

Conspectus
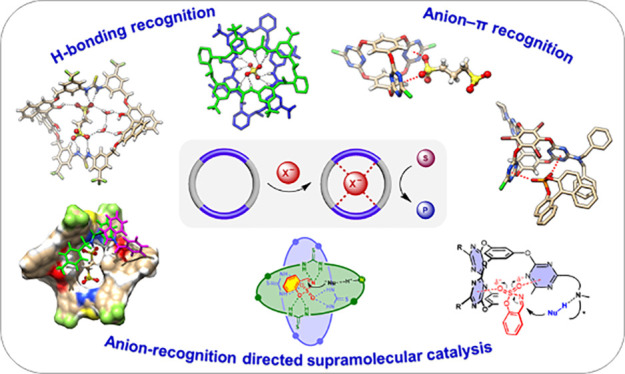
The development of supramolecular chemistry has provided a variety of host molecules and noncovalent tools for boosting catalytic processes, stimulating the emergence and advance of supramolecular catalysis, among which macrocyclic and cage-like compounds have attracted great attention due to their possession of an enzyme-mimetic cavity and recognition ability. While the privileged scaffolds such as crown ethers, cyclodextrins, cucurbiturils, calixarenes, and metal-coordinated cages have been widely used, their skeletons usually do not contain a directional binding site; binding and activation mainly rely on cation-associated interactions or hydrophobic effects. In this context, the recent advance of anion supramolecular chemistry has drawn our attention to developing an anion recognition-directed approach by using tailor-made functionalized macrocycles and cages. Anions are important widely existing species in both biological and chemical systems and play an important role in regulating the structure and function of enzymes. We envisioned that by taking advantage of anions, including their rich variety, diverse geometry, and multiple interaction sites, the sophisticated cooperation of multiple noncovalent interactions can be manipulated in a confined cavity for directing efficient and selective catalysis.
Following this concept, we initiated our study by introducing typical thiourea H-bonding groups to design and synthesize a series of bis-thiourea macrocycles, especially chiral macrocycles, by incorporating chiral linkers. Taking advantage of the obtained strong, cooperative anion binding, a macrocycle-enabled counteranion trapping strategy was developed, which afforded greatly enhanced catalytic efficiency and excellent stereocontrol in acid-catalyzing reactions. Furthermore, inspired by sulfate-induced macrocyclic dimerization assembly, we built a substrate-induced assembly system, enabling an induced-fit cooperative activation network for efficient and enantioselective catalysis. In addition, anion recognition-driven chirality gearing with a more sophisticated trithiourea cage was revealed, which could provide a basis for implementing anion-triggered allosteric catalysis within the induced helical space. Not limited to hydrogen bonding, the emerging anion−π interactions were largely exploited. A series of triazine-based prism cages containing three V-shaped electron-deficient π-cavities were constructed, and their anion−π binding properties were studied. Based on this system, cooperative anion−π activation was established for driving highly efficient and selective catalysis, which paved a way to push anion−π interactions toward more practical and useful catalyst design.
These results demonstrated that the anion-recognition direction can serve as a powerful, versatile approach for boosting highly efficient and selective supramolecular catalysis. It is feasible not only for employing exogenous anions (e.g., counteranion) as a handle but also for recognition and regulation of anionic active intermediates/transition states, from use in conventional H-bonding to emerging anion−π recognition.
Key References
Ning R.; Zhou H.; Nie S.-X.; Ao Y.-F.; Wang D.-X.; Wang Q.-Q.. Chiral Macrocycle-Enabled Counteranion Trapping for Boosting Highly Efficient and Enantioselective Catalysis. Angew. Chem., Int. Ed. 2020, 59, 10894–10898 10.1002/anie.202003673.1A macrocycle-enabled counteranion trapping catalysis strategy was developed. Through tight trapping of the counteranion of an acid catalyst by the designer BINOL-based chiral bis-thiourea macrocycles, greatly enhanced catalytic efficiency and excellent stereocontrol were achieved.
Guo H.; Zhang L.-W.; Zhou H.; Meng W.; Ao Y.-F.; Wang D.-X.; Wang Q.-Q.. Substrate-Induced Dimerization Assembly of Chiral Macrocycle Catalysts Toward Cooperative Asymmetric Catalysis. Angew. Chem., Int. Ed. 2020, 59, 2623–2627 10.1002/anie.201910399.2Inspired by sulfate-induced dimerization assembly of tetraamino-bisthiourea chiral macrocycles, a rare example of a substrate-induced dimerization assembly catalysis system was demonstrated, which enabled a highly cooperative hydrogen-bonding activation network for efficient and enantioselective transformation.
Hu Q.-P.; Zhou H.; Huang T.-Y.; Ao Y.-F.; Wang D.-X.; Wang Q.-Q.. Chirality Gearing in an Achiral Cage Through Adaptive Binding. J. Am. Chem. Soc. 2022, 144, 6180–6184 10.1021/jacs.2c02040 .3A synthetic achiral trithiourea molecular cage was shown to be able to adaptively bind tricarboxylate anions and form gear-like helical complexes. A biotic-resembling sequence of primary binding, conformational adaption, and precise stereocontrol was revealed.
Luo N.; Ao Y.-F.; Wang D.-X.; Wang Q.-Q.. Exploiting Anion−π Interactions for Efficient and Selective Catalysis with Chiral Molecular Cages. Angew. Chem., Int. Ed. 2021, 60, 20650–20655 10.1002/anie.202106509.4A cooperative anion−π catalysis strategy was developed by building chiral molecular cage catalysts with a confined, V-shaped, electron-deficient π-cavity to harness cooperative anion−π interactions for efficient activation and excellent stereoselectivity control.
1. Introduction
The development of supramolecular chemistry has enabled rich host scaffolds and abundant noncovalent tools for boosting catalytic processes, leading to the emergence of supramolecular catalysis.5−8 The continuing progress on this topic not only formulated an important direction for the fabrication of functional supramolecular systems but also provided inspiration and enabled a new path for innovative catalyst design. Since the early studies on using privileged scaffolds such as crown ethers, cyclodextrins, cucurbiturils, and calixarenes7,9−11 to the later widely explored metal-coordinated cages12,13 and H-bonding capsules,14 macrocycle and cage compounds have received considerable interest and played a vital role in this aspect. This was not surprising, as the macrocyclic and cage host molecules usually possess a confined cavity and specific binding ability, which mimic the characteristics of enzyme catalysis. The skeletons of these host molecules, however, typically do not contain a directional binding site. The binding and activation are mainly driven by cation-related interactions or hydrophobic effects, benefiting from a general confinement effect.
Anions are important, widely existing species in both biological and chemical systems. In nature, over 70% of enzyme substrates and cofactors are anions. Anions play important roles in regulating the structure and function of enzymes. Compared with cations, anions are relatively large and occur in a range of shapes and geometries. Despite the inherent challenges posed by the complicated shapes, low charge density, and high solvation free energies, significant progress has been made on anion coordination chemistry over the past few decades through the efforts of many groups.15−17 Inspired by the latest development of anion supramolecular chemistry, we envisioned that by incorporating multiple binding sites in a tailor-made macrocyclic or cage skeleton, efficient recognition toward particular anions could be achieved. Through anion recognition, sophisticated cooperation of multiple noncovalent interactions can be built, which can be manipulated for directing efficient and selective catalysis. Based on this concept, an anion can be not only an exogenous anion such as the counteranion of an acid catalyst which persists in the system but also could be the formed anionic intermediate or developing transition state along the occurring catalytic process. Moreover, when a chiral macrocycle or cage is applied, stereoselective catalysis could operate through the nested noncovalent interaction network within the confined chiral cavity.
In this Account, I describe our efforts in fabricating anion recognition-directed supramolecular catalysis using functional macrocycles and cages since I started working independently in 2015. I will first describe the efforts in developing macrocycle-enabled counteranion trapping, substrate-induced assembly, and anion recognition-driven chirality gearing systems through tailor-made H-bonding macrocycles and cages. Then, I will go across the efforts on exploiting the emerging anion−π interaction to develop a cooperative anion−π catalysis system through constructing triazine-based prism cages with V-shaped electron-deficient π-cavities.
2. Macrocycle-Enabled Counteranion Trapping for Boosting Efficient and Enantioselective Catalysis
The strong binding ability of macrocycles toward particular guests can be exploited to mediate a catalytic process through tightly binding an “indirect” catalytic species (e.g., counterions). In this regard, crown ethers have been widely used as cation-binding catalysts for the solubilization of alkali metal salts in the organic phase and increasing the nucleophilicity of an associated anion (Figure 1a).18,19 When a chiral crown ether was applied, stereoselective induction could be realized through the formation of a tight chiral ion-pairing complex. On the contrary, the opposite strategy by applying a macrocyclic anion receptor to trap a counteranion for tuning the cation-directed catalytic process remained unexplored when we initiated our project (Figure 1b).20 At that stage, anion binding emerged as an appealing approach for implementing asymmetric ion-pairing catalysis, where typical small molecules of anion binders such as ureas and thioureas and some oligomeric helical triazole binders were used.21−25 We envisioned that the use of a macrocyclic anion receptor could provide stronger, tight binding (trapping) and a confined macrocyclic cavity for both catalytic acceleration and stereocontrol.
Figure 1.
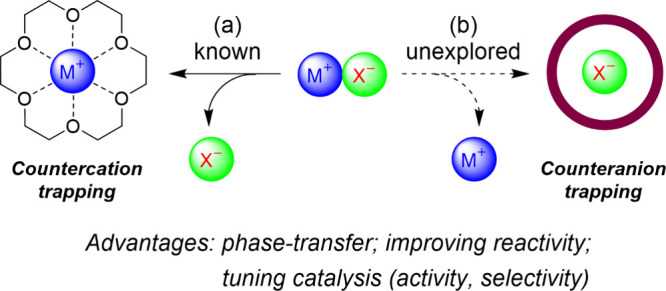
Macrocycle-enabled counterion trapping strategies for tuning the (catalytic) reactivity. Adapted with permission from ref (20). Copyright 2018, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
Following the envisioning, a diarylthiourea group was chosen as a typical binding unit due to its good anion binding property and easy connection with other motifs.26,27 The introduction of CF3 further increased the N–H acidity and led to better structural preorganization (facilitating intramolecular C–H···S H-bonding) as well as better solubility.26 As the initial attempt, two resorcinol linkers were employed to bridge two diarylthiourea groups to form [2 + 2] macrocycle 6 in order to take advantage of the cooperativity of two thiourea groups and other weak C–H sites on the macrocyclic skeleton (Figure 2).20 The synthesis was straightforward, including four high-yielding steps from starting compounds 1 and 2. The key macrocyclization condensation between diamine 4 and diisothiocyanate 5 furnished the macrocycle in around 60% yield and on a gram scale (Figure 2a). As shown by the crystal structure, the macrocycle possessed an overall planar conformation and formed a tight complex with the ethanedisulfonate anion, –O3SCH2CH2SO3– (EDS2–) (Figure 2b,c). The anion was tightly trapped within the cavity through a cooperative H-bonding network, including two pairs of strong bifurcated H-bonds with the two thiourea units, along with some weak C–H H-bonds. The strong binding was confirmed in solution by NMR titration and isothermal titration calorimetry (ITC) experiments. A binding affinity of up to 2.1 × 106 M–1 was determined by ITC (in CH3CN, nBu4N+ was a counterion). The binding toward the methanesulfonate anion, CH3SO3–, was much weaker (7.8 × 103 M–1), showcasing the power of complementary binding by the integration of multiple interaction sites into a macrocyclic skeleton.
Figure 2.

