

Significance
Our data show that histone methyltransferase SETDB1 is critical for preventing transcription factor (TF) activity on nonphysiological binding sites, in part overlapping with mouse endogenous retrovirus sequences. When SETDB1 is lacking, these cryptic enhancers can become overly active, leading to abnormal expression of nearby genes. This disruption coincides with compromised function of stem cells, altered differentiation of blood cell types, and increased hematopoietic stem cell proliferation. Notably, we demonstrate that SETDB1 acts as a guardian, restricting the unwarranted activity of TFs on these cryptic enhancers and maintaining the proper balance and differentiation of mouse fetal liver hematopoietic stem and progenitor cells.
Keywords: epigenetics, heterochromatin, H3K9me3, ERV, hematopoietic stem cells
Abstract
The H3K9me3-specific histone methyltransferase SETDB1 is critical for proper regulation of developmental processes, but the underlying mechanisms are only partially understood. Here, we show that deletion of Setdb1 in mouse fetal liver hematopoietic stem and progenitor cells (HSPCs) results in compromised stem cell function, enhanced myeloerythroid differentiation, and impaired lymphoid development. Notably, Setdb1-deficient HSPCs exhibit reduced quiescence and increased proliferation, accompanied by the acquisition of a lineage-biased transcriptional program. In Setdb1-deficient HSPCs, we identify genomic regions that are characterized by loss of H3K9me3 and increased chromatin accessibility, suggesting enhanced transcription factor (TF) activity. Interestingly, hematopoietic TFs like PU.1 bind these cryptic enhancers in wild-type HSPCs, despite the H3K9me3 status. Thus, our data indicate that SETDB1 restricts activation of nonphysiological TF binding sites which helps to ensure proper maintenance and differentiation of fetal liver HSPCs.
H3K9me3 is a histone modification associated with heterochromatin and transcriptional silencing. H3K9me3 chromatin is thought to prevent binding of activating TFs, thereby inducing transcriptional repression (1). SETDB1 is the histone methyltransferase that induces most of this modification on specific classes of repetitive elements and distinct developmental genes (2, 3). Setdb1 is recruited through interaction with the corepressor Trim28/Kap1 (4), which, in turn, is recruited by KRAB Zinc Finger proteins that bind specific DNA sequences in the genome (5). Establishment of H3K9me3 methylation on these regions is coordinated with additional pathways, such as DNA methylation (6) and Atrx/Daxx/Morc3-mediated histone H3.3 deposition (7–9), needed for full transcriptional repression.
Loss of Setdb1 is fatal for normal development. Setdb1 ko embryos die during implantation (10). In mouse embryonic stem cells, Setdb1 ablation leads to loss of pluripotency and cell death (3, 11). Conditional deletion of Setdb1 in primordial germ cells or during neurogenesis results in developmental impairments (12, 13). During early B cell development, loss of Setdb1 leads to derepression of endogenous retroviruses (ERVs) that triggers apoptosis of pro-B cells (14, 15). Setdb1 loss does not always result in cell death but can also bias lineage decisions. For example, Setdb1-deficient T cells display exacerbated Th1 priming without compromising viability (16).
The consequence of impaired SETDB1-mediated silencing on the transcriptional network of cells and the connection with developmental phenotypes is multifaceted. SETDB1 mediates silencing of lineage-inappropriate genes by establishing H3K9me3 on their promoters (3, 11, 13, 17, 18). Derepression of ERVs upon Setdb1 loss can lead to overexpression of their neighboring genes through the generation of chimeric transcripts (12, 13, 17) or enhancer activity (15, 16). Finally, SETDB1 impacts the topological chromatin organization by preventing inappropriate CTCF binding, mainly to SINE B2 repeats (19). Therefore, the phenotypes of Setdb1 loss and subsequent ERV derepression likely depend on the cell type and developmental stage of the cells.
Here, we demonstrate that conditional deletion of Setdb1 in mouse fetal hematopoietic stem cells impairs maintenance and lineage specification of HSPCs. Transcriptome analysis confirmed loss of hematopoietic stem cell identity and biased differentiation toward enhanced myeloerythroid output. We found that distinct nonhematopoietic genes and ERVs are repressed by SETDB1-mediated H3K9me3 deposition. Finally, we describe an activity of SETDB1, that is to prevent aberrant TF activity on nonphysiological binding sites. This is important for regulating the cell-type-specific transcriptional network in mouse fetal HSPCs and for ensuring their maintenance and balanced differentiation.
Results
Setdb1 Loss Leads to Postnatal Lethality and Impaired Hematopoietic Lineage Differentiation.
To study the role of SETDB1 during hematopoietic development (SI Appendix, Fig. S1A), we deleted the critical exon 4 in Setdb1flox mice using Vav-Cre which is active from early embryonic HSCs onward (20). Setdb1 hematopoietic mutant mice are Setdb1flox/flox; Vav-Cre (hereafter called Setdb1vav), whereas Setdb1flox/+; Vav-cre or Setdb1flox/+; +/+ genotypes were used as control.
Setdb1vav mice display early postnatal lethality (SI Appendix, Fig. S1B). Therefore, we focused our analyses of hematopoietic changes in Setdb1vav mice at the age of 2 wk. Histological analysis revealed hypocellularity of Setdb1vav bone marrow (BM) (Fig. 1A). Setdb1vav mice had enlarged spleen and severe thymus atrophy (Fig. 1 B and C). Consistently, the cellularity of BM and thymus was significantly decreased (Fig. 1D). FACS analysis of thymocyte progenitor stages revealed a striking increase in the frequency of Setdb1vav DN3 (CD44− CD25+) cells, while DN4 (CD44− CD25−) cells were decreased (Fig. 1E). These data are consistent with an impaired development from DN3 to DN4 stage. Thymocyte development appeared further impaired from DP to CD4 and CD8 SP cell transition with ~50% decreased frequency of CD4+ and CD8+ T cells (Fig. 1F). Consistent with defective development, peripheral CD4+ and CD8+ T cells were 10-fold decreased in the spleen of Setdb1vav mice (SI Appendix, Fig. S1C). We previously showed that the deletion of Setdb1 in pro-B cells impairs further B cell development (15). Similarly, Setdb1vav B cells (B220+ CD19+) were absent in the BM and spleen (Fig. 1G and SI Appendix, Fig. S1D), while there was no significant difference in the frequency of common lymphoid progenitors (CLPs, Lin− IL7ra+ Sca-1low ckitlow) (SI Appendix, Fig. S1E). Together, our data demonstrate an overall impaired lymphoid output in Setdb1vav mice.
Fig. 1.
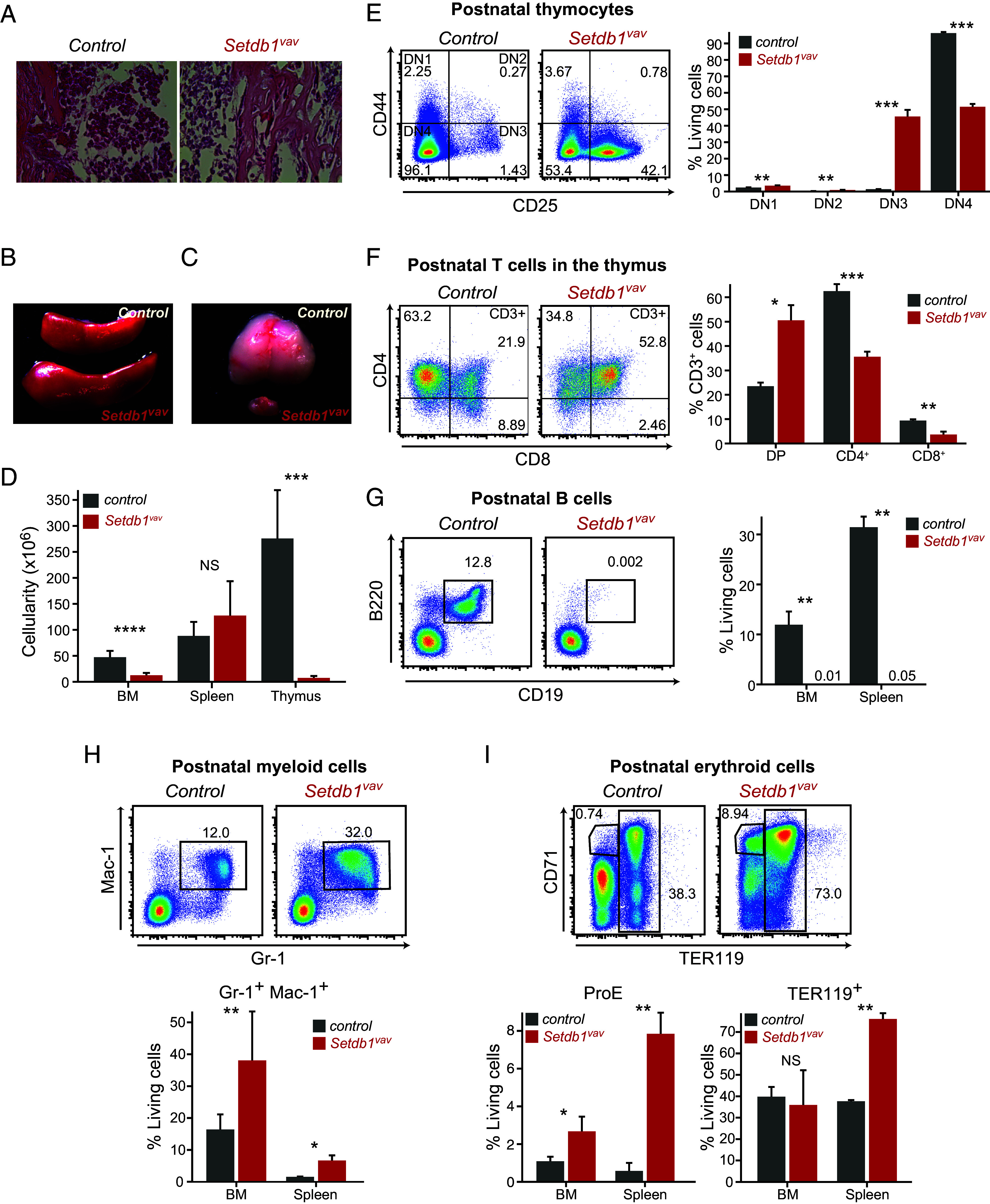
Setdb1 loss leads to postnatal lethality and impaired hematopoietic lineage differentiation. (A) H&E staining of decalcified bone marrow sections from 2-wk-old control and Setdb1vav mice. (B) Spleen, and (C) thymus of 2-wk-old control and Setdb1vav mice. (D) Total cell number of bone marrow (n = 12), spleen (n = 9), and thymus (n = 9) from 2-wk-old control and Setdb1vav mice. (E) Representative FACS plots and bar graphs showing percentages of T cell progenitors (DN1-4) and (F) DP, CD4+, and CD8+ thymocytes in the thymus of control and Setdb1vav mice (n = 3). (G) Representative FACS plots and bar graphs showing percentages of B cells in the bone marrow (n = 4) and spleen of control and Setdb1vav mice (n = 3). (H) Representative FACS plots and bar graphs showing percentages of Gr-1+ Mac-1+ and (I) ProE and TER119+ cells in the bone marrow (n = 8) and spleen (n = 3) of control and Setdb1vav mice. Data are shown as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 (unpaired two-tailed Student’s t test); DN, double negative; DP, double positive; NS, not significant.
