References
- 1.Kang X. Research progress of forest genetics and tree breeding. J Nanjing Forestry University (Natural Sciences Edition). 2020;44:1–10. 10.3969/j.issn.1000-2006.202002033. [Google Scholar]
- 2.Namkoong G, Kang HC, Brouard JS. Tree breeding: principles and strategies. New York, NY, USA: Springer Verlag; 1988. [Google Scholar]
- 3.Bouffier L, Klapste J, Suontama M, Dungey HS, Mullin TJ. Evaluation of forest tree breeding strategies based on partial pedigree reconstruction through simulations: Pinus pinaster and Eucalyptus nitens as case-studies. Can J For Res. 2019;49:1504–15. 10.1139/cjfr-2019-0145. [Google Scholar]
- 4.Kang X. Thoughts on tree breeding strategies. Journal of Beijing Forestry University. 2019;41:15–22. 10.12171/j.1000-1522.20190412.
- 5.Talbert JT. An advanced-generation breeding plan for the N.C. State University-Industry pine tree improvement cooperative. Silvae Genetica. 1979;28:72–75.
- 6.Han Z, Han Q, Xia Y, Geng X, Du K, Yang J, Kang X. Construction of a breeding parent population of Populus tomentosa based on SSR genetic distance analysis. Sci Rep. 2020;10:18573. 10.1038/s41598-020-74941-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grattapaglia D, Mamani EMC, Silva-Junior OB, Faria DA. A novel genome-wide microsatellite resource for species of Eucalyptus with linkage-to-physical correspondence on the reference genome sequence. Mol Ecol Resour. 2014;15:437–48. 10.1111/1755-0998.12317. [DOI] [PubMed] [Google Scholar]
- 8.Jin Y, Ma Y, Wang S, Hu X, Huang L, Li Y, Wang X, Mao J. Genetic evaluation of the breeding population of a valuable reforestation conifer Platycladus orientalis (Cupressaceae). Sci Rep. 2016;6:34821. 10.1038/srep34821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo Q, Cao S, Dong L, Li X, Zhang J, Zhang Y, Zhang Z, Sun Y, Long C, Fan Y, Han C, Han P, Liu X, Li Y. Genetic diversity and population structure of Robinia pseudoacacia from six improved variety bases in China as revealed by simple sequence repeat markers. J For Res. 2022;33:611–21. 10.1007/s11676-021-01356-2. [Google Scholar]
- 10.Xie Y. Research progress on Eucalyptus breeding and its strategy in China. World Forest Res. 2011;24:50–54. 10.13348/j.cnki.sjlyyj.2011.04.015.
- 11.Rezende GDSP, Resende MDV, Assis TF. Eucalyptus breeding for clonal forestry. In: Fenning T, editor. Challenges and opportunities for the world's forests in the 21st century. Dordrecht: Springer, Netherlands. 2014;393–424. 10.1007/978-94-007-7076-8_16.
- 12.Balasaravanan T, Chezhian P, Kamalakannan R, Ghosh M, Yasodha R, Varghese M, Gurumurthi K. Determination of inter- and intra-species genetic relationships among six Eucalyptus species based on inter-simple sequence repeats (ISSR). Tree Physiol. 2005;25:1295–302. 10.1093/treephys/25.10.1295. [DOI] [PubMed] [Google Scholar]
- 13.Cupertino FB, Leal JB, Corrêa RX, Gaiotto FA. Genetic diversity of Eucalyptus hybrids estimated by genomic and EST microsatellite markers. Biol Plant. 2011;55:379–82. 10.1007/s10535-011-0059-x. [Google Scholar]
- 14.Hung TD, Brawner JT, Meder R, Lee DJ, Southerton S, Thinh HH, Dieters MJ. Estimates of genetic parameters for growth and wood properties in Eucalyptus pellita F Muell. to support tree breeding in Vietnam. Ann For Sci. 2015;72:205–217. 10.1007/s13595-014-0426-9.
