

Abstract
Antimicrobial resistance is a significant cause of mortality globally due to infections, a trend that is expected to continue to rise. As existing treatments fail and new drug discovery slows, the urgency to develop novel antimicrobial therapeutics grows stronger. One promising strategy involves targeting bacterial systems exclusive to pathogens, such as the transcription regulator protein GabR. Expressed in diverse bacteria including Escherichia coli, Bordetella pertussis, and Klebsiella pneumoniae, GabR has no homolog in eukaryotes, making it an ideal therapeutic target. Bacillus subtilis GabR (bsGabR), the most studied variant, regulates its own transcription and activates genes for GABA aminotransferase (GabT) and succinic semialdehyde dehydrogenase (GabD). This intricate regulatory system presents a compelling antimicrobial target with the potential for agonistic intervention to disrupt bacterial gene expression and induce cellular dysfunction, especially in bacterial stress responses. To explore manipulation of this system and the potential of this protein as an antimicrobial target, an in‐depth understanding of the unique PLP‐dependent transcription regulation is critical. Herein, we report the successful structural modification of the cofactor PLP and demonstrate the biochemical reactivity of the PLP analog pyridoxal‐5′‐tetrazole (PLT). Through both spectrophotometric and X‐ray crystallographic analyses, we explore the interaction between bsGabR and PLT, together with a synthesized GABA derivative (S)‐4‐amino‐5‐phenoxypentanoate (4‐phenoxymethyl‐GABA or 4PMG). Most notably, we present a crystal structure of the condensed, external aldimine complex within bsGabR. While PLT alone is not a drug candidate, it can act as a probe to study the detailed mechanism of GabR‐mediated function. PLT employs a tetrazole moiety as a bioisosteric replacement for phosphate in PLP. In addition, the PLP‐4PMG adduct observed in the structure may serve as a novel chemical scaffold for subsequent structure‐based antimicrobial design.
Keywords: antibiotic, bioisosterism, external aldimine, GabR, internal aldimine, pyridoxal‐5'‐phosphate, tetrazole
1. INTRODUCTION
Antimicrobial resistance (AMR) continues to advance in pathogenic bacteria of concern through intrinsic, acquired, and adaptive mechanisms highlighting the need to discover novel therapeutics (Christaki et al., 2020). It is estimated that AMR contributes to 700,000 deaths annually with the number expected to approach 10 million by the year 2050 contributing to a potential global health and economic crisis (O'Neill, 2014). Bacteriophages, phage‐encoded enzymes, immunomodulators, and monoclonal antibodies represent some of the innovative agents to combat life‐threatening AMR bacterial infections and exemplify the diversity of therapeutic approaches (Chang et al., 2022). Targeting the transcription regulator protein GabR exemplifies exploitation of a biological system unique to the bacterial cell. Demonstrating the broad‐spectrum relevance of this transcription regulator protein, the GabR gene has been shown to be present in several AMR pathogenic bacteria of interest including Bordetella pertussis, Klebsiella pneumoniae, and Escherichia coli (Ivaska et al., 2022; Poirel et al., 2018; Wang et al., 2020; Figure S1). All genes included in this alignment are confirmed to be located just upstream of the gabT and gabD genes in their respective host genome, likely forming a divergent gabR operon like that of B. subtilis. Furthermore, GabR‐like proteins have no functional homolog in eukaryotic systems. GabR is directly responsible for the transcription of genes of both γ‐aminobutyric acid (GABA) aminotransferase (GabT) and succinic semialdehyde dehydrogenase (GabD), both of which are critical enzymes that help constitute the GABA shunt pathway (Belitsky, 2004; Belitsky & Sonenshein, 2002). Specifically, this pathway is related to the stress response in virulent microorganisms and plays a significant role in pH regulation, osmotic regulation, and nitrogen metabolism (Feehily et al., 2013; Feehily & Karatzas, 2013). One of the main obstacles is that the unique PLP‐dependent molecular mechanism of this type of transcription regulator still lacks an in‐depth understanding. Bacillus subtilis GabR (bsGabR) is the most extensively characterized GabR protein to date and represents an appropriate model for studying the ligand‐initiated transcription regulation of all GabR‐type proteins.
bsGabR is a PLP‐dependent transcription regulator directly involved in the metabolism of GABA (Belitsky & Sonenshein, 2002). The biologically active derivative of vitamin B6, pyridoxal‐5′‐phosphate (PLP), canonically contributes to the catalysis and reaction rate‐enhancement within the enzymes that it modifies. PLP acts to catalyze a variety of reactions including racemization, transamination, decarboxylation, elimination, retro‐aldol cleavage, Claisen condensation and other reactions on amino group containing substrates (Phillips, 2015; Toney, 2011). bsGabR does not catalyze a net reaction with the GABA substrate but rather produces a GAPA‐PLP external aldimine that alters the transcriptional regulatory properties, reversibly culminating in an external aldimine end‐product resulting in gene expression of gabT and gabD (Wu et al., 2017). The head‐to‐tail domain swap homodimer consists of an N‐terminal winged helix‐turn‐helix DNA‐binding domain and a C‐terminal effector binding and oligomerization (Eb/O) domain (Edayathumangalam et al., 2013). Additionally, this Eb/O architectural framework is similar to the composition of Type‐I aminotransferases and binds to both PLP and GABA in an analogous fashion (Bramucci et al., 2011). Biochemically inactive without PLP, bsGabR acts with the synergistic pair of PLP and GABA to regulate three genes: gabT, gabD, and its own transcript gabR. Through interaction with a 49‐bp region including the −35 region of the gabT promoter and the −10, +1 regions of the GabR promoter, the bsGabR dimer overlaps the bsGabR promoter region effectively downregulating its own expression (Belitsky, 2004). When effectors PLP and GABA bind within the bsGabR Eb/O domain, bsGabR is thought to induce a conformational relaxation in the regulated region of DNA, inducing transcription of the gabT and gabD genes (Frezzini et al., 2019; Milano et al., 2017). Both GabT and GabD function together preventing over‐consumption of GABA, a critical bacterial nitrogen source. This system presents an attractive therapeutic target, susceptible to either agonistic or antagonistic modulation, resulting in unrestricted gene upregulation, induced metabolic imbalances, and promotion of cellular catastrophe. Through upregulation of this nonessential pathway with the use of agonistic ligands on GabR, the bacterial stress response can be compromised by disrupting pH homeostasis and ultimately limiting pathogenicity.
The strategy of cofactor modification is relatively unexplored, representing an innovative frontier in the continued effort of antibiotic development. Several isosteric analogs of PLP have been reported and studied for purposes ranging from the development of competitive inhibitors to assessment of the pyridine nitrogen as it influences catalytic function (DeSouza et al., 2020; Griswold & Toney, 2010; Griswold & Toney, 2011). The substituents required for PLP reactivity include the 3′‐carbinol, the formyl head residing at 4′, and the pyridine nitrogen. To our knowledge, structural alteration at the 5′ position of PLP has been under‐explored and represents an innovative frontier. The 5′ phosphate lacks contribution toward reactivity in all aminotransferase type folds aside from type 5 (Eliot & Kirsch, 2004). With the bsGabR Eb/O domain homologous to a type‐1 aminotransferase fold, we expected that the introduction of a tetrazole as a phosphate bioisostere at the 5′ position would increase the drug‐like properties of the molecule with a decrease in ionization while preserving integral hydrogen bonding contacts to maintain cofactor position and reactivity within bsGabR. Herein, we confirm the interaction of pyridoxal‐5′‐tetrazole (PLT) with bsGabR through UV–Vis spectrophotometric and X‐ray crystallographic analyses. A designed GABA analog, 4‐phenoxymethyl‐GABA (4PMG; (S)‐4‐amino‐5‐phenoxypentanoate), has been reported to be a suitable mimic in inducing transcription of gabT and gabD via a similar mechanism to that of the native GABA ligand (Catlin et al., 2020). Here, we present a high‐resolution crystal structure of the PLT & 4PMG external aldimine adduct within the bsGabR Eb/O domain. The molecules' synergistic combination with bsGabR protein represents a novel therapeutic scaffold capable of targeting pathogenic organisms.
