

ABSTRACT
A major factor limiting the biodegradation of organofluorine compounds has been highlighted as fluoride anion toxicity produced by defluorinating enzymes. Here, two highly active defluorinases with different activities were constitutively expressed in Pseudomonas putida ATCC 12633 to examine adaption to fluoride stress. Each strain was grown on α‐fluorophenylacetic acid as the sole carbon source via defluorination to mandelic acid, and each showed immediate fluoride release and delayed growth. Adaptive evolution was performed for each recombinant strain by serial transfer. Both strains adapted to show a much shorter lag and a higher growth yield. The observed adaptation occurred rapidly and reproducibly, within 50 generations each time. After adaption, growth with 50–70 mM α‐fluorophenylacetic acid was significantly faster with more fluoride release than a preadapted culture due to larger cell populations. Genomic sequencing of both pre‐ and postadapted strain pairs revealed decreases in the defluorinase gene content. With both defluorinases, adaption produced a 56%–57% decrease in the plasmid copy number. Additionally, during adaption of the strain expressing the faster defluorinase, two plasmids were present: the original and a derivative in which the defluorinase gene was deleted. An examination of the enzyme rates in the pathway suggested that the defluorinase rate was concurrently optimised for pathway flux and minimising fluoride toxicity. The rapid alteration of plasmid copy number and mutation was consistent with other studies on microbial responses to stresses such as antibiotics. The data presented here support the idea that fluoride stress is significant during the biodegradation of organofluorine compounds and suggest engineered strains will be under strong selective pressure to decrease fluoride stress.
Keywords: bacterium, biodegradation, defluorination, enzyme, fluoride stress, organofluorine, PFAS, Pseudomonas
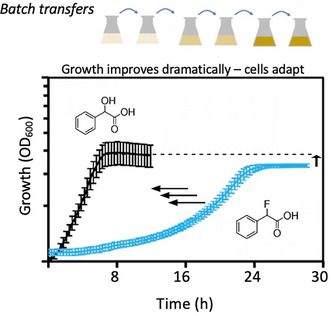
Fluoride is obligately generated during the degradation of organofluorine compounds and is highly cytotoxic. Biodegradation of significant levels of fluorinated compounds led to extensive phenotypic and genomic adaption.
1. Introduction
There is widespread interest in identifying or engineering microbes to biodegrade organofluorine compounds, the most problematic of which are multiply‐fluorinated chemicals known as PFAS. PFAS, per‐ and polyfluorinated alkyl substances, are widespread and persistent in the environment (Sznajder‐Katarzyńska, Surma, and Cieślik 2019; Glüge et al. 2020; Schymanski et al. 2023). Their persistence as a class is typically attributed to the strength of carbon–fluorine (C–F) bonds, but a more compelling reason may be fluoride stress acting as a strong negative selection (Wackett 2024). Fluoride is highly toxic and the biodegradation of anthropogenic fluorinated compounds generates unnaturally high levels of fluoride anion.
In natural environments, prokaryotes are largely exposed to fluoride extracted from minerals by water (McIlwain, Ruprecht, and Stockbridge 2021), but high levels only reach 15–20 ppm fluoride or 1 mM (Amini et al. 2008). The biodegradation of fluoroalkanes exposes Pseudomonas sp. 273 to 10 mM fluoride (Xie et al. 2020). The biodegradation of 2‐fluoropropionic acid by Pseudomonas putida ATCC 12633 (abbreviated throughout as Pp12633) was shown to generate 50 mM fluoride, an amount that led to 90% nonculturability (Dodge et al. 2024). In earlier studies, levels of intracellular fluoride as low as 0.1 mM have been shown to exert toxic effects (Marquis, Clock, and Mota‐Meira 2003; Qin et al. 2006).
The toxicity imposed by intracellular fluoride during biodegradation of C‐F compounds is acute, especially if there is a high metabolic flux with the C–F compound serving as the sole source of carbon. All enzymatic mechanisms of C–F cleavage release fluoride, since fluorine is the most electronegative element and will retain the electrons in the bond (O'Hagan 2008; Wackett 2022). To our knowledge, defluorinating enzymes studied to date are expressed intracellularly. Intracellular fluoride release is expected to impose a severe limitation on PFAS biodegradation. The manifestation of biodegradative‐induced toxicity was previously investigated with Pp12633 expressing a cytoplasmic hydrolytic defluorinase enzyme that reacted with α‐fluorophenylacetic acid (α‐FPhAA) and 2‐fluoropropionic acid. Both substrates were used as sole growth substrates, necessitating fluoride anion release intracellularly. Signs of fluoride toxicity included an extended lag phase and a lower growth yield compared to growth on the defluorinated alcohol that contained the same metabolic energy (Dodge et al. 2024). It would be expected that PFAS, which contain more fluorine substituents and less oxidisable carbon, would impose even more stress during biodegradation (Wackett 2024). In this context, mitigating fluoride stress is likely a major obstacle for natural and engineered microbes to degrade fluorinated molecules, particularly PFAS.
Adaption to toxic metabolites has been achieved with other Pseudomonas strains via laboratory evolution. For example, P. putida KT2440 was adapted to metabolise toxic p‐coumaric and ferulic acids by serial transfer, known as adaptive laboratory evolution (ALE; Mohamed et al. 2020). Adaptive evolution has also been used to enhance growth of P. putida S12 in the presence of high levels of the solvent toluene, which is also known to induce a stress response (Kusumawardhani et al. 2021). Here, we carried out adaptive evolution experiments with recombinant Pseudomonas strains, independently expressing two growth‐supporting defluorinases with different activities, to determine if and how they would adapt to better handle fluoride stress. With both defluorinases, we observed a rapid adaption to improved growth, characterised by a shorter entry into exponential growth and a higher final optical density with limited substrate concentrations. Genomic sequencing revealed that a major adaption was a lowering of the defluorinase gene copy number, which occurred by two different mechanisms. The findings indicate that engineering strains for the biodegradation of organofluorine compounds will require a balance among substrate defluorination rates, overall metabolic rates, and fluoride stress management mechanisms.
2. Experimental Procedures
2.1. Chemicals
Tris base, hydrogen chloride, sodium chloride and glacial acetic acid were purchased from ThermoFisher Scientific (Rockford, IL, USA) and racemic α‐FPhAA from Enamine (Kyiv, Ukraine). Kanamycin sulphate, HEPES and isopropyl‐beta‐D‐thiogalactoside (IPTG) were purchased from GoldBio (Saint Louis, MO, USA). All other chemicals were purchased from MilliporeSigma (Saint Louis, MO, USA). Racemic mandelic acid was used in all experiments.
2.2. Bacterial Strains, Plasmids and Growth Conditions
Pp12633 was purchased from the American Type Culture Collection (Manassas, VA, USA), and Escherichia coli NEB 5‐alpha and BL21(DE3) competent cells were purchased from New England Biolabs (Ipswitch, MA, USA). Plasmid pBBR1MCS‐2 (Kovach et al. 1995) was purchased from Addgene (Watertown, MA, USA). Kanamycin sulphate (50 mg/L) was used as the selection marker for the plasmids in all strains and was added to media when growing recombinant strains in all experiments or as noted. Cultures were grown on shaking platforms (200 rpm) in incubators at 37°C ( E. coli ) or 28°C (Pp12633) or as indicated. Miller lysogeny broth (LB) (BD Biosciences, Franklin Lakes, NJ, USA) was used to grow cells during cloning or for enzyme purifications. Growth experiments were conducted in 125‐mL baffled Erlenmeyer flasks with 10 mL aliquots of 10 mM mandelic acid or 20 mM α‐FPhAA (or as noted) as sole carbon and energy sources in mineral salts broth medium (MSB) (Stanier, Palleroni, and Doudoroff 1966) comprised of a 39 mM sodium/potassium phosphate buffer (pH 6.8), 1 g/L ammonium chloride as the sole nitrogen source, salts of metals and trace elements as described and vitamins added to the following working concentrations: 5 μg/L biotin, 0.5 mg/L nicotinic acid and 0.25 mg/L thiamine hydrochloride. Growth media were sterilised by autoclaving (LB) or by passing through sterile 0.2 μm polyethersulphone (PES) membrane filters (MSB).