- 15.Wang H. On the strategy to develop plantations with eucalypts in southern China. World Forestry Res. 1989;2:48–55. [Google Scholar]
- 16.Ouyang L, Arnold RJ, Chen S, Xie Y, He S, Liu X, Zhang W. Prediction of the suitable distribution of Eucalyptus grandis in China and its responses to climate change. New Forest. 2022;53:81–99. 10.1007/s11056-021-09845-2. [Google Scholar]
- 17.Souza TS, Ramalho MAP, Lima BM, Rezende GDSP. Performance of Eucalyptus clones according to environmental conditions. Sci For. 2017;45:601–610. 10.18671/scifor.v45n116.01.
- 18.Souza TS, Lima BM, Lima JL, Aguiar AM, Dias DC, Rezende GDSP, Ramalho MAP. Selection of eucalypt clones with higher stability in pulp yield. Rev Árvore. 2020;44:1–9. 10.1590/1806-908820200000003. [Google Scholar]
- 19.Ramalho MAP, Marques TL, Lemos RDC. Plant breeding in Brazil: Retrospective of the past 50 years. Crop Breed Appl Biotechnol. 2021;21:e383021S3. 10.1590/1984-70332021v21sa16.
- 20.Toit BD, Dovey SB. Effect of site management on leaf area, early biomass development, and stand growth efficiency of a Eucalyptus grandis plantation in South Africa. Can J For Res. 2005;35:891–900. 10.1139/x04-205. [Google Scholar]
- 21.Santos SAO, Vilela C, Domingues RMA, Oliveira CSD, Villaverde JJ, Freire CSR, Neto CP, Silvestre AJD. Secondary metabolites from Eucalyptus grandis wood cultivated in Portugal, Brazil and South Africa. Ind Crops Prod. 2017;95:357–64. 10.1016/j.indcrop.2016.10.044. [Google Scholar]
- 22.Forrester DI, Baker TG. Growth responses to thinning and pruning in Eucalyptus globulus, Eucalyptus nitens, and Eucalyptus grandis plantations in southeastern Australia. Can J For Res. 2012;42:75–87. 10.1139/x11-146. [Google Scholar]
- 23.Smethurst PJ, Valadares RV, Huth NI, Almeida AC, Elli EF, Neves JCL. Generalized model for plantation production of Eucalyptus grandis and hybrids for genotype-site-management applications. For Ecol Manage. 2020;469: 118164. 10.1016/j.foreco.2020.118164. [Google Scholar]
- 24.Wang H. Research on introduction and cultivation of Australian broad-leaved trees species- China-Australia cooperative research project. For Res. 1988;1:112–3. [Google Scholar]
- 25.Wang H, Yan H, Zhou W. Provenance trials and prediction of suitable planting area based on bioclimatic analysis for Eucalyptus grandis in China. For Res. 1989;2:411–9. [Google Scholar]
- 26.Xie Y. Study on Eucalyptus selection objectives and current situation of genetic resources in China. Eucalypt Sci Technol. 2012;29:33–39. 10.13987/j.cnki.askj.2012.02.008.
- 27.Liu G, Xie Y, Chen H, Wu Z. Genetic diversity among the introduced germplasms of Eucalyptus in China. Acta Botan Boreali-Occiden Sin. 2017;37:2163–71. 10.7606/j.issn.1000-4025.2017.11.2163. [Google Scholar]
- 28.Turnbull JW. Development of sustainable forestry plantations in China: a review. Impact Assessment Series Report No. 45, Australian Centre for International Agricultural Research. 2007.
- 29.Chen YL. Studies on Eucalyptus grandis provenances from Australia. J Fujian Forest Sci Technol. 2000;27:29–31. 10.13428/j.cnki.fjlk.2000.s1.009.
- 30.Hu T, Li X, Zeng P, Feng D, Hou D, Du J. Experimental study on selection of superior germplasm resources of Eucalyptus grandis in Sichuan province. Eucalypt Sci Technol. 2000;48–55. 10.13987/j.cnki.askj.2000.01.011.
