

Abstract
Gaining spatial control over innate immune activation is of great relevance during vaccine delivery and anticancer therapy, where one aims at activating immune cells at draining lymphoid tissue while avoiding systemic off‐target innate immune activation. Lipid‐polymer amphiphiles show high tendency to drain to lymphoid tissue upon local administration. Here, pH‐sensitive, cholesteryl end group functionalized polymers as stimuli‐responsive carriers are introduced for controlled immunoactivation of draining lymph nodes. Methacrylamide‐based monomers bearing pendant 2‐propionic‐3‐methylmaleic anhydride groups are polymerized by Reversible Addition‐Fragmentation Chain Transfer (RAFT) polymerization using a cholesterol chain‐transfer agent (chol‐CTA). The amine‐reactive anhydrides are conjugated with various amines, however, while primary amines afforded irreversible imides, secondary amines provided pH‐responsive conjugates that are released upon acidification. This can be applied to fluorescent dyes for irreversibly carrier labeling or immunostimulatory Toll‐like receptor (TLR) 7/8 agonists as cargos for pH‐responsive delivery. Hydrophilization of remaining anhydride repeating units with short PEG‐chains yielded cholesteryl‐polymer amphiphiles that showed efficient cellular uptake and increased drug release at endosomal pH. Moreover, reversibly conjugated TLR 7/8 agonist amphiphiles efficiently drained to lymph nodes and increased the number of effectively maturated antigen‐presenting cells after subcutaneous injection in vivo. Consequently, cholesteryl‐linked methacrylamide‐based polymers with pH‐sensitive 2‐propionic‐3‐methylmaleic anhydride side groups provide ideal features for immunodrug delivery.
Keywords: 2‐propionic‐3‐methylmaleic anhydride, cholesterol, immunodrug delivery, pH sensitivity, RAFT‐polymerization
Cholesteryl end group functionalized polymeric 2‐propionic‐3‐methyl‐maleic anhydride polymers can reversibly be conjugated with secondary amine functionalized Toll‐like receptor 7/8 agonists, promote their delivery after subcutaneous injection into draining lymph nodes, and subsequently release them upon mild acidification.
1. Introduction
The immune system is a complex organization of different cells and lymphoid organs with the function of protecting the human body from infection as well as possibly preventing tumor growth. Separated into two essential types, the innate immune response on the one hand uses a variety of cells including macrophages, natural killer cells, or dendritic cells that immediately recognize pathogens or abnormal features. The adaptive response, on the other hand, is characterized by the precise activation of T‐ and B‐cell subsets localized in lymphoid organs or tissues and their ability to form long‐lived memory.[ 1 , 2 , 3 , 4 , 5 ] Thereby, the lymphoid organs are further subdivided into the primary organs—thymus and bone marrow—responsible for the T‐ and B‐cell generation, while cell activation occurs in the spleen, Peyer's patches, mucosa‐associated lymphoid tissue, and lymph nodes, summarized as secondary lymphoid organs.[ 6 , 7 , 8 ]
Precisely controlling the activation of those immune components facilitates advanced opportunities for the treatment of infectious diseases and, more recently, also cancer. The application of immune modulators, monoclonal antibodies, or vaccines providing antigens or adjuvants revealed the effective stimulation of innate and adaptive immune cells.[ 9 , 10 ] In particular, adaptive immune responses require well‐orchestrated contributions by the innate immune system, most effectively via addressing pathogen‐associated molecular patterns (PAMPs). For that purpose, small molecules or viral nucleic acids can be applied to get recognized by various pattern‐recognition receptors (PRRs) for triggering necessary inflammatory responses. Toll‐like receptors (TLRs) localized on the plasma membrane of macrophages and dendritic cells, or inside their endosomes constitute an important category of PPRs and can be activated by many diverse agonists.[ 11 , 12 ] Imidazoquinoline derivates, such as 1‐(4‐(aminomethyl)benzyl)‐butyl‐1H‐imidazo[4,5‐c]quinoline‐4‐amine (IMDQ), are very potent activators, stimulating the TLR7/8 signaling pathway.[ 13 , 14 ] However, for effective therapy such types of immune modulators need to reach the target site in sufficient concentration, whereas most of them show a poor selectivity and rapidly diffuse after administration, inducing immune‐related inflammatory toxicities all over the body.[ 14 , 15 , 16 ]
Directing the immune activation to the site of interest by conjugating, encapsulating, or entrapping immune modulatory cues to the matrix of carrier systems can significantly increase the therapeutic window.[ 17 , 18 ] For advancing transport into lymph nodes, a variety of materials, including synthetic polymers,[ 19 ] micelles,[ 20 ] nanoparticles,[ 21 , 22 ] dendrimers[ 23 ] or lipids[ 24 ] demonstrated to improve the pharmacokinetic and biodistribution of potent small molecule immune modulators.[ 25 ] In particular, lipid motives emerged as an auspicious new approach for albumin‐hitchhiking to the lymph node, and cholesterol, mono‐ and diacyl lipids have been characterized for that purpose intensively.[ 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 ] Our group has also recently demonstrated that interaction between water‐soluble polymers and blood plasma compartments occurs when a cholesteryl‐end group modification is present.[ 64 ] Besides, covalent cholesterol conjugation affects the cell membranes’ fluidity and, thus, the permeability, which further advances cell internalization and the activity of intracellularly active immune modulators, too.[ 34 , 35 , 36 ]
However, most drugs show reduced or even no activity when conjugated to a carrier, hence, necessitating the introduction of a cleavable linker.[ 37 , 38 ] In this context, acid‐labile linkers[ 39 , 40 , 41 ] such as ketals,[ 42 ] acetals[ 43 ] and hydrazones[ 44 ] are of greatest interest, as macromolecular drug conjugates typically enter the cells through endocytosis and are transported into endosomal vesicles with gradually decreasing pH values. In this context, 2,3‐dialkymaleic anhydrides have less been explored but offer several advantages.[ 45 , 46 ] They enable a fast release of amine‐containing proteins or drugs under mild acidic conditions (pH 5.5–6.8).[ 45 , 47 ] Furthermore, the hydrolysis rate can be adjusted by varying the substitutes of the cis‐double bond and, thus, the internal angle between the amide and the carboxylic acid group.[ 48 , 49 ] Most importantly, as traceless linkers 2,3‐dialkymaleic anhydride structures release their conjugated molecules in their native form and, consequently, do not affect the drugs’ activity.[ 50 , 51 ]
For that purpose, we here report on the use of cholesteryl‐polymer scaffolds with pendant amine‐reactive 2‐propionic‐3‐methylmaleic anhydrides[ 52 , 53 ] for the design of acid‐labile amphiphile immuno‐drug conjugates that efficiently target lymph nodes (Figure 1 ).[ 25 , 54 ] RAFT polymerization using a cholesteryl‐functionalized CTA enabled the controlled polymerization of methacrylamides with 2‐propionic‐3‐methylmaleic anhydride side groups. Postpolymerization conjugation of IMDQ analogs and amine‐functionalized short hydrophilic PEG chains yielded water‐soluble IMDQ‐conjugated cholesteryl‐polymer amphiphiles. We demonstrated that an IMDQ bearing a primary amine remained irreversibly conjugated to the polymer backbone, whereas an IMDQ bearing a secondary amine could be liberated from the carrier at endosomal pH and, thus, re‐installed its bioactivity in vitro. Moreover, in vivo the amphiphile conjugates efficiently drained to lymph nodes upon local injection and increased the number of effectively maturated antigen‐presenting cells. Altogether, these characteristics demonstrate the advances of cholesteryl‐linked methacrylamide‐based polymers with pendant 2‐propionic‐3‐methylmaleic anhydride units as versatile platforms for effectively localized and pH‐stimulative immunodrug delivery.