(a) Synthesis of the bis-thiourea macrocycle 6 and (b, c) crystal structures of 6•EDS2–. H-bonding distances: N–H···O, 2.87–2.88 Å; C–H···O, 3.40–3.55 Å. Panel c was reproduced with permission from ref (20). Copyright 2018, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
With strong binding toward ethanedisulfonate, the macrocycle was applied in acid-catalyzed reactions, aiming to trap the counteranion of the acid catalyst and improve the “proton” catalytic activity. As a typical example of protonic acid-catalyzed reactions, the Povarov reaction between imine 10 and 2,3-dihydrofuran 11 was investigated (Figure 3a).20 The reaction catalyzed with only ethanedisulfonic acid (EDSA) was sluggish, and the coexistence of as low as 0.25 mol % of the macrocycle significantly improved the catalytic efficiency (Figure 3b). The macrocycle itself, however, did not show any catalytic activity. Different acyclic control compounds 7–9 were subjected for comparison, and an obviously diminished acceleration effect was observed, correlating with their low binding affinity toward EDS2– (Figure 3c). When methanesulfonic acid (MSA) was used, both the macrocycle and acyclic analogues showed little acceleration effects. This can be explained by the results that all of them showed similar and significantly weaker binding toward methanesulfonate. It was worth noting that the acceleration effect of the macrocycle was general as tested for a series of imine substrates, although their intrinsic reactivities varied due to different electronic properties. Further binding experiments suggested that the catalytic acceleration was mainly attributed to an anion complexation-induced acidity enhancement (pKa shifts of −1.6 and −3.0). The presence of the macrocycle shifted the imine protonation equilibrium through tightly trapping the counteranion, leading to the enrichment of the active protonated imine intermediate and thus the observed acceleration effect (Figure 3d).
Figure 3.

Macrocycle acceleration effect on acid-catalyzed Povarov reaction compared with acyclic analogues. (a) Reaction scheme, (b) the monitored reaction progress, (c) binding affinities for EDS2–, and (d) the reaction mechanism. Panels b and d were adapted with permission from ref (20). Copyright 2018, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
Encouraged by the above result, we wondered if chiral macrocycles can be applied for not only boosting high catalytic efficiency but also triggering chiral induction through counteranion trapping. With this in mind, BINOL moieties28 were introduced instead of resorcinol linkers to furnish chiral bis-thiourea macrocycles 13–18 (Figure 4a).1 The choice of 3,3′-linking on BINOL was to maintain a suitable distance between the two thiourea binding units and also leave the internal 2,2′-hydroxyl sites for various modifications so as to provide versatile chiral cavity environments. The synthesis was straightforward by following a similar route to that of the achiral macrocycle. Good yields of 50–66% were retained for the macrocyclization step. As anticipated, these chiral macrocycles also showed very strong binding toward EDS2– (>105 M–1, in CH3CN). For 17 and 18 containing larger internal substituents, a slow host–guest exchange dynamic was observed on the NMR time scale, indicating particularly tight binding. The crystal structure of 13•EDS2– showed that the ethanedisulfonate anion was tightly bound within the macrocyclic cavity through multiple H-bonds with the two thiourea groups and the side hydroxyl groups (Figure 4b). For 18•EDS2–, as suggested by DFT modeling, the anion was also tightly trapped within the cavity through two pairs of parallel H-bonds. The two side bismethylene ethylene groups help clip the anion, which could provide additional confinement (Figure 4c). Upon tight complexation, the macrocycles adopted a saddle-like conformation and formed a large, chiral cleft on both faces.
Figure 4.

