

Abstract
FDA reviews applications that are filed under the PEPFAR program to ensure that these products are manufactured to FDA’s stringent requirements. Dolutegravir is a comparatively recent molecular entity that represents an advance over previous products. The stability behaviors of tablets that contain dolutegravir, lamivudine, and tenofovir disoproxil fumarate and tablets that contain dolutegravir, emtricitabine and tenofovir alafenamide were surveyed and it was found that tenofovir-related degradants increase the most and are the parameters most likely to result in product failure. A desiccant is advantageous and this desiccant should remain in the bottle after it has been opened. In-use studies simulate consumer use. Bottles are stored at 30 °C/75 % RH and opened for about 1 min a day. Water content increased significantly and the rate of degradation was faster than the degradation rate observed during long-term storage. The data predict that most formulations containing TDF will stay within specification over 4 years of long-term storage followed by dispensing one tablet per day. With the current data it appears that some TAF-containing formulations may fail under similar conditions. However, the data are limited and preliminary and it is possible that the situation may improve as more stability data are acquired.
Keywords: PEPFAR, Dolutegravir, Tenofovir disoproxil fumarate, Tenofovir alafenamide, Stability testing, Long-term storage, In-use testing, Desiccant, Fixed-dose combination tablets, Product failure
Introduction
The President’s Plan for AIDS Relief (PEPFAR) must count as one of the public health success stories of the 21st Century. PEPFAR was formulated in response to the growing HIV epidemic in resource-poor countries, notably in Sub Saharan Africa, that had little access to expensive antiretroviral therapies. The program was announced by President George W. Bush in the State of the Union address on January 28, 2003. An account of the origin of PEPFAR has been provided by Harold Varmus.1 Increased funding together with the enormous cost savings that have been achieved through improved manufacturing, procurement, and distribution efficiencies have greatly increased the supply of antiretroviral drugs and during the 21st century the growth in the number of HIV-infected people on treatment through PEPFAR has been rapid. Currently PEPFAR supports more than 17 million people on treatment in resource poor countries around the world.2 Since 2004, support from the U.S. Food and Drug Administration (FDA) has been critical to PEPFAR’s success in making available high quality antiretroviral (ARV) medications from low-cost manufacturers.
Methods
New Drug Applications for dolutegravir-containing formulations intended for PEPFAR were examined and applications with reasonable amounts of long-term stability data obtained at 30 °C/75 % RH were selected for further study. Ten applications described FDC tablets that contained 50 mg dolutegravir, 300 mg lamivudine, and 300 mg tenofovir disoproxil fumarate and 7 applications described FDC tablets that contained 50 mg dolutegravir, 200 mg emtricitabine, and 25 mg tenofovir alafenamide.
To determine the effectiveness of the desiccant after the bottle has been opened a small-scale proof-of-concept study was carried out. Four 1 g cannisters of silica gel desiccant were placed in an otherwise empty white round 250 cc HDPE bottles that had no induction seal. This arrangement represents a worst-case scenario with a maximum headspace:desiccant ratio. These bottles were fitted with screw caps and placed in 75 % RH and 100 % RH desiccators.3 Once per day the bottles were removed, allowed to equilibrate, weighed and allowed to sit in the desiccator for about 1 min without a cap. The experiment continued for 180 days.
FDA’s Role in PEPFAR
FDA reviews applications that are filed under the PEPFAR program to ensure that these products are manufactured to FDA’s stringent requirements. The number of successful PEPFAR submissions continues to increase at a brisk pace. A list of successful PEPFAR applications is found in the PEPFAR Database.4 By World AIDS Day, December 1, 2022, there were 241 successful applications. Many applications describe unique and innovative formulations such as fixed-dose combination (FDC) tablets for adults that can be used alone as complete regimens (one tablet once a day) and formulations that are specifically designed for children. Other applications describe lower dose tablets that allow for flexible dosing across multiple pediatric weight bands.
FDA’s Review Process
A core element of PEPFAR is that medications supplied under the program have been found to be acceptable by the FDA. PEPFAR applications that arrive at FDA are submitted under either Section 505(b)(2) or Section 505(j) of the Food, Drug, and Cosmetic Act (FD&C Act). Applications that describe a drug that has the same route of administration, strength, dosage form and intended use as a product that has already approved for marketing in the United States are reviewed by the Center for Drug Evaluation and Research’s (CDER) Office of Generic Drugs (OGD) as 505(j) applications. For these products there is a Reference Listed Drug (RLD) in the Orange Book (Approved Drug Products with Therapeutic Equivalence Evaluations).5 The application is termed an Abbreviated New Drug Application (ANDA).
If the product has no reference listed drug in the Orange Book the application is reviewed under Section 505(b)(2), and the application is termed a New Drug Application (NDA). The clinical and clinical pharmacology sections of the 505(b)(2) applications (including portions of the labeling) are reviewed by CDER’s Division of Antivirals and the Office of Clinical Pharmacology. For both ANDAs and NDAs, the overall product quality aspects (chemistry, facility inspections, biopharmaceutics, etc.) of the applications are reviewed by CDER’s Office of Pharmaceutical Quality.