In contrast, we observed an increase in the myeloerythroid lineage differentiation. The percentage of myeloid cells (Gr-1+ Mac-1+) showed a marked increase in BM and spleen (Fig. 1H and SI Appendix, Fig. S1F). Further analysis of the myeloid progenitors revealed a slight but not significant decrease in the frequency of common myeloid progenitors (CMPs, c-kithigh CD34+CD16/32-/low) and a significant increase in the proportion of granulocyte-monocyte progenitors (GMPs, c-kithigh CD34+CD16/32+) in the BM (SI Appendix, Fig. S1G). Furthermore, the frequency of Setdb1vav proerythroblasts (ProE, CD71+ TER119-) and TER119+ erythroblasts in the spleen and BM was augmented (Fig. 1I and SI Appendix, Fig. S1H). These data show that Setdb1 ablation leads to a drastically altered hematopoietic lineage differentiation.
Depletion of LT-HSCs and Enhanced Apoptosis In the BM of Setdb1vav Mice.
In postnatal Setdb1vav BM, the frequency of the stem and progenitor cell population LSK (Lin− Sca-1+ ckit+) was comparable to control (Fig. 2A). However, we observed a marked reduction in the percentage of LT-HSCs (LSK CD150+ CD48-), whereas ST-HSCs (LSK CD150− CD48−) and MPPs (LSK CD150− CD48+) remained unchanged (Fig. 2B). Of note, the frequency of AnnV+ apoptotic cells significantly increased in LT- and ST-HSCs, while the MPP population exhibited normal survival (Fig. 2C). Increased apoptosis in stem cell populations could explain overall reduced absolute numbers of hematopoietic stem and progenitor cell (HSPC) populations (SI Appendix, Fig. S1I). These findings highlight the importance of SETDB1 for HSC survival in postnatal stage and are in agreement with a previous study by Koide et al., that showed rapid depletion of LSK cells upon acute SETDB1 depletion (18).
Fig. 2.

Depletion of LT-HSCs and enhanced apoptosis in the BM of Setdb1vav mice. (A) Representative FACS plots and bar graphs showing percentages of LSK cells and (B) LT-HSCs, ST-HSCs, and MPPs in the bone marrow of control and Setdb1vav 2-wk-old mice (n = 4). (C) Representative FACS plots and bar graphs showing percentages of apoptotic cells (Annexin V+) in LT-HSCs, ST-HSCs, and MPPs in 2-wk-old control and Setdb1vav mice (n = 4). Data are shown as mean ± SD. *P < 0.05 and **P < 0.01 (unpaired two-tailed Student’s t test); NS, not significant.
Expansion of Myeloerythroid Cells and HSPCs in the Fetal Liver of Setdb1vav Embryos.
Defective hematopoiesis in postnatal Setdb1vav mice prompted us to investigate the hematopoietic compartment during fetal development. Given that Setdb1 deletion by Vav-Cre occurs during fetal hematopoiesis, we analyzed the fetal livers (FL) from embryos at E13.5. Setdb1vav embryos were indistinguishable from controls and were found at the Mendelian ratio (SI Appendix, Fig. S2A). The cellularity of the fetal liver increased in Setdb1vav embryos (Fig. 3A). Phenotypic changes in lymphoid lineage resembled those in the BM with lack of B cells and relatively unchanged frequency of CLPs (SI Appendix, Fig. S2 B and C). Interestingly, Setdb1vav FLs encompassed a significantly higher frequency of Gr-1+ Mac-1+ and ProE cells (Fig. 3B and SI Appendix, Fig. S2 D and E). Analysis of the myeloerythroid progenitors revealed expanded GMPs at the expense of CMPs (Fig. 3C). The megakaryocyte–erythroid progenitors (MEPs, c-kithigh CD34−CD16/32−) remained unchanged (Fig. 3C), suggesting that the enhanced differentiation toward erythroid lineage happened at later stages of erythroid development. Strikingly, Setdb1-deficient FL cells contained a higher proportion of LSK cells compared to control (Fig. 3D). Accordingly, LT-HSCs, ST-HSCs, and MPP cells demonstrated a consistent expansion (Fig. 3E), and we did not observe signs of enhanced apoptosis in these progenitors (SI Appendix, Fig. S3 A and B). To determine whether enhanced HSPC populations were due to changes in cell proliferation, we analyzed the cell cycle profile by measuring the proliferation marker Ki-67 vs. cellular DNA content. In the absence of Setdb1, the fraction of LT- and ST-HSCs in the G0 phase significantly decreased with a concomitant increase of cells in the G1 phase (Fig. 3F).
Fig. 3.
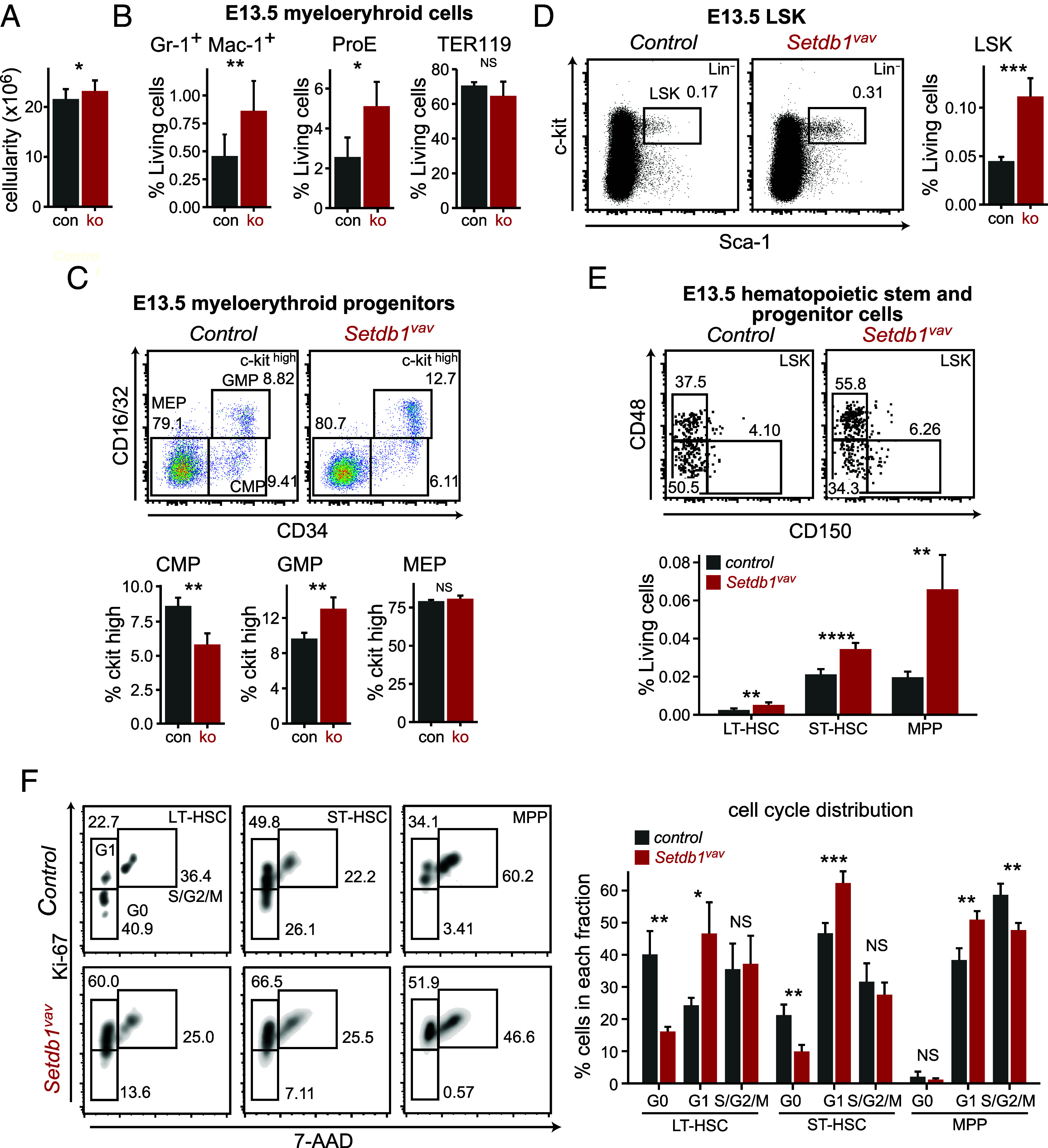
Expansion of myeloerythroid cells and HSPCs in the fetal liver of Setdb1vav. (A) Bar plot showing total cell numbers for control and Setdb1vav E13.5 fetal livers (n = 10). (B) Bar plots showing frequencies of Gr-1+ Mac-1+, ProE, and TER119+ erythroblasts in control and Setdb1vav E13.5 fetal livers (n = 4). (C) Representative FACS plots and bar graphs showing percentages of CMP, GMP, and MEP populations in control and Setdb1vav E13.5 fetal livers (n = 4). (D) Representative FACS plots and bar graphs showing percentages of LSK and (E) LT-HSC, ST-HSC, and MPP populations in control and Setdb1vav E13.5 fetal livers (n = 4). (F) Representative FACS plots and bar graphs showing percentages of LT-HSCs, ST-HSCs, and MPPs in distinct cell cycle phases in control and Setdb1vav E13.5 fetal livers (n = 4). Data are shown as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 (unpaired two-tailed Student’s t test); NS, not significant.