2. MATERIALS AND METHODS
All reagents and solvents were purchased from commercial suppliers and used without any further purification unless specified. The structures of all synthesized compounds were confirmed with 1H NMR, 13C NMR, and high‐resolution mass spectrometry (HRMS). All reactions were maintained under an inert atmosphere of either nitrogen or argon. Reactions were monitored by both TLC and reverse‐phase HPLC. Analytical thin‐layer chromatography (TLC) was carried out on aluminum‐backed silica gel plates (200 μm) and visualized under UV light at 254 nm. Flash column chromatography for sample purification was carried out using a Teledyne ISCO CombiFlash Rf + automated chromatography system using prepacked disposable RediSep Rf flash columns (60 Å, 40–63 μm, 230/400 mesh) coupled to a 200–400 nm variable detector. Melting points were determined using 90 mm capillary tubes in a Mel‐Temp melting point apparatus and are uncorrected. Analytical HPLC was performed on all samples to confirm a purity of ≥95% using an Agilent 1100 series high‐performance liquid chromatography system with a reversed‐phase C18 column, autosampler, and a diode‐array detector (DAD) employing a gradient of 90% A (5% CH3CN in H2O) and 10% B (CH3CN with 0.1% TFA) with a flow rate of 1.0 mL/min. Both 1H and 13C nuclear magnetic resonance (NMR) spectra were conducted on a 500 MHz Brucker spectrometer with 13C spectra determined at 125 MHz. Chemical shifts are reported in parts per million (ppm), and tetramethylsilane (TMS) was used as an internal reference. High‐resolution mass spectra (HRMS) were measured on a time‐of‐flight (TOF) instrument using electrospray ionization (ESI). High‐resolution mass spectra (HRMS) data were obtained at the Integrated Molecular Structure Education and Research Center (IMSERC, Northwestern University, Evanston, IL, USA). HRMS spectra were collected using a Waters Acquity I class UPLC and Xevo G2‐XS QT of mass spectrometer with Waters Acquity BEH C18 column (1.7 μm, 2.1 × 50 mm). Acquired HRMS data were processed using MassHunter software version B.04.00. Mobile phase A was comprised of 0.05% formic acid in water, and mobile phase B was 0.05% formic acid in acetonitrile; a gradient of 5 to 90% B in Mobile phase A over 5 min was applied. The Supporting Information contains 1H NMR, 13C NMR, and high‐resolution mass spectrometry (HRMS) data for PLT (1) and all synthetic intermediates (Figures S2–S13).
3,4‐O‐Isopropylidenepyridoxine (3)

The pyridoxine acetal 3 was prepared according to the literature (Bachmann et al., 2020) with modification. To a stirred suspension of pyridoxine hydrochloride (3.00 g, 14.6 mmol, 1 eq.) in acetone dimethyl acetal (2,2‐DMP, 30 mL) was added anhydrous TsOH (prepared by heating 9.97 g TsOH monohydrate (57.9 mmol, 3.97 eq.) to 100°C under vacuum for 18 h). The reaction mixture immediately turned black and was allowed to stir at room temperature under argon for 22 h. The black solution was quenched with water and extracted with DCM (4×) until the blackish‐purple color dissipated almost completely from the aqueous layer. The aqueous solution was then slowly digested with solid NaHCO3 directly into a separatory funnel until basic. The aqueous suspension was extracted with DCM (5×), and the combined organic extracts were dried over anhydrous Na2SO4 and concentrated in vacuo. The resulting pale yellow solid residue was recrystallized from 160 mL Et2O:pet ether (5:3), vacuum filtered, and the filtride was washed with cold pet ether resulting in the acetonide 3 as white needles (1.87 g, 61%). The physical and spectroscopic data matched those reported in the literature (Bachmann et al., 2020).
2,2,8‐Trimethyl‐4H‐[1,3]dioxino[4,5‐c]pyridine‐5‐carbaldehyde (4)

Compound 4 was prepared according to the procedure of Haque (Haque, 2003) with modifications. A suspension of 3,4‐O‐isopropylidenepyridoxine (1.4 g, 6.67 mmol, 1 eq.) with activated MnO2 (1.76 g, 20.0 mmol, 3 eq.) was allowed to stir in anhydrous toluene (45 mL) under nitrogen at 50°C for 22 h. A second portion of MnO2 (850 mg, 9.73 mmol, 1.46 eq.) was added, and stirring was continued for an additional 4 h. The reaction mixture was allowed to cool to room temperature, and the solution was then filtered through a bed of celite and washed with DCM (2×). The filtrate was concentrated under vacuum to yield the title carbaldehyde 4 (1.24 g, 90%) as a slightly yellow white solid. The physical and spectroscopic data matched those reported in the literature (Haque, 2003).
(E)‐3‐(2,2,8‐Trimethyl‐4H‐[1,3]dioxino[4,5‐c]pyridin‐5‐yl)acrylonitrile (5)

To a flask was added aldehyde 4 (1.23 g, 5.94 mmol, 1 eq.), diethyl cyanomethylphosphonate (1.15 mL, 7.13 mmol, 1.2 eq.), and diethyl ether (3 mL), and then, the ether was removed by a steady stream of nitrogen, yielding a thick yellow oil to which 5.0 M aqueous K2CO3 (2.4 mL, 11.9 mmol, 2 eq.) was added dropwise with vigorous stirring. The reaction mixture was stirred at room temperature under argon for 45 min. Petroleum ether (3 mL) was then added to the resulting suspension, and stirring was continued for an additional 5 min under argon. The precipitate was collected by filtration and washed with minimal amounts of water and petroleum ether to yield a yellow solid which was purified by washing the filtride with diethyl ether resulting in the E‐isomer 5 as a white solid (404.8 mg, 30.0%). MP: 148–150°C (decomposition above 150°C). 1H NMR (500 MHz, CDCl3) δ 8.19 (s, 1H), 7.25 (d, J = 16.6 Hz, 1H), 5.85 (d, J = 16.6 Hz, 1H), 4.86 (s, 2H), 2.44 (s, 3H), 1.57 (s, 6H). 13C NMR (500 MHz, CDCl3) δ 192.0, 150.8, 146.6, 145.9, 143.6, 137.7, 124.3, 123.6, 117.4, 100.1, 99.5, 58.6, 24.6, 18.9. HRMS (APCI): m/z calculated C13H14N2O2 − for [M‐H]−: 229.1021, found 229.0986.
(E)‐5‐(2‐(1H‐Tetrazol‐5‐yl)vinyl)‐2,2,8‐trimethyl‐4H‐[1,3]dioxino[4,5‐c]pyridine (6)

A suspension of acrylonitrile 5 (410.2 mg, 1.78 mmol, 1 eq.), ZnBr2 (406 mg, 1.8 mmol, 1.01 eq), and NaN3 (153 mg, 2.35 mmol, 1.32 eq.) in 4:1 H2O: i PrOH (4.05 mL) was refluxed under argon for 46.5 h. The gray‐white murky solution was allowed to cool to room temperature and 5.0 M NaOH (891 μL, 2.5 eq.) was added, and the reaction mixture was allowed to stir for 30 min to break up the original precipitate. The resulting suspension of Zn(OH)2 was filtered off, and the clear amber solution filtrate was acidified to pH 6.5 with 1.0 M HCl resulting in the formation of a precipitate. The precipitated product was collected by vacuum filtration and washed with water resulting in tetrazole 6 as white‐gray flakes (385.5 mg, 79.2%). MP: >375°C. 1H NMR (500 MHz, DMSO‐d 6) δ 8.45 (s, 1H), 7.51 (d, J = 16.6 Hz, 1H), 7.30 (d, J = 16.6 Hz, 1H), 5.02 (s, 2H), 2.34 (s, 3H), 1.53 (s, 6H). 13C NMR (500 MHz, DMSO‐d 6) δ 154.6, 147.5, 145.6, 138.3, 131.2, 125.6, 125.3, 113.8, 100.1, 58.8, 24.9, 18.9. HRMS (ESI): m/z calculated C13H15N5O2 − for [M‐H]−: 272.1121, found 272.1150.