- 31.Liang J, Li Z, Huang Z, Zhu R, Huang X. Early selection in cold-resistant Eucalyptus II. Analysis on growth of the young forests of Eucalyptus grandis provenances and families. Journal of Central South Forestry University. 2000;20:75–79. 10.14067/j.cnki.1673-923x.2000.03.014.
- 32.Zou X, Liang Y, Hu Y. Correlation analysis of Eucalyptus grandis provenances and families. Acta Agriculturae Universitatis Jiangxiensis. 2003;25:60–64. 10.13836/j.jjau.2003016.
- 33.Huang D. The introduction tests of provenances and families of Eucalyptus grandis. Journal of Fujian Forestry Sci Technol. 2008;35:7–11. 10.13428/j.cnki.fjlk.2008.04.029.
- 34.Zhang J, Chen G, Xu J, Wu S, Zhu Y, Zeng Y, Li L. Comprehensive selection for Eucalyptus grandis provenances and families. J Tropical Subtropical Botany. 2016;24:280–286. 10.11926/j.issn.1005-3395.2016.03.006.
- 35.Wu S, Chen G, Xu J, Li G, Zhu Y, Song P, Guo W. Variation analysis and selection for Eucalyptus grandis provenances and families in multiple-sites. Forest Environ Sci. 2016;32:10–5. [Google Scholar]
- 36.Wang W. Genetic analyses and genotypic selection from an 18 years old half-sib progeny trial of Eucalyptus grandis. Eucalypt Sci Technol. 2018;35:16–22. 10.13987/j.cnki.askj.2018.01.004.
- 37.Zheng S. Introduction of Eucalyptus grandis and superior genotypes selection of Eucalyptus grandis. Chin Forest Sci Technol. 2010;24:101–3. [Google Scholar]
- 38.Zheng Z. Research progress in clonal Eucalyptus urophylla × E. grandis DH32–29 plantation. Protect Forest Sci Technol. 2013;7:64–68. 10.13601/j.issn.1005-5215.2013.07.039.
- 39.Wei C, Li T. Growth regularity of Eucalyptus urophylla × E. grandis DH32–29. Agricultural Res Appl. 2014;4:22–24.
- 40.Chen S, Liao M, Liu X, Chen G, Chen Y, Lin J. Drying characteristics of Eucalyptus grandis × E. urophylla GLGU9 woods with different ages. Guangxi Forest Sci. 2020;49:442–446. 10.19692/j.cnki.gfs.2020.03.023.
- 41.Yang H, Liao H, Zhang W, Pan W. Genome-wide assessment of population structure and genetic diversity of Eucalyptus urophylla based on a multi-species single-nucleotide polymorphism chip analysis. Tree Genet Genomes. 2020;16:47. 10.1007/s11295-020-01441-3. [Google Scholar]
- 42.Thomson MJ, Tai TH, McClung AM, Lai X-H, Hinga ME, Lobos KB, Xu Y, Martinez CP, McCouch SR. Mapping quantitative trait loci for yield, yield components and morphological traits in an advanced backcross population between Oryza rufipogon and the Oryza sativa cultivar Jefferson. Theor Appl Genet. 2003;107:479–93. 10.1007/s00122-003-1270-8. [DOI] [PubMed] [Google Scholar]
- 43.Tiwari G, Singh R, Singh N, Choudhury DR, Paliwal R, Kumar A, Gupta V. Study of arbitrarily amplified (RAPD and ISSR) and gene targeted (SCoT and CBDP) markers for genetic diversity and population structure in Kalmegh [Andrographis paniculata (Burm. f.) Nees]. Ind Crops Prod. 2016;86:1–11. 10.1016/j.indcrop.2016.03.031.
