

Abstract

The asymmetric functionalization of unstrained C(sp3)–C(sp3) bonds could be a powerful strategy to stereoselectively reconstruct the backbone of an organic compound, but such reactions are rare. Although allylic substitutions have been used frequently to construct C–C bonds by the cleavage of more reactive C–X bonds (X is usually an O atom of an ester) by transition metals, the reverse process that involves the replacement of a C–C bond with a C–heteroatom bond is rare and generally considered thermodynamically unfavorable. We show that an unstrained, inert allylic C–C σ bond can be converted to a C–N bond stereoselectively via a designed solubility-control strategy, which makes the thermodynamically unfavorable process possible. The C–C bond amination occurs with a range of amine nucleophiles and cleaves multiple classes of alkyl C–C bonds in good yields with high enantioselectivity. A novel resolution strategy is also reported that transforms racemic allylic amines to the corresponding optically active allylic amine by the sequential conversion of a C–N bond to a C–C bond and back to a C–N bond. Mechanistic studies show that formation of the C–N bond is the rate-limiting step and is driven by the low solubility of the salt formed from the cleaved alkyl group in a nonpolar solvent.
Introduction
The ubiquity of saturated C–C bonds in organic molecules makes the selective functionalization of such bonds an approach that could change and make more efficient synthetic strategies.1−11 However, the kinetic and thermodynamic challenges that confront the mild cleavage of C(sp3)–C(sp3) bonds make reactions at these positions of molecules rare. Even less common are enantioselective reactions at such bonds. Most reported enantioselective reactions occurring at C(sp3)–C(sp3) bonds arise from strain release of small rings.12−34 One report by Zhu et al. described a catalytic process occurring through a rearrangement step and included a single example of enantioselective cleavage of an unstrained C(sp3)–C(sp3) bond with high selectivity, but this reaction occurred in only 27% yield.35 Recently, Zuo et al. described the deracemization of unstrained alcohols by Ti-catalyzed, photochemical cleavage and reformation of C–C σ bonds, but focused on the use of special structures and did not introduce a new bond (Scheme 1A, left).36 Up to now, a protocol to convert inert C–C σ bonds into thermodynamically less stable C–heteroatom bonds stereoselectively remains undeveloped (Scheme 1A, right).
Scheme 1. Asymmetric C(sp3)–C(sp3) Bond Functionalization and Our Strategy.

Asymmetric allylic substitution catalyzed by transition-metal complexes has become a common transformation in organic synthesis.37−51 Generally, a carbon–oxygen bond vicinal to an alkene unit cleaves to release a leaving group and form the critical allyl metal intermediate. The inherent inertness of a C–C bond and relatively high reactivity of C–heteroatom bonds toward transition metals causes conventional studies of this process to focus on the conversion of allylic C–O bonds into allylic C–C bonds (Scheme 1B). Although a few prior studies revealed the cleavage of unstrained allylic C–C bonds, no C–heteroatom bond was constructed from these reactions, and no stereocenter was introduced at the allylic position.52−57 If this general sequence for reactions at allylic C–C bonds could be reversed, then one could cleave unstrained allylic C–C bonds to form C–heteroatom bonds and broaden the scope of C–C bonds that can be considered to be reactive positions of organic molecules.
Recently, we reported the stereoselective replacement of one allylic C–C bond by another C(sp3)–C(sp3) bond by a Pd-catalyzed kinetic resolution and a Pd-catalyzed dynamic kinetic asymmetric transformation.58 The driving force for this exchange of C–C bonds resulted from the difference in thermodynamic stability of the anionic form of the carbon-based nucleophile and the carbon-based leaving group. However, we were unable to achieve the enantioselective synthesis of a C–X (X = heteroatom) bond from a C–C bond by this strategy due to the unfavorable stability of the respective leaving groups and nucleophile. We envisioned that continuous removal of the carbon-based leaving group from the reaction solution could enable the formation of C–X bond from a C–C bond (Scheme 1C).
We report the realization of this strategy as an asymmetric amination that occurs by the cleavage of a C(sp3)–C(sp3) bond and the removal of the carbon-based leaving group. This protocol enables the replacement of an inert C–C σ bond by the C–N bond to a variety of amines, including primary amines, secondary amines, and N-heteroaryl amines in moderate to good yield and with high enantioselectivity. Even a mixture of four stereoisomers of the starting alkenes is converted into a single stereoisomer by this C–C bond amination. This strategy, combined with typical allylation, also provides a novel resolution of racemic allylic amines to a single corresponding enantiomer. Mechanistic studies show that formation of the C–N bonds is likely the turnover-limiting step and is driven by the low solubility of the cleaved carbon salt in nonpolar arene solvents.
Results and Discussion
Reaction Development
We initiated studies of the C–C bond amination by conducting reactions with racemic 1a bearing a malonate as a carbon leaving group and morpholine 2a as the nucleophile, inorganic Cs2CO3 as the base, and MTBE as the solvent with a palladium catalyst. A set of chiral JosiPhos-type bisphosphines were evaluated first as the ligand on palladium (Table 1, entries 1–11). The reaction with L1 as the ligand formed the expected allylic amination product 3a in 37% yield but with no enantioselectivity (entry 1). Reactions with catalysts containing other JosiPhos ligands provided similar results with varying yields and enantioselectivities (less than 81:19 er, entries 2–10 and see SI for more details) of 3a. The reaction with L11 formed 3a in only 10% yield, but it did occur with a high 94:6 er (entry 11). Increasing the amount of nucleophile 2a increased the conversion to form 3a in 22% yield with 95:5 er (entry 12).
Table 1. Reaction Development for Asymmetric C–C Amination.

