

Abstract
Acute myeloid leukemia (AML) is a devastating disease initiated and maintained by a rare subset of cells called leukemia stem cells (LSCs). LSCs are responsible for driving disease relapse, making the development of new therapeutic strategies to target LSCs urgently needed. The use of mass spectrometry–based metabolomics profiling has enabled the discovery of unique and targetable metabolic properties in LSCs. However, we do not have a comprehensive understanding of metabolite differences between LSCs and their normal counterparts, hematopoietic stem and progenitor cells (HSPCs). In this study, we used an unbiased mass spectrometry–based metabolomics analysis to define differences in metabolites between primary human LSCs and HSPCs, which revealed that LSCs have a distinct metabolome. Spermidine was the most enriched metabolite in LSCs compared with HSPCs. Pharmacological reduction of spermidine concentrations decreased LSC function but spared normal HSPCs. Polyamine depletion also decreased leukemic burden in patient-derived xenografts. Mechanistically, spermidine depletion induced LSC myeloid differentiation by decreasing eIF5A-dependent protein synthesis, resulting in reduced expression of a select subset of proteins. KAT7, a histone acetyltransferase, was one of the top candidates identified to be down-regulated by spermidine depletion. Overexpression of KAT7 partially rescued polyamine depletion–induced decreased colony-forming ability, demonstrating that loss of KAT7 is an essential part of the mechanism by which spermidine depletion targets AML clonogenic potential. Together, we identified and mechanistically dissected a metabolic vulnerability of LSCs that has the potential to be rapidly translated into clinical trials to improve outcomes for patients with AML.
INTRODUCTION
Acute myeloid leukemia (AML) is the most common acute leukemia in adults and has about a 30% 5-year survival rate (1). Most patients with AML who receive intensive therapy achieve a clinical response; however, a subset of cells called leukemia stem cells (LSCs) persist through treatment and ultimately drive disease relapse (2). To improve outcomes of patients with ML, it is critically important to develop new therapies to target the LSC population.
Dysregulated cellular metabolism is a hallmark of cancer and has been described as a critical vulnerability in LSCs (3, 4). The development of metabolomics analysis methods that use relatively low cell numbers (5, 6) has led to a rapid increase in our understanding of LSC metabolism. Metabolomics profiling of LSCs compared with the more mature leukemic blasts revealed that the metabolite abundances between these cell populations are distinct (7, 8), highlighting the importance of examining metabolism specifically in LSCs. AML cells harboring different genetic drivers also display unique metabolic characteristics (9–12), demonstrating that metabolic heterogeneity exists between patients with AML. Last, metabolite profiling of therapy-resistant LSCs uncovered metabolic pathways responsible for driving therapy resistance (9, 13, 14). However, we lack a comprehensive understanding of metabolite differences between LSCs and their healthy counterparts, hematopoietic stem and progenitor cells (HSPCs).
Given that the ideal therapy would target LSCs and not harm HSPCs, we sought to fill in this critical knowledge gap. Here, we measured metabolite abundance in LSCs and HSPCs enriched from human AML specimens and healthy bone marrow donors, which revealed a notable enrichment of the polyamine spermidine in LSCs. Polyamines (putrescine, spermidine, and spermine) are cationic compounds containing at least two amino groups that have pleiotropic effects on cellular function (15). Polyamines are important for the growth of several cancers, including neuroblastoma, glioma, melanoma, hepatocellular, colorectal, prostate, lung, lymphoma, and breast cancers (16–26). Spermidine metabolism has not been explored in LSCs. We demonstrate that spermidine is essential for enriched LSC populations by maintaining stemness through the selective regulation of protein synthesis. Overall, these data demonstrate that spermidine depletion represents a promising strategy to target LSCs.
RESULTS
Arginine is essential for LSC viability by mediating spermidine synthesis
To identify metabolic properties of LSCs that differ from HSPCs, we quantified metabolite abundance using an unbiased metabolomics analysis. We enriched LSCs from cryopreserved AML cells isolated from 18 patients and HSPCs from five normal bone marrow (NBM) specimens and performed mass spectrometry–based metabolomics analysis (Fig. 1A). LSCs were enriched using relative reactive oxygen species (ROS) abundance as previously described (27). HSPCs were enriched from NBM specimens using cluster of differentiation marker 34 (CD34). The use of ROS-low sorting enabled us to include a wider range of AML specimens, including those with CD34− LSCs, which have been shown to have different metabolic properties and response to metabolic targeting therapies (28, 29). Consistent with previous studies, ROS-low–sorted cells had a higher colony-forming potential compared with ROS-high cells, indicating an increase in stem and progenitor cells within the ROS-low compartment (fig. S1A). Our metabolomics analysis revealed that enriched LSCs and HSPCs displayed distinct metabolite profiles (Fig. 1B). To identify potential functional metabolic differences between enriched LSCs and HSPCs, we performed pathway analysis, which identified arginine biosynthesis as the most enriched pathway in ROS-low LSCs compared with CD34+ HSPCs (Fig. 1C). Arginine and proline metabolism was also among the most highly enriched pathways in ROS-low LSCs. Moreover, enriched LSCs displayed a higher arginine abundance compared with enriched HSPCs (Fig. 1D). Arginine has been previously shown to be important for AML survival (30). To determine whether arginine was essential for leukemic clonogenic potential, we cultured ROS-low–enriched LSCs in medium lacking arginine for 24 hours and then measured colony-forming potential by plating an equal number of viable cells in methylcellulose medium containing normal amounts of arginine (fig. S1B). Short-term arginine deprivation resulted in reduced colony-forming potential of all primary AML specimens examined (fig. S1C). In contrast, arginine deprivation did not affect the colony-forming potential of an NBM specimen (fig. S1C), suggesting that a therapeutic window exists to target arginine metabolism in enriched LSCs without harming HSPCs. Clinical trials evaluating safety and efficacy of arginine-depleting therapies in AML have been conducted (NCT02899286, NCT02732184, and NCT01910012) and, in contrast with preclinical studies, have shown limited efficacy (31). This is likely because cancer cells up-regulate arginine synthesis upon arginine depletion therapy (30, 32).
Fig. 1. Arginine metabolism is enriched in LSCs compared with HSPCs.
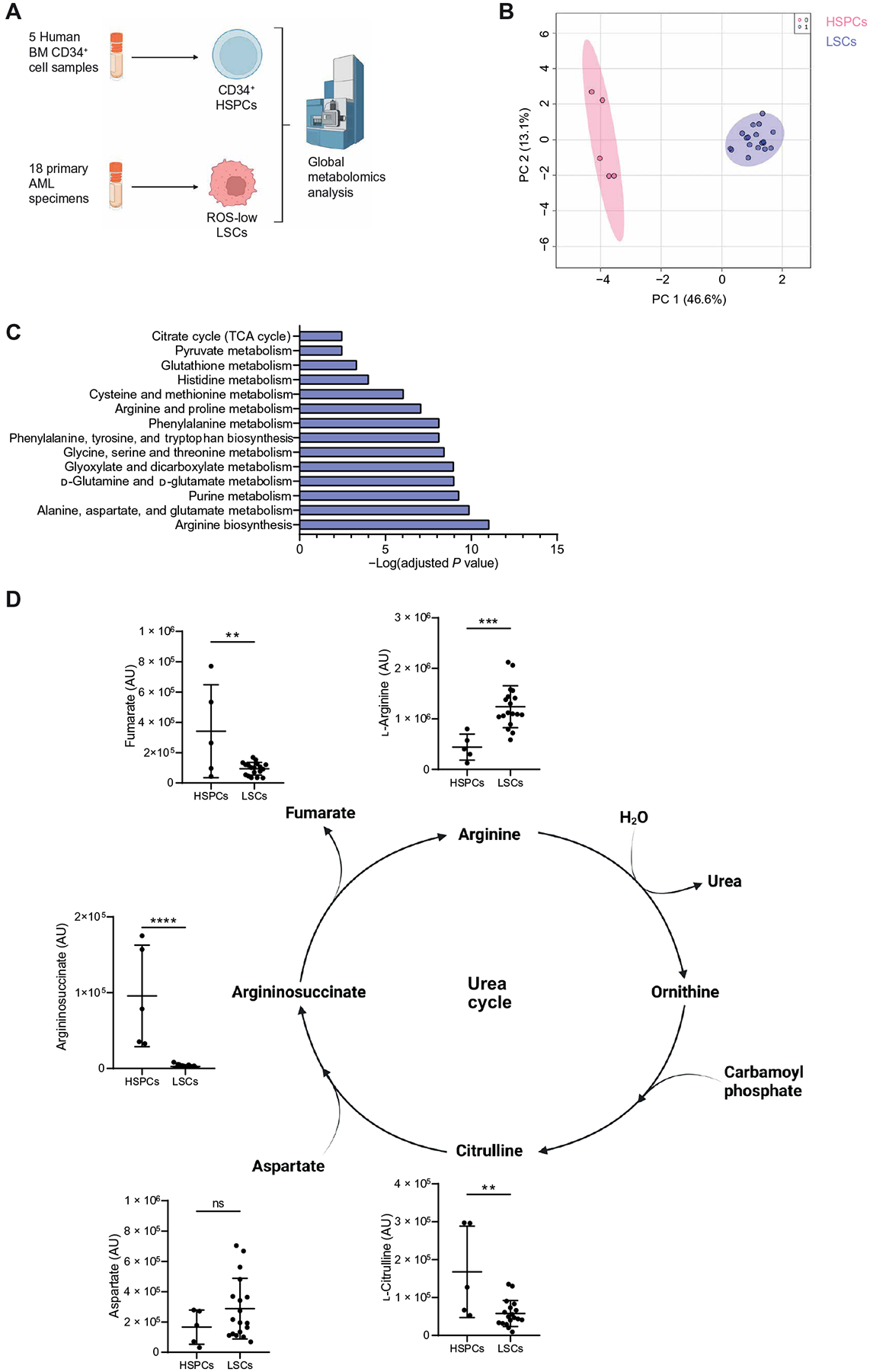
(A) Experimental design of metabolomics analysis. LSCs were enriched from cryopreserved AML specimens isolated from 18 patients with AML (AML1 to AML18) using relative ROS abundance. HSPCs were enriched using five CD34-enriched NBM samples. Metabolite abundance was determined by mass spectrometry. Created with BioRender.com. (B) Principal components (PC) analysis of ROS-low LSCs and CD34+ HSPCs based on metabolomics data. (C) Pathway analysis of metabolites enriched in ROS-low LSCs compared with CD34+ HSPCs determined using Metaboanalyst 4.0. (D) Metabolite abundance detected by steady-state metabolomics analysis in ROS-low LSCs and CD34+ HSPCs. Significance was determined using a paired t test. AU, arbitrary unit. All error bars represent SD. **P < 0.01, ***P < 0.005, and ****P < 0.001; ns, not significant. TCA, tricarboxylic acid.