2.3. Gene Cloning and Creation of Expression Strains
Reagents and enzymes used for cloning were from New England Biolabs. Creation of the Pp12633 strain that constitutively expressed the Delftia acidovorans (formerly Moraxella sp. strain B) strain B defluorinase (defluorinase one, DEF1) was described previously (Dodge et al. 2024). New gene constructs were synthesised by Twist Biosciences (San Francisco, CA, USA). The translated sequence of the gene encoding a known defluorinase from Dechloromonas aromatica RCB (GenBank Locus 8SDC_A and Protein Data Bank accession 8SDC) (Khusnutdinova et al. 2023) (defluorinase two, DEF2) was used to generate codon‐optimised gene sequences for expression in E. coli or P. putida (Figure S1) using the Twist BioSciences or Integrated DNA Technologies (IDT, Coralville, IA, USA) codon optimisation tools, respectively. The synthetic codon‐optimised gene for E. coli was inserted into a pET28a vector by the vendor so that a six‐histidine tag would be added to the N‐terminus of the expressed enzyme, and this construct was used to transform chemically competent E. coli BL21(DE3) cells to create the expression strain. The DEF2 gene optimised for P. putida was synthesised with the same 5′ and 3′ additions as previously described for the DEF1 gene (Dodge et al. 2024) (codons for six histidine residues, a start codon, a ribosomal binding site, the T5 promoter sequence and sequences complementary to the upstream and downstream vector arms) to create an expression cassette that could be inserted into the pBBR1‐MCS2 backbone using an NEB HiFi assembly kit. Plasmid pBBR1‐MCS2 was cut with restriction enzymes AgeI and NsiI, purified using a QIAquick PCR purification kit (QIAGEN, Hilden, Germany) and combined with the synthetic expression cassette in an assembly reaction per the manufacturer's instructions, which was then used to transform high‐efficiency NEB 5‐alpha competent E. coli cells. The plasmid was isolated from kanamycin‐resistant colonies using a QIAGEN QiaPrep Spin Miniprep kit and the entire sequence was verified by whole‐plasmid nanopore sequencing (Plasmidsaurus, Eugene, OR, USA). Purified plasmid DNA was then used to transform Pp12633 cells via electroporation as previously described (Choi, Kumar, and Schweizer 2006). Cells were grown overnight in LB to stationary phase, washed twice with 300 mM sucrose and then resuspended in 0.05 mL of 300 mM sucrose per ml of the harvested culture. Plasmid DNA (0.2 μg) was combined with 0.1 mL aliquots of concentrated cells and transferred into Fisher Scientific (Pittsburgh, PA, USA) 2‐mm‐gap electroporation cuvettes. Cells were pulsed at 2.5 kV in a Bio‐Rad (Hercules, CA, USA) MicroPulser electroporator, recovered from the cuvettes with 0.9 mL of SOC medium into 14 mL Falcon tubes (Corning, Reynosa, Mexico) and then incubated with shaking for 2 h. Selection for transformants was done by spreading recovered cells onto LB + kanamycin agar plates and incubating for 48 h.
2.4. Enzyme Purification and Activity Assays
DEF2 was purified essentially as previously described for DEF1 (Dodge et al. 2024) via immobilised‐metal affinity chromatography (IMAC) using a GE Healthcare (Cytiva, Marlborough, MA, USA) ÄKTA fast liquid protein chromatography (FPLC) system. To purify DEF2, a 1.5 L aliquot of LB + kanamycin in a 4‐L Erlenmeyer flask was inoculated to a starting OD600 of 0.1 with an overnight culture of the E. coli BL21(DE3) DEF2 expression strain grown in the same medium. The cells were incubated at 37°C until the OD600 reached 0.5, then the culture was cooled on a shaker at 16°C for 30 min and recombinant protein expression was induced by adding IPTG to 0.6 mM. The induced culture was incubated at 16°C for another 18 h, after which the cells were harvested by centrifugation at 4100 g for 15 min. Cell pellets were resuspended in 15 mL of buffer A (50 mM Tris–HCl + 0.2 M NaCl, pH 7.5) and lysed with a French pressure cell (three cycles at 140 MPa). The resulting crude lysate was centrifuged at 19,000 g for 90 min and the supernatant (cleared lysate) was decanted and injected onto a HisTrap HP 5‐mL column (Cytiva) charged with Ni2+. The column was washed with 98% buffer A and 2% buffer B (buffer A + 500 mM imidazole, pH 7.5) until unbound proteins were removed and the chromatogram returned to baseline. Proteins with low affinity were eluted from the column with 90% buffer A + 10% buffer B (50 mM imidazole), and then the bound defluorinase was eluted during a linear gradient increasing from 10% buffer B to 50% buffer B (250 mM imidazole). Purity of the eluted fractions was determined via SDS‐PAGE using a 12.5% polyacrylamide gel. Fractions of equivalent high purity were pooled, and the elution buffer was exchanged with 20 mM HEPES + 0.2 M NaCl, pH 7.5 and the protein was concentrated using an Amicon Ultra 30 kDa MWCO concentrator (MilliporeSigma). The purified DEF2 remained in solution during several concentration/dilution cycles and aliquots were frozen in liquid nitrogen and stored at −80°C at a concentration of 9 mg/mL as determined with the Bradford assay using the Bio‐Rad Protein Assay Dye Reagent Concentrate and a Pierce bovine serum albumin as standard (ThermoFisher).
The activity of the purified DEF2 enzyme on α‐FPhAA was determined using a fixed time point fluoride release assay, essentially as previously described for DEF1 (Dodge et al. 2024). Reactions were conducted with 1 μg enzyme added to 0.5 mL aliquots of 30 mM α‐FPhAA in 100 mM Tris buffer (pH 9.0) at 30°C. Independent triplicate reactions were stopped at each of four time points from 2.5 to 10.0 min by adding 0.5 mL of total ionic strength adjustment buffer (TISAB: 1 M glacial acetic acid, 1 M NaCl, 10 mM CDTA, pH 5) to each reaction. Total fluoride was then measured in each reaction with a fluoride ion electrode integrated with an Orion Star A214 pH/ISE meter (Thermo Fisher) that was calibrated with sodium fluoride standards. Net fluoride release by the enzyme was then determined by subtracting background fluoride measured in aliquots of the substrate solution that were incubated in parallel without enzyme added. Reaction rates were derived from the slope of a plot of the average net fluoride release at each time point versus incubation time (R 2 = 0.9995).
2.5. Adapting Defluorinase Strains to Growth on α‐FPhAA
Successive passaging in MSB + α‐FPhAA was performed with recombinant Pp12633 strains individually expressing either DEF1 or DEF2. The initial inocula were stationary‐phase cells that had been grown overnight in MSB + 25 mM mandelic acid from single colonies that were picked from LB + kanamycin plates. Aliquots of cells were harvested in a microcentrifuge (5 min at 8600 g ), washed twice and resuspended with carbon‐free MSB and then used to inoculate MSB + 20 mM α‐FPhAA to a starting OD600 = 0.1 as measured with a Beckman DU‐640 spectrophotometer (Beckman‐Coulter, Indianapolis, IN, USA). Transfer cultures were started by harvesting stationary phase cells as above, removing the supernatant and then adding the cells in the pellet to a fresh aliquot of MSB + α‐FPhAA medium in a new flask.