| Entrya | L | Solvent | Yield (%) | er |
|---|---|---|---|---|
| 1 | L1 | MTBE | 37 | 50:50 |
| 2 | L2 | MTBE | 4 | 55:45 |
| 3 | L3 | MTBE | 10 | 30:70 |
| 4 | L4 | MTBE | 32 | 75:25 |
| 5 | L5 | MTBE | 34 | 19:81 |
| 6 | L6 | MTBE | 30 | 72:28 |
| 7 | L7 | MTBE | 41 | 69:31 |
| 8 | L8 | MTBE | 76 | 40:60 |
| 9 | L9 | MTBE | 12 | 75:25 |
| 10 | L10 | MTBE | 72 | 58:42 |
| 11 | L11 | MTBE | 10 | 94:6 |
| 12 | L11 | MTBE | 22 | 95:5 |
| 13 | L11 | cyclohexane | 61 | 90:10 |
| 14 | L11 | mesitylene | 69 | 82:18 |
| 15 | L11 | MeOH | 18 | 53:47 |
| 16b, | L11 | PhEt | 56 | 91:9 |
| 17b,c, | L11 | PhEt | 86 | 95:5 |
The reaction was carried out in 0.10 mmol scale. The yield was determined by 1H NMR. The er was determined by HPLC analysis.
2a (3 equiv) was used.
PhEt (0.5 M) was used as the solvent.
KOtBu was used instead of Cs2CO3. Isolated yield.
Because the cleaved malonate leaving group is also a nucleophile and the allylic C–C bond is thermodynamically more stable than the allylic C–N bond, the formation of a C–C bond over the C–N bond by the allyl intermediate is favored and accounts for the low yields.59−63 For this reason, we considered that a strategy to override the thermodynamic preferences by controlling the concentrations might provide a solution to this challenge. A reduction in the solvent polarity could reduce the solubility of the malonate salt produced by the reaction, thereby favoring the amination process. Thus, we conducted the reaction in several nonpolar solvents and found that the yield of 3a increased to over 60% in both cyclohexane and mesitylene solvents with only a slight erosion of enantioselectivity (entries 13–14). In contrast, reactions in MeOH as a solvent generated 3a in only 18% yield (entry 15). The reaction in MeOH also occurred with low enantioselectivity, presumably because of the facile racemization of the allylic amine product in protic solvents in the presence of the palladium catalyst. Ultimately, the reaction in ethylbenzene provided 3a in good yield and with good er (entry 16), and reactions with different bases (see Supporting Information for details) showed that those with KOtBu as base formed 3a in 86% yield and with 95:5 er (entry 17).
Scope of the Unstrained C–C σ Bond Amination
Having identified conditions for enantioselective C–C bond amination in high yield and er, the scope of the reaction was examined, and the results are summarized in Scheme 2. A series of secondary amines, including morpholine, piperazine, and tetrahydroisoquinoline, underwent the C–C bond amination smoothly, affording the corresponding products 3a–3c in 52–86% yield and 93:7–95:5 er (Scheme 2A). An excess of amine as nucleophile was not always necessary. The reaction with 1.0 equiv of morpholine 2a at the allylic C–C bond formed 3a in a yield (62%) that was only slightly lower than that with 3.0 equiv and with the same 95:5 er. The reaction also occurred with N–H bonds of aromatic heterocycles to form the corresponding amination products 3d–3e in reasonable yields and with good enantioselectivity (Scheme 2B). In addition, a set of primary amines containing arenes, ethers, acetals, and small rings reacted to replace the malonate moiety in the alkenes and form the corresponding chiral allylic amines in moderate to good yields and with high enantioselectivities (Scheme 2C, 3f–3l).
Scheme 2. Scope for C–C σ Bond Amination.
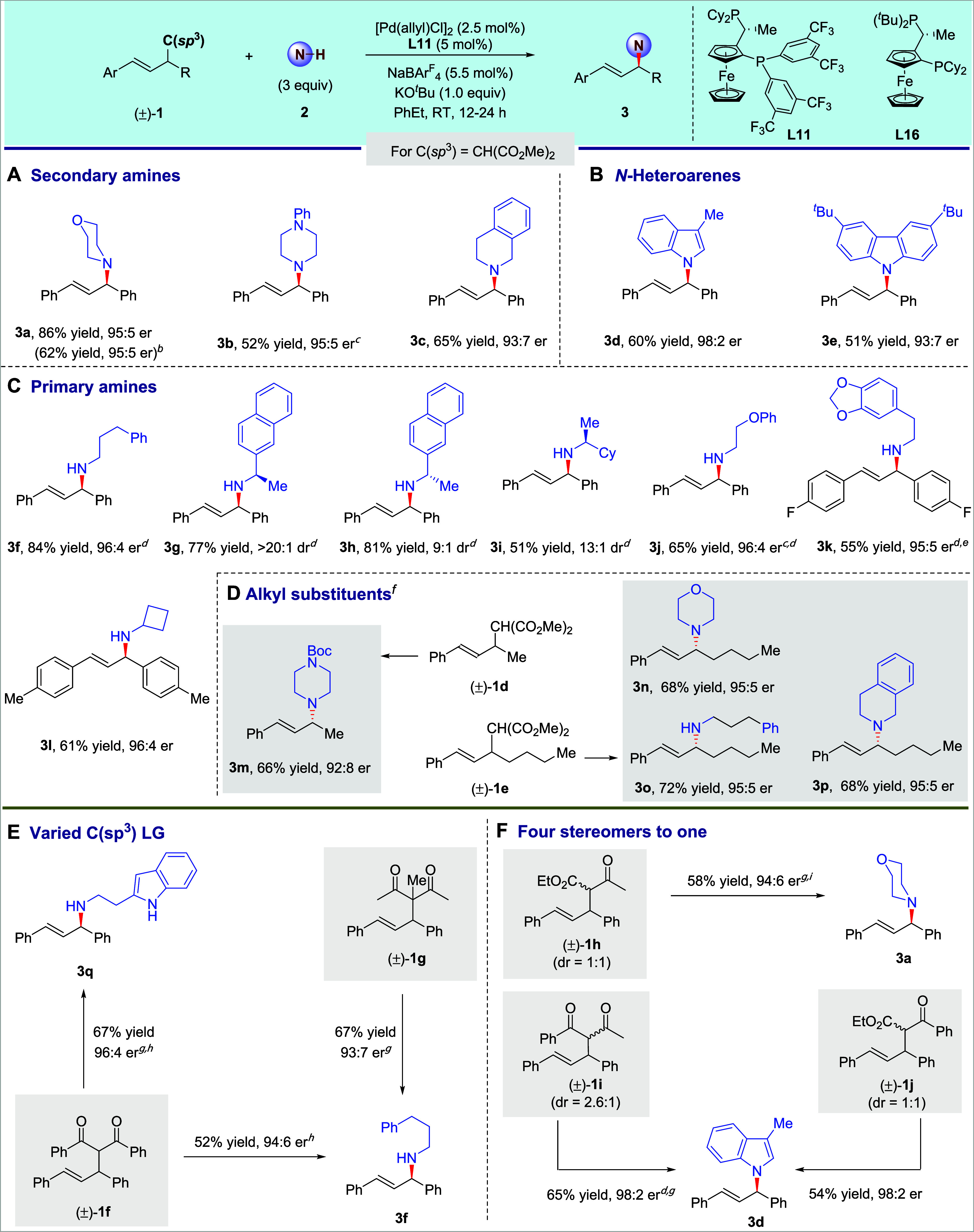
Isolated yield. The ee was determined by HPLC analysis.
Morpholine 2a (1.0 equiv) as the nucleophile was used.