Figure 1.
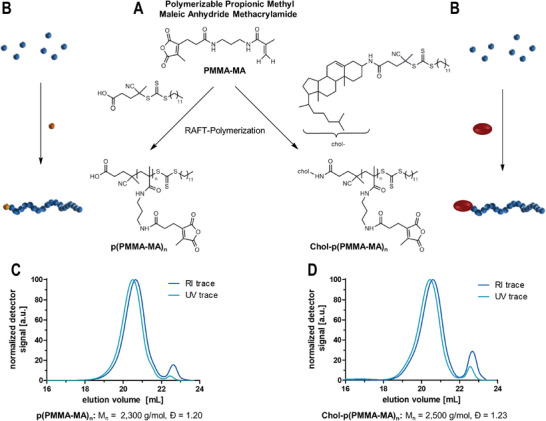
2‐Propionic‐3‐methyl maleic anhydride methacrylamides (PMMA‐MA) as RAFT polymerizable monomers for the formulation of pH‐sensitive polymers. A) Reaction scheme for polymerizations of PMMA‐MA with a small molecular trithiocarbonate chain transfer agent (TTC‐CTA) or a cholesterol chain transfer agent (chol‐TTC) and the resulting polymers. B) Schematic reaction overview. C) SEC traces of the polymers obtained by the reaction with the TTC‐CTA (analyzed in HFIP (hexafluoroisopropanol) and calibrated with PMMA standards). D) SEC analysis of the polymer obtained by the reaction with the chol‐CTA.
2. Results and Discussion
2.1. Synthesis and Characterization of Methacrylamide‐Based Polymers with Pendant 2‐Propionic‐3‐Methmaleic Anhydride Groups
We previously identified the propionic‐methylmaleic anhydride methacrylamide monomer PMMA‐MA (Figures S22–S24, Supporting Information) as a suitable monomer for Reversible Addition‐Fragmentation Chain Transfer (RAFT) polymerization yielding well‐defined polymers suitable for the design of pH‐labile drug conjugates.[ 52 ] A trithiocarbonate as chain transfer agent (TTC‐CTA) with azobisisobutyronitrile (AIBN) as initiator provided well‐defined narrowly distributed homopolymers p(PMMA‐MA)n (Figure 1A,B; Figure S29, Supporting Information) with degrees of polymerization around DP = 33 (determined by 1H NMR spectroscopy‐compare Supporting Information). Via size‐exclusion chromatography (SEC) with hexafluoroisopropanol (HFIP) as eluent and poly(methyl methacrylate) (PMMA) as calibration standard, a number‐average molecular weight (Mn) of 2300 g mol−1 and a narrow polydispersity (Ð) of 1.20 were obtained (Figure 1C). Noteworthy, under these conditions the PMMA‐MA monomer solely polymerizes by the methacrylamide function, while its maleic anhydride group is not affected.[ 55 ]
To extend this polymer concept to lipid‐polymer amphiphiles, a cholesteryl‐functionalized chain transfer agent can be synthesized in order to combine the albumin‐hitchhiking properties[ 25 , 29 , 35 ] with the polymeric 2‐propionic‐3‐methylmaleic anhydride. By conjugating a cholesteryl motif to chain transfer agents, they ensure that each polymer chain will be equipped with cholesteryl functionality during the RAFT polymerization. For that purpose, we first converted the hydrophilic 3‐hydroxy headgroup of cholesterol into a primary amine (Figures S1–S11, Supporting Information) and then treated it with a pentafluorophenyl ester‐activated trithiocarbonate chain transfer agent[ 42 ] (Figures S12–S14, Supporting Information) affording an amide‐conjugated cholesteryl‐CTA (Figures S15–S21, Supporting Information). Of note, we opted for the amide‐conjugation rather than an ester‐linked structure, due to the hydrolytic resistance of the amide bond during later applications. The resulting chemical structure of the amide‐conjugated cholesteryl‐CTA can be found in Figure 1A on the right, while its non‐cholesteryl functionalized CTA analog is provided on the left.
Subsequent RAFT polymerizations of PMMA‐MA by the obtained cholesteryl‐CTA and initiated by AIBN in methanol (Figure 1A,B; Figure S25, Supporting Information) yielded well‐defined chol‐p(PMMA‐MA)n polymers at similar degrees of polymerization, e.g., with a number‐average molecular weight (Mn) of 2500 g mol−1 and a narrow Ð of 1.23 (Figure 1D) (determined by SEC in HFIP with PMMA calibration‐note that the minor peaks appearing at 22.5 mL elution volume correspond to remaining traces of monomer and diethyl ether from the purification by precipitation, compare Figure S26, Supporting Information). 1H diffusion‐ordered NMR spectroscopy (1H DOSY NMR) revealed one diffusing species at high diffusing units, giving proof of a single macromolecular entity with cholesteryl end groups (Figures S27 and S28, Supporting Information). The homopolymers p(PMMA‐MA)n obtained from the parent CTA served as a control lacking the cholesteryl end group (Figure 1C; Figures S29–S31, Supporting Information).
2.2. pH‐Reversible or Irreversible Chol‐Polymer Modification by Amidation with Primary and Secondary Amines
We then verified the accessibility of the anhydride groups for pH‐reversible conjugation of different amines (Figure 2A). For this purpose, chol‐p(PMMA‐MA)30 was dissolved in DMSO, mixed with triethylamine (TEA), and incubated with benzylamine or dibenzylamine (Figure 2B; Figure S32, Supporting Information). Similar to our previous studies on p(PMMA‐MA)38, also chol‐p(PMMA‐MA)30 could successfully be conjugated with secondary amines under pH‐responsive release conditions.[ 52 ] 1H NMR spectroscopy confirmed the covalent quantitative attachment of dibenzylamine, as evidenced by the broadening of specific dibenzylamine signals. Due to the formation of two ring‐opened 2‐propionic‐3‐methylmaleic anhydride regioisomers, the related α‐positioned methyl signal splits into two signals (Figure S33, Supporting Information). By contrast, the modification with benzylamine resulted in the formation of one single sharp peak (Figure S35, Supporting Information), representing the pH‐resistant imide structure.
Figure 2.