To circumvent this compensatory mechanism, we sought to identify and target the downstream metabolic pathways supported by arginine metabolism in ROS-low–enriched LSCs. Arginine metabolism has been widely studied in cancer, including AML, and is commonly studied for its role in the urea cycle (30, 32). In contrast with that of arginine, the abundances of other urea cycle intermediates—such as citrulline, argininosuccinate, or fumarate—were lower in enriched LSCs compared with enriched HSPCs (Fig. 1D). Further, other amino acids, including aspartate, that contribute to the urea cycle had similar abundances between enriched LSCs and HSPCs, demonstrating that not all amino acids are elevated in ROS-low–enriched LSCs relative to CD34+-enriched HSPCs. These data suggest that the elevated arginine abundance observed in ROS-low–enriched LSCs may be important beyond contributions to the urea cycle. Therefore, we sought to identify which metabolic pathways arginine was used for in ROS-low–enriched LSCs by performing a stable isotope–labeled (SIL) tracing analysis using [13C6,15N4]–arginine (Fig. 2A). We observed that arginine was metabolized into the polyamines, putrescine, and spermidine in ROS-low–enriched LSCs (Fig. 2B). On the basis of these findings, we hypothesized that arginine is essential for LSC survival by mediating polyamine biosynthesis. To test this hypothesis, we supplemented arginine-depleted medium with the polyamines spermidine and spermine. The addition of spermidine but not spermine completely rescued the viability of ROS-low LSCs enriched from four AML patient specimens cultured in the absence of arginine, demonstrating that spermidine is required for enriched LSC survival (Fig. 2C).
Fig. 2. Arginine metabolism supports spermidine biosynthesis in LSCs.
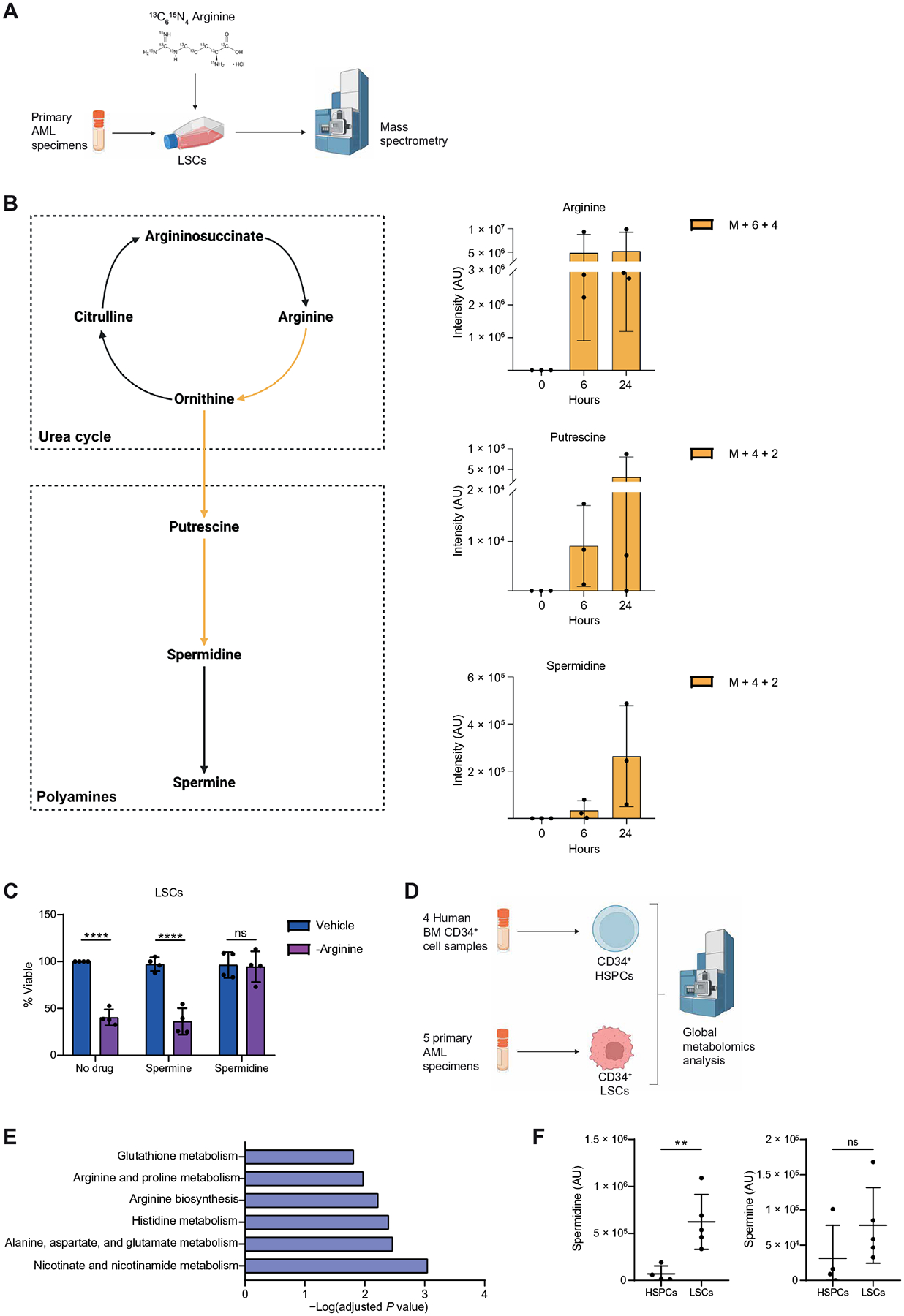
(A) Experimental design of stable isotope tracing experiments. [13C615N4] Arginine was pulsed into ROS-low LSCs enriched from three primary samples (AML27 to AML29) for 6 or 24 hours. Enrichment of 13C and 15N was determined by mass spectrometry. Created with BioRender.com. (B) Graphs show enrichment of heavy atoms (13C and 15N) from the stable isotope from arginine into putrescine and spermidine in ROS-low LSCs. M + 6 + 4 indicates metabolites with six 13C and four 15N atoms. M + 4 + 2 indicates metabolites with four 13C and two 15N atoms. Schematic created with BioRender.com. (C) Viability of LSCs enriched from four primary samples (AML27 and AML30 to AML32) cultured in arginine-depleted medium ±10 μM spermidine, 10 μM spermine, or their combination for 24 hours. Statistical significance was determined using two-way ANOVA. (D) Experimental design of metabolomics analysis. Created with BioRender.com. LSCs were enriched from five primary AMLs (AML6, AML19, and AML33 to AML35) on the basis of CD34+ expression. HSPCs were enriched using four CD34-enriched NBM samples. Metabolite abundance was determined by mass spectrometry. (E) Pathway analysis of metabolites enriched in CD34+ LSCs compared with CD34+ HSPCs determined using Metaboanalyst 4.0. (F) Spermidine and spermine abundance in CD34+ LSCs and CD34+ HSPCs. Significance was determined using a paired t test. All error bars represent SD. **P < 0.01 and ****P < 0.001; ns, not significant.
Last, we sought to validate metabolic changes in LSCs through a second metabolomics characterization in which both LSCs and HSPCs were enriched on the basis of CD34+ expression (Fig. 2D). This validation experiment was critical because it allowed us to verify that our findings were not an artifact of the ROS-low–sorting approach. Arginine biosynthesis and arginine and proline metabolism were once again among the top differentially abundant pathways in CD34+ LSCs compared with CD34+ HSPCs (Fig. 2E). Further, spermidine was the most enriched metabolite in CD34+ LSCs compared with CD34+ HSPCs (Fig. 2F). Consistent with the polyamine rescue experiments (Fig. 2C), spermine was not elevated in enriched LSCs compared to HSPCs (Fig. 2F). Overall, these data suggest that elevated arginine metabolism and its downstream metabolite spermidine are increased in enriched LSCs compared with enriched HSPCs and are required for LSC survival.