(E)‐5‐(2‐(1H‐Tetrazol‐5‐yl)vinyl)‐4‐(hydroxymethyl)‐2‐methylpyridin‐3‐ol (7)

To a vial was added acetonide 6 (374.5 mg, 1.37 mmol) followed by the addition of 1.0 M HCl (3.75 mL). The reaction mixture was backfilled with argon and was allowed to stir at reflux for 3 h. The reaction mixture was cooled to room temperature and a suspension formed which was then cooled to 4°C by placing in a refrigerator for 4 h to promote further crystallization. The suspension was vacuum filtered and washed with water resulting in tetrazole 7 as fine brown needle crystals (287.9 mg, 90.1%). MP: deflagration above 260°C. 1H NMR (500 MHz, DMSO‐d 6) δ 8.37 (s, 1H), 7.88 (d, J = 16.5 Hz, 1H), 7.24 (d, J = 16.6 Hz, 1H), 4.76 (s, 2H), 2.41 (s, 3H). 13C NMR (500 MHz, DMSO‐d 6) δ 154.6, 149.6, 148.0, 138.2, 133.7, 132.7, 129.2, 112.8, 55.6, 20.1. HRMS (ESI): m/z calculated C10H11N5O2 − for [M‐H]−: 232.0821, found 232.0839.
(E)‐5‐(2‐(1H‐Tetrazol‐5‐yl)vinyl)‐3‐hydroxy‐2‐methylisonicotinaldehyde (PLT, 1)

A vial was charged with alcohol 6 (40 mg, 0.17 mmol, 1.0 eq.), water (1.0 mL), and 1.0 M NaOH (378 μL) to promote full dissolution of the alcohol. Activated MnO2 (100 mg, 1.15 mmol, 6.76 eq.) was then added, and the reaction mixture was backfilled with argon and allowed to stir at room temperature for 2 h. Three additional 100 mg portions of MnO2 were added after 2, 5, and 6 h of stirring. After stirring for 18 h, the resulting solution was filtered with applied pressure over a cotton plug containing a small bed of celite and the yellow clear filtrate was collected then 1 M HCl (324 μL) was added which yielded a precipitate. The suspension was vacuum filtered and washed with generous amounts of water resulting in PLT (1) as a yellow‐green solid (18 mg, 44%). MP: deflagration above 290°C. 1H NMR (500 MHz, DMSO‐d 6) δ 10.52 (s, 1H), 8.49 (s, 1H), 8.19 (d, J = 16.7 Hz, 1H), 7.30 (d, J = 16.5 Hz, 1H). The CH3 peak is unresolvable under the DMSO peak. 13C NMR (500 MHz, DMSO‐d 6) δ 195.3, 154.5, 153.0, 151.3, 138.2, 132.3, 128.8, 123.1, 115.3, 19.7. HRMS (ESI): m/z calculated C10H9N5O2 − for [M‐H]−: 230.0721, found 230.0697.
2.1. Expression and purification of bsGabR Eb/O domain
Truncation of the bsGabR gene (UNIPORTP:94426) and subsequent subcloning was performed following a procedure previously described (Wu et al., 2017). HI‐control BL21 (DE3) chemically competent Escherichia coli (Lucigen) cells (SOLOs) underwent transformation to introduce the modified pETite C‐His vector containing the bsGabR Eb/O gene, which is a truncated variant (88–479 residues) without the DNA‐binding domain. Frozen cell stock was added to Luria‐Bertani (LB) media containing 50 μg/mL kanamycin. The culture was grown under agitation (200 rpm) at 37°C for 4 h. The culture grew to an optical density (OD600) of 0.8 at which point the culture flasks were placed on ice for approximately 60 min as preparation for induction. Recombinant gene expression was induced with 0.5 mM isopropyl‐B‐thiogalactopyranoside (IPTG) and incubated for 18 h under agitation (200 rpm) at 18°C for 18 hrs. Following the overnight incubation, the cells were harvested, resuspended, and lysed in binding buffer (50 mM TAPS pH 8.5, 500 mM NaCl, and 50 mM imidazole) prior to purification. bsGabR Eb/O domain was then purified by application to a HisTrap FF Ni‐NTA affinity column (Cytiva Life Sciences) and elution utilizing a 15‐column volume linear gradient of elution buffer (50 mM TAPS pH 8.5, 500 mM NaCl, and 450 mM imidazole). Fractions containing bsGabR Eb/O domain were pooled and concentrated to approximately 5 mL before being applied to a gel filtration column (HiLoad 16/600 Superdex 200 pg.; GE Healthcare) previously equilibrated with gel filtration buffer (50 mM TAPS pH 8.5, 500 mM NaCl). Fractions containing bsGabR Eb/O domain were confirmed via SDS‐PAGE, pooled, concentrated, and flash frozen in liquid nitrogen prior to storage at −80°C.
2.2. Removal and dialysis of PLP cofactor from purified GabR Eb/O domain
Removal of the PLP cofactor from the bsGabR Eb/O domain was performed by following a similar procedure previously published (Du et al., 2016; Watanabe et al., 1999). Approximately 20 mg of purified bsGabR Eb/O domain was thawed and dispensed into 3.5 kDa snakeskin dialysis tubing (ThermoFisher). The protein solution within the dialysis tubing was submerged into 500 mL of buffer containing 50 mM potassium phosphate monobasic pH 7.5, 500 mM NaCl, and 50 mM hydroxylamine (NH3OH), incubated at 4°C for 12–16 h. Following incubation, the dialysis tubing containing bsGabR Eb/O domain was transferred into 500 mL of buffer absent of the 50 mM hydroxylamine (50 mM potassium phosphate monobasic pH 7.5, 500 mM NaCl) and incubated at 4°C for an additional 6 h. The stripped bsGabR Eb/O domain was concentrated to approximately 5 mL and applied to a gel filtration column (HiLoad 16/600 Superdex 200 pg.; GE Healthcare) previously equilibrated with buffer containing 50 mM potassium phosphate monobasic pH 7.5 and 500 mM NaCl. Following gel filtration, apo‐bsGabR Eb/O domain was immediately used for crystallization or flash frozen in liquid nitrogen and stored at −80°C.
2.3. UV–vis spectrophotometric analysis of bsGabR Eb/O domain, PLT, and 4PMG
Confirmation of apo‐bsGabR Eb/O domain followed by reconstitution of PLT and a reconstituted PLP control was done using a Shimadzu UV‐2450 spectrophotometer. The instrument temperature was set to 20°C with a spectrum being collected from 700 to 250 nm. Complete spectra were collected of both 100 μM PLP and PLT free in reaction buffer (100 μM TAPS pH 8.5, 500 mM NaCl). A spectrum of bsGabR Eb/O domain was collected separately in apo form and following the supplementation of 100 μM PLP or PLT in reaction buffer monitored for 60 min. Spectrophotometric changes were monitored upon the addition of 1 mM 4PMG to a previously incubated sample of bsGabR Eb/O domain with 100 μM PLT for 40 min. 1 mM 4PMG was mixed with 100 μM PLT absent bsGabR Eb/O domain in solution and monitored for 60 min under identical experimental conditions. Data analysis and figure generation were done using Kaleidagraph software.
2.4. Crystallization and X‐ray data collection of the GabR Eb/O Apo protein and (PLT‐4PMG) external Aldimine
Crystallization was performed via hanging drop vapor diffusion method with a 2 μL drop and a 500 μL reservoir volume. The reservoir solution of apo‐bsGabR Eb/O domain consisted of 6% PEG 6000 and 100 mM HEPES pH 7.0. The reservoir solution of the reconstituted bsGabR Eb/O domain with 100 μM PLT consisted of 3% PEG 6000 and 100 mM HEPES pH 7.0. The apo‐bsGabR Eb/O domain drop mother liquor consisted of 1 μL reservoir solution and 1 μL 4.0 mg/mL bsGabR Eb/O domain in buffer containing 50 mM potassium phosphate monobasic pH 7.5, and 500 mM NaCl. The co‐crystallization hanging drop consisted of 1 μL reservoir solution, 1 μL 4.0 mg/mL bsGabR Eb/O reconstituted with 100 μM PLT in identical buffer, and 0.2 μL 100 mM 4‐phenoxymethyl‐GABA (4PMG). Crystals grew within 24 h and were left to grow for an additional 7 days. After 7 days, the crystals were submerged in cryo‐protectant consisting of 20% PEG 6000, 100 mM HEPES pH 7.0, and 35% glycerol. After application to the cryo‐protectant, the crystals were submerged and stored in liquid nitrogen. Diffraction data were collected at 100 K at the beamline 21‐ID‐D of the Advanced Photon Source at Argonne National Laboratory. The beamline was equipped with a Dectris Eiger 9 M detector. Data were collected using an oscillation angle of 0.5° over a range of 240° and an exposure time of 0.125 s per frame. The wavelength was fixed at 1.12718 Å. Data processing and reduction, including indexing, integration, and scaling was done using autoPROC (Vonrhein et al., 2011).