During incubation, readings were taken to determine the time to the first doubling of the initial OD600 (T d ' ) and then the OD600 at stationary phase. Cultures were transferred until these parameters were stable in several subsequent transfers. Experiments were conducted by transferring single cultures in five separate experiments with the DEF1 strain and two separate experiments with the DEF2 strain. One additional experiment was done with the DEF1 strain in which OD600 of triplicate cultures was monitored from inoculation through stationary phase in all transfers until a stable adapted phenotype was confirmed.
2.6. Growth Assays
Data to generate growth curves for unadapted (preadapted) and adapted (postadapted) strains were acquired using a Tecan (Männedorf, Switzerland) Infinite M Nano Plus shaking and heating UV–Vis microplate reader. Each strain was first grown overnight in shake flasks to stationary phase in MSB + 25 mM mandelic acid that was inoculated directly from frozen stocks. Cells were washed once and resuspended as above, and then cells of each strain were used to inoculate separate 5 × 0.2 mL aliquots of MSB + 10 mM mandelic acid or 20 mM α‐FPhAA in a Corning Costar (Kennebunk, ME, USA) 96‐well flat‐bottom cell culture plate to a starting OD600 = 0.1. All strains were grown separately on each carbon source. The plates were incubated at 30°C and OD600 measurements were taken in each well at 20 min intervals until all cultures reached stationary phase. Plates were shaken constantly between the OD600 readings at amplitude = 1 mm, with linear shaking for 197 s and then orbital shaking for 1000 s; conditions found to the favourable for growth on mandelic acid. Lag phase, maximum growth rate (μ max) and maximum OD600 were derived from the OD600 data with a linear regression fit model (R 2 = 0.95 to the raw data) using QurvE (Wirth et al. 2023).
To test maintenance of the lower T d ' and higher maximum OD600 phenotypes in the absence of fluoride, cells from a postadapted Pp12633 DEF1 strain were serially passaged in MSB + mandelic acid for seven successive transfers in duplicate. Cells were harvested from each transfer culture at stationary phase, transferred into MSB + α‐FPhAA and then T d ' and maximum OD600 were determined in the resulting duplicate cultures as above.
To further characterise fluoride tolerance in pre‐ and postadapted strains, cells were grown in triplicate in 50, 60 and 70 mM α‐FPhAA in MSB. OD600 measurements were taken during incubation using an Agilent (Santa Clara, CA, USA) Cary 60 UV–Vis spectrophotometer. Total fluoride was also measured with the fluoride electrode in culture supernatants that were combined 1:1 with TISAB as described above.
2.7. Genome Sequencing and Analysis
Combined Illumina and Nanopore genomic sequencing and assembly was done by SeqCenter (Pittsburgh, PA). Genomic DNA was isolated from cells of DEF1 or DEF2 preadapted strains and one each of the DEF1 or DEF2 postadapted strains using a QIAGEN DNeasy Blood and Tissue Kit. Cells were grown in MSB media + mandelic acid (preadapted) or α‐FPhAA (postadapted) inoculated directly from −80°C freezer stocks of each culture.
The Illumina sequencing reads were used to determine overall and relative sequencing coverage and to look for mutations in the chromosome or defluorinase expression plasmids. The Pp12633 genome (NCBI assembly ASM2450811v1) was used as the reference genome for all analyses. Fastp was used for trimming paired ends (v0.23.4) (Chen 2023). Bowtie2 (v2.5.2) (Langmead and Salzberg 2012) was used to create Sequence Alignment Map files by mapping forward and reverse reads to the indexed reference genome in an end‐to‐end fashion. SAMtools (v1.19) (Danecek et al. 2021) was used to compress the ‘.sam’ files into ‘.bam’ files. After compression, the sequence alignment maps were filtered to remove sequences with low alignment using a MapQ20 filter. Finally, the sequence alignment maps were sorted and indexed using SAMtools. Coverage analyses were done in RStudio (v2024.4.2.764) (RStudio Team 2020) using the software tools described below. The coverage of the sequence alignment maps was analysed using bamsignals (v1.34.0) in R (v4.3.3) (R Core Team 2021). gt (v0.10.1) (Pikounis and Oleynick 2013) was used to visualise data and SnapGene (McGuffie and Barrick 2021) was used to create plasmid maps.
The open‐source tool breseq (v0.35.0) (Deatherage and Barrick 2014) was used for mutation, deletion and junction identification. TrimGalore (v0.6.0) (Martin 2011) was used on Illumina short read sequences to trim adaptor sequences and ensure sequence quality prior to processing with breseq.
Plasmid populations in the pre‐ and postadapted strains were further characterised by whole‐plasmid DNA sequencing as described above and agarose gel electrophoresis. Cells were grown as described above for genomic DNA isolation and then plasmid DNA was isolated using the miniprep procedure described above. Separate plasmid aliquots were submitted for sequencing or were linearised by digestion with 20 U of restriction enzyme AgeI (one recognition site in each plasmid sequence) per μg of DNA for 1 h. Aliquots containing 300 ng of DNA from each digest were loaded into a 0.7% agarose gel and run at 95 V for 50 min in 1 × TAE buffer. DNA fragments in the gel were visualised via ethidium bromide staining with a LI‐COR Odyssey Fc Imager and Image Studio Lite Ver 5.2 (Lincoln, NE, USA).
3. Results
3.1. Expression of Two Different Defluorinases
The genes encoding two defluorinases were each synthesised and separately cloned into a moderate‐to‐high copy number plasmid with constitutive expression in Pp12633. One defluorinase, from a Delftia sp., had been described previously (Dodge et al. 2024) and will be referred to here as DEF1. The second defluorinase was from a Dechloromonas sp. and it was described previously (Khusnutdinova et al. 2023), but had never been tested with the fluorinated growth substrate used in this study, α‐FPhAA. The Dechloromonas enzyme is referred to here as DEF2. Both enzymes were purified to homogeneity (Figure S2) and assayed with α‐FPhAA. DEF1 showed a specific activity of 14.5 μmol/min per mg enzyme, giving an apparent k cat of 8 s−1 per subunit. DEF2 had a specific activity of 32 μmol/min per mg enzyme, giving an apparent k cat of 18 s−1 per subunit.
3.2. Defluorination With DEF1 and DEF2 In Vivo
Previously, when Pp12633 expressing DEF1 from a strong promoter on plasmid pBBR1 was grown on α‐FPhAA as the sole carbon source, growth inhibition was observed as a result of fluoride stress (Dodge et al. 2024). The recombinant Pp12633 strain carrying the plasmid was shown to grow rapidly on mandelic acid, the product of α‐FPhAA defluorination.
Here, Pp12633 was transformed with the same pBBR1 plasmid and the same promoter as used for DEF1 but carrying the DEF2 gene instead. The genes are nearly identical in size, 882 bp for DEF1 versus 891 bp for the DEF2 gene. Protein gels showed similar levels of expression of DEF1 and DEF2. Each recombinant strain grew overnight on mandelic acid similarly to the wild‐type, indicating that carrying the plasmids did not cause significant stress (Figure S3). Fluoride determinations showed release of fluoride into the medium within 1 h when the recombinant strains were grown on α‐FPhAA (Figure S4). Both strains showed signs of stress during growth, which had previously been demonstrated with DEF1 to be due to fluoride toxicity (Dodge et al. 2024). Most notably, the time to first doubling (T d' ) for the strain expressing DEF2 was ~15 h, similar to what had been observed previously with the DEF1 strain.