PEPFAR applications are reviewed to exactly the same standards as those used for any other NDA or ANDA application. Review of an original PEPFAR NDA or ANDA typically takes 6-10 months depending on the nature of the application.
Approval or Tentative Approval
To receive approval to market any drug, whether an NDA or an ANDA, in the United States, an applicant submits detailed technical information to the FDA. The type and amount of this information is specified by the Food, Drug, and Cosmetic Act and Title 21 of the Code of Federal Regulations. FDA will review this information in detail and will either grant approval to market the product or issue a Complete Response letter requesting additional information. Under PEPFAR similar information is submitted for antiretrovirals even though there may be no intention of marketing this product in the United States. FDA will review these applications using the same stringent standards as for any other product.
If an application (ANDA or NDA) for a drug product meets the requirements for approval under the Federal Food, Drug, and Cosmetic Act, but it cannot be granted final approval by FDA because of unexpired patents or exclusivities on the brand/listed drug, FDA will grant a Tentative Approval for the ANDA/NDA. Under section 505 of the FD&C Act, a drug product that is tentatively approved is not an approved drug and may not be marketed in the US without the Agency’s final approval (per 21 CFR 314.105(d)). However, tentatively approved ARVs are eligible for purchase by the U.S. government for distribution outside of the U.S. under the PEPFAR program. A formal definition of “tentative approval” can be found under 21 CFR 314.3.
Antiretroviral drugs that have received final approval, or that have received tentative approval under a PEPFAR application, are eligible for purchase through the PEPFAR program.
Dolutegravir-Containing Formulations
Dolutegravir is a comparatively recent molecular entity that is more effective, easier to take and has fewer side effects than alternative drugs that are currently used. It also has a high genetic barrier to developing drug resistance.6 Compared to the previous efavirenz-containing regimens dolutegravir-containing regimens are more potent, more durable with a higher drug resistance barrier, more convenient being a smaller tablet, better tolerated and have fewer drug interactions. Like the efavirenz-containing regimens dolutegravir-containing regimens are dosed at one tablet once a day.7 According to WHO guidelines the preferred regimen for adults and adolescents is dolutegravir, lamivudine (or emtricitabine), and tenofovir disoproxil fumarate (TDF).8 The similar regimen of dolutegravir, emtricitabine (or lamivudine), and tenofovir alafenamide (TAF) is recommended for use under special circumstances. The structures of these compounds are shown in Fig. 1. Because dolutegravir has unexpired exclusivity such applications are eligible for Tentative Approval reflecting the fact that they cannot be legally marketed in the United States.
Fig. 1:
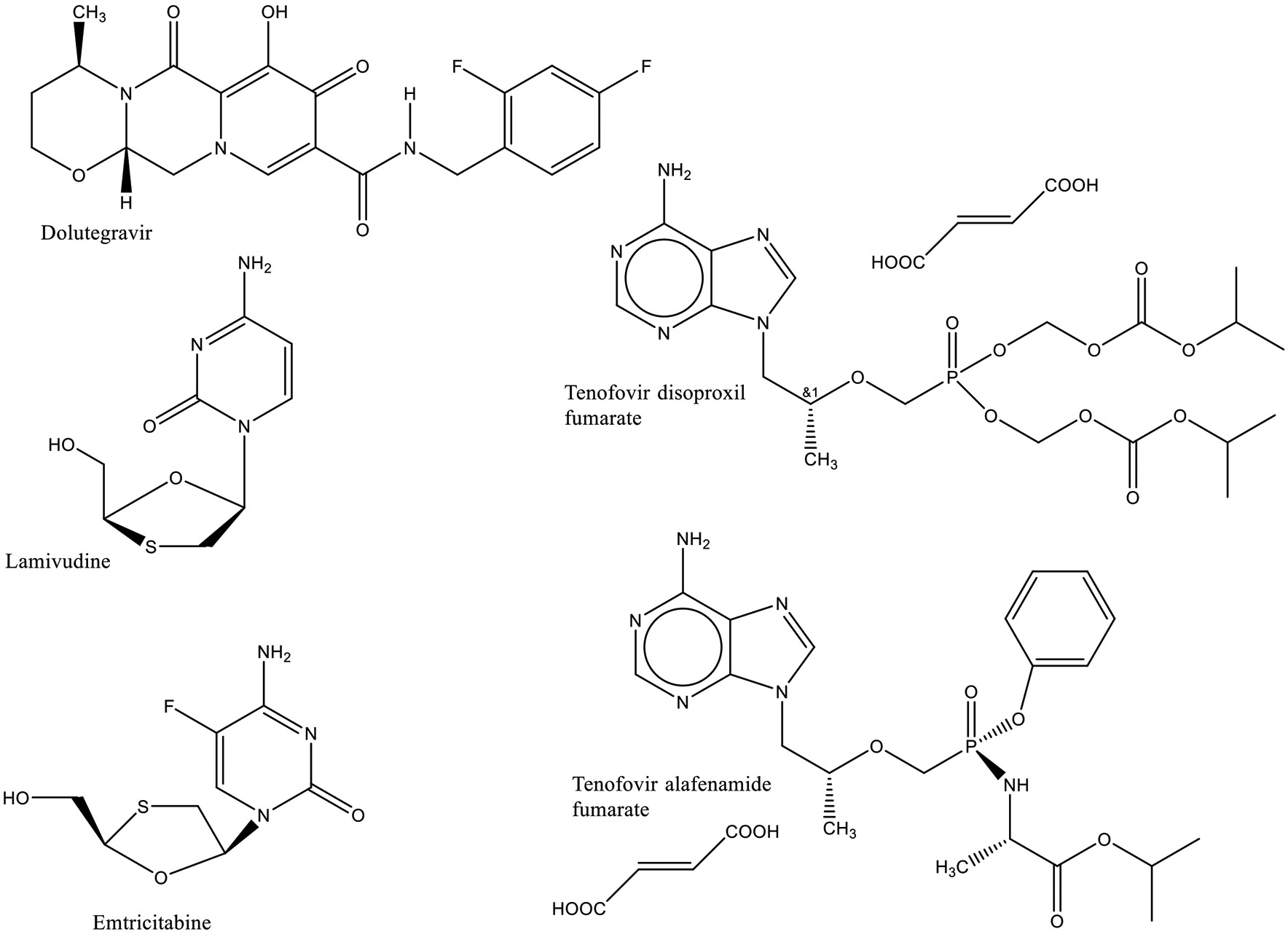
Structures of Dolutegravir, Emtricitabine, Lamivudine, Tenofovir Alafenamide Fumarate, and Tenofovir Disoproxil Fumarate.