2.5. Phasing, model building, and refinement
A structural solution was obtained via molecular replacement with PHASER (McCoy et al., 2007), making use of a known structure PDB 5T4K (Wu et al., 2017) as the search model. Subsequent rounds of model building and refinement were accomplished utilizing Coot (Emsley & Cowtan, 2004) and PhenixRefine (Adams et al., 2002).
2.6. Structural analysis and figure making
All structural analyses were conducted in Coot and UCSF Chimera (Pettersen et al., 2004). All structural figures were made in UCSF Chimera.
3. RESULTS AND DISCUSSION
3.1. Design and synthesis of PLP analog PLT
Our abiding interest in PLP‐dependent enzymes and in new antibiotics to address the growing threat of antibiotic resistance prompted us to consider functional isosteres of PLP that can better penetrate cell membranes. A severe limitation of phosphate‐containing molecules is the dianionic charge of the phosphate, which we determined to replace with a mono‐anionic tetrazole. We now report the synthesis and functional incorporation of a new pyridoxal 5′‐phosphate (PLP) analog referred to as pyridoxal‐5′‐tetrazole (PLT) wherein the phosphate is replaced by a tetrazole side chain.
Bioisosterism, defined as chemical groups that have physical and chemical similarities to moieties being replaced and employed in making drug candidate analogs is a fundamental advancement in improving druggable properties (Lipinski, 1986). Phosphate isosteres have been reviewed (Rye & Baell, 2005), and the Protein Data Bank has been searched for isosteric replacements of phosphates that exist in different binding site environments but have not necessarily been specifically identified or exploited in medicinal chemistry by comparing the binding environments of reference proteins complexed with AMP, ADP, ATP, or pyrophosphate (Zhang et al., 2017). A major limitation of phosphate analogs is poor cellular penetration due to the highly charged dianionic phosphorus species. The evolution of mono‐anionic inhibitors of protein tyrosine phosphatase 1B (PTP1B) has been pursued where mono‐anionic carboxylate isosteres of dianionic phosphate were prepared which successively evolved into tetrazole derivatives, aiming to capture phosphate‐like electrostatic interactions and improving cell penetration, with much greater cellular bioavailability with the tetrazole derivatives (Liljebris et al., 2002; Xin et al., 2003). It has been emphasized that in the process of drug development, a weaker binding compound having favorable cellular permeability can be preferred over a more potent compound having poor cellular permeability (Lima & Barreiro, 2005). Pyridoxal and pyridoxine analogs containing neutral, un‐ionizable tetrazole moieties lacking the acidic NH moiety in place of the phosphate have been claimed as inhibitors of platelet aggregation (Haque, 2003).
Engineering of PLT was pursued in hopes to modulate and exploit the activity of the target regulator protein. This includes the alteration of the native PLP pharmacophore to improve its lipophilicity for enhanced membrane permeability and to preserve both the functional reactivity and binding interactions with bsGabR. As illustrated in Figure 1, the insertion of a vinyl tetrazole isostere on the PLP cofactor agonist was envisioned which would improve membrane permeability, retain adequate interaction with R319, and would provide greater conformational rigidity of the proposed PLT target due to the internal olefin. Additionally, preservation of both the pyridoxine electron sink and the C4 formyl electrophilic moiety were necessary to form the reversible but covalent external aldimine with bsGabR in the presence of 4‐phenoxymethyl‐GABA as previously reported (Catlin et al., 2020).
FIGURE 1.

Pharmacophore modification of pyridoxal‐5′‐phosphate (PLP) to pyridoxal‐5′‐tetrazole (PLT). Classical representation of the formation of the internal aldimine via nucleophilic attack of the bsGabR Eb/O K312 residue and subsequent external aldimine formation via nucleophilic attack of the 4‐phenoxymethyl‐GABA (4PMG) γ‐amino group and elimination of the K312 amine.
The synthesis of the target PLT 1 is illustrated in Scheme 1 which uses a scalable process requiring no chromatography. The synthesis commenced by subjecting pyridoxine hydrochloride 2 to acetal protection. This was executed by treating the 1,3‐diol pyridoxine scaffold with neat 2,2‐dimethoxypropane (2,2‐DMP) in the presence of acid to form protected diol 3. Inclusion of the olefin linker provides conformational rigidity and increases conjugation, causing a red shift in the absorption maximum. The acetonide was subjected to benzylic oxidation to provide aldehyde 4. Preparation of the E‐isomeric acrylonitrile was achieved by subjecting aldehyde 4 to a base‐mediated Horner‐Wadsworth‐Emmons olefination employing diethyl cyanomethylphosphonate affording E‐acrylonitrile 5. The nitrile motif was further transposed by subjecting the acrylonitrile dipolarophile 5 to a zinc‐catalyzed 1,3‐dipolar cycloaddition employing sodium azide providing the vinyl tetrazole 6. Acid‐catalytic cleavage of the acetonide liberated carbinol 7. Final installation of the C4 formyl group on the pyridinium core was accomplished through benzylic oxidation of carbinol 7 resulting in the target PLT 1.
SCHEME 1.

Synthesis of pyridoxal‐5′‐tetrazole 1 (PLT). Reagents and conditions: (a) 2,2‐DMP, PTSA, rt., 22 h; (b) MnO2, PhMe, 50°C, 26 h; (c) diethyl cyanomethylphosphonate, 5.0 M K2CO3, rt., 45 min; (d) NaN3, ZnBr2, 4:1 H2O:i‐PrOH, reflux, 46 h; (e) 1.0 M HCl, reflux, 3 h; (f) MnO2, 1.0 M NaOH, H2O, rt., 18 h.
3.2. Incorporation of PLT into Apo‐ bsGabR Eb/O for internal Aldimine formation
Previously, the crystal structure of the full‐length bsGabR has been solved as a homodimer. Each monomer can be described as a chimeric structure of a C‐terminal effector binding/oligomerization domain and an N‐terminal DNA‐binding domain. To study the ligand binding to bsGabR, a truncated construct of effector binding/oligomerization domain (bsGabR Eb/O) can be expressed, purified, and crystallized as a homodimer comparable to the full‐length bsGabR. Since bsGabR Eb/O dimer is readily crystallized and retains the capacity to bind both PLP and GABA in a functional fashion, it was used in this study instead of the full‐length bsGabR (Wu et al., 2017).
PLP is an essential cofactor within the prokaryotic expression system (E. coli) utilized to obtain the bsGabR Eb/O for this study. Escherichia coli can produce ample PLP through a de novo biosynthetic deoxyxylulose 5‐phosphate (DXP)‐dependent pathway during overexpression of the bsGabR Eb/O (Barile et al., 2019; Mittenhuber, 2001). Due to the presence of endogenous PLP, the bsGabR Eb/O domain is expressed as a holo‐protein covalently modified with the PLP prosthetic group as an internal aldimine (Belitsky, 2004; Fu et al., 2001; Wu et al., 2017). A chemical intervention is thus required to acquire purified apo‐bsGabR Eb/O. As detailed in the experimental section, the initial Schiff base linkage between bsGabR Eb/O and PLP was disrupted through introduction of 50 mM hydroxylamine (Watanabe et al., 1999) which forms an oxime. Subsequent dialysis was performed to remove excess NH3OH and the NH3OH‐PLP adduct.
Spectroscopic UV–Vis and X‐ray crystallographic methods were employed to confirm the removal of PLP. As depicted in Figure 2, the presence of the bsGabR Eb/O‐PLP internal aldimine intermediate is evidenced by an intense absorbance peak at approximately 396 nm. Following the treatment with hydroxylamine, the absorbance peak characteristic of the bsGabR Eb/O internal aldimine is no longer observed, with that region of absorbance reduced to baseline accompanied by a single peak at 279 nm demonstrating the presence of apo‐bsGabR Eb/O. To further validate the removal of the bsGabR Eb/O‐PLP cofactor, the apo‐bsGabR Eb/O protein was subjected to X‐ray crystallographic analysis and a 1.85 Å resolution structure was solved. A simple omit map (2Fo‐Fc) was created using the apo‐bsGabR Eb/O structure PDB 8VXK (data collection and refinement statistics are in Table S1) as structural evidence for complete removal of PLP prior to PLT reconstitution. Relative to the previously solved 4N0B structure of the bsGabR‐PLP internal aldimine (Edayathumangalam et al., 2013), three significant structural alterations resulted from successful removal of the PLP cofactor (Figure 3). The evolutionarily conserved K312 residue responsible for Schiff base formation at the 5′ position of PLP is no longer oriented in a position that would demonstrate the cofactor's occupancy within the binding site. The K312 side chain now resides adjacent to the Y281 side chain, displaying weakly defined electron density about the ε carbon and adjoining amine due to a lack of covalent or electronic interaction of the amine at this position, demonstrating a highly mobile moiety. Electron densities of the key residues responsible for binding PLP are still observed. No discernable density within the PLP binding site is detected that would indicate the presence of PLP. This overall lack of electron density within the bsGabR binding site in tandem with the alternative conformation of the K312 and Y281 side chains complements what we observed spectroscopically to further validate successful production of apo‐bsGabR Eb/O.