[Pd(allyl)Cl]2 (4 mol %), L11 (8.8 mol %), NaBArF4 (8.8 mol %), and 2 (5 equiv) were used.
p-Xylene was used as the solvent.
Pd(allyl)Cl]2 (4 mol %), L11 (8.8 mol %), and NaBArF4 (8.8 mol %) were used.
Nucleophile (1.0 equiv), electrophile (2.0 equiv), and ligand L16 were used. See SI for more detailed conditions.
Cs2CO3 was used.
The reaction temperature was 0 °C.
2 (5 equiv) was used.
In addition to these diaryl-substituted alkene electrophiles, a series of unsymmetric arylalkyl-substituted alkenes were suitable for C–C bond amination (Scheme 2D, 3m–3p). In this case, two equivalents of alkene were required to guarantee the observation of reasonable yields for the transformation. For example, with the unsymmetric racemic alkene 1e as the electrophile, product 3o was prepared smoothly in 72% yield with 95:5 er by this C–C σ-bond functionalization. When an alkene bearing two aryl substituents featuring different electronic characters was used as the substrate, the regioselectivity of corresponding allylation favored the site vicinal to the electron-deficient group, but the related enantioselectivity was difficult to be controlled (see Supporting Information for details).
In addition to alkenes containing a malonate group, alkenes bearing other 1,3-dicarbonyl groups (1f, 1g) underwent aminations at the allylic C–C bond (Scheme 2E). A nucleophile containing an amine and a heteroaryl N–H bond reacted exclusively at the amine to form product 3q in 67% yield with 96:4 er. As observed with malonate nucleophiles, a racemic mixture of all four stereoisomers of substrate 1h reacted with an amine to form one enantiomer of chiral amine 3a with high stereoselectivity (Scheme 2F). This conversion of four stereoisomers into one was further highlighted by the conversion of two additional diastereomeric mixtures (1i and 1j) to form the same amination product 3d in around 60% yield with 98:2 er. The similarly high enantioselectivity of 3d from reactions at different allylic C–C bonds (1a, 1i, and 1j) indicates that the same allyl-Pd intermediate forms without the involvement of the cleaved alkyl nucleophiles.
To illustrate the value of this alkyl C–C bond amination reaction in synthetic applications, a series of transformations shown in Scheme 3 were conducted. First, the aminations of alkyl C–C bonds were run in the opposite direction as reactions that form C–C bonds from C–N bonds. The optically active compound 1f was previously prepared from allylic amine 3r by the Tsuji-Trost reaction.59 Now, by our protocol, the reverse reaction forms the C–N bond in 3r by cleavage of the C–C bond in 1f (Scheme 3A). Second, we developed a protocol to transform racemic allyl amines into enantioenriched versions of the same amine by a two-step resolution. For example, rac-3s was converted into rac-1f by allylic substitution and then subject to asymmetric C–C bond amination to regenerate 3s, but in highly enantioenriched form (Scheme 3B). Third, this asymmetric C–C bond cleavage can facilitate the synthesis of medicinally relevant compounds. For example, as shown in Scheme 3C, a histone deacetylase (HDAC) inhibitor was prepared by a two-step sequence in which the substrate for asymmetric C–C bond functionalization rac-1d was prepared from alkenyl halide 4 and unsaturated carbonyl ester 5 by Michael addition, and subsequent asymmetric amination at the C–C bond led to the generation of 3t, which was converted to the enantioenriched HDAC inhibitor 7.64 Previously, 3t was prepared in only 8% yield and in racemic form from enone 8.64
Scheme 3. Applications of C–C σ Bond Amination.