In summary, our data highlight that fetal and postnatal hematopoiesis are differently affected by the loss of Setdb1. In both developmental stages, loss of Setdb1 resulted in enhanced myelo-erythroid differentiation and impaired lymphoid development. However, while Setdb1vav postnatal HSPCs display enhanced apoptosis, Setdb1vav fetal HSPCs did not show apoptosis and were even expanded due to reduced quiescence.
Setdb1vav FL HSPCs Are Defective in Self-Renewal and Differentiation Outside the FL Niche.
Next, we wanted to characterize functional changes between control and Setdb1vav FL HSPCs. First, we assessed their in vitro differentiation potential in colony formation (CFU) assays. In agreement with impaired B cell development in Setdb1vav FL, we did not observe formation of Setdb1vav B cell colonies (Fig. 4A). In myeloerythroid differentiation conditions, Setdb1vav HSPCs could generate colonies, but they were fewer compared to control (Fig. 4B). These data suggested impaired differentiation potential of Setdb1vav FL HSPCs outside of the fetal liver niche. To assess self-renewal and differentiation potential of FL HSPCs we conducted competitive transplantation experiments. We transplanted Setdb1vav or control (Setdb1 sufficient) CD45.2 FL cells with CD45.1 wild-type competitor FL cells at a ratio of 1:1 into lethally irradiated CD45.1/2 recipient mice (6 to 8 wks old). Setdb1vav FL cells did not show any contribution to the peripheral blood at any assessment time points, whereas control FL cells demonstrated a 50% donor contribution (Fig. 4C). Analysis of BM cells from mice 8 wk after transplantation revealed that Setdb1vav donor FL cells had a competitive disadvantage and ultimately failed to reconstitute the hematopoietic compartment in the recipients, as shown by the ratio of CD45.2 (donor) to CD45.1 (competitor) cells (Fig. 4 D and E). Together, our data show that SETDB1 is required for self-renewal and differentiation of FL HSPCs. To test whether compromised reconstitution is also related to defective homing of Setdb1vav cells to the BM, we performed homing assays. Indeed, we observed a trend toward decreased numbers of labeled CFSE+ cells in the BM of reconstituted mice (Fig. 4F). In addition, we detected reduced expression of homing markers (Fig. 4 G and H). Thus, the reconstitution failure could partially be attributed to compromised homing.
Fig. 4.
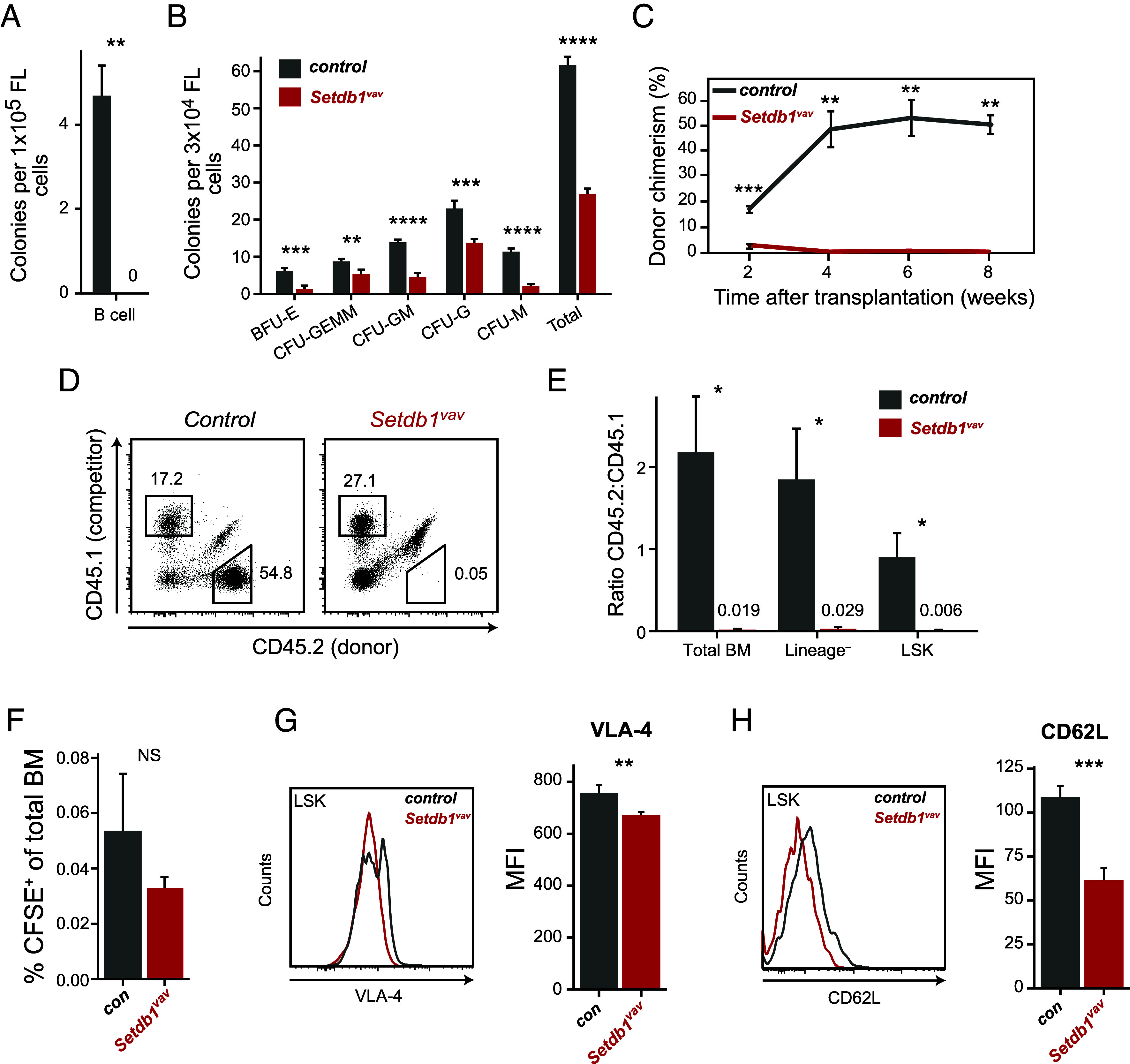
Compromised function of Setdb1vav FL HSPCs. (A) Bar plots showing numbers of B cell and (B) myeloerythroid colonies from control and Setdb1vav FLs in methylcellulose colony-forming assays performed in MethoCult M3630 and MethoCult M3434, respectively (n = 4). (C) Percentages of CD45.2+ donor cells in the peripheral blood of recipient mice at different time points after transplantation (n = 3). (D) Representative FACS plots showing the contribution of CD45.1+ (competitor) and CD45.2+ (donor) cells in the bone marrow of recipient mice, 8 wk after transplantation. (E) Bar plots showing the ratio between control or Setdb1vav and competitor cell frequencies in total BM, lineage–, and LSK cells in the bone marrow of recipient mice, 8 wk after transplantation (n = 3). (F) Frequencies of control and Setdb1vav CFSE+ cells in the BM of lethally irradiated recipient mice 16 h after injection (n = 3). (G) Histogram indicating the expression of VLA-4 homing marker and the comparison of the mean fluorescence intensity (MFI) of VLA-4 expression on control and Setdb1vav fetal liver LSKs (n = 4). (H) Histogram indicating the expression of CD62L homing marker and the comparison of the MFI of CD62L expression on control and Setdb1vav FL LSKs (n = 4). Data are shown as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 (unpaired two-tailed Student’s t test); NS, not significant.
Nonhematopoietic Genes and Retrotransposons Are Derepressed in Setdb1vav LT-HSCs and MPPs.