Polyamine depletion targets LSC function
Polyamine metabolism has been explored as a therapeutic target in several cancer types (16–26); however, the role of this pathway in LSCs has yet to be determined. Approaches to target polyamine metabolism include reducing polyamine biosynthesis and uptake (17) and inducing polyamine export (33). Polyamine export is regulated by spermidine/spermine N1-acetyltransferase 1 (SAT1), an enzyme that acetylates spermidine and spermine, resulting in their cellular export (33). We observed decreased expression of SAT1 in primary AML samples compared with NBM specimens and in enriched LSCs compared with enriched HSPCs, suggesting that AML cells may have reduced baseline polyamine export compared with HSPCs (Fig. 3, A and B). This may contribute to the higher spermidine content in enriched LSCs. Therefore, we first determined the consequences of elevating polyamine export in AML using the SAT1 inducer N1,N11-diethylnorspermine (DENSpm). As expected, we observed the overexpression of SAT1 at mRNA and protein in Molm13 and MV4;11 AML cell lines and enriched LSCs as early as 24 hours after DENSpm treatment (Fig. 3, C and D, and fig. S2, A and B). DENSpm treatment resulted in decreased total polyamines in AML cell lines (fig. S2C) and reduced spermidine in Molm13 cells (fig. S2D), primary AML specimens (Fig. 3E), enriched LSCs (fig. S2, E and F), and CD34+ NBM (fig. S2G), demonstrating that DENSpm treatment is effectively targeting spermidine abundance in both normal and AML cells. As expected, spermine abundance was also decreased in AML cell lines and showed a trend toward a decrease in primary AML specimens, enriched LSCs, and NBM (Fig. 3F and fig. S2, D to G). Last, we measured DENSpm abundance in AML cell lines, primary specimens, enriched LSCs, and normal CD34+ bone marrow (BM) cells, which revealed that DENSpm was imported into all of the different cells analyzed (fig. S2, D to H). Next, we interrogated the consequences of polyamine depletion using DENSpm on other enzymes involved in polyamine metabolism. In contrast with SAT1, we observed no changes in spermine oxidase (SMOX) or polyamine oxidase (PAOX) upon polyamine depletion (fig. S2I). We also evaluated a complementary approach to decrease polyamine abundance in AML cells by inhibiting polyamine biosynthesis and cellular import. Pharmacological inhibition of the rate-limiting enzyme in polyamine biosynthesis, ornithine decarboxylase 1 (ODC1), using difluoromethylornithine (DFMO) has shown promise in preclinical cancer models (17, 23) but lacked cytotoxic effects in clinical trials (21, 34, 35), partly because of cancer cells’ ability to take up polyamines from the microenvironment (17). Therefore, preclinical studies have combined DFMO with AMXT 1501, an inhibitor of SLC3A2, a polyamine transporter. DFMO + AMXT 1501 has shown promising results in preclinical models of pontine glioma (17), which led to the initiation of ongoing clinical trials (NCT03536728 and NCT05500508). DFMO + AMXT 1501 reduced spermidine but not spermine in primary AML specimens (Fig. 3, E and F). Consistent with previous findings, the combination of DFMO + AMXT 1501 reduced spermidine to a greater extent than DFMO or AMXT 1501 as single agents.
Fig. 3. Spermidine metabolism is essential for LSC survival.
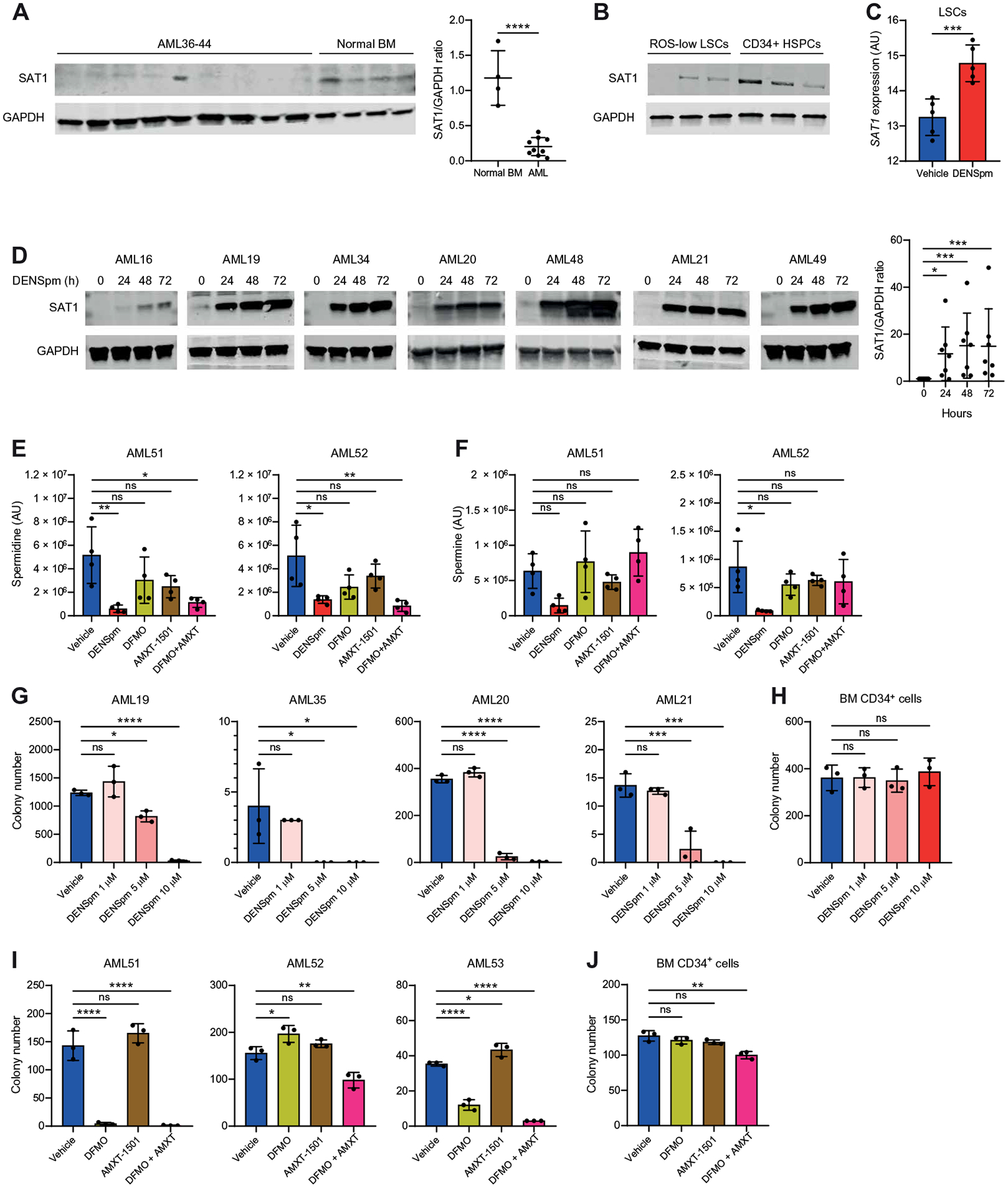
(A) Expression of SAT1 determined by Western blot in nine primary AML specimens (AML36 to AML44) and four NBM specimens. Significance was determined using an unpaired t test. (B) Expression of SAT1 protein was compared by Western blot in ROS-low LSCs from AML19, AML20, and AML35 relative to CD34+-enriched HSPCs. (C) RNA expression from CD34+ HSPCs enriched using four NBM samples and in ROS-low LSCs enriched from five primary AML specimens (AML28, AML29, and AML45 to AML47). HSPCs and LSCs were incubated for 24 hours with 10 μM DENSpm or vehicle. Significance was determined using an unpaired t test. (D) Expression of SAT1 determined by Western blot in seven primary AML specimens (AML16, AML19 to AML21, AML34, AML48, and AML49) incubated for 0, 24, 48, or 72 hours with 10 μM DENSpm. Significance was determined using an unpaired Mann-Whitney test. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) control for AML21 was also used in Fig. 7D, and GAPDH controls for AML16, AML19, AML34, and AML49 were also used in fig. S8B. (E) Spermidine abundance was determined by mass spectrometry in primary AML specimens (AML51 and AML52) incubated for 24 hours with 10 μM DENSpm, 2.5 mM DFMO, 400 nM AMXT-1501, the combination of DFMO + AMXT 1501, or vehicle. Significance was determined using two-way ANOVA. (F) Spermine abundance determined by mass spectrometry in primary AML specimens (AML51 and AML52) incubated for 24 hours with 10 μM DENSpm, 2.5 mM DFMO, 400 nM AMXT-1501, the combination of DFMO + AMXT 1501, or vehicle. Significance was determined using two-way ANOVA. (G) Colony-forming ability of four bulk primary AML specimens (AML19 to AML21 and AML35) after 24-hour incubation with the indicated doses of DENSpm or vehicle. Colony numbers were determined after 1 to 3 weeks. Statistical significance was determined using two-way ANOVA. (H) Colony-forming ability of one CD34-enriched BM sample after 24-hour incubation with the indicated doses of DENSpm or vehicle. Colony numbers were determined after 2 weeks. Statistical significance was determined using two-way ANOVA. (I) Colony-forming ability of three bulk primary AML specimens (AML51 to AML53) after 24-hour incubation with 2.5 mM DFMO, 400 nM AMXT 1501, the combination, or vehicle. Colony numbers were determined after 1 to 3 weeks. Statistical significance was determined using two-way ANOVA. (J) Colony-forming ability of one CD34-enriched BM sample after 24-hour incubation with 2.5 mM DFMO, 400 nM AMXT 1501, the combination, or vehicle. Colony numbers were determined after 2 weeks. Statistical significance was determined using two-way ANOVA. All error bars represent SD. *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001.