(a) BINOL-derived chiral bis-thiourea macrocycles 13–18, (b) crystal structure of 13•EDS2–, and (c) DFT-optimized structure (at the M06-2X/6-311+G(d) level) of 18•EDS2–. H-bonding distances in (b) N–H···O, 2.81–3.09 Å; O–H···O, 2.70–2.94 Å; in (c) N–H···O, 2.77–2.93 Å. Adapted with permission from ref (1). Copyright 2020, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
These chiral macrocycles were then applied in the acid-catalyzed Friedel–Crafts reaction of indoles with imines, which serves as one class of important and widely studied transformations (Figure 5a).1 Among the different macrocycles tested, 18 was found to be the best. The small change in the internal substituents showed a significant effect on chiral induction. In combination with 1 mol % EDSA, the use of 1 mol % 18 afforded product 21 in 99% yield and 82% ee. When only EDSA or 18 was used, no reactivity was observed, suggesting a drastic synergy between the macrocycle and the acid. The use of MSA instead of EDSA afforded only a low yield and selectivity (14% yield, 28% ee), in line with the much weaker binding of the macrocycle toward methanesulfonate. The generality and practical use of this approach was demonstrated for a series of indole and imine substrates, especially for sterically hindered 4- or 7-halo-substituted indoles that were not easily handled by conventional chiral acid catalysis. In most cases, good yields and excellent enantioselectivities (up to 99% ee) were obtained (Figure 5a), and the reaction can be run on a gram scale. The required use of only low loadings of the macrocycle and the acid demonstrated the further advantage of this supramolecular approach. Further theoretical modeling shed more light on the catalytic mode and stereocontrol outcome (Figure 5b,c). In the resting complex, 18•EDS2–•19-H+, the protonated imine substrate was nicely aligned within one side of the cave-like macrocyclic cavity and anchored to the bound counteranion through a strong, ion-pair-mediated H-bonding interaction, exposing its Si face to indole attack. Additional π···π, C–H···π, and dipole–dipole interactions between both substrate components and the cavity surroundings help stabilize the favored transition state.
Figure 5.

(a) Chiral bis-thiourea macrocycle/EDSA-catalyzed Friedel–Crafts reactions, (b) DFT-optimized structure (at the M06-2X/6-311+G(d) level) of the ternary complex 18•EDS2–•19-H+, and (c) proposed transition state leading to the major R-21 product. H-bonding distances in (b) N–H···O, 2.83–2.97 Å (with thiourea groups); (C=)N–H+···O, 2.61 Å (with the substrate iminium group). Panels b and c were adapted with permission from ref (1). Copyright 2020, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
3. Substrate-Induced Assembly of Chiral Macrocycles toward Cooperative Asymmetric Catalysis
Induced fit is one important feature of enzyme catalysis in order to obtain the most favorable interactions between the substrate and active site for maximum transition-state stabilization. In addition to the commonly occurring local induced fit, the global induced fit could also operate through the cooperation of individual protein domains by reciprocal complementation of their active sites. One example is dynamin, a prototypical member of GTPases that undergoes G domain dimerization to trigger catalytic activity.29 While this phenomenon occurs in biological systems, for synthetic catalytic systems, the examples on substrate-induced catalyst assembly and regulation remained scarce, especially for chiral catalysis systems.30−32
Following the project on the fabrication of a BINOL-based chiral bis-thiourea macrocycle catalyst system, a series of chiral diamine units were then chosen to be incorporated into the macrocyclic skeleton (Figure 6a).2 This was on one hand to afford a different chiral environment and on the other hand to furnish bifunctional thiourea-Lewis base sites for dual activation of both electrophile and nucleophile.33 By choosing 1,2-cyclohexanediamine and 1,2-diphenylethylenediamine units and their heterocombination in a different chiral configuration, tetraamino-bisthiourea chiral macrocycles 22–25 were readily synthesized in good yields (39–72% yields for the macrocyclization step). During binding studies, crystals of a sandwich-like complex, (22)2•SO42–, were reproducibly obtained (Figure 6b,c). Sulfate was encapsulated between the two cross-arrayed macrocycles, forming four pairs of complementary bifurcated H-bonds with the four spatially aligned thiourea groups. Upon dimerization, the macrocycle adopted an extended conformation, which was in contrast to the self-folded conformation adopted in the monomeric chloride complex. The sulfate-induced dimerization assembly was also held in solution, as suggested by NMR titrations and ESI-MS experiments.
Figure 6.

(a) Tetraamino-bisthiourea chiral macrocycles 22–25 and (b, c) crystal structure of (22)2•SO42–. H-bonding distances: N–H···O, 2.87–2.93 Å. Adapted with permission from ref (2). Copyright 2020, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
Upon sulfate-induced dimerization assembly, the macrocycle adopted an extroversive conformation and reciprocally complemented the interaction sites, which was reminiscent of the induced-fit regulation of enzymes. This dynamic assembly characteristic was then taken for cooperative catalysis design (Figure 7).2 The decarboxylative Mannich reaction of cyclic N-sulfamidate aldimines with β-ketoacids was investigated. This reaction provides a means for accessing valuable chiral β-amino ketones, but efficient organocatalytic asymmetric transformation was lacking.34 It was speculated that the imine substrate 26 could be activated toward nucleophilic attack through induced dimerization assembly of the chiral macrocycle catalyst, as the generating negative charge on the sulfamate heading group (analogue to sulfate) could be maximumly stabilized through the cooperative H-bonding network formation (Figure 7a). After some screening, it was found that 5 mol % macrocycle 25 could enable quick transformation of 26 and 27 to afford product 28 in a nearly quantitative yield and a high enantioselectivity, 88% ee (Figure 7b). The macrocycles 22–24 gave similar yields but rather low enantioselectivities (<26% ee), suggesting that a subtle structural change of the macrocycle could have a large influence on chiral induction. The transformations could be extended to other substrates regardless of the electronic effect on both imine and β-ketoacid components, affording the desired products in high yields and good to excellent enantioselectivities (up to 95% ee).
Figure 7.

Substrate-induced dimerization assembly catalysis. (a) Design, (b) macrocycle-catalyzed decarboxylative Mannich reaction, (c) dependence of the initial reaction rate of 26 on the concentration of the catalyst, (d) dependence of ee of the product on ee of the catalyst, and (e) DFT modeling (at the B3LYP/3-21G level) of the addition anionic intermediate fitting within the dimeric assembly. Panels a, c, and d were adapted with permission from ref (2). Copyright 2020, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
A collection of experimental evidence supported the proposed dimerization assembly catalytic mechanism. In ESI-MS experiments, 2:1 binding species between the macrocycle and imine substrate as well as between the macrocycle and addition anionic intermediate were detected. A second-order dependence of the initial reaction rate on the concentration of macrocycle catalyst (Figure 7c) as well as the nonlinear effect observed between the reaction enantioselectivity and the enantiomeric purity of the macrocycle catalyst (Figure 7d) further suggested the cooperation of two macrocycle molecules in the catalytic process. By DFT optimization, a structure of the addition anionic intermediate captured within the dimeric assembly was identified, showing the anticipated cooperative H-bonding network (Figure 7e). This type of tetraamino-bisthiourea chiral macrocycle was recently applied in catalyzing the decarboxylative Mannich reaction of isatin-derived ketimines with malonic acid half thioesters, and a remarkable macrocyclic effect was observed.35
4. Anion Recognition-Driven Chirality Gearing with an Adaptive Achiral Cage
In the above work, a series of bis-thiourea macrocycles were fabricated for implementing anion binding and supramolecular catalysis by the use of different dual linkers. To obtain more sophisticated cooperation of multiple binding sites and a more enclosed cavity, trithiourea molecular cage 29 was designed and synthesized by the incorporation of two 1,3,5-triacyloxybenzene caps to link three diarylthiourea arms (Figure 8a).3 The synthesis followed a similar fragment coupling route, in which 1 + 1 macrobicyclization between tripodal tris-amine and tris-isothiocyanate fragments furnished the cage in 44% yield on a gram scale. The free cage adopted a fluxional, averaged D3h-symmetric conformation in solution as suggested by the observation of a set of simple proton and carbon signals on NMR. Due to the existence of three convergent thiourea binding units, the cage showed strong binding to the complementary 1,3,5-benzene tricarboxylate anion guest, 30 (2.4 × 105 M–1 in CH3CN). As shown by the crystal structure of [30 ⊂ 29]3–, the anion was tightly bound in the middle of the cage cavity through three pairs of parallel H-bonds (Figure 8b). Upon binding, the three diarylthiourea arms became more flattened, and the two 1,3,5-triacylbenzene caps were contracted toward the cavity. Due to the structural adaption and conformational lock through binding, the cage formed a twisted helical conformation with a reduced D3 symmetry, producing a pair of racemic complexes. The three carboxyl groups of the guest also formed the same helical sense according to the host helicity in order to accommodate optimal binding.
Figure 8.