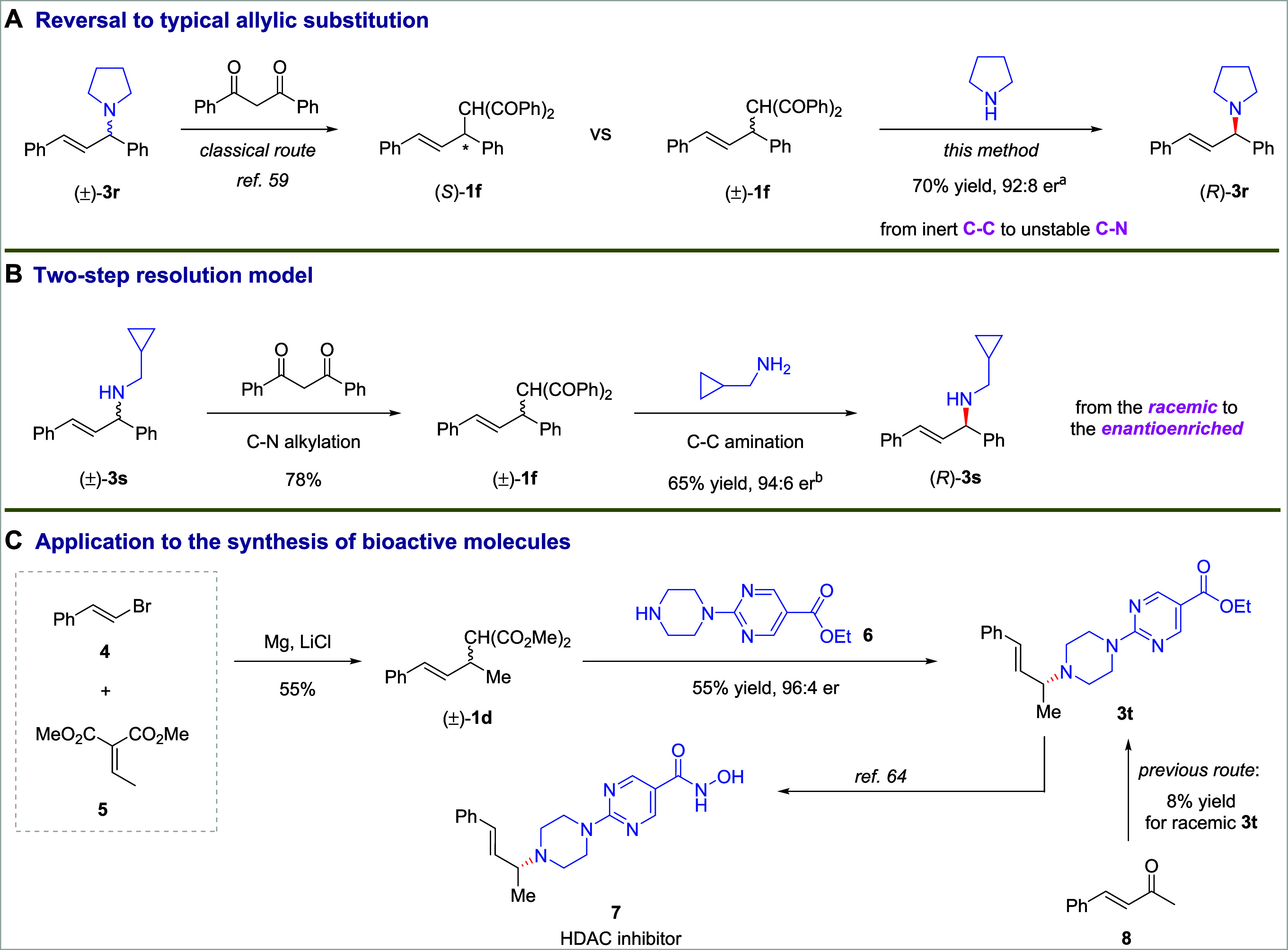
See the Supporting Information for detailed reaction conditions. Isolated yield. The er was determined by HPLC analysis.
Preliminary Mechanistic Studies
To gain experimental information about the mechanism of this C–C bond amination, a series of studies were conducted (Scheme 4). First, the initial rates for the reactions of enantioenriched electrophile 1a with morpholine 2a under the standard conditions were measured. The initial rate of the transformation of matched allyl malonate (S)-1a to form the product (R)-2a is about 4.6-times faster than that of the mismatched malonate (R)-1a (Scheme 4A), suggesting that both enantiomers of 1a are readily converted to product 3a. Meanwhile, when enantioenriched (S)-and (R)-1a were exposed to the catalyst without nucleophile 2a, both enantiomers racemized (Scheme 4B). These results indicate that the amination occurs by an identical π-allyl-Pd intermediate. A full profile of the model reaction between racemic 1a and amine 2a for the preparation of enantioenriched 3a also was obtained and is shown in Scheme 4C. The ee of unreacted substrate 1a increased with the consumption of 1a, while the ee of product 3a was nearly constant during the reaction. These data indicate that both matched and mismatched isomers of 1a react by identical enantiodetermining transition states.
Scheme 4. Mechanistic Studies.

Experiments to probe a relationship between the enantiopurity of L11 and that of product 3a showed that the relationship was within experimental error of linear. This result implies that the reaction occurs with a mononuclear Pd and a chiral ligand as the catalyst (Scheme 4D). The slope of a Hammett plot in Scheme 4E that reveals the effect of the electronic properties of the aryl group in the alkene electrophile on the reaction was negative (ρ = −1.38), indicating that a positive charge accumulates in the allyl group in the intermediate. In addition, the kinetic orders of reactants, catalyst, and base (Scheme 4F) showed that the reaction is first order in the nucleophile 2a and catalyst, but zero order in the electrophile 1a and base. These data imply that the step that forms the amine product from reaction of the Pd-allyl intermediate and the neutral amine nucleophile 2a is rate determining. To further support the C–C activation process, the alkene substrate 1a was treated with 1.0 equiv of the palladium catalyst (Scheme 4G). The in situ 31P NMR spectra showed that the expected allyl-Pd/L11 complex (int-2) was formed as the major species in the reaction.