- 44.Costa R, Pereira G, Garrido I, Tavares-de-Sousa MM, Espinosa F. Comparison of RAPD, ISSR, and AFLP molecular markers to reveal and classify orchardgrass (Dactylis glomerata L.) germplasm variations. PLoS One. 2016;11:e0152972. 10.1371/journal.pone.0152972. [DOI] [PMC free article] [PubMed]
- 45.Taheri S, Abdullah TL, Yusop MR, Hanafi MM, Sahebi M, Azizi P, Shamshiri RR. Mining and development of novel SSR markers using next generation sequencing (NGS) data in plants. Molecules. 2018;23:399. 10.3390/molecules23020399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rasheed A, Hao Y, Xia X, Khan A, Xu Y, Varshney RK, He Z. Crop breeding chips and genotyping platforms: progress, challenges, and perspectives. Mol Plant. 2017;10:1047–64. 10.1016/j.molp.2017.06.008. [DOI] [PubMed] [Google Scholar]
- 47.Carletti G, Cattivelli L, Vietto L, Nervo G. Multiallelic and multilocus simple sequence repeats (SSRs) to assess the genetic diversity of a Salix spp. germplasm collection. J For Res. 2021;32:263–271. 10.1007/s11676-019-00913-0.
- 48.Guo H, Wang Z, Huang Z, Chen Z, Yang H, Kang X. Genetic diversity and population structure of Alnus cremastogyne as revealed by microsatellite markers. Forests. 2019;10:278. 10.3390/f10030278. [Google Scholar]
- 49.Guo R, Xia X, Chen J, An Y, Mi X, Li R, Zhang C, Chen M, Wei C, Liu S. Genetic relationship analysis and molecular fingerprint identification of the tea germplasms from Guangxi Province. China Breed Sci. 2021;71:584–93. 10.1270/jsbbs.21007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nasim N, Sandeep IS, Sahoo A, Das S, Panda MK, Acharya L, RamaRao VV, Nayak S, Mohanty S. Population genetic structure and diversity analysis in economically important Pandanus odorifer (Forssk.) Kuntze accessions employing ISSR and SSR markers. Ind Crops Prod. 2020;143:111894. 10.1016/j.indcrop.2019.111894.
- 51.Liu S, Liu H, Wu A, Hou A, An Y, Wei C. Construction of fingerprinting for tea plant (Camellia sinensis) accessions using new genomic SSR markers. Mol Breed. 2017;37:93. 10.1007/s11032-017-0692-y. [Google Scholar]
- 52.Han Z, Ren Y, Xia Y, Geng X, Du K, Kang X. Construction of polymorphic SSR primer library and germplasm resource fingerprint database of Populus tomentosa. J Beijing Forest Univ. 2019;41:10–18. 10.13332/j.1000−1522.20190040.
- 53.Grattapaglia D, Silva-Junior OB, Kirst M, Lima BM, Faria DA, Pappas GJ. High-throughput SNP genotyping in the highly heterozygous genome of Eucalyptus: assay success, polymorphism and transferability across species. BMC Plant Biol. 2011;11:65. 10.1186/1471-2229-11-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mo X, Long T, Liu X, Yang Y, Zhang H. AFLP analysis of somaclonal variations in Eucalyptus globulus. Biol Plant. 2009;53:741–4. 10.1007/s10535-009-0135-7. [Google Scholar]
- 55.Bradbury D, Smithson A, Krauss SL. Signatures of diversifying selection at EST-SSR loci and association with climate in natural Eucalyptus populations. Mol Ecol. 2013;22:5112–29. 10.1111/mec.12463. [DOI] [PubMed] [Google Scholar]
- 56.Liu G, Xie Y, Zhang D, Chen H. Analysis of SSR loci and development of SSR primers in Eucalyptus. J For Res. 2018;29:273–82. 10.1007/s11676-017-0434-3. [Google Scholar]
- 57.Charmet G, Balfourier F, Ravel, Denis JB. Genotype x environment interactions in a core collection of French perennial ryegrass populations. Theoretical Appl Genet. 1993;86:731–736. 10.1007/BF00222663. [DOI] [PubMed]
- 58.Gepts P. Plant genetic resources conservation and utilization: The accomplishments and future of a societal insurance policy. CSSA Golden Anniversary Symposium. 2006;46:2278–92. 10.2135/cropsci2006.03.0169gas. [Google Scholar]
- 59.Karp A, Kresovich S, Bhat KV, Ayad WG, Hodgkin T. Molecular tools in plant genetic resources conservation: a guide to the technologies. IPGRI TECHNICAL BULLETIN. 1997:1–47.