(a) Trithiourea molecular cage 29 and anion guests 30–32 and (b) crystal structure of [30 ⊂ 29]3– showing a racemic mixture of P- and M-helical complexes. H-bonding distances: N–H···O, 2.70–2.84 Å.
Upon adaptive binding of 1,3,5-benzene tricarboxylate, the flexible, achiral cage was locked in a helical conformation. We envisioned that if a chiral guest was introduced, a dominant helicity could be induced (Figure 9a).3 The deep understanding of the underlying mechanism of chiral induction and transfer in dynamic supramolecular systems is crucial for the implementation of chiral recognition and catalysis.36−38 For this purpose, chiral guests 31–32 with a matched D3 symmetry were synthesized, in which the core tricarboxylate binding motif was maintained and another three aryl arms incorporating six homochiral stereogenic centers were installed (Figure 8a). With such a design, a well-defined, gear-like complex was formed. The two guests showed even stronger binding (6.6 × 105 and 1.7 × 106 M–1), probably due to the favorable interactions gained for the chiral aryl arms fitting within the helical grooves. The generation of the induced helicity was suggested by circular dichroism (CD) titrations. A new set of Cotton signals at 285 and 305 nm corresponding to the cage absorption band and a weak host–guest charge transfer band emerged (Figure 9b). The greater signal intensity induced by 32 compared to that induced by 31 suggested a more efficient chiral induction, although with only a small change in methyl to ethyl. DFT calculations shed more light on the gear-like chirality transmission mechanism (Figure 9c). For both guests, the (S6)-stereoisomer induced the dominance of the P-configured host. In the disfavored complex (e.g., (R6)-32 ⊂ (P)-29), the aryl-to-aryl torsion for the bound guest was severely pushed away from the relaxed status due to the steric hindrance encountered between the ethyl groups (but not methyl) and the helical grooves. This caused a large distortion energy and thus gave greater preference to the favored complex (5.2 vs 1.0 kcal/mol). The primary binding, conformational adaption, and precise stereocontrol resembled a sequence of biotic recognition. Application of this system for anion-triggered allosteric catalysis within the induced helical chiral space is being pursued in the laboratory.
Figure 9.

Anion-induced conformational adaption and gear-like chirality transmission in cage 29. (a) Schematic representation, (b) CD spectra (298 K, CH3CN) of cage 29 upon addition of chiral anions 31 and 32, and (c) DFT-optimized structures (at the B3LYP/6-31G(d) level) of a pair of diastereomeric complexes between 29 (P-configuration adopted) and both enantiomers of 31 and 32.
5. Cooperative Anion−π Catalysis
The understanding and exploiting of noncovalent interactions are crucial for implementing efficient and selective catalysis resembling the way that an enzyme does. The continuing development of supramolecular chemistry has provided inspirations for so doing, as exemplified by the evolution of H-bonding catalysis and the recent emergence of halogen-bonding, chalcogen-bonding, and pnictogen-bonding catalysis.39−43 Anion−π interaction describes the attractive interaction between an anion and an electron-deficient aromatic surface and has been recognized only since the beginning of this century (Figure 10a).44−46 After two decades of both theoretical and experimental effort, it is now considered to be an important driving force and has been applied for anion recognition, sensing, assembly, and ion transport.47−50 Meanwhile, a few studies have implied that anion−π interaction is present in some enzyme active sites and is relevant to their catalytic activity.51,52 However, the exploiting of the anion−π interaction in chemical catalysis (e.g., for the stabilization of anionic intermediates and transition states) only emerged since 2013.53−55 It was originally exemplified by the acceleration of Kemp elimination on naphthalenediimide (NDI) surfaces53 and later expanded to other transformations including enolate chemistry54 and epoxide-opening ether cyclizations.56,57 In addition to the majority use of NDIs, other π-acidic surfaces such as fullerenes58 and carbon nanotubes59 were also explored. While these examples were implemented with a single π-face activation, we envisioned that manipulating two or more π-surfaces for cooperative π-face activation was highly desirable (Figure 10b). The supramolecular cooperation would be beneficial not only for enhancing the activation ability but also for selectivity control, especially considering the rather flexible bonding geometry rendered on an aromatic surface.
Figure 10.

Schematic illustration for (a) the anion−π interaction and (b) two modes for catalysis with the anion−π interaction.
Previously, we constructed a prism-like bis(tetraoxacalix[2]arene[2]triazine) cage 36 from electron-deficient triazine aromatic unit 34 and phloroglucinol 33 (Figure 11).60 The synthesis was straightforward by either two separated steps or two steps in one pot without isolation of the intermediate compound 35.60,61 The yields were quite good considering the formation of many chemical bonds at once, and up to 10 g of cage product could be readily obtained. The cage adopted an overall D3h symmetry, forming three V-shaped electron-deficient cavities as clipped by π-acidic triazine surfaces (Figure 11). The height defined by the distance of the two phloroglucinol caps is 4.5 Å, and the openings of the V-shaped cavities in a symmetrical form are 9.1 Å as determined by the distances between the large-rim carbon atoms of triazine rings.
Figure 11.

Synthesis and crystal structure of triazine-based prism cage 36.
Consisting of two convergent π-acidic triazine surfaces, the V-shaped cavity was found to be able to bind a series of anions through favorable anion−π interactions. As shown by the crystal structure, the cavity bound Br–/H2O simultaneously to form a ternary complex in which the halide anion and water molecule formed anion−π and lone-pair (lp)−π interactions to the two triazine surfaces, respectively (Figure 12a).60 For polyhedral anions (e.g., p-toluenesulfonate (TsO–)), the cavity bound the sulfonate heading group through dual anion−π interactions to the same triazine surface (Figure 12b).4 For 1,3-propanedisulfonate (PDS2–), a cooperative binding motif in which the two triazine faces shrank inward to simultaneously interact with the anion was observed (Figure 12c).62 It was worth noting that while the cage skeleton was overall rigid (the height maintained constant), the V-shaped cavities can self-tune their size by contracting or expanding to a certain level to fit different guests (deviating from 120°, openings ranged from 8.4 to 9.6 Å as observed in different crystal complexes). Directed by the favorable anion−π interactions, diverse intermolecular self-assembly motifs with a series of di- and trisulfonate anions were formed.62 For example, the complexes of 36•PDS2– further interacted with each other by inserting the other sulfonate group into the V-cleft of the adjacent cage and formed an infinite 2D ladder-like self-assembly (Figure 12d).
Figure 12.