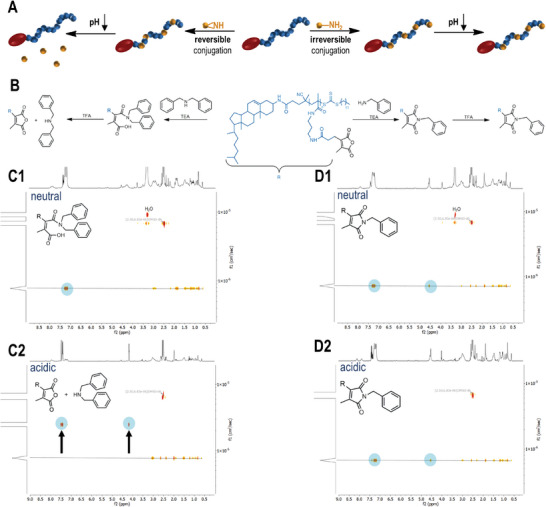
pH sensitivity of chol‐p(PMMA‐MA)30 conjugated with different amines. A) Scheme for the conversion of chol‐polymeric 2‐porpionic‐3‐methylmaleic anhydride groups with primary or secondary amines and their corresponding pH response. B) Amidation of 2‐propionic‐3‐methylmaleic anhydride side groups with benzyl‐ or dibenzylamine affording a pH‐reversible or irreversible polymer system. C1) 1H DOSY NMR spectrum of the chol‐polymer treated with dibenzylamine providing an identical diffusion species under neutral conditions and C2) the related 1H DOSY NMR spectrum upon acidification showing the successful release of the aromatic compound. D1) 1H DOSY NMR spectrum of the chol‐polymer treated with benzylamine under neutral and D2) acidic conditions affording only one diffusion species and no release of the primary amine upon acidification.
We then treated the amine‐modified chol‐p(PMMA‐MA)30 polymers with trifluoroacetic acid (TFA) and measured the release of dibenzylamine and benzylamine, respectively, by 1H diffusion ordered NMR spectroscopy (1H DOSY NMR) (Figure 2C1,D1). For the dibenzylamine‐modified polymers, the aromatic signals of dibenzylamine disappeared at the diffusing polymer species, and faster aromatic diffusion species were immediately recorded (Figure 2C2). By contrast, for the benzylamine‐modified polymer, no signal shift was observed upon acidification (Figure 2D2). This behavior could further be confirmed by 1H NMR measurements (Figures S34 and S36, Supporting Information) emphasizing the complete acid‐triggered cleavage of the secondary amines from the polymer affording the release of dibenzylamine and the regeneration of the disubstituted maleic anhydrides. The primary amines, however, like the conjugated benzylamine, provoked a deprivation of the pH sensitivity by irreversible emergence of the corresponding imide system.
Overall, the obtained results demonstrated the successful synthesis of a cholesteryl‐linked methacrylamide‐based polymer with pH‐sensitive 2‐propionic‐3‐methylmaleic anhydride groups for the conjugation of amines. However, only the use of secondary amines ensured a quantitative acidic‐triggered release from the macromolecular carrier, whereas primary amines formed irreversible imide bonds.
2.3. In Vitro Properties of Dye‐Labeled and PEG‐Modified Methacrylamide‐Based Polymers with Pendant 2‐Propionic‐3‐Methylmaleic Anhydride Groups
Encouraged by the selective reversible amidation results, we aimed to convert chol‐p(PMMA‐MA)30 polymers into lipid‐polymer amphiphiles using short amine‐functionalized polyethylene glycols (PEG) to increase the carriers’ water‐solubility, biocompatibility, as well as low intrinsic affinity to serum proteins and cell membranes.[ 56 , 57 , 58 ]
For that purposes, chol‐p(PMMA‐MA)30 and p(PMMA‐MA)38 were dissolved in DMSO supplemented with TEA and subsequently treated with the fluorescent dye tetramethylrhodamine cadaverine (TMR) followed by mPEG11‐amine (Mn = 0.75 kDa) (Figure 3A; Figure S37, Supporting Information) (note that primary amines were applied yielding two pH‐resistant and completely hydrophilic polymers). Successful quantitative polymer modifications were confirmed by 1H NMR spectroscopy (Figures S38 and S39, Supporting Information) as well as size exclusion chromatography (SEC). In both cases, narrow polymer distributions with increased number‐average molecular weights (Mn) of 26 900 g mol−1 for chol‐p(PMMA‐MA)30 and 28 100 g mol−1 for p(PMMA‐MA)38 were found compared to the unmodified polymers (Figure S40A,B, Supporting Information). In addition, due to intensive purifications by precipitation, covalent conjugation of the fluorescent dye TMR was evidenced by the remaining appearance of a TMR absorbance maximum at the respective wavelength 550 nm, detected by UV–vis spectroscopy measurements for both purified polymers (Figure 3E).
Figure 3.
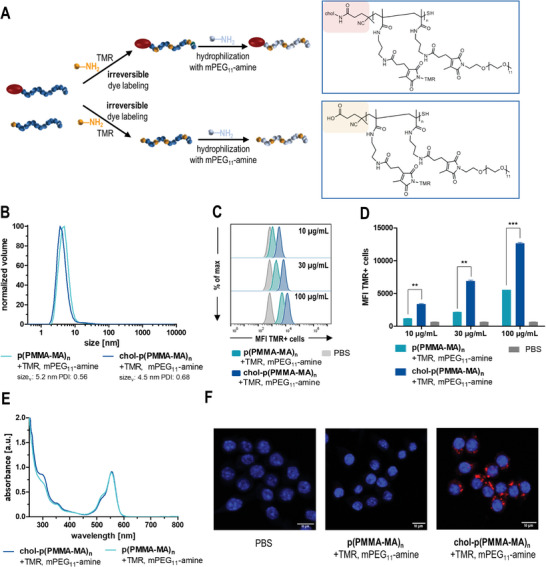
In vitro evaluation of PEGylated 2‐propionic‐3‐methylmaleic anhydride‐containing polymers with and without cholesteryl end groups. A) Synthetic scheme and resulting chemical structure of tetramethylrhodamine cadaverine (TMR)‐labeled and PEGylated p(PMMA‐MA)38 (yellow) and chol‐p(PMMA‐MA)30 (red). B) DLS polymer size distribution shows fully water‐soluble polymers in PBS with a volume mean of 5.21 nm for modified p(PMMA‐MA)38 (light blue) and 4.51 nm for chol‐p(PMMA‐MA)30 (dark blue). C) Flow cytometric histograms of TMR‐labeled, PEGylated p(PMMA‐MA)38 and chol‐p(PMMA‐MA)30 and their concentration‐dependent uptake in RAW‐Dual macrophages. D) Concentration‐dependent mean fluorescence intensity (MFI) of RAW‐Dual macrophages incubated with the respective polymers (n = 3, *: p ≤ 0.05, **: p ≤ 0.01, ***: p ≤ 0.001). E) UV–vis absorbance spectra of the TMR‐labeled polymers revealing similar labeling efficiencies. F) Fluorescent confocal microscopy images of the modified p(PMMA‐MA)38 and chol‐p(PMMA‐MA)30 as well as PBS, confirming an efficient cholesteryl‐induced cellular uptake (red: TMR‐labeled polymer, blue: nuclei stained with Hoechst 33258).