3.3. Adaptive Evolution by Serial Transfer on α‐FPhAA
Adaptive evolution was first conducted with Pp12633 expressing DEF1. The strain was grown in a minimal medium containing a growth‐limiting amount (20 mM) of α‐FPhAA as the sole carbon source and cells were transferred repeatedly into fresh medium after reaching stationary phase. Cultures were monitored for signs of adaption by tracking OD600 to identify changes in T d ' and maximum OD600.
The number of transfers, and hence generations, that gave rise to a shorter T d ' and a higher maximum OD600 was surprisingly small (Figure 1). The initial mandelate‐grown inoculum transferred into α‐FPhAA had a T d ' of 15 h and a maximum OD600 of 1.3. The next culture gave a significantly shorter T d ' of 5 h, but the maximum OD600 did not change. On several subsequent transfers, the T d ' decreased to 2 h and the maximum OD600 increased to > 1.5. While this was still lower than the observed maximum OD600 during growth on molar equivalents of mandelic acid, this suggested that postadapted strains can divert more carbon and energy from the defluorinated product into new biomass. Given the surprisingly low number of transfers that gave rise to the adapted phenotypes, the experiment with Pp12633 DEF1 was repeated five more times (Figure S5) and also conducted with Pp12633 DEF2 twice. The adaption was rapid in all cases. Further transfers, representing ~45 generations, revealed no further improvements in T d' or maximum OD600 (Figure S6).
FIGURE 1.
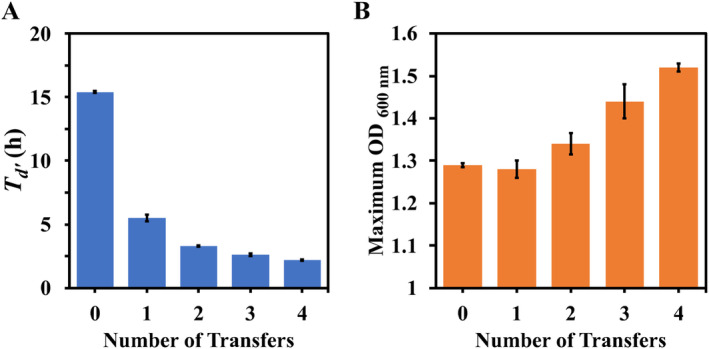
Emergence of (A) shorter T d ' and (B) higher maximum OD600 in successive transfer cultures of Pp12633 DEF1 growing on α‐FPhAA as the sole carbon and energy source. Transfer ‘0’ was started by inoculating α‐FPhAA medium with cells grown on mandelic acid as the sole carbon and energy source. Each transfer represented three to four generations. Further transfers did not lead to substantial changes in Td' or the maximum OD600. Error bars represent the standard errors of three replicate cultures.
We did observe that more transfers were required to attain the shortest T d ' with Pp12633 DEF2, ranging from 11 to 14 transfers, or ~50 generations, versus 4 transfers for Pp12633 DEF1, or ~15 generations. A second independent experiment adapting the DEF2 strain gave similar results. In another separate experiment, the adapted evolution of DEF1 was also conducted without kanamycin in the growth medium, but the results were essentially the same as those from the experiments conducted with kanamycin in the medium.
3.4. Growth Parameters for the Pre‐ Versus Postadapted Cultures
To more rigorously compare growth phenotypes in the postadapted vs. preadapted DEF1 and DEF2 strains, growth curves of all strains growing on α‐FPhAA were generated from quintuplicate cultures in a microwell plate (Figure 2). The curves clearly show the shortened lag phases, faster exponential growth rates and higher maximum OD600 attained in the postadapted versus preadapted strains. The increases in maximum OD600 in the postadapted versus preadapted DEF1 or DEF2 strains were similar: ~17% or ~18% higher, respectively, and in general agreement with the shake flask results above. More striking were the increases in the computationally determined maximum specific growth rates (μ max) in the postadapted versus preadapted strains. For the DEF1 strains, μ max = 0.168 h−1 preadaption and 0.242 h−1 postadaption (144% increase), whereas μ max in the DEF2 strains, increased from 0.126 h−1 preadaption to 0.377 h−1 postadaption (300% increase). While the total amounts of defluorinase in the separate cultures were not determined, the differences in μ max reflect the differences between the kinetic parameters of DEF1 and DEF2 as reported above. The higher catalysis rate of DEF2 may initially be detrimental to growth in preadapted cells due to faster fluoride release, as indicated by no observable increase in OD600 in the DEF2 preadapted strain from 0 to 5 h versus the visible increases apparent in the DEF1 preadapted strain (Figure 2).
FIGURE 2.
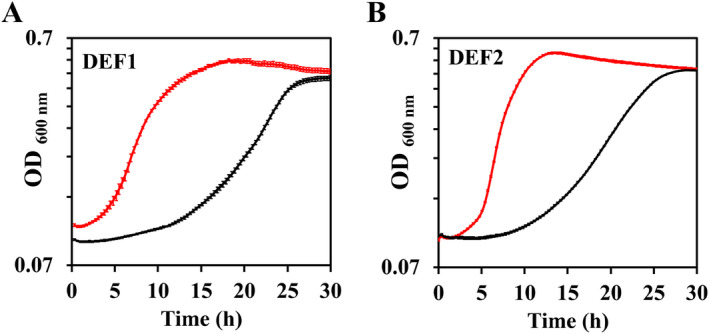
Growth curves of (A) DEF1 or (B) DEF2 preadapted (black) and postadapted (red) strains growing on α‐FPhAA as sole carbon and energy in a 96‐well microplate. Error bars represent the standard errors of five replicates.
3.5. Phenotypic Persistence After Serial Passage on Mandelic Acid
Given the rapid adaption during obligate growth of Pp12633 on α‐FPhAA, we reasoned that reversion to the preadapted phenotype might be similarly rapid if it was largely due to induction of fluoride stress mitigation functions. In that light, a postadapted DEF1 strain was serially passaged in minimal medium with mandelic acid as the sole carbon source for seven transfers. Stationary phase cells from each mandelic acid transfer culture were also transferred into medium with α‐FPhAA and then OD600 was monitored to determine T d ' and maximum OD600 (Figure 3). The initial T d ' of the postadapted culture on α‐FPhAA was ~2 h (Figure 3B). After the first four transfers, the T d ' increased marginally to 3 h but did not change in subsequent transfers. Another signature of the postadapted strain was the increased maximum OD600 of the cultures. This parameter also remained stable after repeated growth on mandelic acid and reintroduction into α‐FPhAA. If adaption was due solely to the induction of fluoride stress proteins (Calero, Gurdo, and Nikel 2022), reversion to the uninduced state would be predicted to occur when the stress was removed for repeated transfers and then the stress was reimposed. However, complete reversion was not observed; in fact, the adapted phenotype was largely retained. Therefore, other mechanisms likely contributed to the postadapted growth phenotypes observed in the DEF1 strain.
FIGURE 3.
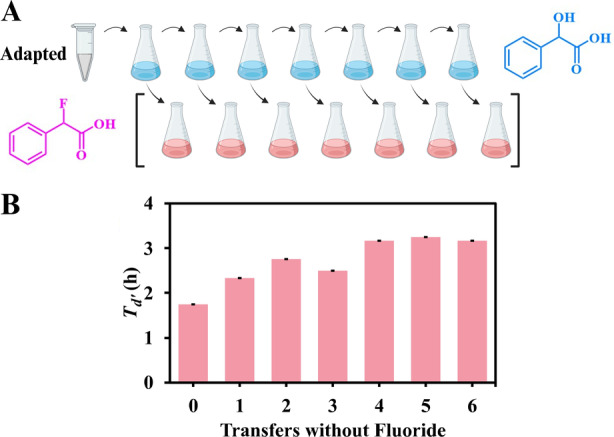
(A) Schematic showing the successive passaging of a postadapted Pp12633 DEF1 strain on mandelic acid (blue) as sole carbon and energy and the successive subculturing of each separate transfer of the mandelic acid‐grown cells on α‐FPhAA (pink) as sole carbon and energy. (B) T d' determined for each α‐FPhAA subculture that was inoculated with cells from each passage on mandelic acid. Maximum OD600 was also measured, but no changes were observed over the duration of the reversion experiment. Transfer ‘0’ was the postadapted Pp12633 DEF1 strain grown only on α‐FPhAA without passaging on mandelic acid and was the inoculum used to start the first passage on mandelic acid. Error bars represent the range of values from duplicate cultures.