To decipher the molecular basis underlying Setdb1-regulated HSPC maintenance and differentiation, we performed high-throughput RNA sequencing (RNA-Seq) on LT-HSCs and MPPs sorted from Setdb1vav and control FL cells. Setdb1 exon 4 was largely deleted in Setdb1vav LT-HSC and MPP cells (SI Appendix, Fig. S4A). Differential expression analysis uncovered 285 dysregulated genes in Setdb1vav LT-HSCs, whereas MPPs exhibited a more pronounced transcriptional shift with 4,271 dysregulated genes, indicating a progressive alteration. Among the dysregulated genes in LT-HSCs, 144 were up-regulated and 141 down-regulated (Fig. 5A and SI Appendix, Tables S5 and S6). In MPPs, 2,249 genes were up-regulated, and 2,022 genes were down-regulated (Fig. 5B and SI Appendix, Tables S7 and S8). Most of the highly up-regulated genes in both Setdb1vav LT-HSCs and MPPs were nonhematopoietic genes (SI Appendix, Fig. S5A), including genes specific to testis (Iqcg, Magea13, Dnah8, M1ap, and Tex19.1), oocytes (Stag3), germ cells (Meioc), and neurons (Akap5). The liver and muscle-specific gluconeogenic enzymes, Fbp1 and Fbp2, respectively, were previously shown to be up-regulated in Setdb1-deficient adult HSPCs (18). The top down-regulated genes include imprinted genes H19/Igf2 and hematopoiesis-related genes, such as chemokine receptors (Ccr2 and Ccr9), adhesion molecule Vcam1, and pattern-recognition receptor Clec7 (Fig. 5 A and B).
Fig. 5.
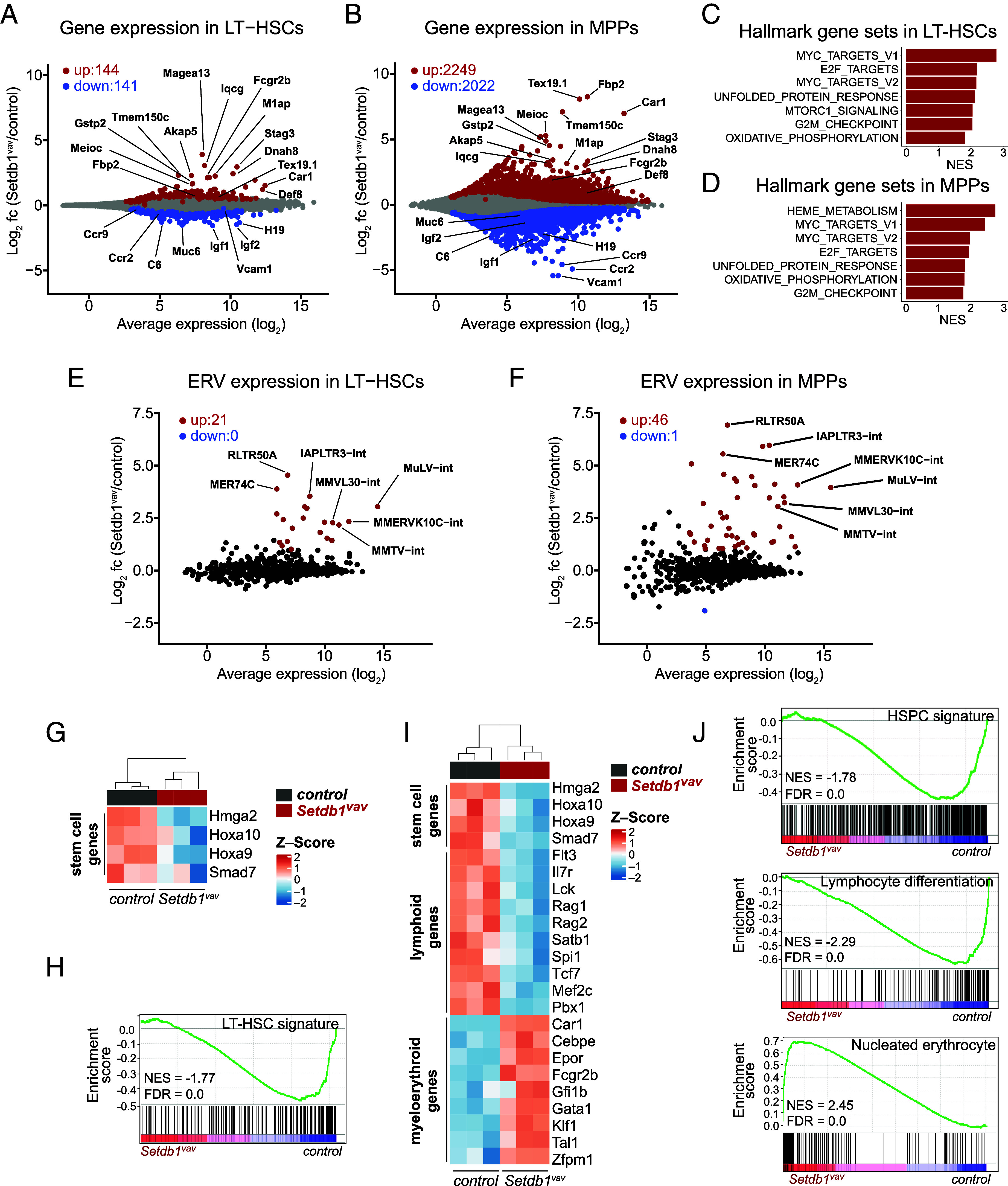
Loss of transcriptional identity and deregulated lineage-specific genes in Setdb1vav FL HSPCs. (A) Average expression of protein-coding genes versus log2 fold change in control compared to Setdb1vav LT-HSCs. Significantly up- (red) and down-regulated (blue) genes are highlighted (Padj < 0.05, n = 3 for each group). (B) Average expression of protein-coding genes versus log2 fold change in control compared to Setdb1vav MPPs (Padj < 0.05, n = 3 for each group). (C) Gene set enrichment analysis (GSEA) of RNA-seq data for hallmark gene sets enriched in Setdb1vav LT-HSCs and (D) MPPs. (E) Scatter plot showing average expression versus log2 fold change of ERV families in control vs. Setdb1vav LT-HSCs, and (F) MPPs. Highlighted are significantly up- (red) and down-regulated (blue) ERVs (Padj < 0.01, log2 fold change > 1, n = 3 for each group). (G) Heatmap showing the expression of selected hematopoietic stem cell-related genes in control and Setdb1vav FL LT-HSCs (Padj < 0.05). (H) GSEA using LT-HSC gene signature (21) on RNA-seq data from control and Setdb1vav FL LT-HSCs. (I) Heatmap showing the expression of selected hematopoietic stem cell-, lymphoid-, and myeloerythroid-associated genes in control and Setdb1vav FL MPPs (Padj < 0.05, log2 fold change > 0.5 or < –0.5). (J) GSEA with HSPC gene signature (21), GO lymphocyte differentiation gene signature, and nucleated erythrocyte gene signature (22) for RNA-seq data from control and Setdb1vav FL MPPs. FL, fetal liver; FDR, false discovery rate; NES, normalized enrichment score.
To assess changes in gene networks more systematically, we performed GSEA for hallmark gene sets. Several gene sets related to proliferation (MYC_TARGETS, E2F_TARGETS, G2M_CHECKPOINT, OXIDATIVE_ PHOSPHORYLATION) were enriched in both Setdb1vav LT-HSCs and MPPs (Fig. 5 C and D), which is consistent with their higher proliferation (Fig. 3F). In MPP, we specifically detected enriched HEME_METABOLISM which indicates a differentiation bias of these cells toward the erythroid lineage that could explain the enhanced generation of proerythroblasts (Fig. 3B).
Further, we detected enrichment of the UNFOLDED_PROTEIN_RESPONSE gene set in both Setdb1vav LT-HSCs and MPPs (Fig. 5 C and D). Interestingly, this gene set was also markedly enriched in Setdb1-deficient pro-B cells, where aberrant expression of endogenous retrovirus proteins led to UPR and apoptosis (15). When we assessed transcription of ERV families in control vs. Setdb1vav LT-HSCs and MPPs, we could indeed detect significant derepression of several ERV classes, such as MuLV and MMVL30 (class I), RLTR50A, MMERVK10C, and IAPLTR3 (class II), and MER74C (class III) (Fig. 5 E and F and SI Appendix, Tables S9 and S10).
Deregulation of Lineage-Specific Genes in Setdb1vav FL HSPCs.
We then investigated more systematically the dysregulation of hematopoietic genes in Setdb1-deficient HSPCs. In Setdb1vav LT-HSCs, we found downregulation of Hoxa10, Hoxa9, Smad7, and Hmga2 genes (Fig. 5G), involved in survival and self-renewal of LT-HSCs (23–26). Consistent with this, the LT-HSC gene signature (21) was depleted in Setdb1vav LT-HSCs (Fig. 5H), suggesting that Setdb1 is important for maintenance of transcriptional identity in LT-HSCs.
In Setdb1vav MPPs we also observed reduced expression of HSC-specific genes and decreased expression of lymphoid-specific genes, while myeloerythroid-specific genes were increased (Fig. 5I). Accordingly, gene signatures related to HSPCs and lymphoid genes were reduced, while a myeloerythroid signature (Nucleated Erythrocytes) was increased in Setdb1vav MPPs (Fig. 5J).
In summary, our transcriptional analysis indicates that Setdb1 is required for the maintenance of FL HSPC identity and for balancing lymphoid vs. myeloid lineage priming.
Loss of H3K9me3 on Up-Regulated Genes and ERVs.