Due to the hydrophilic properties of PEG, the covalently functionalized polymers could easily be redissolved in PBS. To ensure no aggregation during further experiments, the polymer solutions were analyzed by dynamic light scattering (DLS) measurements. Both chol‐p(PMMA‐MA)30 and p(PMMA‐MA)38, conjugated with TMR and mPEG11‐amine, exhibited exclusively soluble polymer chains with volume sizes of 4.5 nm (chol‐p(PMMA‐MA)30) and 5.2 nm (p(PMMA‐MA)38) (Figure 3B). Notably, the cholesteryl moiety itself is insufficient to trigger micellar self‐assembling of the polymers in water. Subsequently, the cellular internalization of TMR‐labeled, hydrophilic polymers was analyzed by flow cytometry and fluorescence confocal microscopy. For that purpose, RAW‐Dual macrophages were incubated with different concentrations of fluorescent dye‐labeled polymers for 24 h at 37 °C. Flow cytometry showed a concentration‐dependent uptake of both polymers (Figure 3C,D; Figure S53, Supporting Information). Interestingly, PEGylated polymers with a cholesteryl end group exhibited a much higher internalization compared to the analogous polymer without cholesterol (Figure 3C,D; Figure S53, Supporting Information). These results were also confirmed by confocal fluorescence microscopy. Only the cholesteryl end group‐containing polymers exhibited sufficient intracellular fluorescence (Figure 3F; Figure S54, Supporting Information) and, thus, evidenced the favorable intracellular delivery features mediated by the cholesteryl end group.
2.4. In Vitro Effect of IMDQ or IMDQ‐Me Loaded and PEG‐Modified Cholesteryl‐Linked Methacrylamide‐Based Polymers with Pendant 2‐Propionic‐3‐Methylmaleic Anhydride Groups
Next, we wanted to combine these two delivery properties to small immune stimulatory imidazoquinoline‐based TLR 7/8 agonists. For that purpose, an imidazoquinoline analog bearing an aliphatic primary amine, IMDQ, was further modified as previously reported[ 52 ] into a secondary amine derivative (2‐butyl‐1‐(4‐((methylamino)methyl)benzyl)‐1H‐imidazo[4,5‐c]quinolin‐4‐amine, IMDQ‐Me) (Figures S41–S43, Supporting Information). Both IMDQ or IMDQ‐Me were conjugated to the chol‐polymers and further treated with TMR and mPEG11‐amine (Figure 4A; Figure S44, Supporting Information), affording water‐soluble lipid‐polymer amphiphiles that either carried the immunostimulatory cue irreversibly (IMDQ) or reversibly (IMDQ‐Me). Successful dye‐labeling and IMDQ‐ or IMDQ‐Me‐loading were verified by UV–vis spectroscopy (Figure 4B) as well as 1H NMR spectroscopy analyses (Figures S45 and S47, Supporting Information). Both drug‐conjugated chol‐polymers revealed an almost similar drug load of 4.4 wt.% (IMDQ) and 4.7 wt.% (IMDQ‐Me) (compare Figures S50 and S51, Supporting Information). Hydrophilization of the chol‐polymers was confirmed by 1H DOSY NMR spectroscopy measurements (Figures S46 and S48, Supporting Information), where all IMDQ‐related aromatic protons provided similar diffusion coefficients as the polymer backbone and the PEG side chains. No unbound IMDQ species were detectable. In addition, SEC analysis showed a significant elution volume shift compared to the unmodified polymer (Figure S49A, Supporting Information), and via DLS measurements in PBS single non‐aggregating polymer chain species were recorded, too (Figure S49B, Supporting Information).
Figure 4.
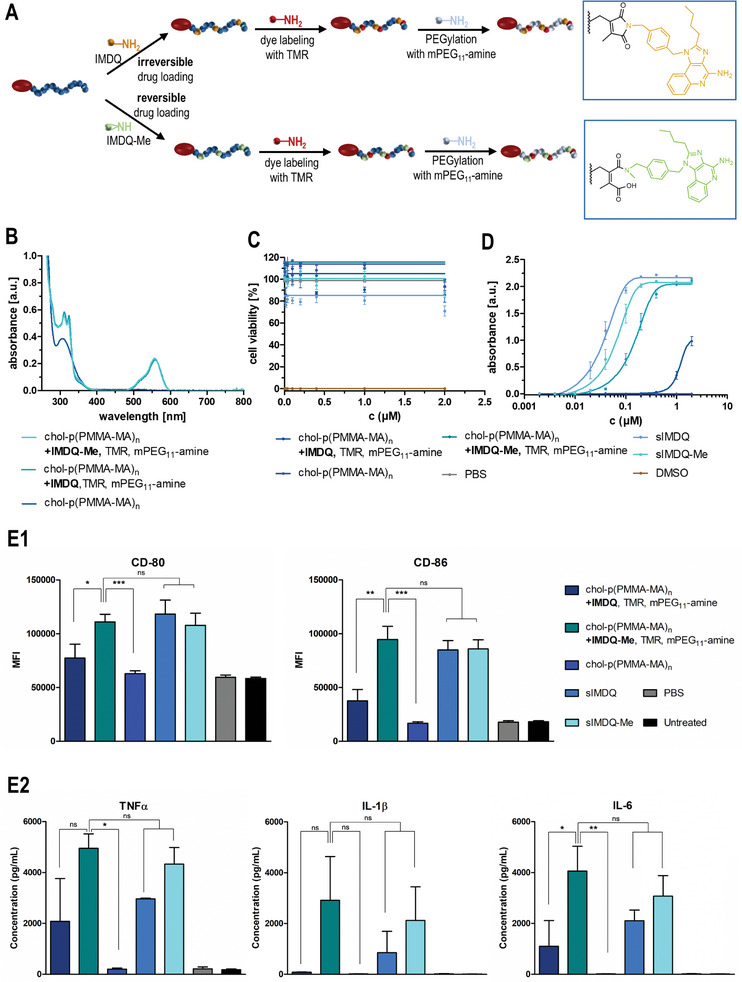
In vitro characterization of drug‐loaded cholesteryl‐linked polymers with 2‐propionic‐3‐methylmaleic anhydride side groups. A) Synthetic strategy for the formulation of a reversibly (IMDQ‐Me) or irreversibly (IMDQ)‐loaded and TMR‐labeled chol‐p(PMMA‐MA)32 hydrophilized with PEG11‐amine. B) UV–vis spectra of the corresponding drug‐loaded and dye‐labeled polymer systems as well as the unmodified chol‐polymer. C) Cell viability assay (MTT) of RAW‐Dual macrophages incubated with the soluble drugs (IMDQ and IMDQ‐Me) and respective drug‐loaded chol‐polymers, the unmodified chol‐polymer as well as PBS (positive control) and DMSO (negative control) (n = 4). D) TLR receptor activation of RAW‐Dual macrophages incubated with the drug‐conjugated chol‐polymers, free drugs, or empty chol‐polymer quantified by RAW blue assay (n = 4). E) Maturation of bone marrow‐derived dendritic cells after incubation with soluble drugs (IMDQ and IMDQ‐Me) and respective drug‐loaded chol‐polymers, the unmodified chol‐polymer as well as PBS. E1) To delineate the cellular activation state, the expression of the costimulatory markers CD80 and CD86 was quantified via flow cytometry by mean fluorescence intensity (MFI) (n = 3, *: p ≤ 0.1, **: p ≤ 0.05, +: p ≤ 0.01). E2) Additionally, the secretion of the proinflammatory cytokines TNFα, IL 1β, and IL‐6 into the cell culture media was quantified by cytometric bead assay (n = 3, *: p ≤ 0.1, **: p ≤ 0.05, ***: p ≤ 0.01).