- 60.Petit R, Abdelhamid EM, Pons OMT. Identifying populations for conservation on the basis of genetic markers. Conserv Biol. 2008;12:844–55. 10.1111/j.1523-1739.1998.96489.x. [Google Scholar]
- 61.Yuan J, Cornille A, Giraud T, Cheng F, Hu Y. Independent domestications of cultivated tree peonies from different wild peony species. Mol Ecol. 2014;23:82–95. 10.1111/mec.12567. [DOI] [PubMed] [Google Scholar]
- 62.Liu S, Lin Y, Liu X, Luo J. Genetic diversity and genetic structure of Corymbia citriodora based on SSR analysis. Mol Plant Breed. 2016;14:1923–1929. 10.13271/j.mpb.014.001923.
- 63.Deng Z, Chen J, Guo D, Li C, Lu C. Genetic diversity analysis of Eucalyptus cloeziana F. Muell. Forest Res. 2019;32:41–46. 10.13275/j.cnki.lykxyj.2019.04.006.
- 64.Song Z, Zhang M, Li F, Weng Q, Zhou C, Li M, Li J, Huang H, Mo X, Gan S. Genome scans for divergent selection in natural populations of the widespread hardwood species Eucalyptus grandis (Myrtaceae) using microsatellites. Sci Rep. 2016;6:34941. 10.1038/srep34941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Torres-Dini D, Delgado-Cerrone L, Luna L, Resquin F, Aguiar AV, Sebbenn AM. The traceability of Eucalyptus clones using molecular markers. Silvae Genet. 2021;70:217–25. 10.2478/sg-2021-0019. [Google Scholar]
- 66.Acuña CV, Villalba P, Hopp HE, Marcucci Poltri SN. Transferability of microsatellite markers located in candidate genes for wood properties between Eucalyptus species. For Syst. 2014;23:506. 10.5424/fs/2014233-05279. [Google Scholar]
- 67.Botstein D, White RL, Skolnick M, Davis RW. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet. 1980;32:314–31. [PMC free article] [PubMed] [Google Scholar]
- 68.Lv J, Li C, Zhou C, Chen J, Li F, Weng Q, Li M, Wang Y, Chen S, Chen J, Gan S. Genetic diversity analysis of a breeding population of Eucalyptus cloeziana F. Muell. (Myrtaceae) and extraction of a core germplasm collection using microsatellite markers. Ind Crops Prod. 2020;145:112157. 10.1016/j.indcrop.2020.112157.
- 69.Hong J, Liu F, Huang D, Chen X. Investigation on cultivation techniques of Brazilian Eucalyptus plantation. J West Chi Forest Sci. 1996:57–76. 10.16473/j.cnki.xblykx1972.1996.s1.008.
- 70.Mo X. Analysis of technical countermeasures for introduction and domestication of Eucalyptus in China. Eucalypt Sci Technol. 2000:21–25. 10.13987/j.cnki.askj.2000.02.003.
- 71.Chen S. Study on classification of Eucalyptus cultivation area. Eucalypt Sci Technol. 1996;2:1–7. 10.13987/j.cnki.askj.1996.02.001.
- 72.Xiang D, Zheng B, Zhou W, Shen W. Review of research on Eucalyptus breeding in Guangxi. 1999:71–91. 10.19692/j.cnki.gfs.1999.02.004.