Next, we assessed which genes might be directly regulated by SETDB1. Due to the limit in cell numbers, we were unable to map SETDB1 binding in FL LSK cells. Instead, we mapped H3K9me3 in control vs. Setdb1vav FL LSK cells as an important readout for SETDB1 enzymatic activity. Reduction in H3K9me3 coverage around transcriptional start sites (TSS) of coding genes (−4.0 kb to +1.0 kb from TSS) should be indicative of SETDB1 activity. Indeed, we detected reduced H3K9me3 on several genes which were up-regulated in Setdb1vav, mainly nonhematopoietic genes such as Fbp2, Dnah8, Gstp2, and M1ap (SI Appendix, Fig. S5B).
We also tested changes in H3K9me3 coverage on ERV families. Most families which were derepressed displayed reduced levels of H3K9me3 (SI Appendix, Fig. S5C), suggesting that SETDB1-mediated H3K9me3 is important for repression of these families.
Derepressed ERVs might act as aberrant enhancers (27–29) or alternative promoters (17, 30, 31) driving inappropriate gene expression. For example, derepressed MuLV elements were connected with up-regulated genes in their vicinity in Setdb1-deficient pro-B cells (15). In Setdb1vav MPPs, we also detected effects of MuLV derepression on genes in their vicinity, such as Def8 and Fcgr2b (SI Appendix, Fig. S5D).
Together, our data show that SETDB1-mediated H3K9me3 is important for silencing nonhematopoietic genes and ERVs. Derepressed ERVs can indirectly affect gene regulation by acting as inappropriate enhancer elements.
SETDB1 Restricts Activity of Inappropriate ERV-Overlapping Enhancers in FL HSPCs.
To better understand how loss of SETDB1-mediated H3K9me3 affects transcriptional regulation in FL HSPCs, we performed ATAC-seq experiments in control vs. Setdb1vav LSK cells. The PCA analysis of ATAC peaks clearly separated control from Setdb1vav LSK cells (SI Appendix, Fig. S6A). Assessment of the differentially accessible regions revealed 952 regions with gained accessibility (ATAC_up) in Setdb1vav LSK cells and only 132 regions with reduced accessibility (ATAC_down) (Fig. 6A). Annotation of the genomic features of ATAC_up peaks unveiled strong enrichment of ERV LTRs (Fig. 6B), mainly from ORR1 and RLTR45 elements (Fig. 6C). In line with the role of SETDB1 in ERV repression, we observed increased accessibility on several ERV families, including ORR1 and RLTR45 families (SI Appendix, Fig. S6B).
Fig. 6.
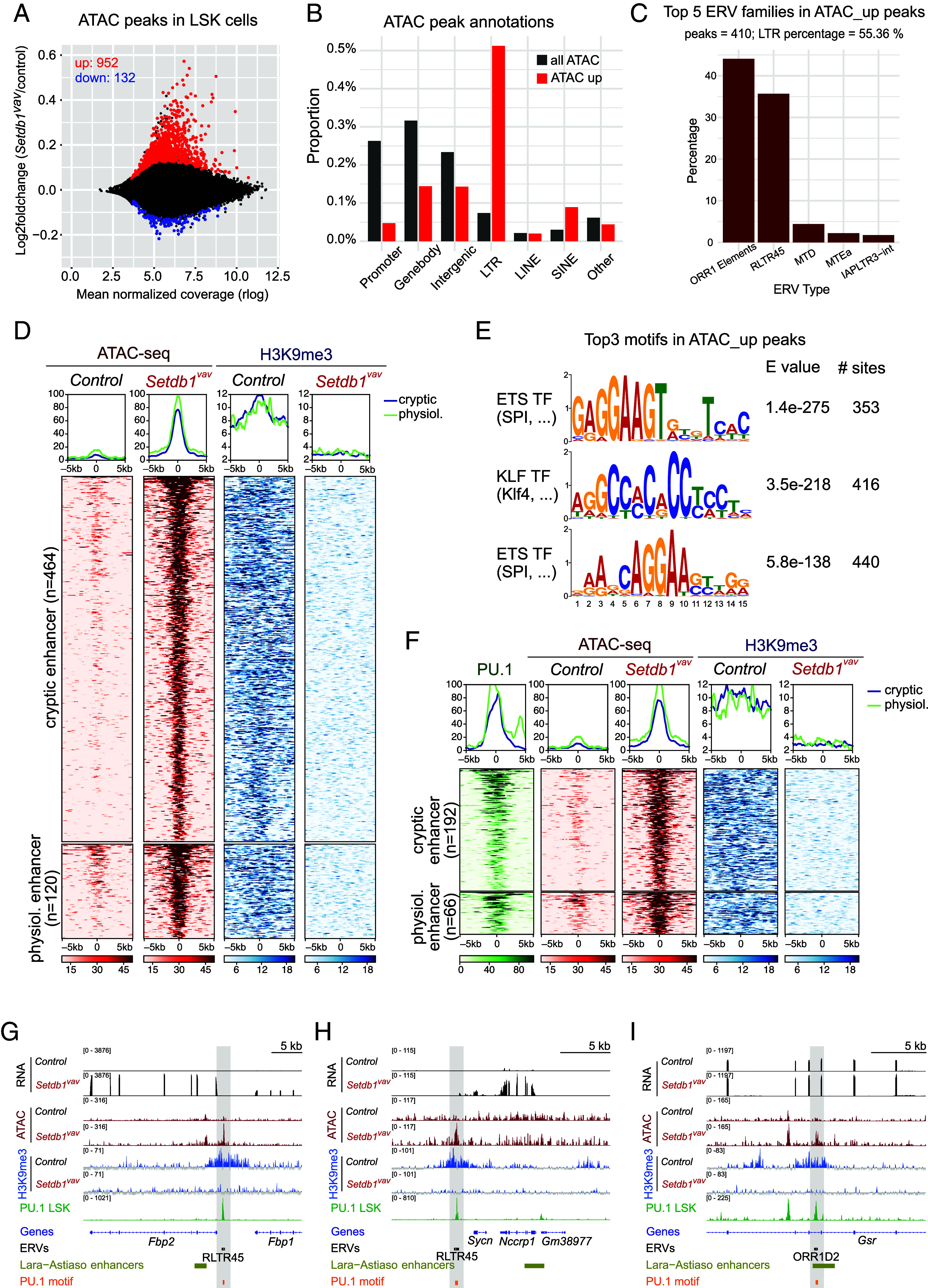
SETDB1 loss alters chromatic landscape at ERVs-overlapping enhancers in FL HSPCs. (A) Scatter plot showing mean normalized coverage vs. log2 fold change of ATAC-seq peaks in Setdb1vav vs. control LSK cells. Significantly up (red) or down-regulated (blue) ATAC peaks (Padj < 0.05, n = 2) in Setdb1vav LSK cells are indicated. (B) Genomic features associated with Setdb1vav up-regulated ATAC peaks in LSK cells. (C) Association of Setdb1vav up-regulated ATAC peaks with ERV families. The bar plot shows the proportion of ATAC_up peaks overlapping with ERVs. (D) Heatmap and cumulative coverage plots showing ATAC and H3K9me3 coverage for H3K9me3 enriched ATAC_up peaks corresponding to cryptic and physiological enhancers. Distance from the peak center is given in bp. (E) MEME motif analysis of Setdb1vav up-regulated ATAC peaks. Top three motif logos are shown. (F) Heatmap and cumulative coverage plots of H3K9me3 enriched ATAC_up peaks overlapping with PU.1 peaks in LSK cells (32) showing PU.1 binding, ATAC-seq, and H3K9me3 ChIP-seq coverage. Distance from the peak center is given in bp. (G–I) Genome browser views of selected Setdb1vav up-regulated ATAC peaks overlapping with PU.1 binding and connected with transcriptional changes of genes in the vicinity.
ATAC-seq peaks are typically generated through binding and activity of specific transcription factors and characteristic features of active enhancers (33). Increased ATAC-seq signal in Setdb1vav cells thus suggests that these regions could represent enhancers which are repressed by SETDB1. We therefore assessed the overlap between ATAC_up peaks with enhancer chromatin modifications during different stages of hematopoiesis. Although a subset of peaks overlapped with regions that display physiological enhancer activity at any stage during hematopoiesis, most peaks did not show signs of physiological activity (SI Appendix, Fig. S6C). We therefore term these regions cryptic enhancers. We then asked which regions are directly regulated by SETDB1-induced H3K9me3 and found that most cryptic enhancers are H3K9me3 target regions (SI Appendix, Fig. S6D) that lose this modification in Setdb1vav LSKs (Fig. 6D and SI Appendix, Fig. S6E).
We then investigated which TFs could act on these putative enhancers. Motif enrichment analysis of ATAC_up peaks revealed strong enrichment of ETS and KLF TFs motifs (Fig. 6E). An important TF for lymphoid and myeloid development is the ETS TF PU.1 (34–37) for which ChIP-seq profiles in wild-type LSK cells were available (32). Using this dataset, we detect PU.1 binding on 258 SETDB1 target ATAC_up peaks (Fig. 6F) and 152 non-SETDB1 target ATAC_up peaks (SI Appendix, Fig. S7A). To identify additional transcription factors which could act on PU.1 vs. non-PU.1 ATAC_up peaks we performed a differential transcription factor motif analysis (SI Appendix, Fig. S7B). This analysis revealed a strong enrichment of CEBPx motifs in PU.1 target regions, consistent with collaboration of these transcription factors for chromatin opening (38, 39). PU.1 binding in ATAC_up peaks is largely constant over myeloid differentiation, indicating that these PU.1 binding sites are not a reflection of an advanced myeloerythroid differentiation state of Setdb1vav LSK cells (SI Appendix, Fig. S7 C and D).