To establish whether polyamines are essential in AML, we evaluated the impact of polyamine depletion on the colony-forming potential of AML cells and LSCs using DENSpm and DFMO + AMXT 1501. Polyamine depletion using DENSpm decreased the colony-forming potential of Molm13 and MV4;11 cells (fig. S3A). Next, we pretreated primary AML specimens and NBM with DENSpm or DFMO + AMXT 1501 for 24 hours before plating in a methylcellulose medium that did not contain the compounds and evaluated colony numbers 2 to 3 weeks after plating. Polyamine depletion with either DENSpm or DFMO + AMXT 1501 decreased colony-forming potential of all AML specimens evaluated but had only a minimal impact on colony-forming potential of NBM (Fig. 3, G to J, and fig. S3B). We observed no decrease in viability upon DENSpm or DFMO + AMTX 1501 pretreatment (fig. S3C), demonstrating that the change in colony numbers was not because of cell death at the time of plating. Last, we measured the consequences of polyamine depletion using DENSpm on the colony-forming ability of enriched LSCs. LSCs enriched using ROS-low or CD34+ sorting had a decrease in colony-forming potential upon polyamine depletion similar to that of bulk AML specimens (fig. S3, D and E), further supporting the concept that polyamine depletion targets LSCs. Overall, these data point to a potential therapeutic window to target polyamines in AML.
To determine whether the consequences of DENSpm and DFMO + AMXT 1501 were due to decreased polyamines, we measured polyamine abundance and colony-forming potential upon the addition of exogenous spermidine. Using three additional AML specimens, we observed a decrease in spermidine upon DENSpm and DFMO + AMXT 1501, which was rescued by the addition of exogenous spermidine (fig. S4A). This resulted in the rescue of colony-forming ability in each of the three primary AML specimens (fig. S4B). The addition of spermidine did not interfere with the mechanism of action of DFMO + AMXT 1501. However, it is possible that spermidine competes with DENSpm for cellular import. Therefore, we measured DENSpm abundance upon spermidine supplement. DENSpm was still detected, albeit at a lower abundance, when compared with non–spermidine-exposed cells (fig. S4C). Exogenous spermidine treatment rescued both spermidine abundance and colony-forming potential of primary AML specimens, suggesting that the polyamine-depleting agents are targeting AML cells through reduced polyamine abundance (fig. S4, A and B). The addition of spermidine to culture medium in the presence of bovine serum results in spermidine oxidation by a bovine serum amine oxidase (36). Therefore, we investigated whether spermidine supplementation still rescued the colony-forming ability of AML cells in the presence of amine oxidase inhibitor aminoguanidine. The addition of spermidine in the presence of aminoguanidine at 50 or 500 μM still resulted in the normalization of the colony-forming ability of DENSpm-treated Molm13 cells (fig. S4D). Together with our mass spectrometry data, which show that addition of spermidine to the culture medium led to a notable increase in spermidine (fig. S4A), our results suggest that the effects observed after spermidine supplementation are not due to metabolization of spermidine by diamine oxidase but to the rescue of spermidine abundance.
Next, we sought to determine the consequences of polyamine depletion on LSC and HSPC function, which is measured by quantifying engraftment of human cells into immunodeficient mice (37). For these assays, AML and NBM specimens were transplanted into NOD-scid IL2Rgnull-3/GM/SF (NSG-SGM3) mice after ex vivo treatment with DENSpm (Fig. 4A). Polyamine depletion resulted in decreased engraftment for eight of the nine primary AML specimens tested ex vivo (Fig. 4B). Six of the nine AML specimens examined, including all responding specimens, were enriched from patients with AML who had relapsed on conventional cytotoxic chemotherapy (data file S1). Further, we evaluated two AML specimens for venetoclax sensitivity and observed that both specimens that were sensitive to DENSpm treatment were resistant to venetoclax (fig. S5A). These data demonstrate that polyamine depletion targets LSC function even in primary AML specimens that do not respond to, or have previously failed on, commonly used AML therapies. Spermidine depletion did not impair the engraftment or lineage output of NBM cells isolated from three healthy donors (Fig. 4C and fig. S5B). The most rigorous examination of LSC function is serial engraftment assays. Therefore, to confirm that polyamine depletion was targeting functional LSCs, we performed secondary engraftment using AML54. For this experiment, we transplanted an equal number of viable cells from primary recipient mice into secondary recipient mice and measured engraftment by flow cytometry. This experiment revealed a decrease in secondary engraftment potential in the DENSpm-treated condition (Fig. 4D), demonstrating a reduction in LSC function upon polyamine depletion. Last, we examined whether DENSpm reduced the function of enriched LSC populations. We enriched LSCs using ROS-low sorting; cells were then treated ex vivo with DENSpm and transplanted into immunedeficient mice, and engraftment was measured by flow cytometry (Fig. 4E). DENSpm treatment resulted in reduced engraftment of enriched LSCs (Fig. 4F), demonstrating that polyamine depletion decreases LSC function. Overall, these data demonstrate that polyamine depletion targets functionally defined LSCs while sparing normal HSPCs.
Fig. 4. Polyamine depletion targets functional LSCs.
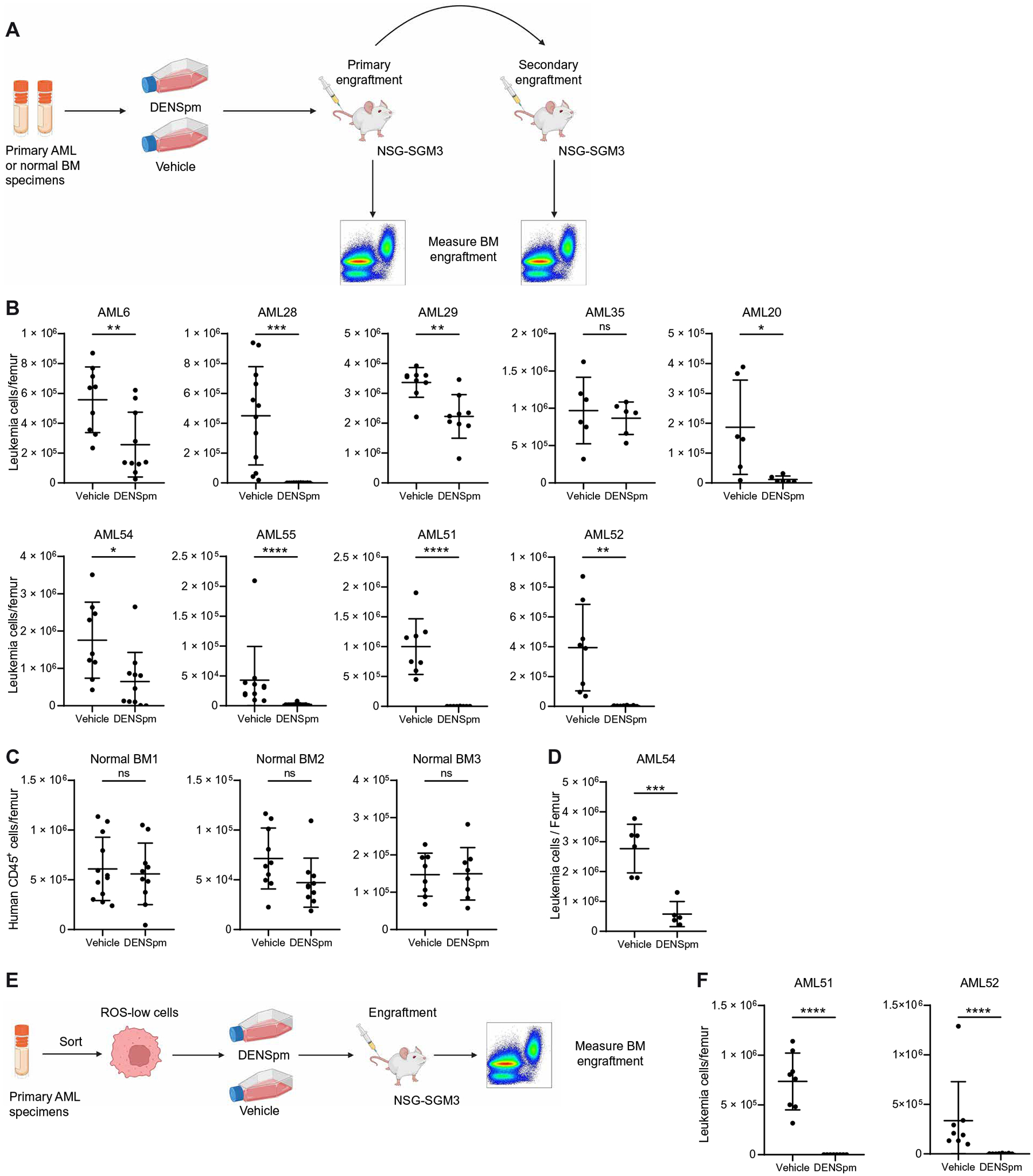
(A) Experimental design of engraftment assays. Nine primary AML specimens (AML6, AML20, AML28, AML29, AML35, AML51, AML52, AML54, and AML55) and three NBM specimens were incubated for 72 hours with 10 μM DENSpm or vehicle before injection into NSG-SGM3 mice. Created with BioRender.com. (B) Engraftment of nine primary AML specimens in NSG-SGM3 mice after ex vivo treatment with DENSpm or vehicle. Each point represents a single mouse. Statistical significance was determined using an unpaired t test (AML28, AML35, and AML52) or using an unpaired Mann-Whitney test (AML6, AML29, AML20, AML54, AML55, and AML51). Vehicle condition for AML20 is also used in fig. S5A. (C) Engraftment of three NBM specimens in NSG-SGM3 mice after ex vivo treatment with DENSpm or vehicle. Each point represents a single mouse. Statistical significance was determined using an unpaired t test. (D) Engraftment of one primary AML specimen (AML54) into secondary NSG-SGM3 recipient mice after ex vivo treatment with DENSpm or vehicle. Each point represents a single mouse. Statistical significance was determined using an unpaired t test. Schematic created with BioRender.com. (E) Experimental design of engraftment assays on ROS-low–enriched LSCs. (F) Engraftment of ROS-low LSCs enriched from two primary AML specimens (AML51 to AML52) in NSG-SGM3 mice after ex vivo treatment with DENSpm or vehicle. Each point represents a single mouse. Statistical significance was determined using an unpaired t test (AML51) or an unpaired Mann-Whitney test (AML52). All error bars represent SD. *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001.