- 73.Keller LF, Waller DM. Inbreeding effects in wild populations. Trends Ecol Evol. 2002;17:230–41. 10.1016/S0169-5347(02)02489-8. [Google Scholar]
- 74.Hendry AP, Kinnison MT, Heino M, Day T, Smith TB, Fitt G, Bergstrom CT, Oakeshott J, Jørgensen PS, Zalucki MP, Gilchrist G, Southerton S, Sih A, Strauss S, Denison RF, Carroll SP. Evolutionary principles and their practical application. Evol Appl. 2011:159–183. 10.1111/j.1752-4571.2010.00165.x. [DOI] [PMC free article] [PubMed]
- 75.Hoffmann AA, Miller AD, Weeks AR. Genetic mixing for population management: From genetic rescue to provenancing. Evol Appl. 2021;14:634–52. 10.1111/eva.13154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gutiérrez-Reinoso MA, Aponte PM, García-Herreros M. A review of inbreeding depression in dairy cattle: current status, emerging control strategies, and future prospects. J Dairy Res. 2022:3–12. 10.1017/S0022029922000188. [DOI] [PubMed]
- 77.Bonnet T, Morrissey MB, Villemereuil P, Alberts SC, Arcese P, Bailey LD, Boutin S, Brekke P, Brent LGN, Camenisch G, Charmantier A, Clutton-Brock TH, Cockburn A, Coltman DW, Courtiol A, Davidian E, Evans SR, Ewen JG, Festa-Bianchet M, Franceschi C, Gustafsson L, Höner O, Houslay TM, Keller LF, Manser M, McAdam AG, McLean E, Nietlisbach P, Osmond HL, Pemberton JM, Postma E, Reid JM, Rutschmann A, Santure AW, Sheldon BC, Slate J, Teplitsky C, Visser ME, Wachter B, Kruuk LEB. Genetic variance in fitness indicates rapid contemporary adaptive evolution in wild animals. Science. 2022. 10.1126/science.abk0853. [DOI] [PubMed]
- 78.Riina J, Thong HL, Leong LS, Judy L, Laura S. Integrating genetic factors into management of tropical Asian production forests: A review of current knowledge. Forest Ecol Manag. 2014:191–201. 10.1016/j.foreco.2013.12.011.
- 79.Lowe AJ, Boshier D, Ward M, Bacles CFE, Navarro C. Genetic resource impacts of habitat loss and degradation; reconciling empirical evidence and predicted theory for neotropical trees. Heredity. 2005:255–273. 10.1038/sj.hdy.6800725. [DOI] [PubMed]
- 80.Wright SI, Kalisz S, Slotte T. Evolutionary consequences of self-fertilization in plants. Proc R Soc B. 2013. 10.1098/rspb.2013.0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu J, Li M, Zhang Q, Wei X, Huang X. Exploring the molecular basis of heterosis for plant breeding. J Integr Plant Biol. 2019:287–298. 10.1111/jipb.12804. [DOI] [PubMed]
- 82.Curik I, Ferenčaković M, Sölkner J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livestock Science. 2014:26–34. 10.1016/j.livsci.2014.05.034.