The formulations discussed in this paper are:
FDC tablets that contain 50 mg of dolutegravir (equivalent to 52.6 mg of dolutegravir sodium), 300 mg of lamivudine, and 300 mg tenofovir disoproxil fumarate (equivalent to 245 mg of tenofovir disoproxil), referred to in this paper as DL-TDF
and
FDC tablets that contain 50 mg of dolutegravir (equivalent to 52.6 of dolutegravir sodium), 200 mg of emtricitabine, and 25 mg of tenofovir alafenamide (equivalent to 28.0 mg of tenofovir alafenamide fumarate), referred to in this paper as DE-TAF.
Multi-Month Dispensing
For a number of reasons9 multi-month dispensing, i.e., dispensing more than one month’s supply during each visit to the clinic, has become desirable. Fewer visits to clinics reduce the stress on the healthcare system and may also be desirable for social reasons. In turn this means that purchasers of PEPFAR products are asking for larger bottles, typically 90 or 180 count. Both regimens described in this paper feature one tablet once a day dosing. The tablets are packaged in HDPE bottles with induction seals and desiccant. The desiccant reflects the need to limit the amount of moisture that is present and hence control the predominantly hydrolytic degradation of the tenofovir-containing moieties.
In certain packaging configurations for products containing moisture-sensitive tenofovir with other active ingredients, FDA has asked for in-use testing to simulate consumer use and demonstrate that repeated opening and closing over the period of use by consumer does not cause assay and degradant values to fall outside the product specification.
Typically, this is one time testing and does not need to be repeated for future batches. This paper discusses the long-term and in-use stability behavior of DL-TDF and DE-TAF products that have been submitted to the FDA. Some of these applications are still waiting for a Tentative Approval decision. The reader will appreciate that, for reasons of confidentiality, proprietary data cannot be revealed but general trends shown by the aggregated data can be discussed.
General Characteristics of the Products
Dosing is one tablet once a day and the tablets are typically packaged 30, 90, and 180 count in HDPE bottles fitted with child-resistant screw caps with induction seals and desiccant. The amount of desiccant varies widely between applications and is typically either silica gel or molecular sieves.
About half the formulations are bilayer and the remainder are monolayer.
Excipients are conventional and not novel (see the PEPFAR database mentioned above). For the most part common excipients such as croscarmellose sodium, lactose monohydrate, magnesium stearate, mannitol, microcrystalline cellulose, povidone, and sodium starch glycolate are used. Tablets are film coated and are identifiable through unique combinations of color and markings.
Generally, tablet specifications are conventional and include tests for appearance, identity, assay, degradants, uniformity (USP <905>), dissolution, water, and microbial contamination (USP <61> and <62>). The specifications follow the recommendations of ICH and USP. Elemental impurities conform to USP <232>/ICH Q3D and residual solvents conform to USP <467>. Conformity may be shown by detailed knowledge of each component of the drug product.
Product must meet its regulatory specification throughout its shelf life. In some cases, tighter release specifications are advantageous, particularly for parameters that are known to change on stability.
Tenofovir Degradation
Product specifications generally group degradants as those that are dolutegravir related, lamivudine related, and tenofovir related. Tenofovir-related degradants were found to be the ones that increase the most and are the parameters most likely to result in product failure. Given the structures this is unsurprising.
The main degradation product from tenofovir disoproxil fumarate is tenofovir monoester10 also frequently called Impurity A or Mono-POC PMPA (see Fig. 2 for structures). For this paper tenofovir monoester has been used as the metric for assessing the comparative degradation of different DL-TDF formulations containing tenofovir disoproxil fumarate. The levels of other degradants are minor compared to the amount of tenofovir monoester formed as the product ages.