Finally, evaluation of the solubility of the potassium salt of the malonate, which would be generated from reaction of the cleaved malonate and KOtBu, showed that its solubility in the ethylbenzene solvent was low (S < 1.6 × 10–5 M). This value is consistent with our observation that the solution gradually turned from clear to cloudy as the reaction progressed (Scheme 4H). To confirm the identity of the precipitate, the model reaction was conducted under standard conditions, and the corresponding malonate potassium salt was isolated and identified and collected in about 80% yield (Scheme 4I). Thus, such an intriguing concentration-control pathway facilitated the formation of the C–N bond.
These data are all consistent with the mechanism shown in Scheme 5. The alkene 1a first binds to the low-valent Pd(0) containing the chiral ligand to provide the Pd-alkene complexes int-1 and int-1’, which then undergo oxidative addition to form the same matched Pd-allyl complex int-2. In this process, exchange of the malonate anion with the anion of KBArF4 produces the potassium salt of the malonate, which precipitates from the nonpolar reaction solvent. Rate-limiting nucleophilic attack of the allyl complex int-2 by morpholine affords the allyl ammonium int-3, which forms the product after deprotonation, and regenerates the Pd(0) catalyst.
Scheme 5. Proposed Mechanism.

Conclusion
In conclusion, an asymmetric amination of a set of unstrained C(sp3)–C(sp3) bonds has been achieved. In this process, an unstrained alkyl C–C bond is replaced by a thermodynamically less stable C–N bond in moderate to good yields with high enantioselectivities driven by the exclusion of the malonate anion from solution. The C–C amination occurs with several classes of amines, and its synthetic value is illustrated by its application to the concise stereoselective synthesis of an HDAC inhibitor. With the combination of a Tsuji-Trost reaction and a C–C σ bond amination, a simple two-step resolution of a racemic allyl amine to the corresponding enantioenriched allylic amine was also achieved. Mechanistic studies show that the transformation occurs by rate-limiting C–N bond formation and is driven thermodynamically by the low solubility of the cleaved alkyl salt in a nonpolar solvent.
Acknowledgments
Z.-T.H acknowledges the National Natural Science Foundation of China (22071262, 22371292), Ningbo Natural Science Foundation (2023J036), Science and Technology Commission of Shanghai Municipality (22ZR1475200), Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0610000), State Key Laboratory of Organometallic Chemistry, and Shanghai Institute of Organic Chemistry for financial support. J.F.H acknowledges financial support from NIH grant (1R35GM130387).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c11802.
Experimental procedures for all reactions and characterization data for all products, including 1H NMR, 13C NMR, 19F NMR, and 31P NMR spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Rybtchinski B.; Milstein D. Metal insertion into C-C bonds in solution. Angew. Chem., Int. Ed. 1999, 38, 870–883. . [DOI] [PubMed] [Google Scholar]
- Souillart L.; Cramer N. Catalytic C–C bond activations via oxidative addition to transition metals. Chem. Rev. 2015, 115, 9410–9464. 10.1021/acs.chemrev.5b00138. [DOI] [PubMed] [Google Scholar]
- Fumagalli G.; Stanton S.; Bower J. F. Recent methodologies that exploit C–C single-bond cleavage of strained ring systems by transition metal complexes. Chem. Rev. 2017, 117, 9404–9432. 10.1021/acs.chemrev.6b00599. [DOI] [PubMed] [Google Scholar]
- Xia Y.; Dong G. Temporary or removable directing groups enable activation of unstrained C–C bonds. Nat. Rev. Chem. 2020, 4, 600–614. 10.1038/s41570-020-0218-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald T. R.; Mills L. R.; West M. S.; Rousseaux S. A. L. Selective carbon–carbon bond cleavage of cyclopropanols. Chem. Rev. 2021, 121, 3–79. 10.1021/acs.chemrev.0c00346. [DOI] [PubMed] [Google Scholar]
- Wang J.-H.; Blaszczyk S. A.; Li X.-X.; Tang W.-P. Transition metal-catalyzed selective carbon–carbon bond cleavage of vinylcyclopropanes in cycloaddition reactions. Chem. Rev. 2021, 121, 110–139. 10.1021/acs.chemrev.0c00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen Y.; Cohen A.; Marek I. Creating stereocenters within acyclic systems by C–C bond cleavage of cyclopropanes. Chem. Rev. 2021, 121, 140–161. 10.1021/acs.chemrev.0c00167. [DOI] [PubMed] [Google Scholar]
- Murakami M.; Ishida N. Cleavage of carbon–carbon σ-bonds of four-membered rings. Chem. Rev. 2021, 121, 264–299. 10.1021/acs.chemrev.0c00569. [DOI] [PubMed] [Google Scholar]