FIGURE 2.

Spectrophotometric characterization of 100 μM pyridoxal‐5′‐phosphate (PLP; red‐dotted line) and pyridoxal‐5′‐tetrazole (PLT; purple‐dotted line) cofactors. Characterization of 20 μM apo‐bsGabR Eb/O domain (black solid line) in the presence of 100 μM PLP (orange solid line) or PLT (turquoise solid line) following a 60 min incubation. Spectral changes of bsGabR and PLT over time (100 μM PLT, at 0, 10, 20, 30, and 60 min) (inset).
FIGURE 3.

Model of apo‐bsGabR Eb/O domain binding pocket surrounded by refined 2Fo‐Fc at 1.1σ.
Once apo‐bsGabR Eb/O was obtained, experiments were conducted to distinguish characteristic spectroscopic signals when comparing PLP to PLT in the absence and in the presence of bsGabR Eb/O. Initial assessment of the spectroscopic characteristics of the PLT and PLP cofactors' binding to bsGabR Eb/O was conducted at equivalent ligand concentration (100 μM) under identical experimental conditions. While this concentration is significantly greater than the proposed Kd value of PLP (1.2 μM) (Okuda et al., 2015), it was deemed necessary to monitor spectroscopic alterations upon addition of bsGabR Eb/O or 4PMG as the apparent molar absorptivity of the PLT cofactor is reduced relative to PLP. At 100 μM cofactor concentration, we expected saturation of bsGabR Eb/O with much of the cofactor existing unbound in solution. The absorption maximum of PLP has been well characterized at approximately 390 nm, which correlates with the maxima observed, under somewhat different experimental conditions, at 387 nm (Figure 2; Soda et al., 1969). Modification of the cofactor with introduction of the tetrazole moiety at the 5′ position in combination with the conjugated alkene within the hydrocarbon linker provides a distinct alteration in the chromophore when observed spectroscopically. A bathochromic shift of the PLT spectrum relative to that of PLP is evident with an absorption maxima now at 403 nm. Following characterization of cofactor absent bsGabR Eb/O, a concentration of approximately 20 μM apo‐bsGabR Eb/O was introduced to the solution containing a 100 μM concentration of either PLP or PLT and monitored for spectrophotometric changes over a 60‐min period. Reactions containing PLP or PLT displayed a comparative trend having a collective hypochromic and bathochromic shift demonstrating the likely formation of the initial Schiff base intermediate defined as the internal aldimine. A 9 nm red shift in the absorbance maxima representative of the K312 nucleophilic attack on the aldehyde moiety at the 4′ position of the pyridine scaffold proceeds through the formation of the carbinolamine intermediate and commences with the complete covalent modification of the apo‐bsGabR Eb/O. While unsuccessful in obtaining an X‐ray crystal structure of the PLT internal aldimine, further spectroscopic investigation of the interaction of bsGabR Eb/O, PLT, and the agonist 4‐phenoxymethylGABA (4PMG) provides confidence in the conclusion that the formation of the external aldimine adduct proceeds through the internal aldimine intermediate.
3.3. Incorporation of 4PMG and PLT into Apo‐ bsGabR Eb/O forming the external Aldimine
To elucidate further spectrum alterations following the introduction of the PLT cofactor and the 4PMG agonist to bsGabR Eb/O, the background spectroscopic signals from the aforementioned 4PMG molecule (Figure S14) and from bsGabR Eb/O were removed to minimize influence in the spectral changes of the PLT cofactor. Following the mixing of 100 μM PLT with approximately 20 μM apo‐bsGabR Eb/O, the initial spectral signal maximizes at 404 nm with a gradual decrease and shift in absorptivity over 40 min to 413 nm (Figure 4a). While this trend remains consistent with the initial spectra collected including absorbance intensity of bsGabR Eb/O at 279 nm, we observed a secondary maximum at 323 nm. The observation of this secondary peak can be attributed to the tautomerization of the internal aldimine, specifically the ketoenamine (413 nm) and enolamine (323 nm) tautomers resulting from the protonation of the Schiff base nitrogen or 3′ oxygen of the pyridine, respectively (Figure 4g; Tramonti et al., 2018). These changes are further exemplified by the complete difference spectrum (spectrum at 40 min minus the spectrum at 0 min; Figure 4b). Maximum hypochromicity is observed at 400 nm which can be assigned as the loss of free PLT in solution and formation of the covalent ketoenamine (Olmo et al., 2002) internal aldimine intermediate. Hyperchromic changes occur over a range of 300–323 nm indicating the presence and accumulation of the covalent enolamine internal aldimine intermediate. Upon achieving an approximate reaction equilibrium after a 40‐min time period, a 1 mM concentration of 4PMG was mixed with the PLT‐bsGabR Eb/O internal aldimine. The reaction was monitored for 30 min with spectroscopic changes being tracked periodically (Figure 4c). An additional hypochromic shift maximizing at 417 nm and simultaneous hyperchromicity maximizing at 344 nm (Figure 4d) is indicative of the PLT‐4PMG ketoenamine and the enolamine external aldimine tautomer, respectively (Figure 4h; Olmo et al., 2002).
FIGURE 4.
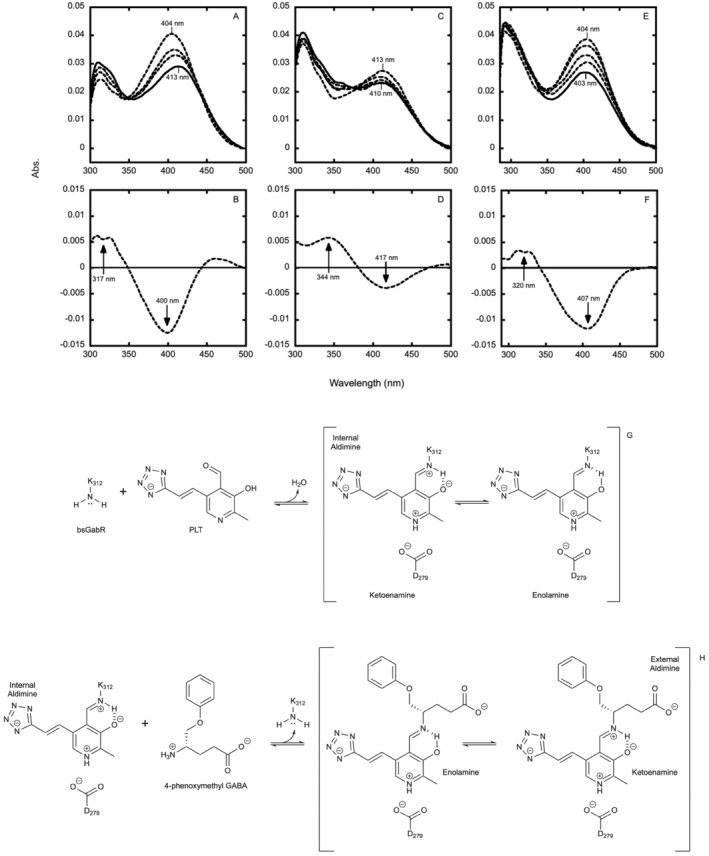
Spectrophotometric characterization of the bsGabR Eb/O‐PLT internal aldimine, bsGabR Eb/O‐PLT‐4PMG external aldimine, and PLT‐4PMG reaction free of bsGabR Eb/O domain. Spectral changes over time (at 0.5, 5, 10, and 40 min) and complete difference spectrum of the internal aldimine (A, B). Spectral changes over time (at 0.5, 2, 4, 8, and 30 min) and complete difference spectrum of the external aldimine (C, D). Spectral changes over time (at 0.5, 4, 16, 32, and 64 min) of PLT‐4PMG mixture absent the bsGabR Eb/O domain (E, F). Reaction scheme for the proposed formation of the internal aldimine intermediate between bsGabR and PLT (G). Reaction scheme for the proposed condensation of the external aldimine intermediate PLT‐4PMG facilitated by bsGabR (H).