Fig. 2:
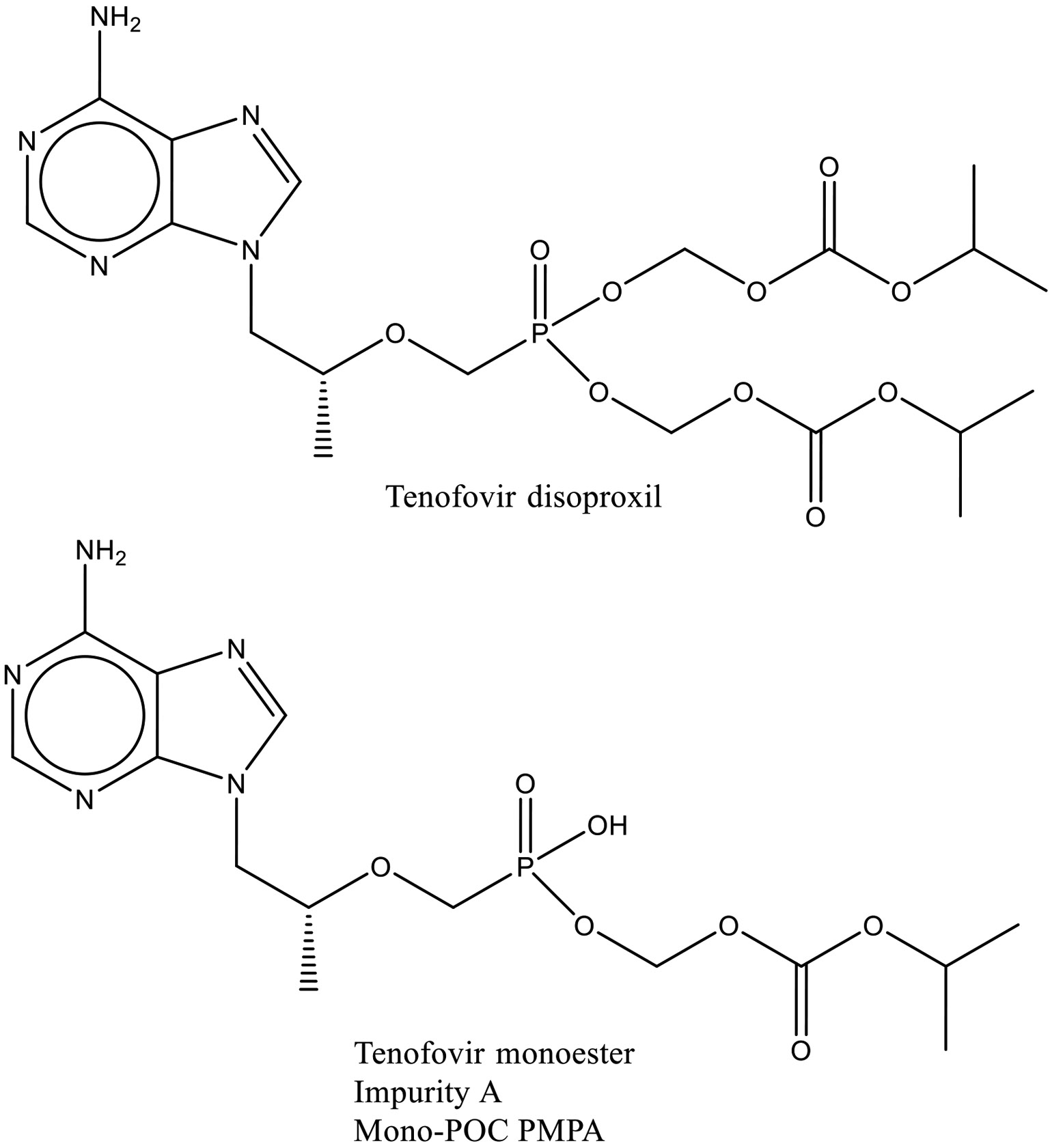
Structures of Tenofovir Disoproxil and Impurity A.
In contrast, tenofovir alafenamide gives rise to a number of degradants11 as shown in Fig. 3. The names beginning with DP are those provided in the citation and the other names are those that are commonly used in the industry.