Crystal structures of anion−π complexes with the triazine-base cage. (a) 36•Br–•H2O, (b) 36•TsO–, (c) 36•PDS2–, and (d) intermolecular self-assembly motif formed in 36•PDS2–.
The remaining three chlorine reactive sites rendered cage 36 a diverse functionalization platform. By nucleophilic substitution with three equivalents of amine containing two terminal olefin arms, followed by a 3-fold ring-closing metathesis reaction, a sophisticated molecular barrel bearing an additional enclosed loop was efficiently constructed.61 The crystal structure showed that each of the V-shaped cavities accommodated a solvent CHCl3 molecule through lp−π interactions between chlorine atoms and triazine surfaces. With intrinsic cavities, the molecular barrel exhibited good CO2 uptake with an exceptionally large isosteric enthalpy due to the existence of favorable lp−π interactions. Meanwhile, by introducing three aromatic aldehyde groups onto the cage skeleton and following condensation with diamine linkers, the first two cage-based covalent organic frameworks were constructed, which exhibited a larger CO2 adsorption capacity.63
In order to probe chiral recognition based on anion−π interactions, novel inherently chiral cages were constructed through a hierarchical desymmetrization strategy from the parent D3h-symmetric cage (Figure 13).64 The strategy referred to first varying the two caps to introduce perpendicular dissymmetry and then varying the three arms to an ABC pattern, leading to inherently chiral cages with C1 symmetry (Figure 13a). The term “inherent chirality” was originally used to describe calixarenes with a typical AABC or ABCD substitution pattern which are devoid of conventional stereogenic elements.65 It was later expanded to other macrocyclic and acyclic concave scaffolds, but higher-level cage inherent chirality was not accessed.66,67 The C3v precursor 38 was first synthesized by introducing two different phloroglucinol caps in a stepwise fashion (Figure 13b). The subsequent progressive substitutions on the triazine arms by different N- and O-nucleophiles afforded inherently chiral cages 39–46 and 47–55 with rich structural diversity. Resolution of the racemic cages was achieved by chiral HPLC. From the enantiopure crystal of (−)-47, the absolute configuration was determined, and a chirality descriptor was provided to define the cage inherent chirality (Figure 13c). With three unequal, chiral electron-deficient π-cavities and minimum interference of other interactions, the inherently chiral cages provided a unique platform for probing regio- and enantioselective anion−π binding. As exemplified with (−)-39 and chiral phosphate (CPA–), NMR titration studies suggested that the anion was preferentially bound within the most electron-deficient cavity consisting of chlorine- and dibenzylamino-substituted triazines, which was further supported by DFT calculations (Figure 13d). Moreover, (−)-39 preferred to bind (S)-CPA– rather than (R)-CPA– with moderate chiral selectivity (12.9 vs 6.9 M–1). As shown in the optimized structure of the favored complex, the phosphate anion was tightly bound in the cavity by cooperative anion−π interactions with the two triazines. The primary anion−π binding along with the asymmetric array of the surrounding π surfaces played a vital role in the chiral discrimination.
Figure 13.

Construction of inherently chiral cages and enantioselective anion−π binding. (a) Hierarchical desymmetrization strategy, (b) synthetic route, (c) crystal structure of (−)-47 with a chirality descriptor presented for cage-inherent chirality, and (d) DFT-optimized structure (at the M06-2X/6-31G(d) level) of the favored complex of (−)-39 toward (S)-CPA–.
As demonstrated, this cage system featured easy synthesis and functionalization and good anion−π and lp−π binding properties, especially the ability to cooperatively bind multidentate anions by the confined yet self-tunable V-shaped electron-deficient cavity. These advantages enabled the cage motif to serve as a good scaffold for the cooperative anion−π catalysis design. It was anticipated that the V-shaped cavity was suitable to bind an electrophilic substrate through cooperative lp−π interactions in the ground state and transit to more favorable anion−π binding to stabilize the developing anionic transition state upon nucleophilic attack (Figure 14).4 An additional base site could be installed on the periphery of the cavity for initiating a nucleophilic attack.
Figure 14.
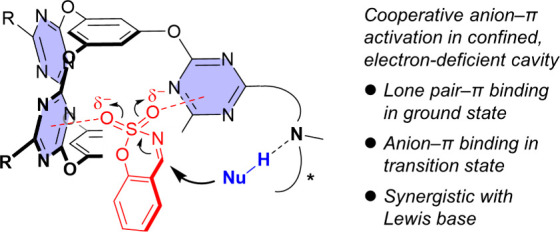
Design of molecular cage catalysts for cooperative anion−π activation. Adapted with permission from ref (4). Copyright 2021, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
Following the design, a series of chiral molecular cage catalysts were readily synthesized in high yields from cage 36 by a one-step reaction with different chiral amines.4 Based on the anticipated activation mode, a decarboxylative Mannich reaction between cyclic N-sulfamidate aldimines and malonic acid half thioesters (MAHTs) was investigated (Figure 15). This type of reaction provided a means for accessing valuable β-amino ester products, and the development of an efficient asymmetric organocatalytic approach was appealing.68,69 After some simple screening, cinchonidine-pendant cage 56 was found to be able to catalyze the reactions in an efficient and highly selective fashion. The use of only a 2 mol % cage enabled the transformation of 57 and 58 to afford 59 in a nearly quantitative yield and 95% ee. The study of substrate scope showed that for imines bearing electron-rich substituents reacted with various MAHTs, excellent yields and enantioselectivities (up to 97% ee) were obtained. For imines bearing electron-withdrawing substituents, the enantioselectivities progressively decreased (Figure 15). This observation was in line with the proposed activation mode where the chiral discrimination was enhanced due to the more favorable anion−π binding gained in the transition state with the higher negative charge density on the sulfamate heading group. The essential role of the V-shaped electron-deficient cavity was further demonstrated by a series of control and competitive experiments (Figure 15). The fragment compound 60 with only one triazine surface was less active and led to very low enantioselectivity (16% ee). The single cinchonidine component 61, although more active, led to a poor and opposite selectivity (−21% ee). On the other hand, the progressive addition of the competitive binder, the p-toluenesulfonate anion, caused a gradual loss of the enantioselectivity. In addition, the in situ detection of a series of cage-associated reaction species, including the key addition anionic intermediate, was obtained by ESI-MS.
Figure 15.

Cage-catalyzed decarboxylative Mannich reaction of cyclic aldimines and MAHTs.
Based on the above experimental results, optimized structures for substrate binding and the reaction transition state were identified. For the cage-substrate complex, the sulfamate heading group was bound within the V-shaped cavity through triple lp−π interactions (Figure 16a). In the favored transition state, the imine component flipped upright and exposed its Re face to accept the attack of enolate, which was positioned by the protonated quinuclidine moiety. The developing sulfamate anion was clipped by the V-shaped cavity and stabilized through cooperative anion−π interactions with both triazines (Figure 16b). Such a noncovalent network nested in the confined π-acidic cavity was accounted for the efficient and selective catalysis.
Figure 16.