- Nanda T.; Fastheem M.; Linda A.; Pati B. V.; Banjare S. K.; Biswal P.; Ravikumar P. C. Recent advancement in palladium-catalyzed C–C bond activation of strained ring systems: Three- and four-membered carbocycles as prominent C3/C4 building blocks. ACS Catal. 2022, 12, 13247–13281. 10.1021/acscatal.2c02667. [DOI] [Google Scholar]
- Song F.-J.; Wang B.-Q.; Shi Z.-J. Transition-metal-catalyzed C–C bond formation from C–C activation. Acc. Chem. Res. 2023, 56, 2867–2886. 10.1021/acs.accounts.3c00230. [DOI] [PubMed] [Google Scholar]
- Liang Y.-F.; Bilal M.; Tang L.-Y.; Wang T.-Z.; Guan Y.-Q.; Cheng Z.-R.; Zhu M.-H.; Wei J.-L.; Jiao N. Carbon–carbon bond cleavage for late-stage functionalization. Chem. Rev. 2023, 123, 12313–12370. 10.1021/acs.chemrev.3c00219. [DOI] [PubMed] [Google Scholar]
- Pirenne V.; Muriel B.; Waser J. Catalytic enantioselective ring-opening reactions of cyclopropanes. Chem. Rev. 2021, 121, 227–263. 10.1021/acs.chemrev.0c00109. [DOI] [PubMed] [Google Scholar]
- Bi X.; Zhang Q.; Gu Z. Transition-metal-catalyzed carbon carbon bond activation in asymmetric synthesis. Chin. J. Chem. 2021, 39, 1397–1412. 10.1002/cjoc.202000591. [DOI] [Google Scholar]
- Yan H.; Smith G. S.; Chen F.-E. Recent advances using cyclopropanols and cyclobutanols in ring-opening asymmetric synthesis. Green Synth. Catal. 2022, 3, 219–226. 10.1016/j.gresc.2022.05.007. [DOI] [Google Scholar]
- Matsumura S.; Maeda Y.; Nishimura T.; Uemura S. Palladium-catalyzed asymmetric arylation, vinylation, and allenylation of tert-cyclobutanols via enantioselective C–C bond cleavage. J. Am. Chem. Soc. 2003, 125, 8862–8869. 10.1021/ja035293l. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Haustedt L. O.; Lim J.; Love J. A.; Williams T. J.; Yoon J.-Y. Asymmetric catalysis of the [5 + 2] cycloaddition reaction of vinylcyclopropanes and π systems. J. Am. Chem. Soc. 2006, 128, 6302–6303. 10.1021/ja058590u. [DOI] [PubMed] [Google Scholar]
- Matsuda T.; Shigeno M.; Makino M.; Murakami M. Asymmetric synthesis of 3,4-dihydrocoumarins by rhodium-catalyzed reaction of 3-(2-hydroxyphenyl)cyclobutanones. J. Am. Chem. Soc. 2007, 129, 12086–12087. 10.1021/ja075141g. [DOI] [PubMed] [Google Scholar]
- Seiser T.; Cramer N. Enantioselective C–C bond activation of allenyl cyclobutanes: Access to cyclohexenones with quaternary stereogenic centers. Angew. Chem., Int. Ed. 2008, 47, 9294–9297. 10.1002/anie.200804281. [DOI] [PubMed] [Google Scholar]
- Kleinbeck F.; Toste F. D. Gold(I)-catalyzed enantioselective ring expansion of allenylcyclopropanols. J. Am. Chem. Soc. 2009, 131, 9178–9179. 10.1021/ja904055z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani R.; Nakatsu H.; Takatsu K.; Hayashi T. Rhodium-Catalyzed Asymmetric [5+2] Cycloaddition of Alkyne–Vinylcyclopropanes. Chem.–Eur. J. 2009, 15, 8692–8694. 10.1002/chem.200901463. [DOI] [PubMed] [Google Scholar]
- Moran J.; Smith A. G.; Carris R. M.; Johnson J. S.; Krische M. J. Polarity inversion of donor–acceptor cyclopropanes: Disubstituted δ-lactones via enantioselective iridium catalysis. J. Am. Chem. Soc. 2011, 133, 18618–18621. 10.1021/ja2090993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waibel M.; Cramer N. Desymmetrizations of meso-tert-norbornenols by rhodium(I)-catalyzed enantioselective retro-allylations. Chem. Commun. 2011, 47, 346–348. 10.1039/C0CC01950J. [DOI] [PubMed] [Google Scholar]
- Lin M.; Kang G.-Y.; Guo Y.-A.; Yu Z.-X. Asymmetric Rh(I)-catalyzed intramolecular [3 + 2] cycloaddition of 1-yne-vinylcyclopropanes for bicyclo[3.3.0] compounds with a chiral quaternary carbon stereocenter and density functional theory study of the origins of enantioselectivity. J. Am. Chem. Soc. 2012, 134, 398–405. 10.1021/ja2082119. [DOI] [PubMed] [Google Scholar]
- Xiong H.; Xu H.; Liao S.; Xie Z.; Tang Y. Copper-catalyzed highly enantioselective cyclopentannulation of indoles with donor–acceptor cyclopropanes. J. Am. Chem. Soc. 2013, 135, 7851–7854. 10.1021/ja4042127. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Dong G. Nickel-catalyzed chemo- and enantioselective coupling between cyclobutanones and allenes: Rapid synthesis of [3.2.2] bicycles. Angew. Chem., Int. Ed. 2016, 55, 15091–15095. 10.1002/anie.201609489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; Bai W.-J.; Hohn C.; Bai Y.; Cregg J. J. Palladium-catalyzed asymmetric allylic alkylation of 3-substituted 1H-indoles and tryptophan derivatives with vinylcyclopropanes. J. Am. Chem. Soc. 2018, 140, 6710–6717. 10.1021/jacs.8b03656. [DOI] [PubMed] [Google Scholar]