Upon dialysis against 100 mM NaOAc buffer at pH 5.5 mimicking physiologically acidified conditions inside endolysosomes only the IMDQ‐Me‐loaded water‐soluble lipid‐polymer amphiphiles revealed a gradual decrease of imidazoquinoline absorbance at 324 nm, while for the imide‐bound IMDQ it remained constant for up to 125 h (Figure S52, Supporting Information). These observations suggest that upon endosomal cell internalization, only the IMDQ‐Me can get liberated from the carrier, while IMDQ remains attached irreversibly.
We then tested the ability of the IMDQ(‐Me) conjugates to trigger improved innate immune stimulations. For that purpose, we applied a RAW‐Dual macrophage reporter cell line in which an NF‐κB activation by TLR7/8 triggering is coupled to the expression of the alkaline embryonic phosphatase (SEAP). The immune stimulation can readily be quantified by colorimetric Quanti Blue Assay (Figure 4D), and cell viability can subsequently be determined by MTT assay (Figure 4C). Upon RAW‐Dual macrophage incubation with our samples, both the native IMDQ and IMDQ‐Me as unbound drugs demonstrated effective TLR 7/8 stimulation at sub‐micromolar concentrations, albeit methylation of IMDQ resulted in a slight reduction in activity (Figure 4D). While unmodified polymer (chol‐p(PMMA‐MA)n) did not affect TLR activation, only a very low stimulation could be detected for the irreversibly conjugated IMDQ‐chol‐polymer (chol‐p(PMMA‐MA)n, +IMDQ, TMR, mPEG11‐amine) (Figure 4D). By contrast, the pH‐reversible IMDQ‐Me conjugate (chol‐p(PMMA‐MA)n, +IMDQ‐Me, TMR, mPEG11‐amine) exhibited a significantly increased activity and the observed stimulation was only marginally lower when compared to the native IMDQ‐Me (Figure 4D). This underlines again the potential of an intracellular release of the TLR agonist exclusively for the secondary amine derivative, restoring its full receptor activity upon cellular delivery. Besides, no relevant reduction in cellular viability could be observed for all samples at the given concentrations, either (Figure 4C).
Based on these highly promising results, we further investigated the pH‐sensitive immune stimulatory potency of the PEGylated chol‐polymer conjugated with IMDQ or IMDQ‐Me on primary dendritic cells (DC) derived from murine bone marrow (BMDC). DCs are considered one of the most effective cellular sensors of antigen‐presenting cells (APCs) and represent an essential link between adaptive and innate immune responses.[ 59 ] After bone marrow differentiation in GM‐CSF‐supplemented media, the affording BMDCs were incubated with soluble IMDQ or IMDQ‐Me and the corresponding drug‐loaded chol‐polymers, as well as empty chol‐polymer and PBS. Via flow cytometric analysis, the subsequent immunostimulatory phenotype of the incubated BMDCs could be determined after 24 h by quantification of the surface‐expressed co‐stimulatory molecules CD80 and CD86. Interestingly, IMDQ that was irreversibly conjugated to the chol‐p(PMMA‐MA)n polymer barely triggered any maturation, neither for CD80 nor CD86. Only the chol‐polymers with the reversibly conjugated IMDQ‐Me were able to induce BMDC maturation in analogy to the native sIMDQ and sIMDQ‐Me, while the carrier polymer chol‐p(PMMA‐MA)n alone was immunologically silent. Additionally, the supernatants of the cell cultures were collected and quantified for the cells’ secretion of the proinflammatory cytokines TNFα, IL‐1β, and IL‐6, as further markers for BMDCs’ immunostimulatory phenotype maturation. Again, the polymers with the irreversibly conjugated IMDQ were hardly able to induce the secretion of all three cytokines. To the same extent as the native sIMDQ and sIMDQ‐Me, only the polymers with reversibly conjugated IMDQ‐Me were able to trigger the sufficient secretion of TNFα, IL‐1β, and IL‐6 (the latter even most significantly). These data support once more the ability of the chol‐p(PMMA‐MA)n platform to exclusively release the secondary amine TLR agonists intracellularly, and thereby restore their full receptor activity.
2.5. In Vivo Performance of IMDQ‐ or IMDQ‐Me‐Loaded and PEG‐Modified Cholesteryl‐Linked Methacrylamide‐Based Polymers with Pendant 2‐Propionic‐3‐Methylmaleic Anhydride Groups
Based on the remarkable in vitro results, we finally investigated whether chol‐polymer conjugated IMDQ and IMDQ‐Me could induce a controlled immune activation in vivo upon local administration in draining lymph nodes (Figure 5A).[ 21 , 60 , 61 , 62 ] To monitor the biodistribution of innate immune activation, we injected the respective test samples subcutaneously in the footpad of IFN‐β +/Δ β ‐luc reporter mice, a transgenic mouse model that provides luciferase co‐expression upon expression of the type I interferon IFN‐β.[ 16 , 63 ] Bioluminescence imaging 4 and 24 h postinjection revealed a spatially resolved innate immune activation (Figure 5B; Figure S56, Supporting Information). For the injected native TLR 7/8 agonists IMDQ and IMDQ‐Me a widespread systemic innate activation was observed immediately, indicative of a rapid diffusion of the injected TLR 7/8 agonists from the site of injection into the circulation. Similar effects had been reported in earlier studies that were accompanied by severe acute toxic inflammatory responses.[ 14 , 21 , 42 , 60 , 61 ] Irreversible IMDQ‐chol‐p(PMMA‐MA)n conjugates had negligible activity, confirming our in vitro observation. Reversible IMDQ‐Me‐chol‐p(PMMA‐MA)n conjugates, by contrast, did show activation in the draining popliteal lymph node at 4 h postinjection and resulted in a strongly reduced systemic response (Figure 5B; Figure S56, Supporting Information) (as well as a subtle higher background activity, which increases after 24 h‐Figure 5B1). Corresponding luminescence quantification of full‐body activity and targeted lymph node activity confirm the improved immunostimulation for the reversibly conjugated IMDQ‐Me agonist (Figure 5B2) and, consequently, evidence the in vivo release of the molecule from its chol‐p(PMMA‐MA)n carrier, too.
Figure 5.