Fig. 3:
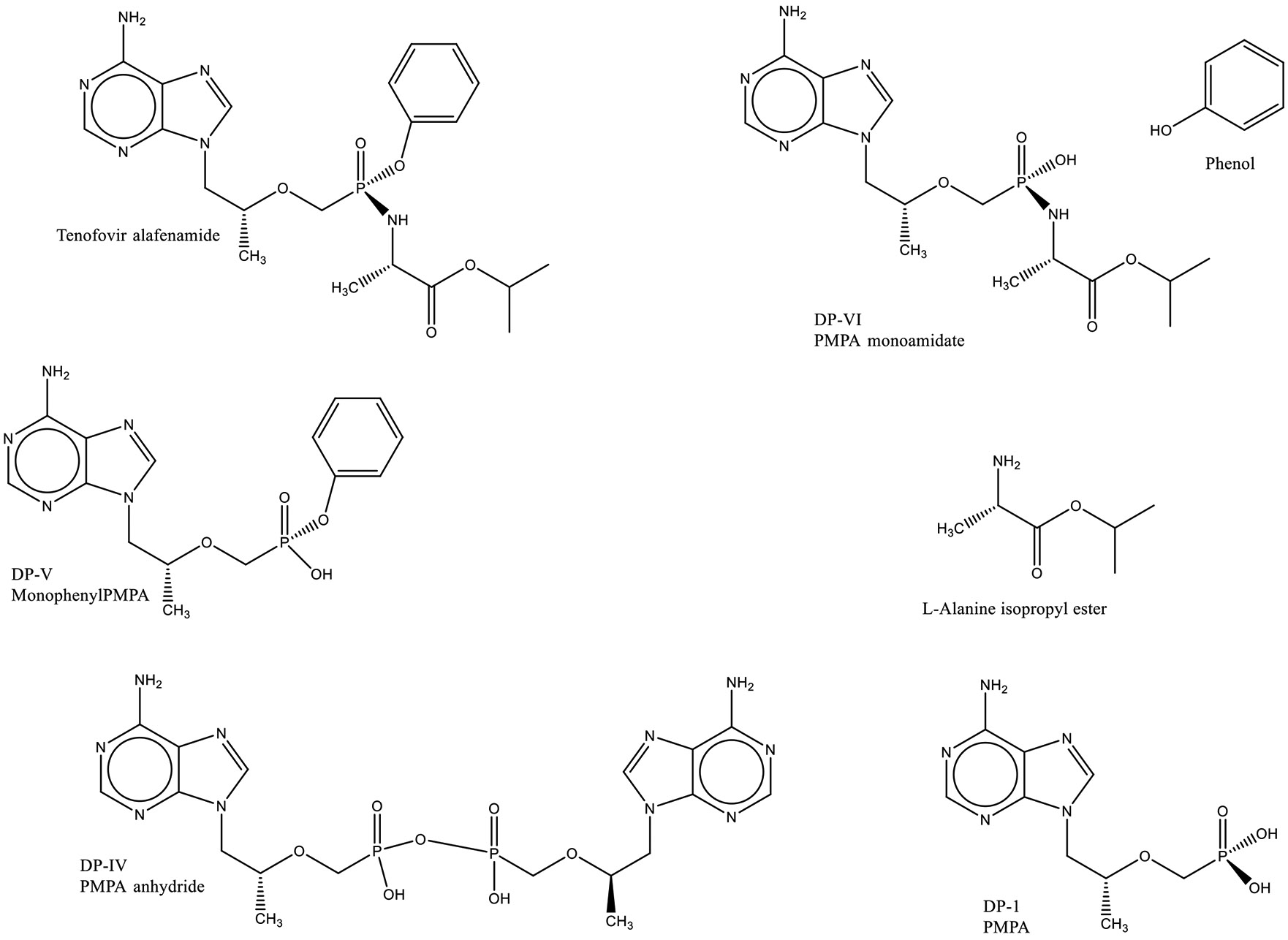
Structures of Tenofovir Alafenamide and Its Degradants.
It is noticeable that the degradation profiles vary between formulations with some degradants being intermediates in the formation of other degradants. For this reason, the degradant specification also varies between formulations. In some formulations a degradant may be a specified impurity and in others it may be covered by the unspecified impurity test. For this paper the total TAF degradants value was used as the metric for assessing the comparative degradation of different DE-TAF formulations containing tenofovir alafenamide (see below).
Stability Studies
PEPFAR products are likely to be used in countries that have hot humid climates and as the program has evolved manufacturers have settled on long-term testing conditions of 30 °C/75 % RH for both the long-term and in-use studies. This supports label statements such as “Store below 30 °C”.
We examined the long-term (30 °C/75 % RH) and in-use stability data of a number of PEPFAR DL-TDF products using Impurity A as the key indicator of product quality, as described above.
In a similar fashion the long-term (30 °C/75 % RH) and in-use stability data of a number of PEPFAR DE-TAF products were examined using Total Tenofovir-related Degradants as the key indicator of product quality, as described above.
Key questions for each dosage form were as follows:
Is the desiccant necessary?
How is in-use testing carried out?
How do the degradation rates of TDF and TAF compare? For a given formulation how do the degradation rates change as the packaging changes? Does the rate of degradant formation accelerate with time, decelerate, or stay the same?
How does the water content of the tablets change during long-term stability and in-use testing?
Is the rate of degradant formation higher during in-use testing? If so, by how much when compared with the long-term stability testing?
Can the available stability data be used to predict the stability behavior of the formulations over the long-term? Will the last tablet removed from the bottle conform to its specification?
Note that these conclusions are based on limited data that have not been subject to a statistical analysis. For the most part these PEPFAR applications arrive with 6 months of long-term (30 °C/75 % RH) stability data for 3 batches. A 12 month stability data update is provided by the time an action is taken. In some cases, 18 and 24 month data are available. Frequently a certain amount of analytical variation is observed making interpretation of the trends more challenging. Twelve months of stability data mean 15 datapoints (0, 3, 6, 9, 12 months test data times 3 batches). Additionally, although the active ingredients stay the same the excipients, manufacturing process, and container-closure system may vary between NDAs.
Results
Is the desiccant necessary?