Polyamine depletion promotes LSC differentiation
Polyamines have pleiotropic effects on cellular function (15, 33). To begin to understand the molecular mechanisms by which polyamines regulate LSC function, we enriched LSCs (defined as ROS-low) from five primary AML specimens and HSPCs (defined as CD34+) from three NBM specimens and performed RNA sequencing (RNA-seq) (Fig. 5A). Gene set enrichment analysis (GSEA) revealed an enrichment of hematopoietic cell differentiation signatures, including in the myeloid lineage in DENSpm-treated ROS-low LSCs, suggesting that polyamine depletion is associated with increased LSC differentiation (Fig. 5B). The same gene signatures were not enriched in DENSpm-treated CD34+ HSPCs, suggesting that polyamine depletion does not affect normal HSPC differentiation (Fig. 5B). To confirm that polyamine depletion induced myeloid cell differentiation in AML, we incubated Molm13 and MV4;11 cells with a low dose of DENSpm for 24 hours, performed colony-forming assays, and measured the differentiation status of the colonies by assessing CD15 and CD11b myeloid-associated marker expression. We observed increased expression of CD15 but not CD11b when Molm13 and MV4;11 cells were treated with DENSpm compared with vehicle (fig. S6A). We then examined the effect of polyamine depletion in primary AML differentiation. Consistent with the cell line data, primary AML specimens had increased CD15 or CD11b expression, confirming that AML differentiation results from polyamine depletion (Fig. 5C). To determine whether enriched LSCs differentiated upon polyamine depletion, we measured CD11b and CD15 expression in ROS-low and CD34+-enriched LSC populations upon colony formation as described above. In enriched LSCs, we observed an increase in CD11b or CD15, consistent with the increased myeloid markers observed in bulk samples (Fig. 5, D and E). Together, these data support that polyamine depletion results in LSC differentiation.
Fig. 5. Polyamine depletion induces myeloid differentiation in LSCs.
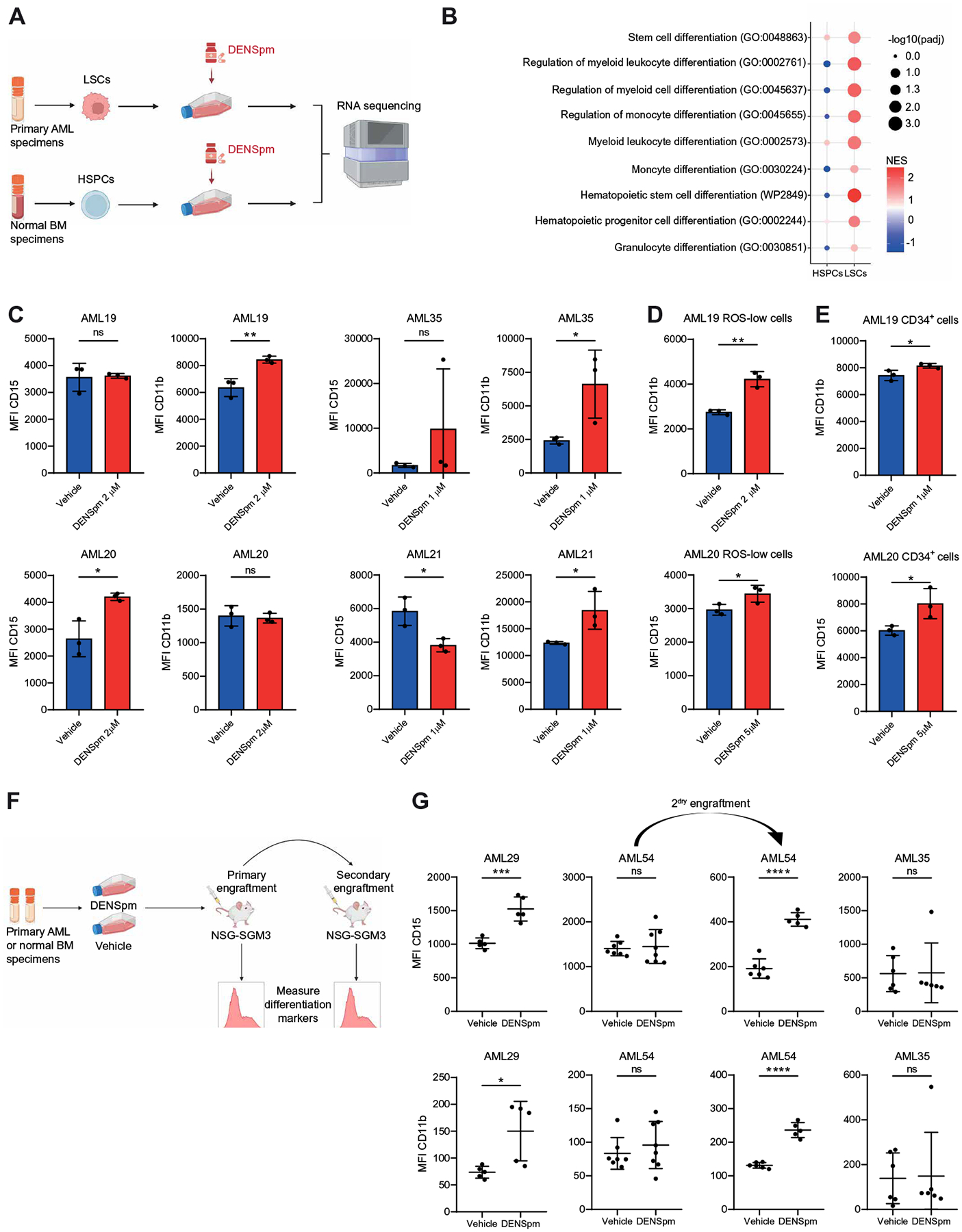
(A) Experimental design of transcriptomic analyses by RNA-seq. LSCs were enriched from five primary AMLs (AML28, AML29, and AML45 to AML47) on the basis of relative ROS abundance. HSPCs were enriched using three CD34-enriched NBM samples. ROS-low LSCs and CD34+ HSPCs were treated with vehicle or 10 μM DENSpm for 24 hours before RNA extraction and RNA-seq. Schematic created with BioRender.com. (B) Dotplot visualization of top gene ontology (GO) related to hematopoietic differentiation determined by GSEA in ROS-low LSCs and CD34+ HSPCs. NES, normalized enrichment score; Padj, adjusted P value. (C) Four primary AML specimens (AML19 to AML21 and AML35) were incubated for 24 hours with DENSpm or vehicle before colony-forming assays. After 14 days, the mean fluorescence intensities (MFI) for CD15 and CD11b in colonies were determined by flow cytometry. Statistical significance was determined using an unpaired t test. (D) LSCs were enriched using ROS-low sorting from two primary AML specimens (AML19 and AML20) and were incubated for 24 hours with DENSpm or vehicle before colony-forming assays. After 14 days, MFI for CD15 and CD11b in colonies were determined by flow cytometry. Statistical significance was determined using an unpaired t test. (E) LSCs were enriched by sorting for CD34+ cells from two primary AML specimens (AML19 and AML20) were incubated for 24 hours with DENSpm or vehicle before colony-forming assays. After 14 days, MFI for CD15 and CD11b in colonies was determined by flow cytometry. Statistical significance was determined using an unpaired t test. (F) Experimental design of engraftment assays. Schematic created with BioRender.com. Three primary AML specimens (AML29, AML35, and AML54) and three NBM specimens were incubated for 72 hours with 10 μM DENSpm or vehicle before injection into NSG-SGM3 mice. For AML54, equal numbers of human leukemic cells from vehicle- and DENSpm-treated mice were injected into secondary recipient mice. (G) MFI for CD15 and CD11b on CD45+ human cells in NSG-SGM3 recipient mice after ex vivo treatment with DENSpm or vehicle for three primary AML specimens. For AML54, MFI for CD15 and CD11b in secondary recipients is shown. Each point represents a single mouse. Statistical significance was determined using an unpaired t test. All error bars represent SD. *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001.
We next investigated the in vivo impact of polyamine depletion on LSC differentiation by engrafting DENSpm-treated primary AML specimens in NSG-SGM3 mice and assessing the expression of myeloid-associated markers in their progeny. For these assays, we used two DENSpm-responding patient specimens (AML29 and AML54), one resistant specimen (AML35), and three NBM specimens. AML29 and AML54 were chosen for this analysis because these were the only specimens without a high percentage of CD11b and CD15 at baseline with sufficient residual cells in the DENSpm-treated condition (from Fig. 4) to quantify the expression of myeloid markers. Primary specimens were transplanted into NSG-SGM3 mice after ex vivo treatment with DENSpm (Fig. 5F). DENSpm treatment increased the expression of CD15 and CD11b in cells derived from primary AML29 specimens (Fig. 5G). There was no change in CD15 or CD11b in AML54 upon primary transplant; however, CD15 and CD11b expression were higher in the polyamine-depleted condition upon secondary transplantation. Last, no changes in CD15 or CD11b were observed in AML35, which was consistent with this AML specimen not exhibiting decreased engraftment upon DENSpm treatment (Fig. 5G). Polyamine depletion did not result in HSPC myeloid differentiation (fig. S6B). Together, these data demonstrate that polyamine depletion promotes the differentiation of LSCs in polyamine-dependent specimens from patients with AML but does not affect HSPC differentiation.