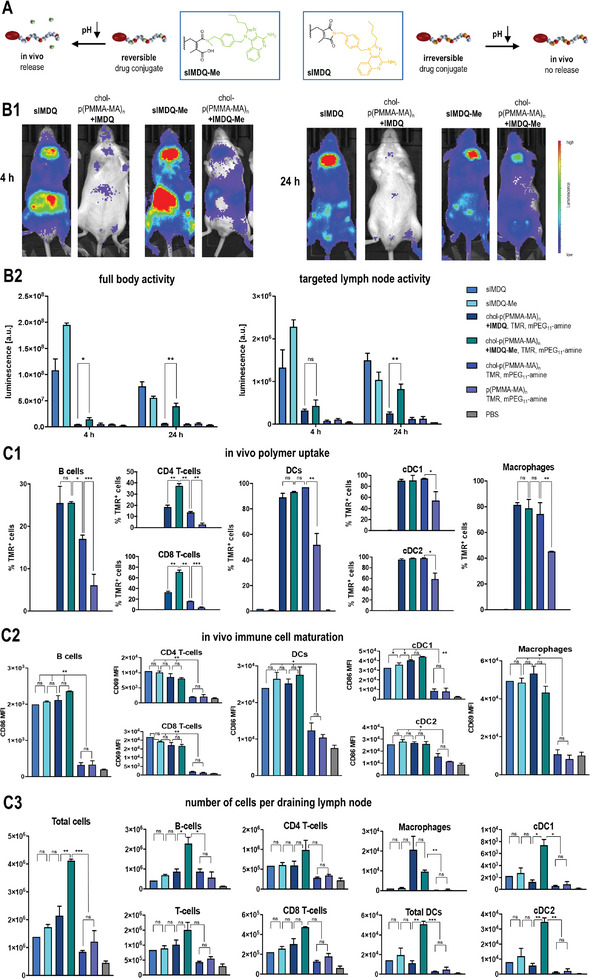
In vivo performance of chol‐polymer with pendant 2‐propionic‐3‐methylmaleic anhydride groups covalently attached with the TLR 7/8 agonist IMDQ or methylated IMDQ‐Me. A) Scheme for the exclusive acid‐triggered drug release for IMDQ‐Me compared to IMDQ, mediated by the dye‐ and drug‐loaded chol‐p(PMMA‐MA)32 polymer, further PEGylated with mPEG11‐amine. B) Immune stimulatory properties of those samples after footpad injection into BALB/c IFN‐β (IFN‐β +/Δ β ‐luc) luciferase reporter. B1) Representative luminescence images of the respective samples after 4 and 24 h and B2) corresponding luminescence quantification of full‐body activity and targeted lymph node activity, which is only fully restored for the mice treated with IMDQ‐Me‐loaded chol‐p(PMMA‐MA)32 (n = 3 or 4, *: p ≤ 0.1, **: p ≤ 0.05, ***: p ≤ 0.01). C) Flow cytometric analyses of the draining popliteal lymph nodes 24 h after footpad injection (n = 2 or 3): C1) The uptake of the (chol)‐p(PMMA‐MA)32 polymer was followed by TMR fluorescence and confirmed effective cholesteryl end group mediated delivery, C2) the maturation of the respective immune cell subpopulations was evaluated by mean fluorescence intensity of the respective maturation markers CD86 or CD69 and confirmed the immunostimulatory activity of the TLR 7/8 agonists, C3) the number of cells in the lymph node was determined in relation to count beads and revealed effective immunodrug delivery properties for chol‐p(PMMA‐MA)n carrying the pH‐releasable TLR 7/8 agonist IMDQ‐Me (n = 3, *: p ≤ 0.1, **: p ≤ 0.05, ***: p ≤ 0.01).
Flow cytometry analysis (Figure 5C‐for detailed gating procedures compare Figure S57, Supporting Information) of the TMRs signal in dissected popliteal lymph nodes, 24 h postinjection, revealed that all chol‐p(PMMA‐MA)n conjugates exhibited greatly enhanced lymphatic drainage compared to p(PMMA‐MA)n conjugates that lacked the cholesteryl motif (Figure 5C1). For all immune cell subpopulations, the amount of TMR+ cells was at least twice as high for the cholesteryl‐functionalized p(PMMA‐MA)n carriers than without cholesterol. While for DCs (cDC1s and cDC2s) as well as macrophages almost all recorded cells were TMR+ when the mice were treated with cholesteryl‐functionalized p(PMMA‐MA)n, for B cells it was only more effective, if the carrier was also equipped with IMDQ or IMDQ‐Me. These observations confirm the improved delivery capacities mediated by the cholesteryl end group modification for enhanced lymph node accumulation and cell interaction. Remarkably, for T‐cells, only the IMDQ‐Me‐carrying chol‐p(PMMA‐MA)n triggered the highest cell uptake significantly (Figure 5C1).
Next, the expression of typical co‐stimulatory surface markers like CD69 or CD86 on those immune cells was examined by flow cytometry, too (Figure 5C2). Given the strong immune stimulatory response that the native TLR 7/8 agonists sIMDQ and sIMDQ‐Me triggered all over the body, the expression of CD69 or CD86 could be found here in all immune subpopulations, too. Despite the IMDQ‐ and IMDQ‐Me‐carrying chol‐p(PMMA‐MA)n polymers not exhibiting systemic activity in the IFN‐β reporter mice (Figure 5B), they still provided an efficient lymph node‐focused expression of CD69 on T lymphocytes (CD4 and CD8) and macrophages, as well as CD86 on B lymphocytes, cDC1s and cDC2s, all to a similar extent as the soluble derivatives (Figure 5C2). Interestingly, for cDC1s‐renowned for their ability to cross‐present cellular antigens to CD8+ T cells and elicit antitumor immunity‐the CD86 expression levels were almost the highest after treatment with the reversibly conjugated polymeric IMDQ‐Me.
With that in mind, we finally analyzed the absolute cellularity of those immune populations in the draining lymph nodes, which is finally required to initiate effective immune responses (for that purpose, the number of cells was counted during flow cytometry analysis in relation to a defined number of added beads). Remarkably, the chol‐p(PMMA‐MA)n polymers with reversibly conjugated IMDQ‐Me featured the highest influx of all immune cells compared to all samples (Figure 5C3). A significantly strong increase of particularly B cells and dendritic cells (cDC1s and cDC2s) was found exclusively upon treatment with chol‐p(PMMA‐MA)n carrying the pH‐releasable TLR 7/8 agonist IMDQ‐Me (the number of T‐cells (CD4+ and CD8+) also increased, but less significant). The lymph node targeting and pH‐responsive IMDQ‐Me releasing polymers were most efficient in recruiting those relevant immune cell populations which are required for effective vaccination or cancer immunotherapy.
Altogether, our results support the successful in vivo release of the secondary amines from polymeric 2‐propionic‐3‐methylmaleic anhydrides, thereby, restoring the drugs’ bioactivity, which can potentially be applied for further immunotherapeutic delivery scenarios.
3. Conclusion
In this study, we herein reported the successful introduction of a cholesteryl‐functionalized pH‐sensitive polymeric delivery system derived from 2‐propionic‐3‐methylmaleic anhydride amide‐based methacrylamides. The resulting polymers can serve as a controlled pH‐responsive polymer carrier platform for delivering small immune stimulatory cues into draining lymph nodes after subcutaneous injection and, thereby, restoring their full bioactivity in vivo.