The presence of desiccant is clearly important for DL-TDF and DE-TAF products. However, the amount of desiccant varies quite widely across different manufacturer’s versions of these products, and there was no obvious correlation between the amount of desiccant and the stability or water content of the drug product. In small scale experiments silica gel desiccant was placed in HDPE bottles that had no induction seal. These bottles were fitted with screw caps and placed in 75 % RH and 100 % RH desiccators. Once per day the bottles were weighed and allowed to sit in the desiccator for about 1 min without a cap. It was found that there was a slow increase in weight with time for about 90 days suggesting that the desiccant continues to exert a protective effect even after the induction seal is removed. However, after about 90 days the weight gain levels off suggesting that the desiccant is no longer effective. The data show that the desiccant cannisters continue to offer protection from water for about 90 days and justify statements such as “Do not discard desiccant” and “Keep tightly closed”.
The graph obtained on testing at 100 % RH is shown in Fig. 4..
Fig. 4:
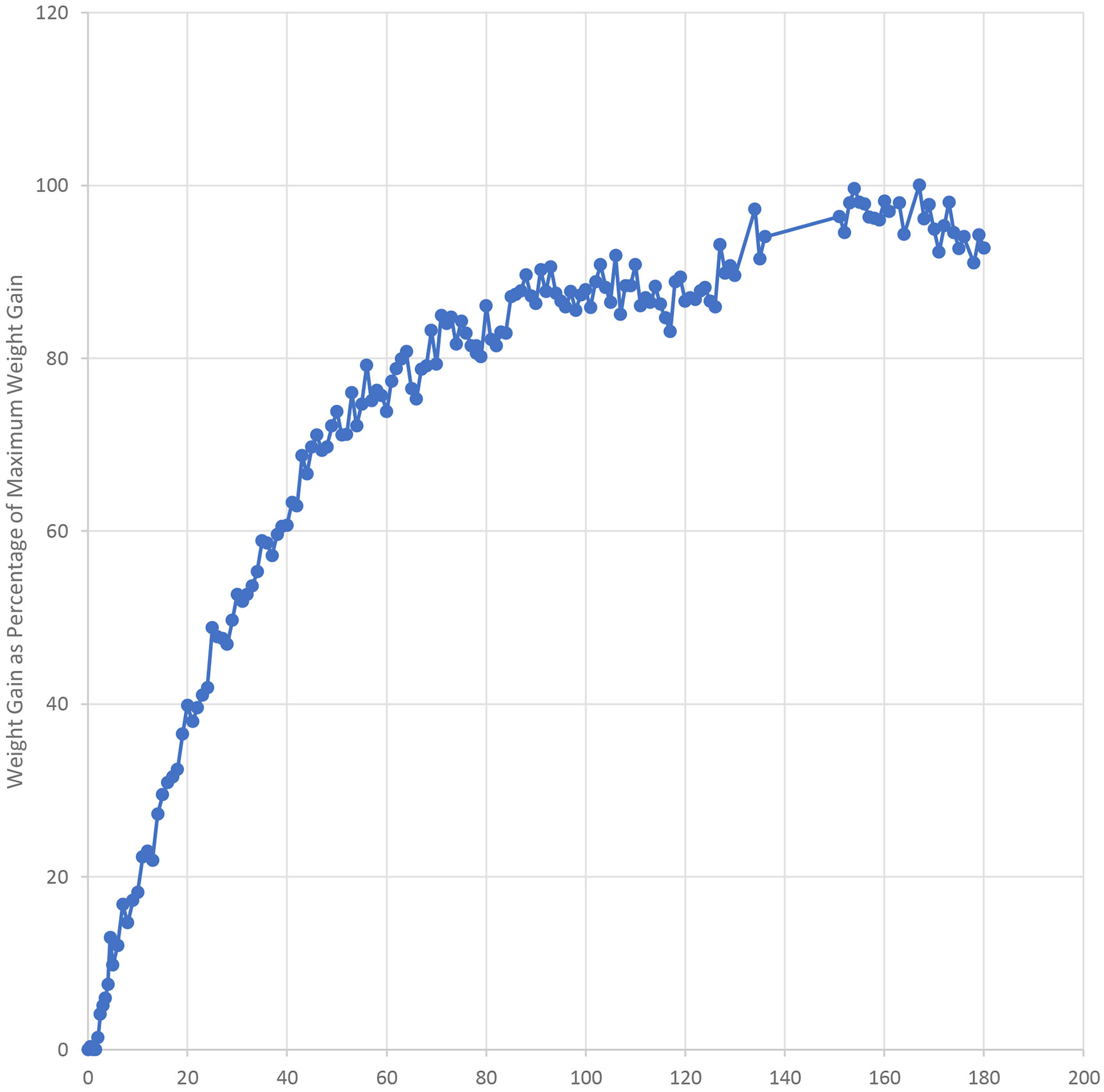
Weight Gain at 100% RH (bottle open for 1 minute each day) as Percentage of Maximum Observed Weight Gain
How is in-use testing carried out?
There are variations among the manufacturers but in-use studies are generally carried out with the desiccant being retained in the bottle after it has been opened and the induction seal removed. The experiments aim to simulate actual consumer use and generally involve the bottles being stored at 30 °C/75 % RH and being opened for about 1 min a day (but not on weekends). In some cases, small (e.g., 20 %) or large (e.g., 80 %) amounts of the tablets are removed at the start and no other changes are made to the contents and in other cases a tablet is removed each day. Experiments are mostly carried out for 90 count bottles over 90 days with some formulations being tested in 180 count bottles over 180 days. Tablets are tested to their specification at the end of the experiment and sometimes at intermediate time points.