(a) DFT-optimized structure (at the M06-2X/6-31G level) of cage-substrate complex 56•57 and (b) identified catalytic transition state leading to the major R-59 product. Adapted with permission from ref (4). Copyright 2021, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
6. Conclusions and Perspectives
Over the past decade, our research group has been devoted to the design and synthesis of functional macrocycles and molecular cages for building efficient supramolecular catalysis systems through shaping an anion recognition-directed approach. As demonstrated, this approach can serve as a powerful, versatile tool and source of inspiration, enabling many possibilities for boosting highly efficient and selective catalysis. It was not only possible for employing exogenous anions such as the counteranion of an acid catalyst as a handle to exert control on cationic intermediates but also competent for the direct recognition and regulation of anionic reactive intermediates/transition states. Not only the conventional H-bonding but also the emerging anion−π interaction can be exploited on design for driving efficient catalysis. Due to the sophisticated cooperation of multiple noncovalent interactions nested in a confined cavity enabled by the tailor-made macrocycles and cages, significant catalytic acceleration and excellent stereocontrol were achieved. The superior performance was distinguished from the acyclic counterparts that in many cases led to only sluggish conversions, showcasing the power of the macrocyclic effect and supramolecular cooperation.
The developed anion recognition-directed supramolecular catalysis shows many prospects but is just in an early stage. As anion recognition itself is a long-standing challenge, the further integration of recognition and catalysis into a system is even more challenging and requires a good understanding of the structure and properties of the involved anion species for a sophisticated design. On the other hand, chiral control typically deals with very subtle spatial structure discrimination, and rational design in this regard is often difficult and sometimes requires trial and error attempts, particularly for the transfer of the relatively large-scale structural chirality of the macrocycles and cages to the small reaction components. The approach is currently mainly applied in boosting transformations of ionic-type imine addition reactions in the demonstrated examples.
As anionic species are abundant and associated with a variety of chemical transformations, we foresee that the formulated concept can be extended for boosting the reactivity of many other catalytic species or intermediates. For example, not limited to Brønsted acid catalysis, the counteranion trapping strategy can be extended for tuning Lewis acid catalysis and transition-metal catalysis, such as gold catalysis where large counterion effects exist.70 It would also be feasible for boosting transformations undergoing active intermediates such as carbocations through manipulating counteranion binding, especially for chiral transformations. In addition to persistent anions, other reactive anions such as high-valence, oxidative oxyanions can be also manipulated for enhancing their solubilities in organic media and for exerting chiral control. On the other hand, besides the traditional chirality elements, the exploration of supramolecular chirality such as the induced helicity and the novel inherent chirality for triggering stereocontrol would be fascinating. The further exploiting of the emerging anion−π interaction as a driving force for novel catalysis design and reaction development is highly attractive, one direction of which could be aqueous anion−π catalysis.71 Last but not least, the transfer of the obtained knowledge for other type of supramolecular catalysis system design would also be acknowledged.72,73
Acknowledgments
The work described in this Account includes the collective efforts of past and present co-workers, whose names appear in the cited references. We express our sincere gratitude to all of them. Financial support from the Natural Science Foundation of China (21502200, 21871276, 22022112, and 21521002), Chinese Academy of Sciences (QYZDJ-SSW-SLH023), and National Key R&D Program of China (2021YFA1501600) is gratefully acknowledged.
Biography
Qi-Qiang Wang received his B.S. degree from Wuhan University, China, in 2003, and his Ph.D. degree from the Institute of Chemistry, Chinese Academy of Sciences (ICCAS), in 2008. After working as a postdoctoral fellow at University of Kansas, USA (2008–2013) and at University of Amsterdam, The Netherlands (2014–2015), he joined ICCAS as a full professor in 2015. His research interests include molecular recognition, assembly, and supramolecular catalysis, with an emphasis on exploiting novel noncovalent interaction modes and functional macrocycle and cage hosts.
The author declares no competing financial interest.
References
- Ning R.; Zhou H.; Nie S.-X.; Ao Y.-F.; Wang D.-X.; Wang Q.-Q. Chiral Macrocycle-Enabled Counteranion Trapping for Boosting Highly Efficient and Enantioselective Catalysis. Angew. Chem., Int. Ed. 2020, 59, 10894–10898. 10.1002/anie.202003673. [DOI] [PubMed] [Google Scholar]
- Guo H.; Zhang L.-W.; Zhou H.; Meng W.; Ao Y.-F.; Wang D.-X.; Wang Q.-Q. Substrate-Induced Dimerization Assembly of Chiral Macrocycle Catalysts Toward Cooperative Asymmetric Catalysis. Angew. Chem., Int. Ed. 2020, 59, 2623–2627. 10.1002/anie.201910399. [DOI] [PubMed] [Google Scholar]
- Hu Q.-P.; Zhou H.; Huang T.-Y.; Ao Y.-F.; Wang D.-X.; Wang Q.-Q. Chirality Gearing in an Achiral Cage Through Adaptive Binding. J. Am. Chem. Soc. 2022, 144, 6180–6184. 10.1021/jacs.2c02040. [DOI] [PubMed] [Google Scholar]
- Luo N.; Ao Y.-F.; Wang D.-X.; Wang Q.-Q. Exploiting Anion−π Interactions for Efficient and Selective Catalysis with Chiral Molecular Cages. Angew. Chem., Int. Ed. 2021, 60, 20650–20655. 10.1002/anie.202106509. [DOI] [PubMed] [Google Scholar]
- Meeuwissen J.; Reek J. N. H. Supramolecular Catalysis beyond Enzyme Mimics. Nat. Chem. 2010, 2, 615–621. 10.1038/nchem.744. [DOI] [PubMed] [Google Scholar]
- Raynal M.; Ballester P.; Vidal-Ferran A.; van Leeuwen P. W. N. M. Supramolecular Catalysis. Part 1: Non-covalent Interactions as a Tool for Building and Modifying Homogeneous Catalysts. Chem. Soc. Rev. 2014, 43, 1660–1733. 10.1039/C3CS60027K. [DOI] [PubMed] [Google Scholar]
- Raynal M.; Ballester P.; Vidal-Ferran A.; van Leeuwen P. W. N. M. Supramolecular Catalysis. Part 2: Artificial Enzyme Mimics. Chem. Soc. Rev. 2014, 43, 1734–1787. 10.1039/C3CS60037H. [DOI] [PubMed] [Google Scholar]
- van Leeuwen P. W. N. M.; Raynal M.. Supramolecular Catalysis: New Directions and Developments; Wiley-VCH GmbH, Weinheim, 2022. [Google Scholar]
- Breslow R.; Dong S. D. Biomimetic Reactions Catalyzed by Cyclodextrins and Their Derivatives. Chem. Rev. 1998, 98, 1997–2011. 10.1021/cr970011j. [DOI] [PubMed] [Google Scholar]