- Cheng Q.; Xie J.-H.; Weng Y.-C.; You S.-L. Pd-catalyzed dearomatization of anthranils with vinylcyclopropanes by [4 + 3] cyclization reaction. Angew. Chem., Int. Ed. 2019, 58, 5739–5743. 10.1002/anie.201901251. [DOI] [PubMed] [Google Scholar]
- Yang J.-F.; Sekiguchi Y.; Yoshikai N. Cobalt-catalyzed enantioselective and chemodivergent addition of cyclopropanols to oxabicyclic alkenes. ACS Catal. 2019, 9, 5638–5644. 10.1021/acscatal.9b00655. [DOI] [Google Scholar]
- Jiang C.; Wang L.; Zhang H.; Chen P.; Guo Y.-L.; Liu G. Enantioselective copper-catalyzed trifluoromethylation of benzylic radicals via ring opening of cyclopropanols. Chem 2020, 6, 2407–2419. 10.1016/j.chempr.2020.07.003. [DOI] [Google Scholar]
- Faltracco M.; van de Vrande K. N. A.; Dijkstra M.; Saya J. M.; Hamlin T. A.; Ruijter E. Palladium-catalyzed cascade to benzoxepins by using vinyl-substituted donor–acceptor cyclopropanes. Angew. Chem., Int. Ed. 2021, 60, 14410–14414. 10.1002/anie.202102862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitai J.; Nimmo A. J.; Slawin A. M. Z.; Smith A. D. Cooperative palladium/isothiourea catalyzed enantioselective formal (3 + 2) cycloaddition of vinylcyclopropanes and α,β-unsaturated esters. Angew. Chem., Int. Ed. 2022, 61, e202202621 10.1002/anie.202202621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikari A. S.; Pandit S.; Kant R.; Majumdar N. Iridium-catalyzed enantioselective allylic substitution of vinylcyclopropanes by carboxylic acids. ACS Catal. 2023, 13, 6261–6267. 10.1021/acscatal.3c00959. [DOI] [Google Scholar]
- Xiao Y.-Q.; Li M.-M.; Zhou Z.-X.; Li Y.-J.; Cao M.-Y.; Liu X.-P.; Lu H.-H.; Rao L.; Lu L.-Q.; Beauchemin A. M.; et al. Taming chiral quaternary stereocenters via remote H-bonding stereoinduction in palladium-catalyzed (3 + 2) cycloadditions. Angew. Chem., Int. Ed. 2023, 62, e202212444 10.1002/anie.202212444. [DOI] [PubMed] [Google Scholar]
- Wang T.-H.; Wang M.-Y.; Wang Y.-D.; Li M.-J.; Zheng Y.; Chen Q.-W.; Zhao Y.; Shi Z.-Z. Ligand cooperativity enables highly enantioselective C–C σ-bond hydroboration of cyclopropanes. Chem 2023, 9, 130–142. 10.1016/j.chempr.2022.09.014. [DOI] [Google Scholar]
- Cao J.; Wu H.; Wang Q.; Zhu J. C–C bond activation enabled by dyotropic rearrangement of Pd(IV) species. Nat. Chem. 2021, 13, 671–676. 10.1038/s41557-021-00698-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L.; Ding J.; Duan L.; Wang S.; An Q.; Wang H.; Zuo Z. Multiplicative enhancement of stereoenrichment by a single catalyst for deracemization of alcohols. Science 2023, 382, 458–464. 10.1126/science.adj0040. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Van Vranken D. L. Asymmetric transition metal-catalyzed allylic alkylations. Chem. Rev. 1996, 96, 395–422. 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]
- Helmchen G.; Pfaltz A. Phosphinooxazolines-a new class of versatile, modular P,N-ligands for asymmetric catalysis. Acc. Chem. Res. 2000, 33, 336–345. 10.1021/ar9900865. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Crawley M. L. Asymmetric transition-metal-catalyzed allylic alkylations: Applications in total synthesis. Chem. Rev. 2003, 103, 2921–2943. 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]
- Lu Z.; Ma S. Metal-catalyzed enantioselective allylation in asymmetric synthesis. Angew. Chem., Int. Ed. 2008, 47, 258–297. 10.1002/anie.200605113. [DOI] [PubMed] [Google Scholar]
- Springer Transition metal catalyzed enantioselective allylic substitution in organic synthesis in Topics in organometallic chemistry Kazmaier U.. Ed.; Springer, Heidelberg: 2012. Vol. 38 [Google Scholar]
- Lumbroso A.; Cooke M. L.; Breit B. Catalytic asymmetric synthesis of allylic alcohols and derivatives and their applications in organic synthesis. Angew. Chem., Int. Ed. 2013, 52, 1890–1932. 10.1002/anie.201204579. [DOI] [PubMed] [Google Scholar]
- Butt N. A.; Zhang W. Transition metal-catalyzed allylic substitution reactions with unactivated allylic substrates. Chem. Soc. Rev. 2015, 44, 7929–7967. 10.1039/C5CS00144G. [DOI] [PubMed] [Google Scholar]
- Huang H.-M.; Bellotti P.; Glorius F. Transition metal-catalysed allylic functionalization reactions involving radicals. Chem. Soc. Rev. 2020, 49, 6186–6197. 10.1039/D0CS00262C. [DOI] [PubMed] [Google Scholar]
- Pàmies O.; Margalef J.; Cañellas S.; James J.; Judge E.; Guiry P. J.; Moberg C.; Bäckvall J.-E.; Pfaltz A.; Pericàs M. A.; Diéguez M. Recent advances in enantioselective Pd-catalyzed allylic substitution: From design to applications. Chem. Rev. 2021, 121, 4373–4505. 10.1021/acs.chemrev.0c00736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig J. F.; Stanley L. M. Mechanistically driven development of iridium catalysts for asymmetric allylic substitution. Acc. Chem. Res. 2010, 43, 1461–1475. 10.1021/ar100047x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q.; Tu H. F.; Zheng C.; Qu J. P.; Helmchen G.; You S. L. Iridium-catalyzed asymmetric allylic substitution reactions. Chem. Rev. 2019, 119, 1855–1969. 10.1021/acs.chemrev.8b00506. [DOI] [PubMed] [Google Scholar]