How do the degradation rates of TDF and TAF compare? For a given formulation how do the degradation rates change as the packaging changes? Does the rate of degradant formation accelerate with time, decelerate, or stay the same?
In long term stability studies (with the induction seal intact) over the first 12 months the average increase in degradants for all TAF packaging configurations is 1.15% which is greater than the average increase in degradants of TDF (0.41% for all packaging configurations). However, the results are preliminary and there is considerable variation in the data.
In long term stability studies (with the induction seal intact) the degradation of TDF increases slightly as package size increases. After 24 months the average increase in degradants is 0.51% for the 30 count bottles, 0.74% for the 90 count bottles, and 0.82% for the 180 count bottles. In contrast, there is a significant increase in TAF degradation as package size increases. After 12 months (less stability data are available for TAF-containing formulations) the average increase in degradants is 0.64% for the 30 count bottles, 1.16% for the 90 count bottles, and 2.05% for the 180 count bottles. These average values are averages of all the data from all the applications that were examined. The values for the individual applications behaved in a similar fashion.
The rate of degradation of the TDF-containing formulations generally decreases with time. In some cases, the nature of the trend cannot be discerned. For TAF-containing formulations the trends cannot yet be discerned. These formulations are newer than the TDF-containing formulations and so less data are available.
How does the water content of the tablets change during long-term stability and in-use testing?
There is no obvious increase in the water content of tablets stored with intact seals under the long-term stability conditions of 30 °C/75 % RH. There is sometimes an early fall in water content followed by random variations.
In contrast to the long-term data obtained with seals intact the average increase in water content for all sizes during the in-use testing is 1.1 % for TDF formulations and 1.2 % for TAF formulations. This compares with water content values of 1.3-3.0 % at release.
Is the rate of degradant formation higher during in-use testing? If so, by how much when compared with the long-term stability testing?
Expressed on an annual basis, i.e., amount of Impurity A increase per year, TDF-containing products had an average increase of 0.38% for tablets stored at 30°C/75% RH with the seal intact and an average increase of 1.58% for tablets undergoing in-use testing at 30°C/75% RH with no seal and daily opening and closing. Similarly, expressed on an annual basis, i.e., amount of Total Impurities per year, TAF-containing products had an average increase of 1.11% for tablets stored at 30°C/75% RH with the seal intact and an average increase of 6.84% for tablets undergoing in-use testing at 30°C/75% RH with no seal and daily opening and closing.
Thus, expressed on an increase per year basis, TDF-containing products used in the in-use study degraded 4 times faster than tablets stored with their seals intact and TAF-containing products used in the in-use study degraded 6 times faster than tablets stored with their seals intact. However, these numbers represent annual values and the in-use studies were carried out for 90 or 180 days so the actual amounts of degradation would be proportionally less.
Can the available stability data be used to predict the stability behavior of the formulations over the long-term? Will the last tablet removed from the bottle conform to its specification?
Again, it is important to note that these observations are based on limited data and the conclusions that were drawn may be subject to change as more data arrive. However, some useful preliminary conclusions can be arrived at. For TDF-containing formulations the formation of Impurity A was investigated and for TAF-containing formulations the Total Degradant values were monitored. The formulations were found to vary greatly in their stability behavior for no obvious reason. Estimates of the amount of degradation for the last tablet out of the bottle were evaluated using the three following scenarios.
Scenario A Typical release value + increase over 24 months (estimated in some cases) + increase during in-use testing
Scenario B Typical release value + increase over 48 months (estimated in each case) + increase during in-use testing
Scenario C The worst-case scenario. Release at the release acceptance criterion + increase over 48 months (estimated) + increase during in-use testing
For the TDF-containing formulations failures were sometimes predicted. However, it was found that if the release acceptance criterion for Impurity A was set at NMT 1.5 % and the stability acceptance criterion was NMT 5.0 % then most formulations were predicted to stay within specification for 48 months at 30 °C/75 % RH followed by in-use testing. In other words, the last tablet out of the bottle will meet its specification.
A hypothetical example is as follows:
Impurity A value at release = 0.58%
Estimated degradation over 48 months long-term stability conditions (twice the 24 month data) = 1.37%
Degradation during the in-use study = 0.63%
Estimated value of Impurity A after long-term storage and in-use testing = 0.58 + 1.37 + 0.63 = 2.58%
Given a typical Impurity A acceptance criterion of ≤ 5.0% this example indicates that the last tablet out of the bottle should meet its specification. (The values used in the above example are the average values from 10 NDAs that were investigated.)
Failures were predicted for some TAF-containing formulations with failures increasing on moving from Scenario A to Scenario C. For these products changing the release and stability acceptance criteria are unlikely to make any difference.
However, the scenarios above assume that degradation will continue at the previously observed rates and there is some evidence that the rate of degradation flattens out with time. Additionally, it should be noted that relatively less information is available for the TAF-containing formulations, which tend to be newer, and degradation rates may decline at later time points.