- Assaf K. I.; Nau W. M. Cucurbiturils: From Synthesis to High-affinity Binding and Catalysis. Chem. Soc. Rev. 2015, 44, 394–418. 10.1039/C4CS00273C. [DOI] [PubMed] [Google Scholar]
- Wang Q.-Q.Supramolecular Catalysis Using Organic Macrocycles. Handbook of Macrocyclic Supramolecular Assembly; Liu Y., Chen Y., Zhang H.-Y., Eds.; Springer: Singapore, 2019. [Google Scholar]
- Yoshizawa M.; Klosterman J. K.; Fujita M. Functional Molecular Flasks: New Properties and Reactions within Discrete, Self-Assembled Hosts. Angew. Chem., Int. Ed. 2009, 48, 3418–3438. 10.1002/anie.200805340. [DOI] [PubMed] [Google Scholar]
- Brown C. J.; Toste F. D.; Bergman R. G.; Raymond K. N. Supramolecular Catalysis in Metal–Ligand Cluster Hosts. Chem. Rev. 2015, 115, 3012–3035. 10.1021/cr4001226. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Catti L.; Tiefenbacher K. Catalysis inside the Hexameric Resorcinarene Capsule. Acc. Chem. Res. 2018, 51, 2107–2114. 10.1021/acs.accounts.8b00320. [DOI] [PubMed] [Google Scholar]
- Bianchi A.; Bowman-James K.; García-España E.. Supramolecular Chemistry of Anions; Wiley-VCH: New York, 1997. [Google Scholar]
- Sessler J. L.; Gale P. A.; Cho W.-S.. Anion Receptor Chemistry; The Royal Society of Chemistry: Cambridge, U.K., 2006. [Google Scholar]
- Bowman-James K.; Bianchi A.; García-España E.. Anion Coordination Chemistry; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Gokel G.Crown Ethers and Cryptands; The Royal Society of Chemistry: Cambridge, 1991. [Google Scholar]
- Oliveira M. T.; Lee J.-W. Asymmetric Cation-Binding Catalysis. ChemCatChem. 2017, 9, 377–384. 10.1002/cctc.201601441. [DOI] [Google Scholar]
- Ning R.; Ao Y.-F.; Wang D.-X.; Wang Q.-Q. Macrocycle-Enabled Counteranion Trapping for Improved Catalytic Efficiency. Chem.—Eur. J. 2018, 24, 4268–4272. 10.1002/chem.201800326. [DOI] [PubMed] [Google Scholar]
- Phipps R. J.; Hamilton G. L.; Toste F. D. The Progression of Chiral Anions from Concepts to Applications in Asymmetric Catalysis. Nat. Chem. 2012, 4, 603–614. 10.1038/nchem.1405. [DOI] [PubMed] [Google Scholar]
- Mahlau M.; List B. Asymmetric Counteranion-Directed Catalysis: Concept, Definition, and Applications. Angew. Chem., Int. Ed. 2013, 52, 518–533. 10.1002/anie.201205343. [DOI] [PubMed] [Google Scholar]
- Brak K.; Jacobsen E. N. Asymmetric Ion-Pairing Catalysis. Angew. Chem., Int. Ed. 2013, 52, 534–561. 10.1002/anie.201205449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García Mancheño O., Eds. Anion-Binding Catalysis; Wiley-VCH: Weinheim, 2022. [Google Scholar]
- Aleksiev M.; García Mancheño O. Enantioselective Dearomatization Reactions of Heteroarenes by Anion-Binding Organocatalysis. Chem. Commun. 2023, 59, 3360–3372. 10.1039/D2CC07101K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp A.; Schreiner P. R. Metal-Free, Noncovalent Catalysis of Diels-Alder Reaction by Neutral Hydrogen Bond Donors in Organic Solvents and in Water. Chem.—Eur. J. 2003, 9, 407–414. 10.1002/chem.200390042. [DOI] [PubMed] [Google Scholar]
- Li A.-F.; Wang J.-H.; Wang F.; Jiang Y.-B. Anion Complexation and Sensing Using Modified Urea and Thiourea-based Receptors. Chem. Soc. Rev. 2010, 39, 3729–3745. 10.1039/b926160p. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Hu Y.; Ning C.; Shi W.; Yang A.; Zhao Y.; Cao Z.-Y.; Xu Y.; Du P. BINOL-Based Chiral Macrocycles and Cages. Angew. Chem., Int. Ed. 2024, 63, e202407034 10.1002/anie.202407034. [DOI] [PubMed] [Google Scholar]
- Chappie J. S.; Acharya S.; Leonard M.; Schmid S. L.; Dyda F. G Domain Dimerization Controls Dynamin’s Assembly-Stimulated GTPase Activity. Nature 2010, 465, 435–441. 10.1038/nature09032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G.; Gupta A. K.; Huang R. H.; Mukherjee M.; Wulff W. D. Substrate-Induced Covalent Assembly of a Chemzyme and Crystallographic Characterization of a Chemzyme–Substrate Complex. J. Am. Chem. Soc. 2010, 132, 14669–14675. 10.1021/ja1070224. [DOI] [PubMed] [Google Scholar]
- Fanlo-Virgós H.; Alba A.-N. R.; Hamieh S.; Colomb-Delsuc M.; Otto S. Transient Substrate-Induced Catalyst Formation in a Dynamic Molecular Network. Angew. Chem., Int. Ed. 2014, 53, 11346–11350. 10.1002/anie.201403480. [DOI] [PubMed] [Google Scholar]
- Muñana P. S.; Ragazzon G.; Dupont J.; Ren C. Z.-J.; Prins L. J.; Chen J. L.-Y. Substrate-Induced Self-Assembly of Cooperative Catalysts. Angew. Chem., Int. Ed. 2018, 57, 16469–16474. 10.1002/anie.201810891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto Y. Recognition and Activation by Ureas and Thioureas: Stereoselective Reactions Using Ureas and Thioureas as Hydrogen-Bonding Donors. Org. Biomol. Chem. 2005, 3, 4299–4306. 10.1039/b511216h. [DOI] [PubMed] [Google Scholar]
- Zhang H.-X.; Nie J.; Cai H.; Ma J.-A. Cyclic Aldimines as Superior Electrophiles for Cu-Catalyzed Decarboxylative Mannich Reaction of β-Ketoacids with a Broad Scope and High Enantioselectivity. Org. Lett. 2014, 16, 2542–2545. 10.1021/ol500929d. [DOI] [PubMed] [Google Scholar]
- Guo H.; Ao Y.-F.; Wang D.-X.; Wang Q.-Q. Bioinspired Tetraamino-Bisthiourea Chiral Macrocycles in Catalyzing Decarboxylative Mannich Reactions. Beilstein J. Org. Chem. 2022, 18, 486–496. 10.3762/bjoc.18.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos-Timoneda M. A.; Crego-Calama M.; Reinhoudt D. N. Supramolecular Chirality of Self-Assembled Systems in Solution. Chem. Soc. Rev. 2004, 33, 363–372. 10.1039/b305550g. [DOI] [PubMed] [Google Scholar]
- Liu M.; Zhang L.; Wang T. Supramolecular Chirality in Self-Assembled Systems. Chem. Rev. 2015, 115, 7304–7397. 10.1021/cr500671p. [DOI] [PubMed] [Google Scholar]
- Yashima E.; Ousaka N.; Taura D.; Shimomura K.; Ikai T.; Maeda K. Supramolecular Helical Systems: Helical Assemblies of Small Molecules, Foldamers, and Polymers with Chiral Amplification and Their-Functions. Chem. Rev. 2016, 116, 13752–13990. 10.1021/acs.chemrev.6b00354. [DOI] [PubMed] [Google Scholar]
- Benz S.; Poblador-Bahamonde A. I.; Low-Ders N.; Matile S. Catalysis with Pnictogen, Chalcogen, and Halogen Bonds. Angew. Chem., Int. Ed. 2018, 57, 5408–5412. 10.1002/anie.201801452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z.; Wang Y. Chalcogen Bonding Catalysis with Phosphonium Chalcogenide (PCH). Acc. Chem. Res. 2023, 56, 608–621. 10.1021/acs.accounts.3c00009. [DOI] [PubMed] [Google Scholar]
- Jovanovic D.; Mohanan M. P.; Huber S. M. Halogen, Chalcogen, Pnictogen, and Tetrel Bonding in Non-Covalent Organocatalysis: An Update. Angew. Chem., Int. Ed. 2024, 63, e202404823 10.1002/anie.202404823. [DOI] [PubMed] [Google Scholar]
- Sekar G.; Naira V. V.; Zhu J. Chalcogen Bonding Catalysis. Chem. Soc. Rev. 2024, 53, 586–605. 10.1039/D3CS00503H. [DOI] [PubMed] [Google Scholar]
- Renno G.; Chen D.; Zhang Q.-X.; Gomila R. M.; Frontera A.; Sakai N.; Ward T. R.; Matile S. Pnictogen-Bonding Enzymes. Angew. Chem., Int. Ed. 2024, 63, e202411347 10.1002/anie.202411347. [DOI] [PubMed] [Google Scholar]