Polyamine depletion decreases eIF5A hypusination in LSCs
To determine the mechanism by which polyamine depletion induces LSC differentiation, we further examined DENSpm-induced transcriptomic changes in ROS-low–enriched LSCs. RNA-seq analysis revealed an enrichment of gene signatures associated with mRNA translation in vehicle-treated–enriched LSCs compared with DENSpm-treated–enriched LSCs, but not in HSPCs (Fig. 6A). To validate whether polyamine depletion reduced protein synthesis in AML and enriched LSCs, we measured puromycin incorporation into newly synthesized proteins upon DENSpm treatment in Molm13 and MV4;11 cell lines, primary human enriched LSCs, and HSPCs (both defined as CD34+ cells). Polyamine depletion using DENSpm resulted in a modest decrease in protein synthesis in AML cell lines and primary enriched LSCs but not in HSPCs (Fig. 6B and fig. S7A). DFMO + AMXT 1501 treatment also reduced protein synthesis in enriched LSCs (Fig. 6C). These data suggest that AML cells and LSCs are dependent on polyamines for protein synthesis.
Fig. 6. Polyamine depletion reduces protein synthesis and eIF5A hypusination in LSCs.
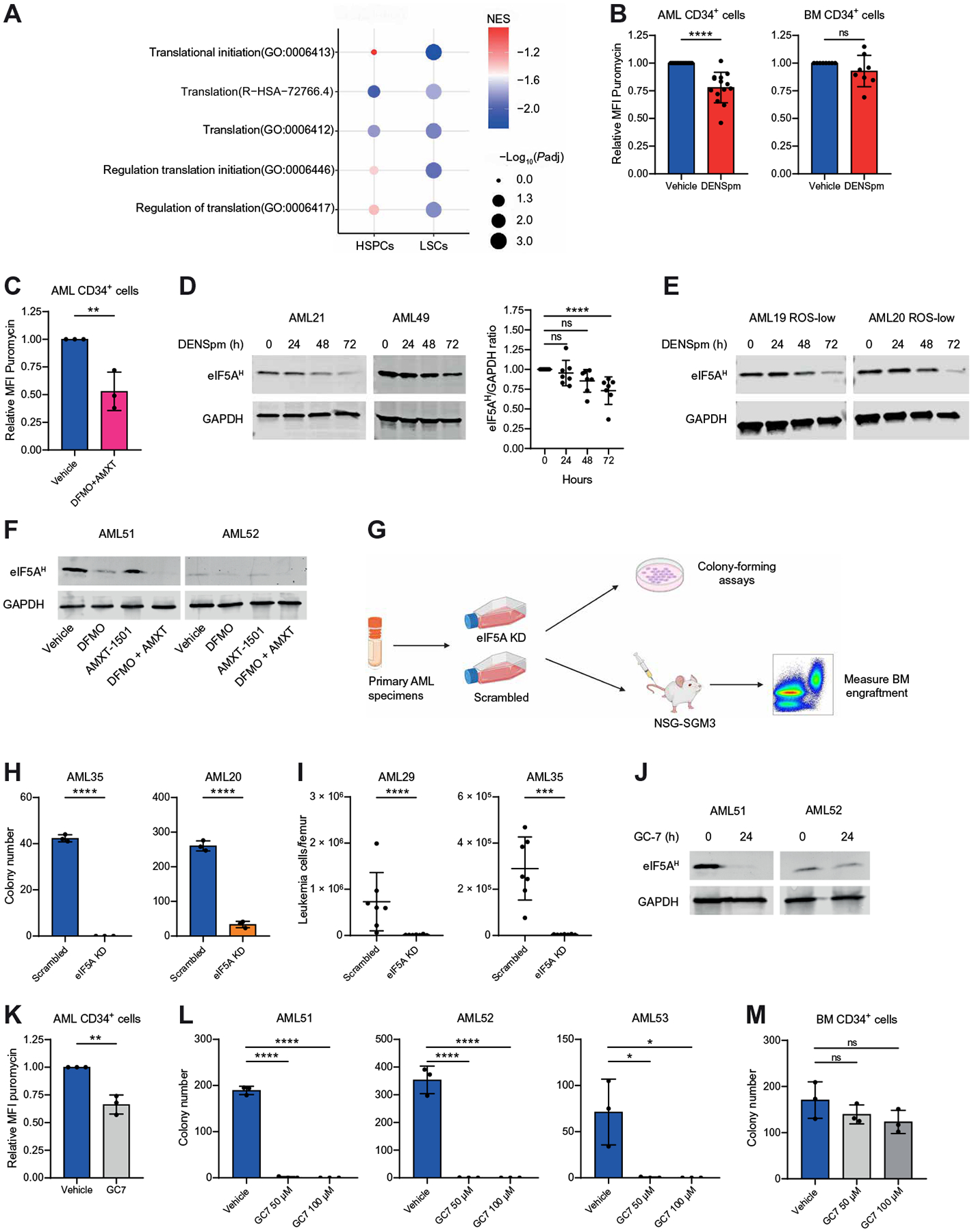
(A) Dotplot visualization of top GO related to mRNA translation determined by GSEA in ROS-low LSCs and CD34+ HSPCs. (B) MFI for puromycin determined by flow cytometry on CD34+ LSCs enriched from 14 primary AML specimens (AML6, AML20, AML21, AML34, AML35, AML45, AML47, AML49, AML51 to AML53, AML55, AML56, and AML58) and CD34+ HSPCs enriched from eight NBM specimens. Cells were incubated for 24 hours with 10 μM DENSpm or vehicle. Statistical significance was determined using an unpaired t test. (C) MFI for puromycin determined by flow cytometry on CD34+ LSCs enriched from three AML specimens (AML51 to AML53) after incubation for 24 hours with 2.5 mM DFMO and 400 nM AMXT 1501 or vehicle. Statistical significance was determined using an unpaired t test. (D) Expression of eIF5AH determined by Western blot in bulk primary AML specimens (AML16, AML19 to AML21, AML34, AML48, and AML49; two are shown in the main figure, and others are in fig. S7F) incubated for 0, 24, 48, or 72 hours with 10 μM DENSpm. Statistical significance was determined using an unpaired Mann-Whitney test. (E) Expression of eIF5AH determined by Western blot in ROS-low–enriched LSCs (AML19 and AML20) incubated for 0, 24, 48, or 72 hours with 10 μM DENSpm. (F) Expression of eIF5AH determined by Western blot in primary AML specimens (AML51 and AML52) incubated for 24 hours with 2.5 mM DFMO, 400 nM AMXT 1501, or the combination. (G) Primary AML specimens were transfected with scrambled or eIF5A-targeting small interfering RNA (siRNA). Twenty-four hours after transfection, cells were seeded in methocult medium for colony-forming assays or injected into NSG-SGM3 mice for engraftment assays. Schematic created with BioRender.com. (H) Colony-forming potential of two primary AML specimens (AML20 and AML35) after scrambled or eIF5A-targeting siRNA transfection. Statistical significance was determined using an unpaired t test. (I) Engraftment of two primary AML specimens (AML29, 35) in NSG-SGM3 mice after scrambled or eIF5A-targeting siRNA transfection. Each point represents a single mouse. Statistical significance was determined using an unpaired t test (AML35) or an unpaired Mann-Whitney test (AML29). (J) Expression of eIF5AH determined by Western blot in primary AML specimens (AML51 and AML52) incubated for 24 hours with 50 μM GC7. (K) MFI for puromycin determined by flow cytometry on CD34+ LSCs enriched from three primary AML specimens (AML51 to AML53) after incubation for 24 hours with 50 μM GC7 or vehicle. Statistical significance was determined using an unpaired t test. (L) Colony-forming ability of three bulk primary AML specimens (AML51 to AML53) after 24-hour incubation with GC7 or vehicle. Colony numbers were determined after 1 to 3 weeks. Statistical significance was determined using one-way ANOVA. (M) Colony-forming ability of one CD34-enriched BM sample after 24-hour incubation with GC7 or vehicle. Colony numbers were determined after 2 weeks. Statistical significance was determined using one-way ANOVA. All error bars represent SD. *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001.
Given that protein synthesis is tightly linked to cell cycle progression, it is essential to uncouple the effect of polyamine depletion on protein synthesis and potential changes in the cell cycle. Polyamines have been shown to promote cell proliferation (15). Moreover, DENSpm has been previously shown to induce cell cycle arrest in cancer cells (18). Accordingly, when AML cell lines were treated with DENSpm for 24 hours, we observed an increased proportion of cells in the G0-G1 phase, whereas the percentage of cells in the S-G2-M phases was decreased (fig. S7B). Decreased protein synthesis in AML cell lines was independent of their cell cycle status (fig. S7C). By contrast, LSCs enriched from 11 primary AML specimens did not show any modulation of cell cycle after incubation with DENSpm for 24 hours (fig. S7D). The absence of impact on LSC cell cycle status might be explained by the observation that these cells are already highly quiescent at steady state. Overall, these data suggest that the impact of polyamine depletion on protein translation is independent from its effect on the cell cycle.
Spermidine is known to regulate protein synthesis through its role as a precursor of hypusine, a posttranslational addition to eukaryotic initiation factor 5A (eIF5A) (38). eIF5A is a highly conserved protein that is thought to regulate about 30% of global translation (39, 40). eIF5A is the only known hypusinated protein. Hypusination of eIF5A helps facilitate protein elongation, especially in proteins containing amino acid sequences that are not efficient at forming peptide bonds, such as poly-proline regions (40). To determine whether polyamine depletion decreases protein synthesis through reducing hypusination of eIF5A, we assessed the abundance of hypusinated eIF5A (eIF5AH) in AML cell lines, primary AML cells, and enriched LSCs after polyamine depletion. DENSpm treatment decreased eIF5AH in Molm13 and MV4;11 cells. Reduced eIF5AH was rescued by cotreatment with spermidine (fig. S7E), demonstrating that the DENSpm treatment alters eIF5AH in a spermidine-dependent manner. Polyamine depletion also decreased eIF5AH abundance in most primary AML specimens (Fig. 6D and fig. S7F). DENSpm treatment resulted in decreased eIF5AH in ROS-low–enriched LSCs (Fig. 6E), often to a greater extent than in the bulk specimen (fig. S7F). Moreover, DFMO + AMXT 1501 treatment also reduced eIF5AH in primary AML specimens (Fig. 6F). Together, these data point to changes in eIF5AH as a potential mechanism by which polyamines regulate protein synthesis in LSCs.