Via RAFT polymerization by a cholesteryl‐amide functionalized chain transfer agent, well‐defined propionic‐methylmaleic anhydride methacrylamide polymers were obtained that could sequentially be post‐modified with primary or secondary amines, albeit only the secondary amines revealed the desired pH‐responsive release upon acidification. Further conversion of the remaining anhydrides with short PEG‐amine yielded fully water‐soluble lipid‐polymer amphiphiles. Exemplified for imidazoquinoline TLR7/8 agonists, only the secondary amine drug derivative could reversibly be released from the carrier in its native form, thereby, preserving its biological activity in contrast to its primary amine derivative. The cholesteryl motif exhibited efficient lymphatic translocation in vivo upon local subcutaneous administration. Whereas native IMDQs induced systemic innate immune activation, the administration of IMDQ‐Me‐chol‐p(PMMA‐MA)n provoked a potent, but localized activation in draining lymph nodes. Notably, irreversible IMDQ‐chol‐p(PMMA‐MA)n neither induced innate immune activation in vitro, nor sufficiently in vivo, thereby, underscoring the key contribution of the reversible bond formed between the IMDQ‐Me, bearing a secondary amine, and the polymer. Overall, our findings demonstrate the potential of the cholesteryl‐linked pH‐sensitive carrier system for lymph node‐targeted immune activation and suggest future applications in vaccination or cancer immunotherapy.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting information
Supporting Information
Acknowledgements
This work was gratefully supported by the DFG through the Emmy Noether program and the CRC/SFB 1006 Projects B03 and B04 (both to L.N.). Stephan Türk, Manfred Wagner, Stefan Spang, and Detlev‐Walter Scholdei are acknowledged for technical assistance during the analytical measurements, and Tanja Weil for providing access to excellent laboratory facilities. The graphical abstract has partially been created with BioRender.com.
Open access funding enabled and organized by Projekt DEAL.
Heck A. G., Medina‐Montano C., Zhong Z., Deswarte K., Eigen K., Stickdorn J., Kockelmann J., Scherger M., Sanders N. N., Lienenklaus S., Lambrecht B. N., Grabbe S., De Geest B. G., Nuhn L., PH‐Triggered, Lymph Node Focused Immunodrug Release by Polymeric 2‐Propionic‐3‐Methyl‐maleic Anhydrides with Cholesteryl End Groups. Adv. Healthcare Mater. 2024, 13, 2402875. 10.1002/adhm.202402875
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Delvers P. J., Roitt I. M., N. Engl. J. Med. 2000, 343, 37. [DOI] [PubMed] [Google Scholar]
- 2. Andersen M. H., Schrama D., Thor Straten P., Becker J. C., J. Invest. Dermatol. 2006, 126, 32. [DOI] [PubMed] [Google Scholar]
- 3. Moon J. J., Huang B., Irvine D. J., Adv. Mater. 2012, 24, 3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Irvine D. J., Hanson M. C., Rakhra K., Tokatlian T., Chem. Rev. 2015, 115, 11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sallusto F., Lanzavecchia A., Araki K., Ahmed R., Immunity 2010, 33, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Randall T. D., Carragher D. M., Rangel‐Moreno J., Ann. Rev. Immunol. 2008, 26, 627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ruddle N. H., Akirav E. M., J. Immunol. 2009, 183, 2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim S., Shah S. B., Graney P. L., Singh A., Shah S. B., Graney P. L., Nat. Rev. Mater. 2019, 4, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lynn G. M., Laga R., Jewell C. M., Cancer Lett. 2019, 459, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shields C. W., Wang L. L. W., Evans M. A., Mitragotri S., Adv. Mater. 2020, 32, 1901633. [DOI] [PubMed] [Google Scholar]
- 11. Mancini R. J., Stutts L., Ryu K. A., Tom J. K., Esser‐Kahn A. P., ACS Chem. Biol. 2014, 9, 1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iwasaki A., Medzhitov R., Haven N., Nat. Immunol. 2015, 16, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Van Herck S., De Geest B. G., Acta Pharmacol. Sin. 2020, 41, 881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nuhn L., Vanparijs N., De Beuckelaer A., Lybaert L., Verstraete G., Deswarte K., Lienenklaus S., Shukla N. M., Salyer A. C. D., Lambrecht B. N., Grooten J., David S. A., De Koker S., De Geest B. G, Proc. Natl. Acad. Sci. USA 2016, 113, 8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cabral H., Miyata K., Osada K., Kataoka K., Chem. Rev. 2018, 118, 6844. [DOI] [PubMed] [Google Scholar]
- 16. Bhagchandani S., Johnson J. A., Irvine D. J., Adv. Drug Delivery Rev. 2021, 175, 113803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shi J., Kantoff P. W., Wooster R., Farokhzad O. C., Nat. Rev. Cancer 2017, 17, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sur S., Rathore A., Dave V., Reddy K. R., Chouhan R. S., Sadhu V., Nano‐Struct. Nano‐Obj. 2019, 20, 100397. [Google Scholar]
- 19. Lynn G. M., Chytil P., Francica J. R., Lagová A., Kueberuwa G., Ishizuka A. S., Zaidi N., Ramirez‐Valdez R. A., Blobel N. J., Baharom F., Leal J., Wang A. Q., Gerner M. Y., Etrych T., Ulbrich K., Seymour L. W., Seder R. A., Biomacromolecules 2019, 20, 854. [DOI] [PubMed] [Google Scholar]
- 20. Van Herck S., Deswarte K., Nuhn L., Zhong Z., Portela Catani J. P., Li Y., Sanders N. N., Lienenklaus S., De Koker S., Lambrecht B. N., David S. A., De Geest B. G., J. Am. Chem. Soc. 2018, 140, 14300. [DOI] [PubMed] [Google Scholar]
- 21. Nuhn L., De Koker S., Van Lint S., Zhong Z., Catani J. P., Combes F., Deswarte K., Li Y., Lambrecht B. N., Lienenklaus S., Sanders N. N., David S. A., Tavernier J., De Geest B. G., Adv. Mater. 2018, 30, 1803397. [DOI] [PubMed] [Google Scholar]
- 22. Stickdorn J., Nuhn L., Eur. Polym. J. 2020, 124, 109481. [Google Scholar]
- 23. Kaminskas L. M., Kota J., McLeod V. M., Kelly B. D., Karellas P., Porter C. J., J. Controlled Release 2009, 140, 108. [DOI] [PubMed] [Google Scholar]
- 24. De Vrieze J., Louage B., Deswarte K., Zhong Z., De Coen R., Van Herck S., Nuhn L., Frich C. K., Zelikin A. N., Lienenklaus S., Sanders N. N., Lambrecht B. N., David S. A., De Geest B. G, Angew. Chem. – Int. Ed. 2019, 58, 15390. [DOI] [PubMed] [Google Scholar]