- Quiñonero D.; Garau C.; Rotger C.; Frontera A.; Ballester P.; Costa A.; Deya P. M. Anion−π Interactions: Do They Exist?. Angew. Chem., Int. Ed. 2002, 41, 3389–3392. . [DOI] [PubMed] [Google Scholar]
- Mascal M.; Armstrong A.; Bartberger M. D. Anion–Aromatic Bonding: A Case for Anion Recognition by π-Acidic Rings. J. Am. Chem. Soc. 2002, 124, 6274–6276. 10.1021/ja017449s. [DOI] [PubMed] [Google Scholar]
- Alkorta I.; Rozas I.; Elguero J. Interaction of Anions with Perfluoro Aromatic Compounds. J. Am. Chem. Soc. 2002, 124, 8593–8598. 10.1021/ja025693t. [DOI] [PubMed] [Google Scholar]
- Schottel B. L.; Chifotides H. T.; Dunbar K. R. Anion−π Interactions. Chem. Soc. Rev. 2008, 37, 68–83. 10.1039/B614208G. [DOI] [PubMed] [Google Scholar]
- Frontera A.; Gamez P.; Mascal M.; Mooibroek T. J.; Reedijk J. Putting Anion−π Interactions into Perspective. Angew. Chem., Int. Ed. 2011, 50, 9564–9583. 10.1002/anie.201100208. [DOI] [PubMed] [Google Scholar]
- Giese M.; Albrecht M.; Rissanen K. Anion−π Interactions with Fluoroarenes. Chem. Rev. 2015, 115, 8867–8895. 10.1021/acs.chemrev.5b00156. [DOI] [PubMed] [Google Scholar]
- Wang D.-X.; Wang M.-X. Exploring Anion−π Interactions and Their Applications in Supramolecular Chemistry. Acc. Chem. Res. 2020, 53, 1364–1380. 10.1021/acs.accounts.0c00243. [DOI] [PubMed] [Google Scholar]
- Estarellas C.; Frontera A.; Quiñonero D.; Deyà P. M. Relevant Anion−π Interactions in Biological Systems: The Case of Urate Oxidase. Angew. Chem., Int. Ed. 2011, 50, 415–418. 10.1002/anie.201005635. [DOI] [PubMed] [Google Scholar]
- Schwans J. P.; Sunden F.; Lassila J. K.; Gonzalez A.; Tsai Y.; Herschlag D. Use of Anion–Aromatic Interactions to Position the General Base in the Ketosteroid Isomerase Active Site. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 11308–11313. 10.1073/pnas.1206710110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Domoto Y.; Orentas E.; Beuchat C.; Emery D.; Mareda J.; Sakai N.; Matile S. Catalysis with Anion−π Interactions. Angew. Chem., Int. Ed. 2013, 52, 9940–9943. 10.1002/anie.201305356. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Cotelle Y.; Liu L.; López-Andarias J.; Bornhof A.-B.; Akamatsu M.; Sakai N.; Matile S. The Emergence of Anion−π Catalysis. Acc. Chem. Res. 2018, 51, 2255–2263. 10.1021/acs.accounts.8b00223. [DOI] [PubMed] [Google Scholar]
- Luo N.; Ao Y.-F.; Wang D.-X.; Wang Q.-Q. Putting Anion−π Interactions at Work for Catalysis. Chem.—Eur. J. 2022, 28, e202103303 10.1002/chem.202103303. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Hao X.; Liu L.; Pham A.-T.; López-Andarias J.; Frontera A.; Sakai N.; Matile S. Primary Anion-π Catalysis and Autocatalysis. J. Am. Chem. Soc. 2018, 140, 17867–17871. 10.1021/jacs.8b11788. [DOI] [PubMed] [Google Scholar]
- Paraja M.; Matile S. Primary Anion-π Catalysis of Epoxide-Opening Ether Cyclization into Rings of Different Sizes: Access to New Reactivity. Angew. Chem., Int. Ed. 2020, 59, 6273–6277. 10.1002/anie.202000579. [DOI] [PubMed] [Google Scholar]
- López-Andarias J.; Frontera A.; Matile S. Anion-π Catalysis on Fullerenes. J. Am. Chem. Soc. 2017, 139, 13296–13299. 10.1021/jacs.7b08113. [DOI] [PubMed] [Google Scholar]
- Bornhof A.-B.; Vázquez-Nakagawa M.; Rodríguez-Pérez L.; Herranz M. Á.; Sakai N.; Martín N.; Matile S.; López-Andarias J. Anion-π Catalysis on Carbon Nanotubes. Angew. Chem., Int. Ed. 2019, 58, 16097–16100. 10.1002/anie.201909540. [DOI] [PubMed] [Google Scholar]
- Wang D.-X.; Wang Q.-Q.; Han Y.; Wang Y.; Huang Z.-T.; Wang M.-X. Versatile Anion−π Interactions Between Halides and a Conformationally Rigid Bis(tetraoxacalix[2]Arene[2]triazine) Cage and Their Directing Effect on Molecular Assembly. Chem.—Eur. J. 2010, 16, 13053–13057. 10.1002/chem.201002307. [DOI] [PubMed] [Google Scholar]
- Wang Q.-Q.; Luo N.; Wang X.-D.; Ao Y.-F.; Chen Y.-F.; Liu J.-M.; Su C.-Y.; Wang D.-X.; Wang M.-X. Molecular Barrel by a Hooping Strategy: Synthesis, Structure, and Selective CO2 Adsorption Facilitated by Lone Pair−π Interactions. J. Am. Chem. Soc. 2017, 139, 635–638. 10.1021/jacs.6b12386. [DOI] [PubMed] [Google Scholar]
- Wang X.-Y.; Zhu J.; Wang Q.-Q.; Ao Y.-F.; Wang D.-X. Anion−π-Directed Self-Assembly Between Di- and Trisulfonates and a Rigid Molecular Cage with Three Electron-Deficient V-Clefts. Inorg. Chem. 2019, 58, 5980–5987. 10.1021/acs.inorgchem.9b00295. [DOI] [PubMed] [Google Scholar]
- Ma J.-X.; Li J.; Chen Y.-F.; Ning R.; Ao Y.-F.; Liu J.-M.; Sun J.; Wang D.-X.; Wang Q.-Q. Cage Based Crystalline Covalent Organic Frameworks. J. Am. Chem. Soc. 2019, 141, 3843–3848. 10.1021/jacs.9b00665. [DOI] [PubMed] [Google Scholar]
- Zhou H.; Ao Y.-F.; Wang D.-X.; Wang Q.-Q. Inherently Chiral Cages via Hierarchical Desymmetrization. J. Am. Chem. Soc. 2022, 144, 16767–16772. 10.1021/jacs.2c08591. [DOI] [PubMed] [Google Scholar]
- Böhmer V.; Kraft D.; Tabatabai M. Inherently Chiral Calixarenes. J. Inclusion Phenom. Mol. Recognit. Chem. 1994, 19, 17–39. 10.1007/BF00708972. [DOI] [Google Scholar]
- Dalla Cort A.; Mandolini L.; Pasquini C.; Schiaffino L. Inherent Chirality” and Curvature. New J. Chem. 2004, 28, 1198–1199. 10.1039/B404388J. [DOI] [Google Scholar]
- Szumna A. Inherently Chiral Concave Molecules—from Synthesis to Applications. Chem. Soc. Rev. 2010, 39, 4274–4285. 10.1039/b919527k. [DOI] [PubMed] [Google Scholar]
- Pan Y.; Tan C.-H. Catalytic Decarboxylative Reactions: Biomimetic Approaches Inspired by Polyketide Biosynthesis. Synthesis 2011, 2011, 2044–2053. 10.1055/s-0030-1260607. [DOI] [Google Scholar]
- Jia C.-M.; Zhang H.-X.; Nie J.; Ma J.-A. Catalytic Asymmetric Decarboxylative Mannich Reaction of Malonic Acid Half Esters with Cyclic Aldimines: Access to Chiral β-Amino Esters and Chroman-4-amines. J. Org. Chem. 2016, 81, 8561–8569. 10.1021/acs.joc.6b01750. [DOI] [PubMed] [Google Scholar]
- Lu Z.; Li T.; Mudshinge S. R.; Xu B.; Hammond G. B. Optimization of Catalysts and Conditions in Gold(I) Catalysis—Counterion and Additive Effects. Chem. Rev. 2021, 121, 8452–8477. 10.1021/acs.chemrev.0c00713. [DOI] [PubMed] [Google Scholar]
- Luo N.; Ao Y.-F.; Wang D.-X.; Wang Q.-Q. π-Face Promoted Catalysis in Water: From Electron-Deficient Molecular Cages to Single Aromatic Slides. Chem. Asian J. 2021, 16, 3599–3603. 10.1002/asia.202100920. [DOI] [PubMed] [Google Scholar]
- Zhang L.-W.; Wang X.-D.; Ao Y.-F.; Wang D.-X.; Wang Q.-Q. Chiral Bis-Phosphate Macrocycles for Catalytic, Efficient, and Enantioselective Electrophilic Fluorination. Chem.—Eur. J. 2024, 30, e202400498 10.1002/chem.202400498. [DOI] [PubMed] [Google Scholar]
- Sun H.; Wang X.-Y.; Wang X.-D.; Ao Y.-F.; Wang D.-X.; Wang Q.-Q. Selective Alkane Desaturation Catalyzed by Molecular Cage Copper Complexes Under Mild Conditions. CCS Chem. 2024, 6, 1876–1884. 10.31635/ccschem.024.202403936. [DOI] [Google Scholar]