We were surprised to detect PU.1 binding on H3K9me3 enriched regions (Fig. 6F), as heterochromatin is generally assumed to prevent access to transcription factors (40–42). Since these regions display enrichment for distinct ERV families, we assessed PU.1 binding to ERV LTRs. Genome-wide PU.1 binding sites are not enriched for LTRs (SI Appendix, Fig. S7E); however, PU.1 binding sites in ATAC_up peaks are strongly enriched for LTR elements with preference toward RLTR45 and ORR1 (SI Appendix, Fig. S7F). Increased chromatin accessibility and reduced H3K9me3 in Setdb1vav FL LSK cells suggest that SETDB1 limits PU.1 activity on heterochromatic binding sites (Fig. 6F). Aberrant activity of PU.1 containing ATAC_up peaks could affect transcription of neighboring genes (SI Appendix, Fig. S7G). Examples include Fbp1/Fbp2 and Sycn/Nccrp1 loci where PU.1 binding and H3K9me3 are observed on cryptic enhancers containing RLTR45 elements (Fig. 6 G and H) and the Gsr locus, where PU.1 binding overlaps an ORR1 element in the context of a physiological enhancer (Fig. 6I). In Setdb1vav LSK cells, these and other PU.1 binding sites in ATAC_up peaks show loss of H3K9me3, elevated chromatin accessibility on the PU.1 binding sites and, eventually, enhanced transcription of nearby genes (Fig. 6 G–I and SI Appendix, Fig. S8A).
Discussion
Lineage-specific TFs have many potential binding sites in the genome but, only a subset of these binding sites should become activated enhancers that drive lineage-specific gene expression. Our data show that in LSK cells, hematopoietic TFs, such as PU.1, can bind to regions in the genome that should not become activated during hematopoiesis. These regions, largely overlapping with RLTR45 and ORR1 ERVs, are characterized by H3K9me3 and low chromatin accessibility (Fig. 7). In Setdb1 knock-out LSK cells, loss of the H3K9me3 pathway results in increased chromatin accessibility and enhanced expression of neighboring genes (Fig. 7). This is surprising as heterochromatin is generally assumed to prevent binding of activating transcription factors (40–42). Since binding of PU.1 can be detected on H3K9me3 regions, downstream activities after PU.1 binding might be inhibited by the SETDB1 pathway. This can include the following mechanisms: 1) Cobinding of collaborating TFs. In ATAC_up peaks overlapping with PU.1 binding, we detect motifs for CEBP TFs. PU.1 is known to collaborate with Cebpa in myeloid differentiation and helps in the recruitment of this transcription factor to distinct binding sites (38, 39). Cobinding of TFs is often needed to efficiently recruit or activate chromatin remodelers to induce enhanced chromatin accessibility (43). Thus, low chromatin accessibility of PU.1 target sites in H3K9me3 regions could be linked to the failure of recruiting additional TFs. 2) Recruitment of chromatin remodelers. Recent studies indicate that PU.1 can interact with and activate SWI/SNF-type chromatin remodelers (38, 39, 44). A recent study suggests that hypoacetylated H3K9me3 heterochromatin prevents recruitment or activity of such remodelers, resulting in low chromatin accessibility (45). It is currently unclear whether PU.1 mediated chromatin remodeling is inhibited by H3K9me3 chromatin. Further experiments are needed to assess whether H3K9me3 chromatin inhibits recruitment of chromatin remodelers or whether chromatin remodelers can be recruited but their activities are restricted. 3) Recruitment of chromatin modifiers. Enhancer activation coincides with establishment of active chromatin modifications, such as histone acetylation (e.g., H3K27ac), histone methylation (e.g., H3K4me1), and DNA hydroxymethylation. In this context, SETDB1 was already shown to prevent activity of DNA demethylation enzymes (46, 47); however, it is still unclear whether recruitment or activity of MLL complexes (H3K4me1) or histone acetyltransferases (p300) could be restricted by SETDB1.
Fig. 7.

SETDB1 mediates repression of transcription factor activity on nonphysiological binding sites. In HSPCs, SETDB1 establishes H3K9me3 on diverse classes of retroelements, cryptic enhancers, and promoter regions of genes. A subset of SETDB1 target sites can be bound by hematopoietic transcription factors, for example, PU.1. However, SETDB1 prevents the activity of such transcription factors which is critical to maintain HSPC identity and differentiation. In Setdb1-deficient HSPCs, TF activity on aberrant binding sites is not limited, indicated by increased chromatin accessibility on their binding sites and enhanced transcription of neighboring genes. This contributes to impaired HSPC identity and biased lineage differentiation.
Our data align with phenotypes observed in hematopoietic knock-out models of Daxx and H3.3, such as HSC impairment, enhanced myeloerythroid differentiation, and impaired lymphopoiesis (48, 49). DAXX and H3.3 are important components of the SETDB1/H3K9me3 pathway and are needed for full H3K9me3 establishment and low chromatin accessibility on ERVs (7, 9, 50). In agreement with this, Daxx and H3.3 knock-out mice displayed impaired silencing of retroelements in HSPCs (48, 49). Interestingly, the observed phenotypes in Daxx ko HSPCs were connected with aberrantly high activity of PU.1 and could even be partially rescued by PU.1 knock-out (48). These data strengthen our hypothesis that the SETDB1/H3K9me3 pathway is critical for repressing TF activity on nonphysiological binding sites during hematopoiesis.
Setdb1 knock-out was already analyzed in adult HSCs (18); however, the knock-out resulted in rapid death of Setdb1-deficient HSCs. This phenotype was related to derepression of Fbp1/2 enzymes, enhanced gluconeogenesis, and reduced ATP production (18). We also found derepression of Fbp1 and Fbp2 in Setdb1vav FL HSCs; however, we did not observe any signs of apoptosis. This contrasting phenotype could be attributed to developmental differences between fetal and adult HSCs. In quiescent adult BM HSCs, glycolysis is the main metabolic pathway for energy production (51, 52). In contrast, cycling FL HSPCs are fueled by oxidative phosphorylation (OxPhos) (53). We found the OxPhos gene set even enriched in Setdb1vav FL LT-HSCs and MPPs, supporting their normal survival and increased proliferation despite Fbp1/2 upregulation. However, Setdb1-deficient FL HSPCs are compromised and fail in BM reconstitution assays. This could be due to impaired homing of these cells (Fig. 4) and downregulation of stem cell signature genes (Fig. 5). However, FL HSPCs could also be stressed when taken out of the FL niche. This is suggested by compromised in vitro myeloid differentiation of Setdb1vav HSPCs (Fig. 4B), although myeloid cells are greatly expanded in vivo. The FL is a protective environment for HSPCs, in part due to the production of bile acids that reduce unfolded protein response (UPR) stress (54). Activation of MuLV-derived proteins results in UPR-mediated apoptosis in pro-B cells of adult mice (15). In FL HSPCs, we also detect derepression of MuLV elements, but elevated URP-mediated apoptosis might be prevented through components, such as bile acids, in the FL.
Our analysis provides a basis for assessing the role of the repressive H3K9me3 pathway in differentiation. However, the full mechanistic details are still unclear. For example, we do not know whether only pioneer transcription factors, such as PU.1 (44, 55, 56), can bind to H3K9me3 marked chromatin. In addition, we lack mechanistic understanding of how the SETDB1 pathway antagonizes TF-initiated chromatin remodeling and transactivation. These basic questions deserve further studies, in systems that are biochemically better tractable than FL HSPCs.
Limitations of This Study.
SETDB1 has functions beyond H3K9me3 establishment (57, 58) In this study, we focused on its role in preventing aberrant enhancer activity. However, we cannot exclude that other possible consequences of Setdb1 loss, such as altered topological genome organization, production of long noncoding RNAs, and activation of innate immune signaling, might contribute to the observed phenotypes.
We lack a functional characterization of cryptic enhancers. This would require examining additional chromatin marks, contact maps with target genes, and deletion analyses. We also have no experimental proof for PU.1 mediated transactivation on cryptic enhancers. Although we detect PU.1 binding in wild-type LSK cells, loss of Setdb1 might lead to recruitment of other TFs that induce chromatin accessibility and stimulate transcription of nearby genes.
We cannot easily explain the myeloerythroid expansion through aberrant activation of myeloid enhancers in HSPCs. It is possible that the most strongly up-regulated nonhematopoietic genes and ERVs might be best tolerated in myeloid vs. other cell types. The myeloid-biased transcriptome of MPPs might result from additional factors, such as altered proportion of myeloid vs. lymphoid-primed progenitors, altered communication with niche cells, and selective death of lymphoid-primed progenitors. Myeloerythroid expansion might also be caused by selective expansion after myeloid commitment.
Materials and Methods
Mice.
The Setdb1 floxed mice and Vav-Cre transgenic mice were described previously (6, 59). Housing of mice was performed in the BMC animal core facility which is licensed by local authorities (Az. 5.1-5682/LMU/BMC/CAM, approved on 02-12-2017 by Landratsamt München) following the regulations of German Law (TierSchG, BGBl. I S. 1206, 1313).
Hematoxylin and Eosin (H&E) Staining of the Bone Marrow.
Sterna collected from 2-wk-old control and Setdb1vav mice were fixed in 4% formaldehyde, decalcified in Osteosoft (Merck), embedded in paraffin, sectioned, and stained with hematoxylin and eosin following deparaffinization and rehydration.
Flow Cytometry and Cell Sorting.