3.6. Defluorination Activity and Growth at Higher Substrate Concentrations
In a previous study, growth of Pp12633 at high concentrations of the fluorinated substrate caused very high levels of stress due to high levels of fluoride released by the constitutive defluorinase enzyme (Dodge et al. 2024). In this study, the postadapted strain containing DEF1 grew faster (Table 1). Due to faster initial growth and metabolism, the adapted cell cultures degraded α‐FPhAA to release fluoride more extensively as compared to the preadapted strain. This was particularly acute at the highest level of substrate tested, 70 mM. At that concentration, the preadapted strain grew to a maximum OD600 ~ 1.0, whereas the postadapted strain grew to an OD of ~1.9. The postadapted strain also degraded almost 4× more substrate, as determined by fluoride release. Note that the α‐FPhAA is racemic and a previous study showed that only the (S)‐isomer is defluorinated. Therefore, the amounts of fluoride released by the postadapted strain at 50 mM and 60 mM substrate were essentially quantitative. A parallel experiment with the DEF2 strain gave similar results.
TABLE 1.
Comparing growth and fluoride release of pre‐ and postadapted DEF1 on higher α‐FPhAA concentrations.
| Strain | α‐FPhAA (mM) | Time to max OD600 (h) | Max cell density (OD600) | Fluoride released (mM) |
|---|---|---|---|---|
| Preadapted | 50 | 47 | 2.6 ± 0.1 | 23.7 ± 0.3 |
| Preadapted | 60 | 47 | 1.7 ± 0.2 | 23.0 ± 1.2 |
| Preadapted | 70 | 52 | 1.0 ± 0.1 | 7.8 ± 0.5 |
| Postadapted | 50 | 24 | 2.8 ± 0.1 | 24.3 ± 0.3 |
| Postadapted | 60 | 23 | 2.6 ± 0.1 | 31.0 ± 0.6 |
| Postadapted | 70 | 25 | 1.9 ± 0.0 | 30.0 ± 0.6 |
3.7. Plasmid and Chromosomal DNA Pre‐ and Postadaption
To discover genetic changes that were potentially responsible for the postadapted phenotypes, total DNA was extracted from pre‐ and postadapted strains expressing DEF1 or DEF2 and sequenced with combined short‐ and long‐read methods. The chromosomes and defluorinase expression plasmids were completely assembled for the four strains.
For the preadapted DEF1 strain, the average sequencing coverage of any individual nucleotide was 142× for the chromosome and 8492× for the defluorinase expression plasmid (Table S1). By taking the ratio of plasmid sequence coverage:chromosome sequence coverage, a plasmid copy number of ~60 per cell was inferred (Figure 4A). Plasmids with the pBBR1 origin have been reported to be maintained at a moderate‐to‐high copy number in P. putida KT2440 (Cook et al. 2018) and therefore our estimate of plasmid copy number based on relative sequencing coverage is unsurprising. Sequencing coverage for the postadapted DEF1 strain chromosome was 148×, similar to that of the preadapted strain. However, the sequence coverage of the postadapted DEF1 plasmid was only 3895× (Table S1), and the inferred plasmid copy for this strain was ~26 per cell or only 43% of the inferred plasmid copy number in the preadapted strain. The assembled plasmid sequences from the DEF1 pre‐ and postadapted strains were identical and no point mutations, deletions or insertions were present. Comparison of the chromosome sequences revealed several mutations in the postadapted strain (not present in the preadapted strain), most of which were in intergenic regions not identified as regulatory elements (Table S2). The only mutation considered to be of possible interest was a 12 bp deletion in ftsK. While ftsK is localised in the genome near some fluoride‐responsive genes, the gene itself has not been implicated in the fluoride stress response (Calero, Gurdo, and Nikel 2022). FtsK has been reported to mediate chromosomal separation in Pseudomonas strains, and other bacteria, but plasmid partitioning is not thought to be affected by FtsK (Massey et al. 2006).
FIGURE 4.
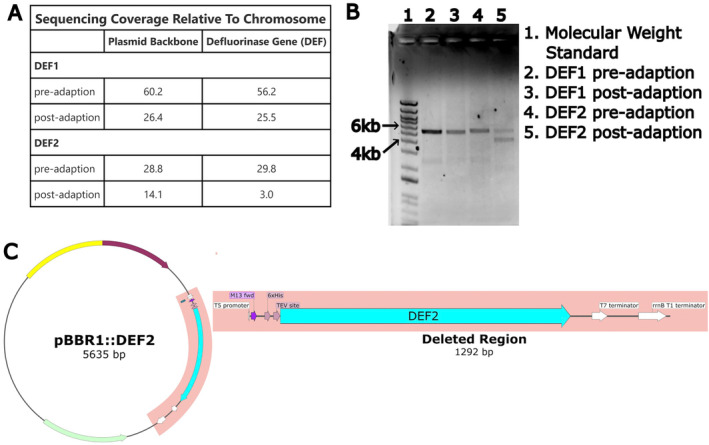
Analysis of DNA extracted from adapted strains of Pp12633 expressing either DEF1 or DEF2. (A) Sequencing coverage of plasmid and defluorinase genes relative to chromosome in preadapted and postadapted DEF1 and DEF2 strains. (B) Agarose gel of plasmids. (C) Schematic of plasmid in DEF2 strain showing region of deletion for the smaller plasmid.
A similar analysis of the DEF2 pre‐ and postadapted genomic data revealed a similar relative decline (49%) in plasmid reads:chromosome reads to that observed in the postadapted DEF1 strain above. However, the sequence coverage of the defluorinase gene in the DEF2 postadapted strain decreased by 90% relative to the DEF2 preadapted strain. Plasmid assembly revealed a mixed population of two plasmids in the postadapted strain that differed in size by 1292 bp. Aligning the two plasmid sequences revealed that the size difference was solely due to deletion of a 1292 bp fragment that included the complete DEF2 gene and T5 promoter sequences. No point mutations or other sequence differences were present in either plasmid. Agarose gel electrophoresis of postadapted DEF2 plasmid miniprep DNA that was linearised with a single restriction enzyme cut revealed two bands of 4–5 kb and 5–6 kb, respectively, whereas similarly, processed plasmid DNA from the preadapted DEF2 strain or the pre‐ or postadapted DEF1 strains had only one visible band of 5–6 kb on the gel (Figure 4B). These observations further support that the mixed plasmid population, implied by the genomic sequence assembly for the postadapted DEF2 strain and attributed to the deletion of a 1292 bp fragment from a portion of the plasmid population, was real and not a sequencing error or artefact (Figure 4C). Similar analysis of plasmid DNA from the second DEF2 postadapted strain showed only a single band of 5–6 kb on the gel (not shown), indicating that in this last adaption, evolution did not lead to a mixed plasmid population. This demonstrates stochasticity of the adaptive process and shows that the adaptive phenotypes can result from different mechanisms in the same strain.