- 25. De Vrieze J., Baptista A. P., Nuhn L., Van Herck S., Deswarte K., Yu H., Lambrecht B. N., De Geest B. G., Adv. Ther. 2021, 4, 2100079. [Google Scholar]
- 26. Schudel A., Francis D. M., Thomas S. N., Nat. Rev. Mater. 2019, 4, 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu H., Moynihan K. D., Zheng Y., Szeto G. L., Li A. V., Huang B., Van Egeren D. S., Park C., Irvine D. J., Nature 2014, 507, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wan D., Que H., Chen L., Lan T., Hong W., He C., Yang J., Wei Y., Wei X., Nano Lett. 2021, 21, 7960. [DOI] [PubMed] [Google Scholar]
- 29. Famta P., Shah S., Jain N., Srinivasarao D. A., Murthy A., Ahmed T., Vambhurkar G., Shahrukh S., Singh S. B., J. Controlled Release 2023, 353, 166. [DOI] [PubMed] [Google Scholar]
- 30. Spada A., Emami J., Tuszynski J. A., Lavasanifar A., Mol. Pharm. 2021, 18, 1862. [DOI] [PubMed] [Google Scholar]
- 31. Hoogenboezem E. N., Duvall C. L., Adv. Drug Deliv. Rev. 2018, 130, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lacroix A., Fakih H. H., Sleiman H. F., J. Controlled Release 2020, 324, 34. [DOI] [PubMed] [Google Scholar]
- 33. Abdallah M., Müllertz O. O., Styles I. K., Mörsdorf A., Quinn J. F., Whittaker M. R., Trevaskis N. L., J. Controlled Release 2020, 327, 117. [DOI] [PubMed] [Google Scholar]
- 34. Raffy S., Teissié J., Biophys. J. 1999, 76, 2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Albuquerque H. M. T., Santos C. M. M., Silva A. M. S., Molecules 2019, 24, 116. [Google Scholar]
- 36. Irby D., Du C., Li F., Mol. Pharm. 2017, 14, 1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Duncan R., Gaspar R., Mol. Pharm. 2011, 8, 2101. [DOI] [PubMed] [Google Scholar]
- 38. Kanamala M., Wilson W. R., Yang M., Palmer B. D., Wu Z., Biomaterials 2016, 85, 152. [DOI] [PubMed] [Google Scholar]
- 39. Tong R., Tang L., Ma L., Tu C., Baumgartner R., Cheng J., Chem. Soc. Rev. 2014, 43, 6982. [DOI] [PubMed] [Google Scholar]
- 40. Schattling P., Jochum F. D., Theato P., Polym. Chem. 2014, 5, 25. [Google Scholar]
- 41. Liu B., Thayumanavan S., J. Am. Chem. Soc. 2017, 139, 2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huppertsberg A., Kaps L., Zhong Z., Schmitt S., Stickdorn J., Deswarte K., Combes F., Czysch C., De Vrieze J., Kasmi S., Choteschovsky N., Klefenz A., Medina‐Montano C., Winterwerber P., Chen C., Bros M., Lienenklaus S., Sanders N. N., Koynov K., Schuppan D., Lambrecht B. N., David S. A., De Geest B. G., Nuhn L., J. Am. Chem. Soc. 2021, 143, 9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bixenmann L., Stickdorn J., Nuhn L., Polym. Chem. 2020, 11, 2441. [Google Scholar]
- 44. Van Driessche A., Kocere A., Everaert H., Nuhn L., Van Herck S., Griffiths G., Fenaroli F., De Geest B. G., Chem. Mater. 2018, 30, 8587. [Google Scholar]
- 45. Fleige E., Quadir M. A., Haag R., Adv. Drug Deliv. Rev. 2012, 64, 866. [DOI] [PubMed] [Google Scholar]
- 46. Jazani A. M., Oh J. K., Polym. Chem. 2020, 11, 2934. [Google Scholar]
- 47. Du J. Z., Li H. J., Wang J., Acc. Chem. Res. 2018, 51, 2848. [DOI] [PubMed] [Google Scholar]
- 48. Su S., Du F. S., Li Z. C., Org. Biomol. Chem. 2017, 15, 8384. [DOI] [PubMed] [Google Scholar]
- 49. Kang S., Kim Y., Song Y., Choi J. U., Park E., Choi W., Park J., Lee Y., Bioorgan. Med. Chem. Lett. 2014, 24, 2364. [DOI] [PubMed] [Google Scholar]
- 50. Lyu M., Yazdi M., Lin Y., Höhn M., Lächelt U., Wagner E., ACS Biomater. Sci. Eng. 2024, 10, 99. [DOI] [PubMed] [Google Scholar]
- 51. Wang Q., Wang C., Li S., Xiong Y., Wang H., Li Z., Wan J., Yang X., Li Z., Chem. Mater. 2022, 34, 2085. [Google Scholar]
- 52. Heck A. G., Stickdorn J., Rosenberger L. J., Scherger M., Woller J., Eigen K., Bros M., Grabbe S., Nuhn L., J. Am. Chem. Soc. 2023, 145, 27424. [DOI] [PubMed] [Google Scholar]
- 53. Heck A. G., Schwiertz D., Lantzberg B., Nguyen H.‐C., Forster R., Scherger M., Opatz T., Van Ginderacher J. A., Nuhn L., Eur. Polym. J. 2024, 214, 113150. [Google Scholar]
- 54. Jangra S., De Vrieze J., Choi A., Rathnasinghe R., Laghlali G., Uvyn A., Van Herck S., Nuhn L., Deswarte K., Zhong Z., Sanders N. N., Lienenklaus S., David S. A., Strohmeier S., Amanat F., Krammer F., Hammad H., Lambrecht B. N., Coughlan L., García‐Sastre A., De Geest B. G., Angew. Chem. – Int. Ed. 2021, 60, 9467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tieke B., Makromolekulare Chemie: Eine Einführung, Wiley‐VCH Verlag GmbH, Weinheim, Germany: 2014, pp. 1–391. [Google Scholar]
- 56. Rabanel J. M., Hildgen P., Banquy X., J. Controlled Release 2014, 185, 71. [DOI] [PubMed] [Google Scholar]
- 57. Fang Y., Xue J., Gao S., Lu A., Yang D., Jiang H., He Y., Shi K., Drug Deliv. 2017, 24, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shyh‐Dar L., Leaf H., J. Controlled Release 2011, 145, 178. [Google Scholar]
- 59. Palucka K., Banchereau J., Nat. Rev. Cancer 2013, 12, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Czysch C., Medina‐Montano C., Zhong Z., Fuchs A., Stickdorn J., Winterwerber P., Schmitt S., Deswarte K., Raabe M., Scherger M., Combes F., De Vrieze J., Kasmi S., Sandners N. N., Lienenklaus S., Koynov K., Räder H. J., Lambrecht B. N., David S. A., Bros M., Schild H., Grabbe S., De Geest B. G., Nuhn L., Adv. Funct. Mater. 2022, 32, 2203490. [Google Scholar]
- 61. Stickdorn J., Stein L., Arnold‐Schild D., Hahlbrock J., Medina‐Montano C., Bartneck J., Ziß T., Montermann E., Kappel C., Hobernik D., Haist M., Yurugi H., Raabe M., Best A., Rajalingam K., Radsak M. P., David S. A., Koynov K., Bros M., Grabbe S., Schild H., Nuhn L., ACS Nano 2022, 16, 4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stickdorn J., Czysch C., Medina‐Montano C., Stein L., Xu L., Scherger M., Schild H., Grabbe S., Nuhn L., Int. J. Mol. Sci. 2023, 24, 15417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lienenklaus S., Cornitescu M., Ziętara N., Łyszkiewicz M., Gekara N., Jabłońska J., Edenhofer F., Rajewsky K., Bruder D., Hafner M., Staeheli P., Weiss S., J. Immunol. 2009, 183, 3229. [DOI] [PubMed] [Google Scholar]
- 64. Scherger M., Pilger Y., Stickdorn J., Komforth P., Schmitt S., Koynov K., Räder H.‐J., Nuhn L., Biomacromolecules 2023, 24, 2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.