For FACS analysis, bone marrow cell suspension was prepared by flushing tibiae and femurs and filtered through a 70 μm cell strainer. To obtain hematopoietic cells from the spleen and thymus, the organs were gently meshed using the thumb rest of a 10 mL plunger on a 70 μm cell strainer. Fetal liver hematopoietic cells were prepared through dissociation of fetal livers using a 1 mL pipette and filtered through 70 μm cell strainer. Single-cell suspensions from different organs were incubated with unconjugated CD16/CD32 Fc-blocking antibody (2.4G2) for 20 min at 4 °C and then stained using antibody conjugates. For myeloid progenitor analysis, CD16/CD32 Fc- unblocked sample was subjected to staining. For fetal liver HSPCs analysis, anti-Mac1 (M1/70) was removed from the lineage+ staining antibody cocktail. All antibodies were purchased from BD Bioscience and Thermo Fischer Scientific (SI Appendix, Table S1). The scheme of the analyzed hematopoietic cell populations is indicated in SI Appendix, Fig. S1A.
For FACS sorting, fetal liver cells were filtered through a 40 μm filter, pretreated with red blood cell lysis buffer (BD Bioscience) following the manufacturer’s instructions, stained with PE-conjugated antibodies for lineage markers, and then enriched for lineage- HSPCs using Anti-PE MicroBeads (Miltenyi) through magnetic separation with the QuadroMACS separator. Cells were then labeled with fluorochrome-coupled antibodies specific for LT-HSCs, MPPs, and LSKs (antibodies, markers to define hematopoietic cell populations, and reagents are listed in SI Appendix, Tables S1–S3, respectively). Based on the purpose of the experiment, the labeled cells were run on BD FACSCanto or BD LSRFortessa for analysis or on FACSAria III or BD FACSAria Fusion for cell sorting. FACS data were analyzed with FlowJo software (Tree Star).
Annexin V Staining.
One million bone marrow or fetal liver cells were first stained for the specific markers to detect LSKs. Labeled cells were washed once with 1 mL 1X PBS and then with 1 mL 1X Annexin V binding buffer (Thermo Fischer Scientific). 5 μL of fluorochrome-conjugated Annexin V was added to the 100 μL cell suspension in Annexin V binding buffer. Cells were incubated at room temperature for 15 min, protected from light. Cells were then washed in 2 mL 1X Annexin V binding buffer and resuspended in 200 μL of 1X Annexin V Binding Buffer. 5 μL of Propidium Iodide Staining Solution was added to discriminate the dead cells. Samples were analyzed within 1 h, while stored at 4 °C in the dark.
Intracellular Staining for Cell Cycle Analysis.
For intracellular Ki-67 staining, RBC-lysed fetal liver cells were subjected to MACS cell separation to enrich for lineageneg cells. To eliminate dead cells, 1.5 million cells were first stained with Zombie Aqua Fixable Viability Dye (BioLegend) according to the manufacturer’s instructions. After the last wash, cells were labeled for the specific markers to detect LSKs. Fixation and permeabilization were carried out using Foxp3/Transcription Factor Staining Buffer Set (Thermo Fischer Scientific) following the manufacturer’s protocol. After fixation/permeabilization, intracellular staining was performed with Ki-67 (SolA15) antibody. Samples were run on BD LSRFortessa for analysis.
Transplantation Assay.
For transplantation, fetal liver cells were collected from CD45.1 wild-type (competitor) and CD45.2 Setdb1vav or Setdb1-sufficient control E13.5 embryos. One million competitor fetal liver cells were mixed with either Setdb1vav (Setdb1flox/flox; Vav-cre) or control (Setdb1flox/+; Vav-cre or Setdb1flox/+; +/+) donor fetal liver cells at a ratio of 1:1 and a total of two million cells were transplanted into lethally irradiated (9.5 Gy) recipient mice (CD45.1/2) by tail vein injection. To analyze donor-derived chimerism, peripheral blood was collected from tail veins of recipients every 2 wk after transplantation. The percentage of the chimerism in the bone marrow of recipient mice was assessed 8 wk after transplantation. CD45.1 and CD45.2 antibodies were added to measure the percentage of contribution from each donor.
Colony Formation Assay.
For methylcellulose assays, 3 × 104 cells were plated in methylcellulose medium supporting myeloerythroid colonies, MethoCult M3434 (STEMCELL Technologies), in 60 mm dishes, in duplicate. For B cell colony forming assay, 1 × 105 fetal liver cells were seeded in MethoCult M3630 (STEMCELL Technologies). Plates were incubated at 37 °C in 5% CO2 with ≥95% humidity. Myeloerythroid and B cell colonies were counted on day 10 and day 7 after culture, respectively.
Homing Assay.
Fetal liver cells were collected from Setdb1vav and control embryos, pretreated with red blood cell lysis buffer (BD Biosciences), and enriched for lineage- cells, as described for FACS sorting. Lineage- cells were labeled with CFSE (Thermo Fischer Scientific) for 10 min at 37 °C at the final concentration of 10 µM, and 10 million CFSE-labeled lineage- cells were injected into the lethally irradiated (9.5 Gy) recipient mice via tail vein. The frequency of CFSE+ cells was analyzed in the bone marrow, 16 h after injection by flow cytometry.
RNA Sequencing.
RNA-seq was performed from total RNA, isolated from E13.5 Setdb1vav and control fetal liver LT-HSCs and MPPs using the PicoPure RNA Isolation Kit (Thermo Fischer Scientific). Details are provided in SI Appendix.
Paired-end reads were mapped to version mm10 of the mouse genome using STAR (60). Normalization of read counts and differential expression analysis were performed using DESeq2 (61). Heatmaps were plotted with pheatmap either using rlog-normalized expression values from RSEM-normalized data. GSEA was performed with GSEA software (62). The expression levels of repeat classes were assessed with Homer using the analyzeRepeats.pl program with “repeats” function, following loading the repeat definitions from UCSC.
ULI-NChIP Sequencing.
ChIP-seq experiments for H3K9me3 were performed using the ultra-low-input micrococcal nuclease-based native ChIP (ULI-NChIP) protocol with minor modifications (63) starting from 5,000 LSK cells, sorted from E13.5 Setdb1vav and control fetal livers. The detailed protocol is in SI Appendix.
Reads were aligned to version mm10 of the mouse genome using bowtie (64), excluding multimapped reads. For repeat analysis, multimapped reads were included. Enrichment of H3K9me3 on repeats was assessed by Homer (55) using the analyzeRepeats.pl program with “repeats” function, following loading the repeat definitions from UCSC. Coverage heatmaps were generated using deeptools (65).
Omni-ATAC Sequencing.
ATAC-seq was performed as previously described (66) from 10,000 LSK cells sorted from E13.5 Setdb1vav and control fetal livers. The detailed protocol is in SI Appendix.
ATAC-seq reads were mapped to version mm10 of the mouse genome using Bowtie. ATAC peaks over input background were identified with Homer using the findPeaks.pl program with option “-style factor.” Peaks from all samples were merged using mergePeaks. The unified Peak list was filtered for promoter-associated peaks (distance to TSS < 1,000 bp) with bedtools. ATAC coverage counts were then calculated with Homer using the annotatePeaks.pl from corresponding tag directories. ATAC peaks with log2 fold change >2 were defined as differentially accessible regions. PCA analysis was performed on ATAC peak coverage data with plotPCA function in R. Transcription factor motif prediction for differential ATAC peak was done with Homer using findMotifsGenome.pl program. Differential TF motif analysis was performed with monaLisa (67).
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
Work in the G.S. lab is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), Project-ID 213249687–SFB1064. L.K. receives funding through the DFG (German Research Foundation) under SFB 1054/3 Project A01 (210592381), SFB-TRR 355/1 Project B01 (490846870), and Project number 456882036. We acknowledge the Flow Cytometry Core Facility at the Biomedical Center, Ludwig Maximilian University, Munich, for assistance with generating the flow cytometry data. Portions of the paper were developed from the dissertation of M.K.
Author contributions
M.K. and G.S. designed research; M.K., F.C., A.P., M.H., and A.N. performed research; L.K. contributed new reagents/analytic tools; M.K. and G.S. analyzed data; L.K. provided support for transplantation experiments; and M.K. and G.S. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
Data are presented as mean ± SD calculated from “n” number of independent experiments. Statistical analysis was performed in R. NGS data (SI Appendix, Table S4) were deposited on the NCBI GEO database with Accession Nos. GSE266387 (68), GSE266388 (69), and GSE266392 (70).