To determine whether LSCs are dependent on eIF5A for their function, we used small interfering RNA (siRNA) to knockdown eIF5A in primary AML cells (Fig. 6G) and measured LSC function using colony-forming assays and engraftment assays. eIF5A knockdown (fig. S7G) resulted in decreased colony-forming potential (Fig. 6H) and engraftment (Fig. 6I) of primary AML specimens. In contrast with AML specimens, which had a greater than 90% reduction in colony-forming potential upon eIF5A knockdown, eIF5A reduction resulted in about a 50% decrease in colony-formation potential in NBM specimens, suggesting that AML cells are more reliant on eIF5A relative to HSPCs (fig. S7H). To examine the importance of hypusination of eIF5A for AML and HSPC clonogenic potential, we measured the colony-forming potential of AML cell lines, primary AML specimens, and NBM upon treatment with GC7, a tool compound that inhibits deoxyhypusine synthase, which is an enzyme essential for hypusination of eIF5A. GC7 treatment decreased colony-forming potential of AML cell lines and resulted in elevated expression of myeloid differentiation marker CD15, similar to what was observed upon polyamine depletion (fig. S7, I and J). In primary specimens, GC7 treatment resulted in reduced eIF5AH, decreased protein synthesis, and completely abrogated colony-forming potential of AML specimens without harming NBM (Fig. 6, J to M). Overall, these results demonstrate that polyamine depletion impairs eIF5AH and that eIF5AH is essential for AML survival and LSC function.
Polyamine depletion alters the expression of KAT7
On the basis of the data above, we hypothesized that polyamine depletion targets LSCs by decreasing protein expression of one or several proteins crucial for maintaining stemness in LSCs. To identify these proteins, we performed mass spectrometry–based proteomics analysis on Molm13 cells treated with DENSpm (Fig. 7A). Overall, polyamine depletion affected the expression of 213 proteins in Molm13 cells [false discovery rate (FDR) > 0.1 and log2FC > 2]. One hundred fifty-one and 62 proteins were down- and up-regulated after DENSpm treatment, respectively. To determine which proteins identified in our proteomics analysis may help maintain stemness in LSCs, we overlapped our hits with genes whose knockouts have been shown to promote AML differentiation in an unbiased CRISPR screen (41). Because polyamine depletion targets LSCs and not HSPCs, we removed any potential candidate genes identified to be pan-essential in the cancer dependency map project (DepMap). This analysis revealed five candidate genes/proteins: KAT7, APLP2, CDK13, AFTPH, and EHMT1. Of the five genes identified, KAT7 had the greatest dependency score across 24 AML cell lines (DepMap) (Fig. 7B). KAT7, also known as HBO1, is one of the five members of the MYST family of histone acetyltransferases previously reported to function as a major transcriptional regulator, primarily through its role in histone acetylation (42). Loss of KAT7 in AML and LSCs results in increased myeloid differentiation gene expression, loss of LSC stemness, and ultimately cell death (43, 44).
Fig. 7. Polyamine depletion targets LSCs through reduced expression of KAT7.
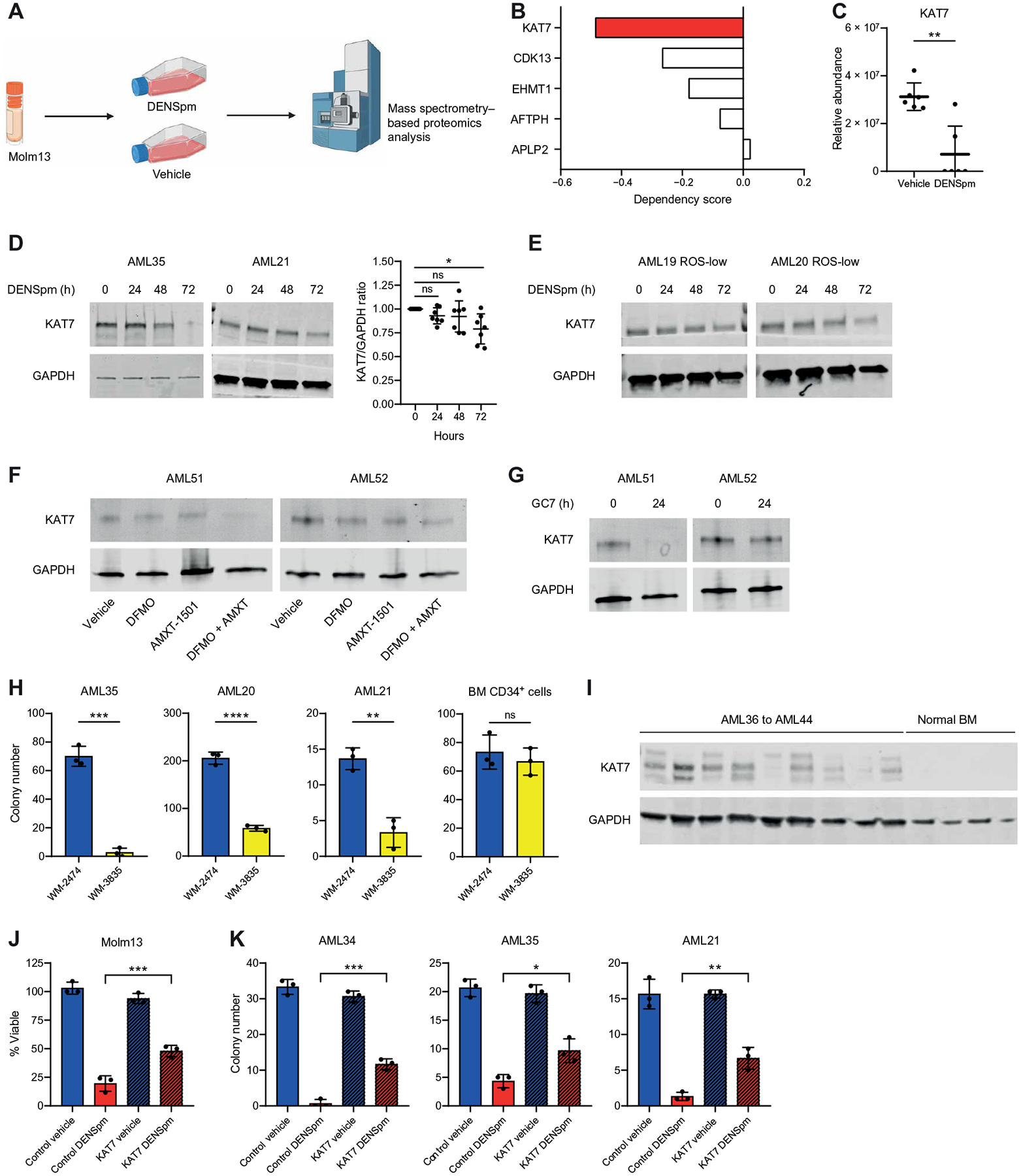
(A) Experimental design of proteomics analysis. Protein abundance in Molm13 cells was determined by mass spectrometry 24 hours after treatment with 10 μM DENSpm. Schematic created with BioRender.com. (B) Dependency scores for KAT7, CDK13, EHMT1, AFTPH, and APLP2 were determined using DepMap. (C) KAT7 abundance detected by mass spectrometry proteomics in Molm13 cells. Significance was determined using an unpaired Mann-Whitney test. (D) Expression of KAT7 determined by Western blot in eight bulk primary AML specimens (AML16, AML19 to AML21, AML34, AML35, AML48, and AML49 shown in the main figure and others shown in fig. S8B) incubated for 0, 24, 48, or 72 hours with 10 μM DENSpm. Statistical significance was determined using an unpaired t test. (E) Expression of KAT7 determined by Western blot in ROS-low–enriched LSCs (AML19 and AML20) incubated for 0, 24, 48, or 72 hours with 10 μM DENSpm. (F) Expression of KAT7 determined by Western blot in two bulk primary AML specimens (AML51 and AML52) incubated for 24 hours with 2.5 mM DFMO and 400 nM AMXT1501 or the combination. (G) Expression of KAT7 determined by Western blot in two bulk primary AML specimens (AML51 and 52) incubated for 24 hours with 50 μM GC7. (H) Colony-forming potential of three bulk AML samples (AML20, AML21, and AML35) and one CD34-enriched bone marrow sample treated with 1 μM KAT7 inhibitor WM-3835 or inactive analog WM-2474. Colony numbers were determined after 1 to 3 weeks. Statistical significance was determined using an unpaired t test. (I) Expression of KAT7 determined by Western blot in nine primary AML specimens (AML36 to AML44) and four NBM specimens. (J) Viability of Molm13 cells incubated for 48 hours with 10 μM DENSpm or vehicle and transfected or not with KAT7 recombinant. Statistical significance was determined using two-way ANOVA. (K) Colony-forming ability of three primary AML specimens (AML21, AML34, and AML35) incubated for 24 hours with 10 μM DENSpm or vehicle and transfected or not with KAT7 recombinant. Colony numbers were determined after 1 to 3 weeks. Statistical significance was determined using two-way ANOVA. All error bars represent SD. *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001.