- 83.Dobeš C, Konrad H, Geburek T. Potential population genetic consequences of habitat fragmentation in central European forest Ttrees and associated understorey species An Introductory Survey. Diversity. 2017. 10.3390/d9010009. [Google Scholar]
- 84.Song W, Cao L, Wang Y, Li B, Wei S. Novel microsatellite markers for the oriental fruit moth Grapholita molesta (Lepidoptera: Tortricidae) and effects of null alleles on population genetics analyses. Bullet Entomological Res. 2016;1–10. 10.1017/S0007485316000936. [DOI] [PubMed]
- 85.Hedgecock D, Li G, Hubert S, Bucklin K, Ribes V. Wide-spread null alleles and poor cross-species amplification of microsatellite DNA loci cloned from the Pacific oyster, Crassostrea gigas. J Shellfish Res. 2004;23:379–85. 10.1016/j.seares.2003.11.002. [Google Scholar]
- 86.Zhan A, Hu J, Hu X, Zhou Z, Hui M, Wang S, Peng W, Wang M, Bao Z. Fine-scale population genetic structure of Zhikong scallop (Chlamys farreri): do local marine currents drive geographical differentiation. Mar Biotechnol. 2009;11:223–35. 10.1007/s10126-008-9138-1. [DOI] [PubMed] [Google Scholar]
- 87.Sanderson MJ, Wojciechowski MF. Improved bootstrap confidence limits in large-scale phylogenies, with an example from Neo-Astragalus (Leguminosae). Syst Biol. 2000;49:671–85. 10.2307/2585287. [DOI] [PubMed] [Google Scholar]
- 88.Klie M, Menz I, Linde M, Debener T. Lack of structure in the gene pool of the highly polyploid ornamental chrysanthemum. Mol Breeding. 2013;32:339–48. 10.1007/s11032-013-9874-4. [Google Scholar]
- 89.Xu C, Lu X, Ma L, Liu X, Liu H, Su H, Lin X, Cai Q. Phenotypic traits and genetic diversity of Erianthus arundinaceum germplasm. Agricultur Sci Technol. 2014. 10.13331/j.cnki.jhau.2014.02.002.
- 90.Wang L, Wei J, Ge C, Yu H, Jiang Y, Yang S, Zeng Q, Tian L. Analysis on genetic diversity and population genetic structure of open-pollinated progenies of Vaccinium corymbosum ‘Lanmei 1’ based on SSR marker. J Plant Resource Environ. 2022;31:35–43. 10.3969/j.issn.1674-7895.2022.03.05. [Google Scholar]
- 91.Rakshit A, Rakshit S, Santhy V, Gotmare VP, Mohan P, Singh VV, Singh S, Singh J, Balyan HS, Gupta PK, Bhat SR. Evaluation of SSR markers for the assessment of genetic diversity and fingerprinting of Gossypium hirsutum accessions. J Plant Biochem Biotech. 2010;19:153–60. 10.1007/BF03263335. [Google Scholar]
- 92.Taniguchi F, Kimura K, Saba T, Ogino A, Yamaguchi S, Tanaka J. Worldwide core collections of tea (Camellia sinensis) based on SSR markers. Tree Genet Genomes. 2014;10:1555–65. 10.1007/s11295-014-0779-0. [Google Scholar]
- 93.Matsumoto S, Kiriiwa Y, Takeda Y. Differentiation of Japanese green tea cultivars as revealed by RFLP analysis of phenylalanine ammonia-lyase DNA. Theor Appl Genet. 2002;104:998–1002. 10.1007/s00122-001-0806-z. [DOI] [PubMed] [Google Scholar]
- 94.Zhou C, He X, Li F, Weng Q, Yu X, Wang Y, Li M, Shi J, Gan S. Development of 240 novel EST-SSRs in Eucalyptus L’Hérit. Mol Breed. 2014;33:221–5. 10.1007/s11032-013-9923-z. [Google Scholar]
- 95.Schuelke M. An economic method for the fuorescent labeling of PCR fragments. Nat Biotechnol. 2000;18:233–4. [DOI] [PubMed] [Google Scholar]
- 96.Han Z, Geng X, Du K, Xu C, Yao P, Bai F, Kang X. Analysis of genetic composition and transmitted parental heterozygosity of natural 2n gametes in Populus tomentosa based on SSR markers. Planta. 2018;247:1407–21. 10.1007/s00425-018-2871-4. [DOI] [PubMed] [Google Scholar]
- 97.Wang X, Chen W, Luo J, Yao Z, Yu Q, Wang Y, Zhang S, Liu Z, Zhang M, Shen Y. Development of EST-SSR markers and their application in an analysis of the genetic diversity of the endangered species Magnolia sinostellata. Mol Genet Genomics. 2019;294:135–47. 10.1007/s00438-018-1493-7. [DOI] [PubMed] [Google Scholar]
- 98.Holland MM, Parson W. GeneMarker® HID: A reliable software tool for the analysis of forensic STR data. J Forensic Sci. 2011;56:29–35. 10.1111/j.1556-4029.2010.01565.x. [DOI] [PubMed] [Google Scholar]