Supporting Information
References
- 1.Becker J. S., Nicetto D., Zaret K. S., H3K9me3-dependent heterochromatin: Barrier to cell fate changes. Trends Genet. 32, 29–41 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matsui T., et al. , Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 464, 927–931 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Bilodeau S., Kagey M. H., Frampton G. M., Rahl P. B., Young R. A., SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev. 23, 2484–2489 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schultz D. C., Ayyanathan K., Negorev D., Maul G. G., Rauscher F. J., SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes. Dev. 16, 919–932 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ecco G., Imbeault M., Trono D., KRAB zinc finger proteins. Development 144, 2719–2729 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z., et al. , Dominant role of DNA methylation over H3K9me3 for IAP silencing in endoderm. Nat. Commun. 13, 5447 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sadic D., et al. , Atrx promotes heterochromatin formation at retrotransposons. EMBO Rep. 16, 836–850 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voon H. P. J., et al. , ATRX plays a key role in maintaining silencing at interstitial heterochromatic loci and imprinted genes. Cell Rep. 11, 405–418 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groh S., et al. , Morc3 silences endogenous retroviruses by enabling Daxx-mediated histone H3.3 incorporation. Nat. Commun. 12, 5996 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dodge J. E., Kang Y., Beppu H., Lei H., Histone H3–K9 methyltransferase ESET is essential for early development histone H3–K9 methyltransferase ESET is essential for early development. Mol. Cell. Biol. 24, 2478–2486 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan P., et al. , Eset partners with Oct4 to restrict extraembryonic trophoblast lineage potential in embryonic stem cells. Genes Dev. 23, 2507–2520 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu S., et al. , Setdb1 is required for germline development and silencing of H3K9me3-marked endogenous retroviruses in primordial germ cells. Genes Dev. 28, 2041–2055 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan S.-L., et al. , Essential roles of the histone methyltransferase ESET in the epigenetic control of neural progenitor cells during development. Development (Cambridge, England) 139, 3806–16 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Collins P. L., Kyle K. E., Egawa T., Shinkai Y., Oltz E. M., The histone methyltransferase SETDB1 represses endogenous and exogenous retroviruses in B lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 112, 8367–8372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pasquarella A., et al. , Retrotransposon derepression leads to activation of the unfolded protein response and apoptosis in pro-B cells. Development 143, 1788–1799 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Adoue V., et al. , The histone methyltransferase SETDB1 controls T helper cell lineage integrity by repressing endogenous retroviruses. Immunity 50, 629–644.e8 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Karimi M. M., et al. , DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mescs. Cell Stem Cell 8, 676–687 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koide S., et al. , Setdb1 maintains hematopoietic stem and progenitor cells by restricting the ectopic activation of nonhematopoietic genes. Blood 128, 638–649 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Gualdrini F., et al. , H3K9 trimethylation in active chromatin restricts the usage of functional CTCF sites in SINE B2 repeats. Genes Dev. 36, 414–432 (2022), 10.1101/gad.349282.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogilvy S., et al. , Promoter elements of vav drive transgene expression in vivo throughout the hematopoietic compartment. Blood 94, 1855–63 (1999). [PubMed] [Google Scholar]
- 21.Ivanova N. B., et al. , A stem cell molecular signature. Science 298, 1–45 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Chambers S. M., et al. , Hematopoietic fingerprints: An expression database of stem cells and their progeny. Cell Stem Cell 1, 578–591 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Magnusson M., et al. , HOXA10 is a critical regulator for hematopoietic stem cells and erythroid/megakaryocyte development. Blood 109, 3687–3696 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Lawrence H. J., et al. , Loss of expression of the Hoxa-9 homeobox gene impairs the proliferation and repopulating ability of hematopoietic stem cells. Blood 106, 3988–3994 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blank U., et al. , Smad7 promotes self-renewal of hematopoietic stem cells. Blood 108, 4246–4254 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Copley M. R., et al. , The Lin28b–let-7–Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat. Cell Biol. 15, 916–925 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Chuong E. B., Rumi M. A. K., Soares M. J., Baker J. C., Endogenous retroviruses function as species-specific enhancer elements in the placenta. Nat. Genet. 45, 325–329 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ivancevic A., et al. , Endogenous retroviruses mediate transcriptional rewiring in response to oncogenic signaling in colorectal cancer. Sci. Adv. 10, eado1218 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deniz Ö., et al. , Endogenous retroviruses are a source of enhancers with oncogenic potential in acute myeloid leukaemia. Nat. Commun. 11, 3506 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dunn C. A., Medstrand P., Mager D. L., An endogenous retroviral long terminal repeat is the dominant promoter for human β1,3-galactosyltransferase 5 in the colon. Proc. Natl. Acad. Sci. U.S.A. 100, 12841–12846 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Babaian A., Mager D. L., Endogenous retroviral promoter exaptation in human cancer. Mobile DNA 7, 24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pundhir S., et al. , Enhancer and transcription factor dynamics during myeloid differentiation reveal an early differentiation block in cebpa null progenitors. Cell Rep. 23, 2744–2757 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Grandi F. C., Modi H., Kampman L., Corces M. R., Chromatin accessibility profiling by ATAC-seq. Nat. Protoc. 17, 1518–1552 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeKoter R. P., Singh H., Regulation of B lymphocyte and macrophage development by graded expression of PU.1. Science 288, 1439–1441 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Nerlov C., Graf T., PU.1 induces myeloid lineage commitment in multipotent hematopoietic progenitors. Genes Dev. 12, 2403–2412 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKercher S. R., et al. , Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. 15, 5647–5658 (1996). [PMC free article] [PubMed] [Google Scholar]
- 37.Scott E., Simon M., Anastasi J., Singh H., Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science 265, 1573–1577 (1994). [DOI] [PubMed] [Google Scholar]
- 38.Frederick M. A., et al. , A pioneer factor locally opens compacted chromatin to enable targeted ATP-dependent nucleosome remodeling. Nat. Struct. Mol. Biol. 30, 31–37 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lian T., Guan R., Zhou B.-R., Bai Y., Structural mechanism of synergistic targeting of the CX3CR1 nucleosome by PU.1 and C/EBPα. Nat. Struct. Mol. Biol. 31, 633–643 (2024). [DOI] [PubMed] [Google Scholar]
- 40.Soufi A., Donahue G., Zaret K. S., Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 151, 994–1004 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Methot S. P., et al. , H3K9me selectively blocks transcription factor activity and ensures differentiated tissue integrity. Nat. Cell Biol. 23, 1163–1175 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L., et al. , Chromatin landscape instructs precise transcription factor regulome during embryonic lineage specification. Cell Rep. 43, 114136 (2024). [DOI] [PubMed] [Google Scholar]
- 43.Cernilogar F. M., et al. , Pre-marked chromatin and transcription factor co-binding shape the pioneering activity of Foxa2. Nucleic Acids Res. 47, 9069–9086 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Minderjahn J., et al. , Mechanisms governing the pioneering and redistribution capabilities of the non-classical pioneer PU.1. Nat. Commun. 11, 402 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sahu R. K., et al. , Nucleosome remodeler exclusion by histone deacetylation enforces heterochromatic silencing and epigenetic inheritance. Mol. Cell 84, 3175–3191.e8 (2024), 10.1016/j.molcel.2024.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leung D., et al. , Regulation of DNA methylation turnover at LTR retrotransposons and imprinted loci by the histone methyltransferase Setdb1. Proc. Natl. Acad. Sci. U.S.A. 111, 6690–6695 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deniz Ö., de la Rica L., Cheng K. C. L., Spensberger D., Branco M. R., SETDB1 prevents TET2-dependent activation of IAP retroelements in naïve embryonic stem cells. Genome Biol. 19, 6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gerber J. P., et al. , Aberrant chromatin landscape following loss of the H3.3 chaperone Daxx in haematopoietic precursors leads to Pu.1-mediated neutrophilia and inflammation. Nat. Cell Biol. 23, 1224–1239 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo P., et al. , Histone variant H3.3 maintains adult haematopoietic stem cell homeostasis by enforcing chromatin adaptability. Nat. Cell Biol. 24, 99–111 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elsässer S. J., Noh K.-M., Diaz N., Allis C. D., Banaszynski L. A., Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 522, 240–244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suda T., Takubo K., Semenza G. L., Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem. Cell 9, 298–310 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Klimmeck D., et al. , Proteomic cornerstones of hematopoietic stem cell differentiation: Distinct signatures of multipotent progenitors and myeloid committed cells. Mol. Cell. Proteomics 11, 286–302 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Manesia J. K., et al. , Highly proliferative primitive fetal liver hematopoietic stem cells are fueled by oxidative metabolic pathways. Stem Cell Res. 15, 715–721 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Sigurdsson V., et al. , Bile acids protect expanding hematopoietic stem cells from unfolded protein stress in fetal liver. Cell Stem Cell 18, 522–532 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Heinz S., et al. , Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–89 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fernandez Garcia M., et al. , Structural features of transcription factors associating with nucleosome binding. Mol. Cell 75, 921–932.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Delaney C. E., et al. , SETDB1-like MET-2 promotes transcriptional silencing and development independently of its H3K9me-associated catalytic activity. Nat. Struct. Mol. Biol. 29, 85–96 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Warrier T., et al. , SETDB1 acts as a topological accessory to Cohesin via an H3K9me3-independent, genomic shunt for regulating cell fates. Nucleic Acids Res. 50, 7326–7349 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Georgiades P., et al. , VavCre transgenic mice: A tool for mutagenesis in hematopoietic and endothelial lineages. Genesis 34, 251–256 (2002). [DOI] [PubMed] [Google Scholar]
- 60.Dobin A., et al. , STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Subramanian A., et al. , Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brind’Amour J., et al. , An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nat. Commun. 6, 6033 (2015). [DOI] [PubMed] [Google Scholar]
- 64.Langmead B., Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinf. Chapter 11, Unit 11.7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramírez F., et al. , deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Corces M. R., et al. , An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Machlab D., et al. , monaLisa: An R/Bioconductor package for identifying regulatory motifs. Bioinformatics 38, 2624–2625 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kazerani M., et al. , Histone methyltransferase SETDB1 safeguards mouse fetal hematopoiesis by suppressing activation of cryptic enhancers [ATAC-seq]. Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE266387. Deposited 1 May 2024. [DOI] [PMC free article] [PubMed]
- 69.Kazerani M., et al. , Histone methyltransferase SETDB1 safeguards mouse fetal hematopoiesis by suppressing activation of cryptic enhancers [RNA-seq]. Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE266388. Deposited 1 May 2024. [DOI] [PMC free article] [PubMed]
- 70.Kazerani M., et al. , Histone methyltransferase SETDB1 safeguards mouse fetal hematopoiesis by suppressing activation of cryptic enhancers [ChIP-seq]. Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE266392. Deposited 1 May 2024. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
Data are presented as mean ± SD calculated from “n” number of independent experiments. Statistical analysis was performed in R. NGS data (SI Appendix, Table S4) were deposited on the NCBI GEO database with Accession Nos. GSE266387 (68), GSE266388 (69), and GSE266392 (70).