Discussion
Preliminary examination of the stability data for dolutegravir-containing formulations shows that the limiting parameter is the stability of the tenofovir-containing components. Retaining the desiccant in the bottle after opening should help and a statement to that effect could be added to the container.
In-use testing that involves the bottles being stored at 30 °C/75 % RH and being opened for about 1 min a day can be used to simulate actual consumer use. The water content and the rate of degradant formation increase during in-use testing.
Examining possible scenarios it appears that stability failures are possible for formulations that contain TDF. However, with a tight release acceptance criterion for Impurity A coupled with a more accommodating stability acceptance criterion most formulations were predicted to stay within specification for 48 months at 30 °C/75 % RH followed by in-use testing. The last tablet will meet its specification.
With the current data it appears that some TAF-containing formulations may fail under similar conditions. However, the data are limited and preliminary and it is possible that the situation may improve as more stability data are acquired.
Acknowledgments
Dr. Stephen P. Miller contributed to the data analysis used to inform the conclusions reported in this paper. Dr. Miller has since retired from the FDA. Numerous other FDA colleagues are thanked for helpful discussions. This paper is dedicated to the memory of Dr. Balajee Shanmugam who was the Branch Chief for many of these PEPFAR applications. Dr. Shanmugam sadly passed away in December 2020.
This article reflects the views of the author and should not be construed to represent FDA’s views or policies. This work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Abbreviations:
- ANDA
Abbreviated New Drug Application
- ARV
Antiretroviral
- CDER
Center for Drug Evaluation and Research
- CFR
Code of Federal Regulations
- DE-TAF
Dolutegravir, emtricitabine, and tenofovir alafenamide tablets
- DL-TDF
Dolutegravir, lamivudine, and tenofovir disoproxil fumarate tablets
- FDC
Fixed-dose combination
- ICH
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use
- NDA
New Drug Application
- OGD
Office of Generic Drugs
- PEPFAR
The President’s Plan for AIDS Relief
- RLD
Reference Listed Drug
- TAF
tenofovir alafenamide
- TDF
tenofovir disoproxil fumarate
- USP
United States Pharmacopeia
Footnotes
Declaration of Competing Interest: The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Submission declaration: The work described has not been published previously.
References
- 1.Varmus H Making PEPFAR: A triumph of medical diplomacy. Science & Diplomacy [Online] 2013; 2, Article 4. Available at: http://www.sciencediplomacy.org/article/2013/making-pepfar Accessed December 25, 2022. [Google Scholar]
- 2.The United States President’s Emergency Plan for AIDS Relief 2021. Annual Report to Congress. Available at: https://www.state.gov/wp-content/uploads/2021/02/PEPFAR2021AnnualReporttoCongress.pdf); Accessed December 25, 2022. [Google Scholar]
- 3.Quincot G, Azenha M, Barros J, Faria R. Use of salt solutions for assuring constant relative humidity conditions in contained environments. https://studylib.net/doc/18269015/use-of-salt-solutions-for-assuring-constant-relative-humi... Accessed December 25, 2002. [Google Scholar]
- 4.Quick Reference Guide for PEPFAR Database. Available at: https://www.fda.gov/international-programs/presidents-emergency-plan-aids-relief-pepfar/quick-reference-guide-pepfar-database Accessed December 25, 2022. [Google Scholar]
- 5.Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations. Available at: http://www.accessdata.fda.gov/scripts/cder/ob/default.cfm Accessed December 25, 2022. [Google Scholar]
- 6.WHO recommends dolutegravir as preferred HIV treatment option in all populations. Available at: WHO recommends dolutegravir as preferred HIV treatment option in all populations Accessed December 25, 2022. [Google Scholar]
- 7.Tenofovir, Lamivudine, and Dolutegravir (TLD) Transition. General Information for Clients, Clinicians, Counselors, and other Service Providers (August 2019). Available at: fhi360.org/sites/default/files/media/documents/linkages-tld-transition-information.pdf; Accessed December 25, 2022. [Google Scholar]
- 8.Consolidated guidelines on HIV prevention, testing, treatment, service delivery and monitoring: recommendations for a public health approach. Available at: https://www.who.int/publications/i/item/9789240031593 (pp. 131 and 174). Accessed December 25, 2022. [PubMed] [Google Scholar]
- 9.Two times a year is better than four: people spend less time off ART if they only visit their clinic every six months, African study shows. Available at: https://www.aidsmap.com/news/may-2021/two-times-year-better-four-people-spend-less-time-art-if-they-only-visit-their-clinic Accessed December 25, 2022. [Google Scholar]
- 10.Ashenafi D, Chintam V, van Veghel D, Dragovic S, Hoogmartens J, Adams E. Development of a validated liquid chromatographic method for the determination of related substances and assay of tenofovir disoproxil fumarate. Journal of Separation Science 2010; 33(12): 1708–1716. [DOI] [PubMed] [Google Scholar]
- 11.Golla VM, Kurmi M, Shaik K, Singh S. Stability behaviour of antiretroviral drugs and their combinations. 4: Characterization of degradation products of tenofovir alafenamide fumarate and comparison of its degradation and stability behaviour with tenofovir disoproxil fumarate. Journal of Pharmaceutical and Biomedical Analysis 2016; 131: 146–155. [DOI] [PubMed] [Google Scholar]