- 99.Liu K, Muse SV. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics. 2005;21:2128–9. 10.1093/bioinformatics/bti282. [DOI] [PubMed] [Google Scholar]
- 100.Glaubitz JC. Convert: A user-friendly program to reformat diploid genotypic data for commonly used population genetic software packages. Mol Ecol Notes. 2004;4:309–10. 10.1111/j.1471-8286.2004.00597.x. [Google Scholar]
- 101.Peakall R, Smouse PE. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research--an update. Bioinformatics. 2012;28:2537–2539. 10.1093/bioinformatics/bts460. [DOI] [PMC free article] [PubMed]
- 102.Chapuis MP, Estoup A. Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol. 2007;24:621–31. 10.1093/molbev/msl191. [DOI] [PubMed] [Google Scholar]
- 103.Yeh FC, Boyle T, Yang RC. PopGene32, Microsoft windows-based freeware for population genetic analysis. Version 1.32. Molecular biology and biotechnology centre. University of Alberta, Edmonton. 1999.
- 104.Kalinowski ST. Hp-Rare 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes. 2010;5:187. 10.1111/j.1471-8286.2004.00845.x.
- 105.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59. 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Earl DA, VonHoldt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012;4:359–61. 10.1007/s12686-011-9548-7. [Google Scholar]
- 107.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol. 2005;14:2611–20. 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 108.Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–6. 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- 109.Rosenberg NA. Distruct: a program for the graphical display of population structure. Mol Ecol Notes. 2004;4:137–8. 10.1046/j.1471-8286.2003.00566.x. [Google Scholar]
- 110.Vlad IM, Balaji VS, Vikas CR, Ramani D, Larry SD. Automatic online tuning for fast Gaussian summation. Advances in Neural Information Processing Systems (NIPS). 2008:1113–1120.
- 111.Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–567. 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed]
- 112.Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–91. 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Thachuk C, Crossa J, Franco J, Dreisigacker S, Warburton M, Davenport G. Core Hunter: an algorithm for sampling genetic resources based on multiple genetic measures. BMC Bioinform. 2009;243:1471–2105. 10.1186/1471-2105-10-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kim KW, Chung HK, Cho GT, Ma KH, Chandrabalan D, Gwag JG, Kim TS, Cho EG, Park YJ. PowerCore: a program applying the advanced M strategy with a heuristic search for establishing core sets. Bioinformatics. 2007;23:2155–62. 10.1093/bioinformatics/btm313. [DOI] [PubMed] [Google Scholar]
- 115.Cipriani G, Spadotto A, Jurman I, Gaspero DG, Crespan M, Meneghetti S, Frare E, Vignani R, Cresti M, Morgante M. The SSR-based molecular profle of 1005 grapevine (Vitis vinifera L.) accessions uncovers new synonymy and parentages, and reveals a large admixture amongst varieties of diferent geographic origin. J Appl Genet. 2010;121:1569–1585. 10.1007/s00122-010-1411-9. [DOI] [PubMed]
- 116.Hu J, Zhu J, Xu HM. Methods of constructing core collections by stepwise clustering with three sampling strategies based on the genotypic values of crops. Theor Appl Genet. 2000;101:264–8. 10.1007/s001220051478. [Google Scholar]
- 117.Kalinowski ST, Taper ML, Marshall TC. Corrigendum. Mol Ecol. 2010;19:1512–1512. 10.1111/j.1365-294X.2010.04544.x. [Google Scholar]
- 118.Kalinowski ST, Taper ML, Marshall TC. Revising how the computer program cervus accommodates genotyping error increases success in paternity assignment. Mol Ecol. 2007;16:1099–106. 10.1111/j.1365-294X.2007.03089.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analyzed during the current study are available in the Supplementary Materials.