4. Discussion
To our knowledge, ALE to enhance growth and biodegradation with organofluorine (C–F) compounds had not been conducted previously. A major highlight of this work is that efforts to bioengineer rapid biodegradation of C‐F compounds will likely incur strong negative selective pressure to titrate down defluorinase gene copy. In the present study, some level of gene maintenance was forced by using a minimal medium with α‐FPhAA as the sole carbon source. This also required the strains to catalyse C–F bond cleavage and release fluoride to grow. In eight separate ALE transfer series, adaption as shown by improved growth occurred rapidly, within 50 generations in all experiments. Genomic analysis showed a sharp reduction in defluorinase gene copy post‐adaption.
To better understand the adaptive advantage of the lower defluorinase gene copy, we compared k cat or apparent k cat values of the recombinant defluorinases, determined experimentally, with all enzymes in the mandelate catabolic pathway (Dewanti and Mitra 2003; Kaschabek et al. 2002; Matsumura et al. 2006; Nakai et al. 1980; Ngai, Ornston, and Kallen 1983; Ornston 1970; Polovnikova et al. 2003; Rivard et al. 2015; Whited et al. 1986; Yeh and Ornston 1981; Zahniser et al. 2017) (Table S3). The K M values were not available for some enzymes. It is difficult to know substrate concentrations inside the cell so the utility of K M values is more limited, but the k cat values were instructive to consider possible rate‐determining steps through the mandelate pathway. Figure 5 compares DEF1 with an apparent k cat of 8 s−1 and DEF2 with an apparent k cat of 18 s−1 with the enzymes in the upper part of the mandelate pathway. While the initial enzymes have higher rate constants, the k cat of benzoate 1,2‐dioxygenase is reported as 4 s−1 and that of benzoate 1,2‐dihydrodiol dehydrogenase as 0.1 s−1. With 56–60 copies of the defluorinase gene preadaption and being expressed from a strong promoter, the flux of defluorination was likely to be much higher than needed to drive metabolic flux through the pathway leading to acetyl‐CoA and succinyl‐CoA, which subsequently enter the TCA cycle (Figure 5). The significant lowering of the plasmid copy number, coupled with partial defluorinase gene deletion in the DEF2 strain, would be expected to lower the fluoride stress while not compromising the flux into TCA cycle intermediates.
FIGURE 5.
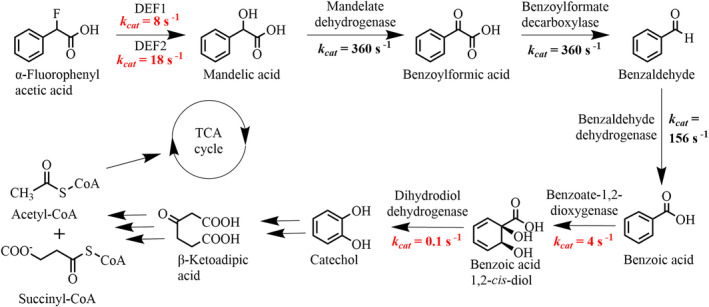
α‐FPhAA is metabolised by the recombinant P. putida as the sole source of carbon and energy via the mandelic acid pathway that feeds intermediate into the TCA cycle. It liberates toxic fluoride in the necessary first step. The reactions and relevant k cat values are highlighted. Key rate constants for the defluorinases and the slower steps of the benzoate 1,2‐dioxygenase and dihydrodiol dehydrogenase are shown in red. The k cat values for the defluorinases are apparent. They are derived from specific activity measurements and may slightly underestimate the true k cat . The mandelate racemase reaction and later steps catabolic are not shown for clarity. All available rate constants are available in the Supplemental Section.
The above explanation for the observed basis of the adaption relating to metabolic flux is also supported by studies with 13C‐mandelic acid and Pp12663 which had shown benzoic acid to be an accumulating metabolite (Halpin, Hegeman, and Kenyon 1981). Also, previous studies with P. putida KT2440 grown on benzyl alcohol led to benzoic acid accumulation as a metabolic intermediate (Rodríguez‐Verdugo, Vulin, and Ackermann 2019). This was consistent with the relatively low level of benzoate 1,2‐dioxygenase activity expected from the rate constants (Figure 5).
Natural evolution is thought to have occurred for the biodegradation of natural product organofluorine compounds such as fluoroacetate (Murphy, Schaffrath, and O'Hagan 2003; Chan et al. 2022). Fluorinated natural products are limited to ~20 and are uncommon in most environments (Walker and Chang 2014). In recent evolutionary history, humans have made more than 20 million fluorinated compounds (Schymanski et al. 2023). Thousands of anthropogenic fluorine compounds have entered commerce (Glüge et al. 2020). Indeed, more than 50% of new pesticides introduced over a 22‐year period contain one or more fluorine substituents (Ogawa et al. 2020). This tsunami of organofluorine entering the environment makes for new challenges for microorganisms (Stockbridge and Wackett 2024).
Microbes often evolve new enzymes rapidly to handle novel anthropogenic chemicals but fluorinated compounds pose a larger challenge (Copley 2009; Busch et al. 2023; Wackett 2024). In addition to evolving enzymes for defluorination, the release of fluoride will impose significant toxicity that will constrain gene maintenance. Given that most fluorinated molecules of concern contain multiple fluorine atoms, fluoride toxicity will be highly acute. This may pose a major challenge for evolving or engineering C‐F bond cleavage in nature or the laboratory.
Pp12633 adapted rapidly to α‐FPhAA, much more rapidly than others have observed adaption to use growth substrates (Mohamed et al. 2020), handle substrate stress (Kusumawardhani et al. 2021) or survive antibiotic stress (Wardell et al. 2019). In those cases, DNA sequencing was carried out to determine some of the features of adaptive evolution, and this was conducted here, too. It was initially perplexing to find no mutations in the defluorinase gene or regulatory elements, and no mutations in known fluoride stress response genes previously demonstrated in Pseudomonas (Calero, Gurdo, and Nikel 2022). However, both the data presented here, and more than a dozen papers on the mandelate pathway in Pp12633 support the idea that a significant reduction in defluorinase gene copy was necessary to bring metabolism into balance and mitigate fluoride toxicity.
A balanced metabolism in the adapted strains enabled both improved growth and more extensive α‐FPhAA biodegradation than unadapted strains (Figure 2 and Table 1). The results were somewhat counterintuitive since a lower copy number of the defluorinase gene gave more overall defluorination. However, slower fluoride release prompted shorter lag phases, higher growth rates and a higher maximum OD600, which led to greater α‐FPhAA biodegradation. These data highlight the importance of balanced metabolism, and rapid adaption to the balanced state could be readily achieved due to plasmid localisation of the defluorinase genes.
The results here are consistent with previous reports showing microbial adaptions can be driven by a change in plasmid copy number (Garoña et al. 2021; Ilhan et al. 2019; Rodríguez‐Beltrán et al. 2021). Moreover, copy number has been reported to decrease as a result of stress imposed on bacterial cells (Rouches et al. 2022; Wegrzyn and Wegrzyn 2002). This is also consistent with the observations here. Here, a lower plasmid copy number was driven by selective pressure to lower the flux of fluoride. In turn, this would allow native fluoride stress functions in Pseudomonas putida (Calero, Gurdo, and Nikel 2022) to better handle fluoride release, mitigate toxicity and allow for more efficient growth and metabolism.
5. Conclusions
In this study, bacteria engineered to grow on α‐FPhAA showed signs of significant stress, much of which was alleviated by adaptive evolution. The adaption was unexpectedly rapid. The mechanism underlying the observed rapid and reproducible adaption was an observed decrease in the defluorinase gene copy number. This was accomplished largely by decreasing the plasmid copy number, but gene deletion was also observed. The lowered fluoride flux provided fluoride stress mitigation without a decrease in carbon assimilation based on kinetic constants. Postadapted cells show superior growth and defluorination activity compared to preadapted cells. In total, this study suggested that engineering Pseudomonas and other bacteria to biodegrade organofluorine compounds at high rates will necessitate enhancing their tolerance to cytoplasmically released fluoride anion via either evolutionary or engineered mechanisms.