An X‐ray crystal structure of the bsGabR Eb/O PLT‐4PMG external aldimine complex was obtained at a 2.24 Å resolution (PDB 8VXL; data collection and refinement statistics are in Table S1). A polder map (Liebschner et al., 2017), an enhanced omit map gained through removal of bulk solvent masking, was generated by omitting the atoms of the PLT‐4PMG external aldimine complex. This unbiased density map supports the formation of the PLT‐4PMG external aldimine complex. We have superimposed the PLT‐4PMG external aldimine complex within the polder map density and present its interaction with surrounding residues in Figure 5.
FIGURE 5.

Polder map of PLT‐4PMG external aldimine complex (3.0σ). Key hydrogen bonding and hydrophobic interactions are presented as dashed lines.
The phosphate bioisostere tetrazole moiety is within H‐binding distances of Ser311, Ser321, as well as Thr181, Thr309, and Arg319, all of which ordinarily interact with the phosphate group of PLP (Figure 6a). The presence of the olefin in the linker may restrict the conformational flexibility of the cofactor's anchor. Reduction of this alkene may allow the tetrazole to adopt an alternative conformation within its binding pocket. The pyridine ring of the PLT is positioned to retain critical interactions between Asp279 and N1 (measured at 2.6 Å) as in PLP; this interaction is necessary to maintain proper conformation and protonation of the pyridine ring facilitating the formation of the external aldimine complex. The carboxylic acid moiety of 4PMG sits in the expected positively charged pocket surrounded by His114, Arg207, and Arg430 (Figure 6b). Designed to take advantage of an observed hydrophobic pocket of bsGabR designated by Met145 and Pro316, the 6‐membered phenoxy ring substituent of the PLT‐4PMG complex appears to be more mobile relative to that of the PLP‐4PMG but occupies roughly the same areas in the ligand binding site based on the previously published PLP‐4PMG structure (6UXZ) (Catlin et al., 2020).
FIGURE 6.

Characteristic structural determinants consistent with the formation of PLT‐4PMG external aldimine. (a) Internal aldimine (PDB Code 4N0B) superimposed onto the PLP‐GABA external aldimine structure (PDB Code 5T4J). (b) PLP‐GABA external aldimine superimposed onto the PLP‐4PMG external aldimine structures (PDB Code 6UXZ). (c) PLP‐4PMG external aldimine superimposed to PLT‐4PMG external aldimine structure. Key structural determinants Y281, K312, and R319 are highlighted in pink. Carboxylate binding residues are labeled with * and highlighted in pink. Key hydrogen bonding distances are indicated as dashed lines. (d) The biological assembly of a bsGabR Eb/o domain dimer is shown as a ribbon diagram. The PLP‐4PMG external aldimine is shown as spheres.
The guanidinium moiety of Arg319 has adopted an alternative conformation due to the tetrazole substituent's inability to maintain a dual salt bridge interaction as in the PLP form of the structure (Figure 7). Due to geometric restraints, the ε nitrogen of Arg319 maintains a single hydrogen bonding interaction with the PLT tetrazole at 2.7 Å. Nardella et al. demonstrated the integral nature of Arg319 in PLP interactions with bsGabR by UV–Vis and thermal stability of an R319A mutant in the absence and in the presence of PLP and GABA (Nardella et al., 2020). A significant decrease in PLP interaction with the R319A mutant relative to wild‐type bsGabR was observed and can be attributed to the beneficial salt bridge interaction due to presence of Arg319. With introduction of the tetrazole at the 5′ position, the dual salt bridge interaction of Arg319 is disrupted which may demonstrate the difficulty in saturating apo‐bsGabR Eb/O with PLT in our attempts to obtain an X‐ray crystal structure of the bsGabR Eb/O‐PLT internal aldimine.
FIGURE 7.

Arginine 319 alternate conformation influenced by presence of the tetrazole or phosphate moiety of PLT or PLP, respectively. R319 surrounding polder map of R319‐PLT interaction (3.75σ) or R319‐PLP interaction (4.0σ).
To ensure the bsGabR Eb/O domain's participation in the formation of the external aldimine intermediate, 1 mM 4PMG agonist was mixed with 100 μM PLT absent bsGabR Eb/O and monitored for spectroscopic changes for a duration of 60 min (Figure 4e,f). Following the initial mixing of the two bsGabR effectors, a similar trend of hypochromicity was observed with the initial maxima at 404 nm, which however lacks a shift to a longer wavelength exemplified in the presence of bsGabR Eb/O. While activity is greatly reduced, if an apoenzyme and organic cofactor become separated, activity may remain with the cofactor alone (Christen & Mehta, 2001). A pyruvoyl group facilitates the amino acid decarboxylation within histidine decarboxylase without the formation of the internal aldimine, proceeding through a de novo external aldimine formation (Recsei & Snell, 1970). This de novo condensation of PLT and 4PMG may progress through nonenzymic nucleophilic attack of the γ‐amino group of 4PMG toward the aldehyde group at the 4′ position of PLT, resulting in a product demonstrating the unique absorbance spectrum obtained from the carbinolamine and/or imine/external aldimine. While we cannot rule out the possibility that a PLT‐4PMG adduct results from the nucleophilic attack of the γ‐amino group of the 4PMG molecule on the reactive aldehyde species of PLT, we can conclude the spectroscopic signal resulting from the PLT and 4PMG mixture is unique and dependent on the presence of the bsGabR Eb/O domain.
3.4. Tetrazole as a Bioisostere for the phosphate group of PLP
In bacteria, MocR‐type regulators such as GabR employ PLP as part of the specific recognition of the GABA metabolite to activate gene expression. The formation of PLP‐GABA external aldimine was confirmed as the transcription activation trigger (Wu et al., 2017). Both PLP and GABA are considered effectors of this type (MocR/GabR subfamily) of bacterial transcription activator (Belitsky, 2004), where ligand design mimicking either or both could provide novel tools for studying bacterial transcription control as well as novel antimicrobial approaches. In a previous ligand design effort, 4PMG was designed as a bulky and more hydrophobic analog of GABA; the designed phenoxy substituent when bound occupies an observed hydrophobic cavity in the ligand binding side. While 4PMG can evade off‐target binding to most of the aminotransferases, it binds and interacts with PLP bound GabR (Catlin et al., 2020) forming a PLP‐4PMG external aldimine. To further explore the ligand design, the tetrazole analog PLT was synthesized. The designed structural change is the substitution of tetrazole for the phosphate group. Tetrazoles are commonly employed as bioisosteres of carboxylic acids (Malik et al., 2014; Zou et al., 2020) and here is employed as a bioisostere of phosphate. There are reported examples of 5‐membered heterocyclic rings such as thiazolidinone or tetronic acid moieties being used as bioiosteres of phosphate (Elliott et al., 2012). To our best knowledge, there is no previous example of using tetrazole as a direct bioisostere for a phosphate group in PLP. Phosphorylation of vitamin B6 converts it into PLP, stabilizing it within cells, and enabling it to participate effectively in vital metabolic pathways. The designed PLT could be more membrane permeable due to the higher lipophilicity of the tetrazole group relative to the phosphate group and the lower overall charge. On the other hand, the designed PLT should retain the reactivity of the pyridine ring, which is necessary in the specific ligand‐triggered transcription control. The results of this study clearly support that tetrazole‐substituted PLT can effectively replace PLP and demonstrates specific binding to prepared apo‐bsGabR Eb/O. It is interesting to note, from a potential druggability perspective, that PLT has a lower total polar surface area (TPSA) than PLP, with 101.75 Å2 versus 132.42 Å2, respectively. The tetrazole‐containing PLT also has a higher clogP than PLP with 0.62 for PLT versus −0.62 for PLP.
3.5. PLT reactivity and functional structural determinants
As previously established, bsGabR can bind to PLP as an internal aldimine or with PLP and GABA as an external aldimine. While the internal aldimine of GabR can autorepress its own transcription, the external aldimine composed of PLP, GABA, and certain GABA analogs can activate the expression of two genes, gabT and gabD, which produce GABA aminotransferase (GabT) and succinic semialdehyde dehydrogenase (GabD) in Bacillus subtilis. The binding of the two effectors PLP and GABA will trigger dynamic changes of functional structural determinants. Examining these structural determinants can be indicative of the functional execution triggered by the specific recognition of the ligand.