- Rössler S. L.; Petrone D. A.; Carreira E. M. Iridium-catalyzed asymmetric synthesis of functionally rich molecules enabled by (phosphoramidite,olefin) ligands. Acc. Chem. Res. 2019, 52, 2657–2672. 10.1021/acs.accounts.9b00209. [DOI] [PubMed] [Google Scholar]
- Turnbull B. W. H.; Evans P. A. Asymmetric rhodium-catalyzed allylic substitution reactions: Discovery, development and applications to target-directed synthesis. J. Org. Chem. 2018, 83, 11463–11479. 10.1021/acs.joc.8b00583. [DOI] [PubMed] [Google Scholar]
- Süsse L.; Stoltz B. M. Enantioselective formation of quaternary centers by allylic alkylation with first-row transition-metal catalysts. Chem. Rev. 2021, 121, 4084–4099. 10.1021/acs.chemrev.0c01115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexakis A.; Bäckvall J. E.; Krause N.; Pamies O.; Diéguez M. Enantioselective copper-catalyzed conjugate addition and allylic substitution reactions. Chem. Rev. 2008, 108, 2796–2823. 10.1021/cr0683515. [DOI] [PubMed] [Google Scholar]
- Nilsson Y. I.; Andersson P. G.; Bäckvall J.-E. Example of thermodynamic control in palladium-catalyzed allylic alkylation. Evidence for palladium-assisted allylic carbon-carbon bond cleavage. J. Am. Chem. Soc. 1993, 115, 6609–6613. 10.1021/ja00068a018. [DOI] [Google Scholar]
- Bricout H.; Carpentier J.-F.; Mortreux A. Further developments in metal-catalyzed C–C bond cleavage in allylic dimethyl malonate derivatives. Tetrahedron Lett. 1997, 38, 1053–1056. 10.1016/S0040-4039(96)02510-5. [DOI] [Google Scholar]
- Nečas D.; Turský M.; Kotora M. Catalytic deallylation of allyl- and diallylmalonates. J. Am. Chem. Soc. 2004, 126, 10222–10223. 10.1021/ja047320t. [DOI] [PubMed] [Google Scholar]
- Sumida Y.; Yorimitsu H.; Oshima K. Nickel-catalyzed arylative ring-opening of 3-methylenecycloalkane-1,1-dicarboxylates. Org. Lett. 2010, 12, 2254–2257. 10.1021/ol100599c. [DOI] [PubMed] [Google Scholar]
- Clavier H.; Giordano L.; Tenaglia A. Palladium-mediated phosphine-dependent chemoselective bisallylic alkylation leading to spirocarbocycles. Angew. Chem., Int. Ed. 2012, 51, 8648–8651. 10.1002/anie.201204629. [DOI] [PubMed] [Google Scholar]
- Higashida K.; Smail V.; Nagae H.; Carpentier J.-F.; Mashima K. Nickel-catalyzed asymmetric allylic alkylation of β-dicarbonyl compounds via C–C bond activation of 2-allylated cyclic 1,3-diketones. ACS Catal. 2023, 13, 2156–2161. 10.1021/acscatal.2c05664. [DOI] [Google Scholar]
- Chen Y.-W.; Qiu Y.; Liu Y.; Lin G.-Q.; Hartwig J. F.; He Z.-T. Intermolecular asymmetric functionalization of unstrained C(sp3)–C(sp3) bonds in allylic substitution reactions. Nat. Synth. 2024, 3, 1011–1020. 10.1038/s44160-024-00555-z. [DOI] [Google Scholar]
- Zhao X. H.; Liu D. L.; Guo H.; Liu Y. G.; Zhang W. B. C–N bond cleavage of allylic amines via hydrogen bond activation with alcohol solvents in Pd-catalyzed allylic alkylation of carbonyl compounds. J. Am. Chem. Soc. 2011, 133, 19354–19357. 10.1021/ja209373k. [DOI] [PubMed] [Google Scholar]
- Li M.-B.; Wang Y.; Tian S.-K. Regioselective and stereospecific cross-coupling of primary allylic amines with boronic acids and boronates through palladium-catalyzed C–N bond cleavage. Angew. Chem., Int. Ed. 2012, 51, 2968–2971. 10.1002/anie.201109171. [DOI] [PubMed] [Google Scholar]
- Li M.-B.; Li H.; Wang J.; Liu C.-R.; Tian S.-K. Catalytic stereospecific alkylation of malononitriles with enantioenriched primary allylic amine. Chem. Commun. 2013, 49, 8190–8192. 10.1039/c3cc44914a. [DOI] [PubMed] [Google Scholar]
- Yu H.; Zhang G.; Huang H. Palladium-catalyzed dearomative cyclocarbonylation by C–N bond activation. Angew. Chem., Int. Ed. 2015, 54, 10912–10916. 10.1002/anie.201504805. [DOI] [PubMed] [Google Scholar]
- Xu Y.-N.; Zhu M.-Z.; Tian S.-K. Chiral α-amino acid/palladium-catalyzed asymmetric allylation of α-branched β-ketoesters with allylic amines: Highly enantioselective construction of all-carbon quaternary stereocenters. J. Org. Chem. 2019, 84, 14936–14942. 10.1021/acs.joc.9b02282. [DOI] [PubMed] [Google Scholar]
- Angibaud P.; et al. Identification of a series of substituted 2-piperazinyl-5-pyrimidylhydroxamic acids as potent histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 294–298. 10.1016/j.bmcl.2009.10.118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.