Author Contributions
Madeline R. O'Connor: formal analysis, investigation and visualisation. Calvin J. Thoma: formal analysis and investigation. Anthony G. Dodge: formal analysis and investigation. Lawrence P. Wackett: conceptualisation, writing and funding acquisition.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Appendix S1.
Acknowledgements
We thank Will Harcombe for helpful discussions. This research was supported by a grant from MnDRIVE Industry and the Environment Program and support from the United States National Science Foundation, grant MCB 2343831.
Funding: This work was supported by the United States National Science Foundation (MCB 2343831).
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article and the Supporting Information.
References
- Amini, M. , Mueller K. I. M., Abbaspour K. C., et al. 2008. “Statistical Modeling of Global Geogenic Fluoride Contamination in Groundwaters.” Environmental Science & Technology 42: 3662–3668. [DOI] [PubMed] [Google Scholar]
- Busch, M. R. , Drexler L., Mahato D. R., Hiefinger C., Osuna S., and Sterner R.. 2023. “Retracing the Rapid Evolution of an Herbicide‐Degrading Enzyme by Protein Engineering.” ACS Catalysis 13: 15558–15571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calero, P. , Gurdo N., and Nikel P. I.. 2022. “Role of the CrcB Transporter of Pseudomonas putida in the Multi‐Level Stress Response Elicited by Mineral Fluoride.” Environmental Microbiology 24: 5082–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, P. W. , Chakrabarti N., Ing C., et al. 2022. “Defluorination Capability of l‐2‐Haloacid Dehalogenases in the HAD‐Like Hydrolase Superfamily Correlates With Active Site Compactness.” Chembiochem 23: e202100414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S. 2023. “Ultrafast One‐Pass FASTQ Data Preprocessing, Quality Control, and Deduplication Using Fastp.” iMeta 2: e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, K. H. , Kumar A., and Schweizer H. P.. 2006. “A 10‐Min Method for Preparation of Highly Electrocompetent Pseudomonas aeruginosa Cells: Application for DNA Fragment Transfer Between Chromosomes and Plasmid Transformation.” Journal of Microbiological Methods 64: 391–397. [DOI] [PubMed] [Google Scholar]
- Cook, T. B. , Rand J. M., Nurani W., Courtney D. K., Liu S. A., and Pfleger B. F.. 2018. “Genetic Tools for Reliable Gene Expression and Recombineering in Pseudomonas putida .” Journal of Industrial Microbiology and Biotechnology 45: 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copley, S. D. 2009. “Evolution of Efficient Pathways for Degradation of Anthropogenic Chemicals.” Nature Chemical Biology 5: 559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Bonfield J. K., Liddle J., et al. 2021. “Twelve Years of SAMtools and BCFtools.” GigaScience 1: giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deatherage, D. E. , and Barrick J. E.. 2014. “Identification of Mutations in Laboratory‐Evolved Microbes From Next‐Generation Sequencing Data Using breseq .” Methods in Molecular Biology 1151: 165–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewanti, A. R. , and Mitra B.. 2003. “A Transient Intermediate in the Reaction Catalyzed by (S)‐mandelate Dehydrogenase From Pseudomonas putida .” Biochemistry 42: 12893–12901. [DOI] [PubMed] [Google Scholar]
- Dodge, A. G. , Thoma C., O'Connor M. R., and Wackett L. P.. 2024. “Recombinant Pseudomonas Growing on Non‐natural Fluorinated Substrates Shows Stress but Overall Tolerance to Cytoplasmically‐Released Fluoride Anion.” MBio 15: e0278523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garoña, A. , Hülter N. F., Romero‐Picazo D., and Dagan T.. 2021. “Segregational Drift Constrains the Evolutionary Rate of Prokaryotic Plasmids.” Molecular Biology and Evolution 38: 5610–5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glüge, J. , Scheringer M., Cousins I. T., et al. 2020. “An Overview of the Uses of Per‐and Polyfluoroalkyl Substances (PFAS).” Environmental Science: Processes & Impacts 22: 2345–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin, R. A. , Hegeman G. D., and Kenyon G. L.. 1981. “Carbon‐13 Nuclear Magnetic Resonance Studies of Mandelate Metabolism in Whole Bacterial Cells and in Isolated, In Vivo Cross‐Linked Enzyme Complexes.” Biochemistry 20: 1525–1533. [DOI] [PubMed] [Google Scholar]
- Ilhan, J. , Kupczok A., Woehle C., et al. 2019. “Segregational Drift and the Interplay Between Plasmid Copy Number and Evolvability.” Molecular Biology and Evolution 36: 472–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaschabek, S. R. , Kuhn B., Muller D., Schmidt E., and Reineke W.. 2002. “Degradation of Aromatics and Chloroaromatics by Pseudomonas Sp. Strain B13: Purification and Characterization of 3‐Oxoadipate:Succinyl‐Coenzyme A (CoA) Transferase and 3‐Oxoadipyl‐CoA Thiolase.” Journal of Bacteriology 184: 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khusnutdinova, A. N. , Batyrova K. A., Brown G., et al. 2023. “Structural Insights Into Hydrolytic Defluorination of Difluoroacetate by Microbial Fluoroacetate Dehalogenases.” FEBS Journal 290: 4966–4983. [DOI] [PubMed] [Google Scholar]
- Kovach, M. E. , Elzer P. H., Hill D. S., et al. 1995. “Four New Derivatives of the Broad‐Host‐Range Cloning Vector pBBR1MCS, Carrying Different Antibiotic‐Resistance Cassettes.” Gene 166: 175–176. [DOI] [PubMed] [Google Scholar]
- Kusumawardhani, H. , Furtwängler B., Blommestijn M., et al. 2021. “Adaptive Laboratory Evolution Restores Solvent Tolerance in Plasmid‐Cured Pseudomonas putida S12: A Molecular Analysis.” Applied and Environmental Microbiology 87: e0004121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , and Salzberg S. L.. 2012. “Fast Gapped‐Read Alignment With Bowtie 2.” Nature Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquis, R. E. , Clock S. A., and Mota‐Meira M.. 2003. “Fluoride and Organic Weak Acids as Modulators of Microbial Physiology.” FEMS Microbiology Reviews 26: 493–510. [DOI] [PubMed] [Google Scholar]
- Martin, M. 2011. “Cutadapt Removes Adapter Sequences From High‐Throughput Sequencing Reads.” EMBnet.Journal 17: 10–12. [Google Scholar]
- Massey, T. H. , Mercogliano C. P., Yates J., Sherratt D. J., and Löwe J.. 2006. “Double‐Stranded DNA Translocation: Structure and Mechanism of Hexameric FtsK.” Molecular Cell 23: 457–469. [DOI] [PubMed] [Google Scholar]