In this study, the structural results support that tetrazole is an effective bioisostere of phosphate in mimicking PLP. At physiological pH, the tetrazole is mono‐anionic and as such can sustain evolutionarily conserved binding interactions with the phosphate binding site in GabR (cf. Figures 6 and 7) including the critically important hydrogen bonds. Concurrently, the tetrazole is planar and more lipophilic and is therefore expected to have a somewhat weakened overall interaction with GabR.
It was previously established that the pyridine ring assumes a different pose after the transition from internal aldimine (Edayathumangalam et al., 2013) to external aldimine (Wu et al., 2017; Figure 6a). Accommodating the newly formed covalent interaction with PLP, the PLP ring moves further away from the conserved lysine that is the covalent anchor for the internal aldimine. This change of pose for PLP causes notable conformational changes in residues Y281 and R319. While R319 maintains interactions with the phosphate group of PLP, a ~90‐degree rotation of the Y281 side change allows the Y281 and Y205 to sandwich the pyridine ring in a new position facilitating the formation of the external aldimine. Such is the case in both PLP‐GABA external aldimine structure and PLP‐4PMG external aldimine structures (Figure 6b). The PLT‐4PMG structure is well superimposed with the PLP‐4PMG structure including K312, and Y281 both assuming the same positions as observed in the PLP‐4PMG external aldimine (Figure 6c). The electron density clearly supports that the carboxylate group of the PLP‐4PMG resides in the conserved carboxylate binding site (Figure 6c) among three positive residues H114, R207, and R430, both supporting the formation of PLT‐4PMG external aldimine.
We cannot eliminate the possibility that 4PMG could bind to the apo‐bsGabR Eb/O first, facilitating the binding of PLT since we do not have a crystal structure of the PLT internal aldimine within the bsGabR Eb/O domain. However, the PLT internal aldimine formation is supported by spectroscopic data in the absence of 4PMG. We can nevertheless conclude that tetrazole is an effective bioisostere of the phosphate group in this case, preserving the reactivity and functional movements to form an external aldimine with a designed bulky GABA analog 4PMG. To advance the long‐term goals of utilizing ligand‐triggered GabR‐mediated bacterial transcription regulation for novel antimicrobial strategies and the study of bacterial transcription activation, the new PLP analog PLT, combined with 4PMG, has demonstrated coordinated functional binding and reactivity.
4. CONCLUSION
Spectrophotometric UV–Vis analysis reveals distinct spectroscopic differences between PLP and PLT in solution and when these species interact with bsGabR. PLT appears to follow a similar initial Schiff base formation mechanism to that of PLP, supported by a bathochromic absorption shift upon bsGabR interaction. Furthermore, the formation of an external aldimine complex between PLT and 4PMG is evident through the observed hypochromic decay of the ketoenamine tautomer (maximum at 417 nm) and the hyperchromic increase of the enolamine tautomer (maximum at 344 nm) upon addition of 4PMG to the bsGabR‐PLT complex. X‐ray crystallographic data validates the successful removal of the native PLP cofactor and provides elucidation of the PLT‐4PMG external aldimine intermediate complex interaction with bsGabR. The unique conformation of the R319 sidechain, characterized by a single hydrogen bond acceptor‐donor interaction with the 5′‐tetrazole indicates the placement of the tetrazole moiety of PLT securely within the bsGabR effector binding site. This differs somewhat from the traditional dual hydrogen bond interaction of the R319 sidechain with the phosphate moiety of PLP. Our study presents evidence for bsGabR‐facilitated condensation of the external aldimine complex. Additionally, we underscore the potential for de novo condensation of PLT with 4PMG, supported by the observed hypochromicity peaking at 407 nm in the PLT cofactor spectra when 4PMG is added in the absence of bsGabR. After successfully creating an artificial mimic of the PLP‐GABA external aldimine, as demonstrated by the PLT‐4PMG complex, we propose that this condensed product may potentially agonize transcription activation of the gabT and gabD genes controlled by bsGabR, which is the effort of ongoing studies.
AUTHOR CONTRIBUTIONS
Nicholas E. Kaley: Investigation; writing – original draft; methodology; validation; visualization; writing – review and editing. Zachary J. Liveris: Investigation; writing – original draft; methodology; validation; visualization; writing – review and editing. Maxwell Moore: Conceptualization; methodology; validation. Cory T. Reidl: Conceptualization; methodology; validation. Zdzislaw Wawrzak: Investigation; methodology; validation. Daniel P. Becker: Conceptualization; funding acquisition; writing – original draft; writing – review and editing; supervision; resources; project administration. Dali Liu: Conceptualization; funding acquisition; writing – original draft; writing – review and editing; project administration; supervision; resources.
Supporting information
Data S1.
ACKNOWLEDGMENTS
The work was supported by NIH NIGMS R15GM113229 to D.L. and D.P.B.
Kaley NE, Liveris ZJ, Moore M, Reidl CT, Wawrzak Z, Becker DP, et al. Bioisosteric replacement of pyridoxal‐5′‐phosphate to pyridoxal‐5′‐tetrazole targeting Bacillus subtilis GabR . Protein Science. 2025;34(1):e70014. 10.1002/pro.70014
Nicholas E. Kaley and Zachary J. Liveris contributed equally to this work.
Review Editor: Aitziber L. Cortajarena
Contributor Information
Daniel P. Becker, Email: dbecke3@luc.edu.
Dali Liu, Email: dliu@luc.edu.
REFERENCES
- Adams PD, Grosse‐Kunstleve RW, Hung L, Ioerger TR, McCoy AJ, Moriarty NW, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. [DOI] [PubMed] [Google Scholar]
- Bachmann T, Schnurr C, Zainer L, Rychlik M. Chemical synthesis of 5′‐β‐glycoconjugates of vitamin B6. Carbohydr Res. 2020;489:107940. [DOI] [PubMed] [Google Scholar]
- Barile A, Tramonti A, di Salvo ML, Nogués I, Nardella C, Malatesta F, et al. Allosteric feedback inhibition of pyridoxine 5′‐phosphate oxidase from Escherichia coli . J Biol Chem. 2019;294:15593–15603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belitsky BR. Bacillus subtilis GabR, a protein with DNA‐binding and aminotransferase domains, is a PLP‐dependent transcriptional regulator. J Mol Biol. 2004;340:655–664. [DOI] [PubMed] [Google Scholar]
- Belitsky BR, Sonenshein AL. GabR, a member of a novel protein family, regulates the utilization of Î3‐aminobutyrate in Bacillus subtilis . Mol Microbiol. 2002;45:569–583. [DOI] [PubMed] [Google Scholar]
- Bramucci E, Milano T, Pascarella S. Genomic distribution and heterogeneity of MocR‐like transcriptional factors containing a domain belonging to the superfamily of the pyridoxal‐5′‐phosphate dependent enzymes of fold type I. Biochem Biophys Res Commun. 2011;415:88–93. [DOI] [PubMed] [Google Scholar]
- Catlin DS, Reidl CT, Trzupek TR, Silverman RB, Cannon BL, Becker DP, et al. (S)‐4‐Amino‐5‐phenoxypentanoate designed as a potential selective agonist of the bacterial transcription factor GabR. Protein Sci. 2020;29:1816–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang RYK, Nang SC, Chan H, Li J. Novel antimicrobial agents for combating antibiotic‐resistant bacteria. Adv Drug Deliv Rev. 2022;187:114378. [DOI] [PubMed] [Google Scholar]
- Christaki E, Marcou M, Tofarides A. Antimicrobial resistance in bacteria: mechanisms, evolution, and persistence. J Mol Evol. 2020;88:26–40. [DOI] [PubMed] [Google Scholar]
- Christen P, Mehta PK. From cofactor to enzymes. The molecular evolution of pyridoxal‐5′‐phosphate‐dependent enzymes. Chem Rec. 2001;1:436–447. [DOI] [PubMed] [Google Scholar]
- DeSouza SR, Olson MC, Tinucci SL, Sinner EK, Flynn RS, Marshall QF, et al. SAR of non‐hydrolysable analogs of pyridoxal 5′‐phosphate against low molecular weight protein tyrosine phosphatase isoforms. Bioorg Med Chem Lett. 2020;30:127342. [DOI] [PubMed] [Google Scholar]
- Du Y, Singh R, Alkhalaf LM, Kuatsjah E, He H, Eltis LD, et al. A pyridoxal phosphate–dependent enzyme that oxidizes an unactivated carbon‐carbon bond. Nat Chem Biol. 2016;12:194–199. [DOI] [PubMed] [Google Scholar]
- Edayathumangalam R, Wu R, Garcia R, Wang Y, Wang W, Kreinbring CA, et al. Crystal structure of Bacillus subtilis GabR, an autorepressor and transcriptional activator of gabT. Proc Natl Acad Sci USA. 2013;110:17820‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliot AC, Kirsch JF. Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu Rev Biochem. 2004;73:383–415. [DOI] [PubMed] [Google Scholar]
- Elliott TS, Slowey A, Ye Y, Conway SJ. The use of phosphate bioisosteres in medicinal chemistry and chemical biology. MedChemCommun. 2012;3:735–751. [Google Scholar]
- Emsley P, Cowtan K. Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. [DOI] [PubMed] [Google Scholar]
- Feehily C, Karatzas KAG. Role of glutamate metabolism in bacterial responses towards acid and other stresses. J Appl Microbiol. 2013;114:11–24. [DOI] [PubMed] [Google Scholar]
- Feehily C, O'Byrne CP, Karatzas KAG. Functional Î3‐aminobutyrate shunt in listeria monocytogenes: role in acid tolerance and succinate biosynthesis. Appl Environ Microbiol. 2013;79:74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezzini M, Guidoni L, Pascarella S. Conformational transitions induced by γ‐amino butyrate binding in GabR, a bacterial transcriptional regulator. Sci Rep. 2019;9:19319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu T, Di Salvo M, Schirch V. Distribution of B6 vitamers in Escherichia coli as determined by enzymatic assay. Anal Biochem. 2001;298:314–321. [DOI] [PubMed] [Google Scholar]
- Griswold WR, Toney MD. Chemoenzymatic synthesis of 1‐deaza‐pyridoxal 5′‐phosphate. Bioorg Med Chem Lett. 2010;20:1352–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griswold WR, Toney MD. Role of the pyridine nitrogen in pyridoxal 5′‐phosphate catalysis: activity of three classes of PLP enzymes reconstituted with deazapyridoxal 5′‐phosphate. J Am Chem Soc. 2011;133:14823–14830. [DOI] [PubMed] [Google Scholar]
- Haque W. Pyridoxine and pyridoxal analogues: novel uses. Us 6,548,519 2003.
- Ivaska L, Barkoff A, Mertsola J, He Q. Macrolide resistance in Bordetella pertussis: current situation and future challenges. Antibiotics. 2022;11:1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Moriarty NW, Poon BK, Sobolev OV, Terwilliger TC, et al. Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr D Struct Biol. 2017;73:148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljebris C, Larsen SD, Ogg D, Palazuk BJ, Bleasdale JE. Investigation of potential bioisosteric replacements for the carboxyl groups of peptidomimetic inhibitors of protein tyrosine phosphatase 1B: identification of a tetrazole‐containing inhibitor with cellular activity. J Med Chem. 2002;45:1785–1798. [DOI] [PubMed] [Google Scholar]
- Lima LM, Barreiro EJ. Bioisosterism: a useful strategy for molecular modification and drug design. Curr Med Chem. 2005;12:23–49. [DOI] [PubMed] [Google Scholar]
- Lipinski CA. Bioisosterism in drug design. Annu Rep Med Chem. 1986;21:283–291. [Google Scholar]
- Malik MA, Wani MY, Al‐Thabaiti SA, Shiekh RA. Tetrazoles as carboxylic acid isosteres: chemistry and biology. J Incl Phenom Macrocycl Chem. 2014;78:15–37. [Google Scholar]
- McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milano T, Gulzar A, Narzi D, Guidoni L, Pascarella S. Molecular dynamics simulation unveils the conformational flexibility of the interdomain linker in the bacterial transcriptional regulator GabR from Bacillus subtilis bound to pyridoxal 5′‐phosphate. PLoS One. 2017;12:e0189270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittenhuber G. Phylogenetic analyses and comparative genomics of vitamin B6 (pyridoxine) and pyridoxal phosphate biosynthesis pathways. J Mol Microbiol Biotechnol. 2001;3:1–20. [PubMed] [Google Scholar]
- Nardella C, Barile A, di Salvo ML, Milano T, Pascarella S, Tramonti A, et al. Interaction of Bacillus subtilis GabR with the gabTD promoter: role of repeated sequences and effect of GABA in transcriptional activation. FEBS J. 2020;287:4952–4970. [DOI] [PubMed] [Google Scholar]
- Okuda K, Kato S, Ito T, Shiraki S, Kawase Y, Goto M, et al. Role of the aminotransferase domain in B acillus subtilis GabR, a pyridoxal 5′‐phosphate‐dependent transcriptional regulator. Mol Microbiol. 2015;95:245–257. [DOI] [PubMed] [Google Scholar]
- Olmo MT, Sanchez‐Jimenez F, Medina MA, Hayashi H. Spectroscopic analysis of recombinant rat histidine decarboxylase. J Biochem. 2002;132:433–439. [DOI] [PubMed] [Google Scholar]
- O'Neill J. Antimicrobial resistance: tackling a crisis for the health and wealth of nations. Rev Antimicrob Resist. 2014;1:1–16. [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF chimera‐a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. [DOI] [PubMed] [Google Scholar]
- Phillips RS. Chemistry and diversity of pyridoxal‐5′‐phosphate dependent enzymes. Biochim Biophys Acta. 2015;1854:1167–1174. [DOI] [PubMed] [Google Scholar]
- Poirel L, Madec J, Lupo A, Schink A, Kieffer N, Nordmann P, et al. Antimicrobial resistance in Escherichia coli . Microbiol Spectr. 2018;6:arba‐2017. 10.1128/microbiolspec [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recsei PA, Snell EE. Histidine decarboxylase of Lactobacillus 30a. VI. Mechanism of action and kinetic properties. Biochemistry (NY). 1970;9:1492–1497. [DOI] [PubMed] [Google Scholar]
- Rye CS, Baell JB. Phosphate isosteres in medicinal chemistry. Curr Med Chem. 2005;12:3127–3141. [DOI] [PubMed] [Google Scholar]
- Soda K, Yorifuji T, Misono H, Moriguchi M. Spectrophotometric determination of pyridoxal and pyridoxal 5′‐phosphate with 3‐methyl‐2‐benzothiazolone hydrazone hydrochloride, and their selective assay. Biochem J. 1969;114:629–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toney MD. Controlling reaction specificity in pyridoxal phosphate enzymes. Biochim Biophys Acta. 2011;1814:1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramonti A, Nardella C, Di Salvo ML, Barile A, Cutruzzola F, Contestabile R. Human cytosolic and mitochondrial serine hydroxymethyltransferase isoforms in comparison: full kinetic characterization and substrate inhibition properties. Biochemistry. 2018;57:6984–6996. [DOI] [PubMed] [Google Scholar]
- Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol Crystallogr. 2011;67:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Zhao G, Chao X, Xie L, Wang H. The characteristic of virulence, biofilm and antibiotic resistance of Klebsiella pneumoniae . Int J Environ Res Public Health. 2020;17:6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe A, Yoshimura T, Mikami B, Esaki N. Tyrosine 265 of alanine racemase serves as a base abstracting α‐hydrogen from l‐alanine: the counterpart residue to lysine 39 specific to d‐alanine. J Biochem. 1999;126:781–786. [DOI] [PubMed] [Google Scholar]
- Wu R, Sanishvili R, Belitsky BR, Juncosa JI, Le HV, Lehrer HJS, et al. PLP and GABA trigger GabR‐mediated transcription regulation in Bacillus subtilis via external aldimine formation. Proc Natl Acad Sci USA. 2017;114:3891–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Z, Liu G, Abad‐Zapatero C, Pei Z, Szczepankiewicz BG, Li X, et al. Identification of a monoacid‐based, cell permeable, selective inhibitor of protein tyrosine phosphatase 1B. Bioorg Med Chem Lett. 2003;13:3947–3950. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Borrel A, Ghemtio L, Regad L, Boije af Gennäs G, Camproux A, et al. Structural isosteres of phosphate groups in the protein data Bank. J Chem Inf Model. 2017;57:499–516. [DOI] [PubMed] [Google Scholar]
- Zou Y, Liu L, Liu J, Liu G. Bioisosteres in drug discovery: focus on tetrazole. Future Med Chem. 2020;12:91–93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.