- Matsumura, E. , Sakai M., Hayashi K., Murakami S., Takenaka S., and Aoki K.. 2006. “Constitutive Expression of catABC Genes in the Aniline‐Assimilating Bacterium Rhodococcus Species AN‐22: Production, Purification, Characterization and Gene Analysis of CatA, CatB and CatC.” Biochemical Journal 393: 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffie, M. J. , and Barrick J. E.. 2021. “pLannotate: Engineered Plasmid Annotation.” Nucleic Acids Research 49: W516–W522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwain, B. C. , Ruprecht M. T., and Stockbridge R. B.. 2021. “Membrane Exporters of Fluoride Ion.” Annual Review of Biochemistry 90: 559–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed, E. T. , Werner A. Z., Salvachúa D., et al. 2020. “Adaptive Laboratory Evolution of Pseudomonas putida KT2440 Improves p‐Coumaric and Ferulic Acid Catabolism and Tolerance.” Metabolic Engineering Communications 11: e00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, C. D. , Schaffrath C., and O'Hagan D.. 2003. “Fluorinated Natural Products: The Biosynthesis of Fluoroacetate and 4‐Fluorothreonine in Streptomyces cattleya .” Chemosphere 52: 455–461. [DOI] [PubMed] [Google Scholar]
- Nakai, C. , Horiike K., Kuramitsu S., Kagamiyama H., and Nozaki M.. 1980. “Three Isoenzymes of Catechol 1,2‐Dioxygenase (Pyrocatechase) Alphaalpha, Alphabeta, and Betabeta From Pseudomonas arvilla C‐1.” Journal of Biological Chemistry 265: 660–665. [PubMed] [Google Scholar]
- Ngai, K. L. , Ornston L. N., and Kallen R. G.. 1983. “Enzymes of the Beta‐Ketoadipate Pathway in Pseudomonas putida : Kinetic and Magnetic Resonance Studies of the Cis,Cis‐Muconate Cycloisomerase Catalyzed Reaction.” Biochemistry 22: 5223–5230. [DOI] [PubMed] [Google Scholar]
- Ogawa, Y. , Tokunaga E., Kobayashi O., Hirai K., and Shibata N.. 2020. “Current Contributions of Organofluorine Compounds to the Agrochemical Industry.” Iscience 23: 101467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hagan, D. 2008. “Understanding Organofluorine Chemistry. An Introduction to the C‐F Bond.” Chemical Society Reviews 37: 308–319. [DOI] [PubMed] [Google Scholar]
- Ornston, L. N. 1970. “Conversion of Catechol and Protocatechuate to Beta‐Ketoadipate ( Pseudomonas putida ).” Methods in Enzymolology 17A: 529–549. [Google Scholar]
- Pikounis, B. , and Oleynick J.. 2013. “The Cg Package for Comparison of Groups.” Journal of Statistical Software 52: 1–27.23761062 [Google Scholar]
- Polovnikova, E. S. , McLeish M. J., Sergienko E. A., et al. 2003. “Structural and Kinetic Analysis of Catalysis by a Thiamin Diphosphate‐Dependent Enzyme, Benzoylformate Decarboxylase.” Biochemistry 42: 1820–1830. [DOI] [PubMed] [Google Scholar]
- Qin, J. , Chai G., Brewer J. M., Lovelace L. L., and Lebioda L.. 2006. “Fluoride Inhibition of Enolase: Crystal Structure and Thermodynamics.” Biochemistry 45: 793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . 2021. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing. https://www.R‐project.org/. [Google Scholar]
- Rivard, B. S. , Rogers M. S., Marell D. J., et al. 2015. “Rate‐Determining Attack on Substrate Precedes Rieske Cluster Oxidation During Cis‐Dihydroxylation by Benzoate Dioxygenase.” Biochemistry 54: 4652–4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez‐Beltrán, J. , DelaFuente J., Leon‐Sampedro R., MacLean R. C., and San Millan A.. 2021. “Beyond Horizontal Gene Transfer: The Role of Plasmids in Bacterial Evolution.” Nature Reviews Microbiology 19: 347–359. [DOI] [PubMed] [Google Scholar]
- Rodríguez‐Verdugo, A. , Vulin C., and Ackermann M.. 2019. “The Rate of Environmental Fluctuations Shapes Ecological Dynamics in a Two‐Species Microbial System.” Ecology Letters 22: 838–846. [DOI] [PubMed] [Google Scholar]
- Rouches, M. V. , Xu Y., Cortes L. B. G., and Lambert G.. 2022. “A Plasmid System With Tunable Copy Number.” Nature Communications 13: 3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RStudio Team . 2020. RStudio: Integrated Development for R. Boston, MA: RStudio, PBC. [Google Scholar]
- Schymanski, E. L. , Zhang J., Thiessen P. A., Chirsir P., Kondic T., and Bolton E. E.. 2023. “Per‐and Polyfluoroalkyl Substances (PFAS) in PubChem: 7 Million and Growing.” Environmental Science & Technology 57: 16918–16928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanier, R. Y. , Palleroni N. J., and Doudoroff M.. 1966. “The Aerobic Pseudomonads a Taxonomic Study.” Microbiology 43: 159–271. [DOI] [PubMed] [Google Scholar]
- Stockbridge, R. B. , and Wackett L. P.. 2024. “The Link Between Ancient Microbial Fluoride Resistance Mechanisms and Bioengineering Organofluorine Degradation or Synthesis.” Nature Communications 15: 4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sznajder‐Katarzyńska, K. , Surma M., and Cieślik I.. 2019. “A Review of Perfluoroalkyl Acids (PFAAs) in Terms of Sources, Applications, Human Exposure, Dietary Intake, Toxicity, Legal Regulation, and Methods of Determination.” Journal of Chemistry 2019: 2717528. [Google Scholar]
- Wackett, L. P. 2022. “Nothing Lasts Forever: Understanding Microbial Biodegradation of Polyfluorinated Compounds, Including PFAS.” Microbial Biotechnology 15: 773–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wackett, L. P. 2024. “Evolutionary Obstacles and Not C‐F Bond Strength Make PFAS Persistent.” Microbial Biotechnology 17: e14463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, M. C. , and Chang M. C.. 2014. “Natural and Engineered Biosynthesis of Fluorinated Natural Products.” Chemical Society Reviews 43: 6527–6536. [DOI] [PubMed] [Google Scholar]
- Wardell, S. J. , Rehman A., Martin L. W., Winstanley C., Patrick W. M., and Lamont I. L.. 2019. “A Large‐Scale Whole‐Genome Comparison Shows That Experimental Evolution in Response to Antibiotics Predicts Changes in Naturally Evolved Clinical Pseudomonas aeruginosa .” Antimicrobial Agents and Chemotherapy 63: 10–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegrzyn, G. , and Wegrzyn A.. 2002. “Stress Responses and Replication of Plasmids in Bacterial Cells.” Microbial Cell Factories 1: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whited, M. G. , McCombie W. R., Kwart L. D., and Gibson D. T.. 1986. “Identification of Cis‐Diols as Intermediates in the Oxidation of Aromatic Acids by a Strain of Pseudomonas putida That Contains a TOL Plasmid.” Journal of Bacteriology 166: 1028–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth, N. T. , Funk J., Donati S., and Nikel P. I.. 2023. “QurvE: User‐Friendly Software for the Analysis of Biological Growth and Fluorescence Data.” Nature Protocols 18: 2401–2403. [DOI] [PubMed] [Google Scholar]
- Xie, Y. , Chen G., May A. L., et al. 2020. “ Pseudomonas Sp. Strain 273 Degrades Fluorinated Alkanes.” Environmental Science & Technology 54: 14994–15003. [DOI] [PubMed] [Google Scholar]
- Yeh, W. K. , and Ornston L. N.. 1981. “Evolutionarily Homologous Alpha 2 Beta 2 Oligomeric Structures in Beta‐Ketoadipate Succinyl‐CoA Transferases From Acinetobacter calcoaceticus and Pseudomonas putida .” Journal of Biological Chemistry 256: 1565–1569. [PubMed] [Google Scholar]
- Zahniser, M. P. D. , Prasad S., Kneen M. M., et al. 2017. “Structure and Mechanism of Benzaldehyde Dehydrogenase From Pseudomonas putida ATCC 12633, a Member of the Class 3 Aldehyde Dehydrogenase Superfamily.” Protein Engineering 30: 273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1.
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article and the Supporting Information.