Because KAT7 has been shown to be essential for LSC maintenance (43), we sought to determine whether DENSpm targets AML cells and LSCs, at least in part, by impairing KAT7 protein expression. Consistent with our proteomics data (Fig. 7C), KAT7 expression was decreased upon polyamine depletion in Molm13 and MV4;11 cells (fig. S8A), the majority of primary AML specimens (Fig. 7D and fig. S8B), and ROS-low–enriched LSCs (Fig. 7E). KAT7 expression was rescued by cotreatment with spermidine (fig. S8A), indicating that KAT7 reduction is spermidine dependent. KAT7 protein expression was also reduced by DFMO + AMXT 1501 and GC7 treatment in primary AML specimens (Fig. 7, F and G). KAT7 mRNA expression was unaffected by DENSpm treatment (fig. S8C), suggesting that KAT7 expression is regulated by polyamines in a posttranscriptional mechanism. Overall, these results suggest that AML cells are dependent on spermidine for the expression of KAT7.
To investigate the requirement of KAT7 for enriched LSC and HSPC function, we performed colony-forming assays with primary AML specimens and normal CD34-enriched BM in the presence of the KAT7 inhibitor WM-3835 as previously described (43). In these experiments, we used the inactive analog of this inhibitor (WM-2474) as a negative control (45). Inhibition of KAT7 using WM-3835 led to a decrease in the colony-forming potential of primary AML but not normal HSPCs (Fig. 7H). Primary AML but not NBM specimens expressed detectable abundance of KAT7 protein (Fig. 7I), which likely contributed to the differential response of LSCs to KAT7 inhibition and polyamine depletion compared with HSPCs. To determine whether the gene expression changes observed upon polyamine depletion contributed to KAT7 loss, we compared genes identified to be differentially expressed upon DENSpm treatment in LSCs with genes differentially expressed upon KAT7 knockout in AML cells (44). This analysis revealed a significant overlap in differentially up-regulated (P value = 1.554 × 10−9) and down-regulated (P value = 2.56 × 10−22) genes (fig. S8D), suggesting that KAT7 loss in part may contribute to the gene expression changes observed upon polyamine depletion.
Last, to determine whether decreased KAT7 protein abundance is a critical part of the mechanism by which polyamine depletion targets AML clonogenic potential, we transfected DENSpm-treated Molm13 cells with recombinant KAT7 and measured the effect on cell viability. Transfection of recombinant KAT7 restored KAT7 abundance upon polyamine depletion (fig. S8E) and partially rescued DENSpm-induced cell death in Molm13 cells (Fig. 7J). Further, transfection of recombinant KAT7 (fig. S8F) into three primary AML specimens resulted in a partial rescue in colony-forming potential upon polyamine depletion (Fig. 7K). Together, these results demonstrate that polyamine depletion targets AML clonogenicity at least in part by impairing KAT7 protein expression.
Polyamine depletion targets AML in vivo
Last, we assessed the therapeutic potential of polyamine inhibition on AML cells in vivo. AML and NBM specimens were transplanted into NSG-SGM3 mice; once leukemic/human cell burden reached at least 10% in the bone marrow, mice were treated with DENSpm (60 mg/kg) 5 days/week for 2 weeks (Fig. 8A). DENSpm treatment decreased leukemic burden in mice transplanted with four of six AML specimens examined (Fig. 8B). Consistent with the engraftment assays (Fig. 4), in vivo DENSpm treatment reduced leukemic burden in mice transplanted with relapsed and de novo AML specimens (data file S1). In addition, DENSpm reduced leukemia burden in mice transplanted with AML specimens determined to be sensitive and resistant to venetoclax in vivo (fig. 9A). DENSpm treatment did not alter the numbers of human cells engrafted or lineage output of NBM cells (Fig. 8C and fig. S9B). Last, to determine whether polyamine depletion targets LSC function in vivo, we transplanted cells from one of the nonresponding primary recipient mice into secondary non–drug-treated mice and measured engraftment potential as a readout of LSC function. Despite not observing an initial response to DENSpm in vivo, secondary engraftment was decreased, demonstrating the polyamine depletion can target LSC in vivo (Fig. 8D). These data demonstrate that DENSpm is a promising therapeutic approach to target AML cells even in patients with AML who are resistant to other commonly used AML therapies.
Fig. 8. Polyamine depletion targets primary AML in vivo.
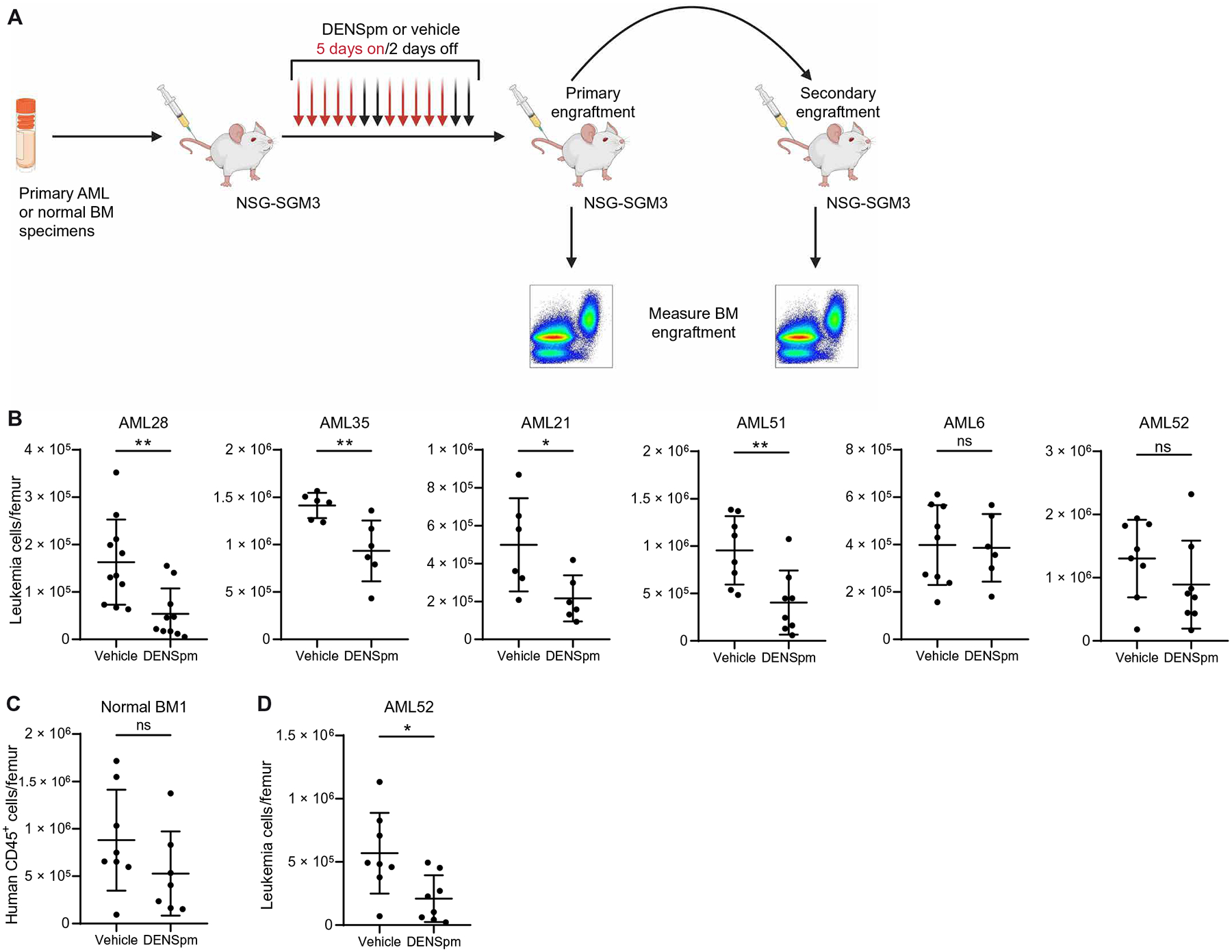
(A) Experimental design of in vivo treatment with DENSpm. Six primary AML specimens (AML6, AML21, AML28, AML35, AML51, and AML52) and one NBM specimen were injected into NSG-SGM3 mice. Once leukemia burden reached 10% in the bone marrow, mice were injected with DENSpm (60 mg/kg) or vehicle 5 days/week for 2 weeks before BM engraftment was measured. For one specimen, an equal number of cells from the primary recipient was transplanted into secondary recipient mice to measure in vivo LSC targeting. Schematic created with BioRender.com. (B) Leukemic burden in six primary AML specimens in NSG-SGM3 mice after in vivo treatment with DENSpm or vehicle. Each point represents a single mouse. Statistical significance was determined using an unpaired t test. Vehicle conditions for AML21 and AML35 are also used in fig. S9A. (C) Engraftment of one NBM specimen in NSG-SGM3 mice after in vivo treatment with DENSpm or vehicle. Each point represents a single mouse. Statistical significance was determined using an unpaired t test. (D) Engraftment of AML52 in NSG-SGM3 mice after secondary transplantation and in vivo treatment with DENSpm or vehicle. Each point represents a single mouse. Statistical significance was determined using an unpaired t test. All error bars represent SD. *P < 0.05 and **P < 0.01.
Together, our data show that polyamines are essential for LSC survival by regulating eIF5A hypusination. Mechanistically, our data suggest that polyamines are required for KAT7 synthesis, which is essential for maintenance of stemness. Loss of polyamines, particularly spermidine, results in reduced KAT7 protein expression, resulting in myeloid cell differentiation.
DISCUSSION
Despite the recent advances in our knowledge of LSC metabolism (3), we lacked a comprehensive understanding of the metabolite differences between human LSCs and HSPCs. Because the optimal AML therapy will effectively eradicate the leukemic cells and only minimally harm the normal HSPCs, understanding such differences is critically important. Metabolic profiling revealed that enriched LSCs and HSPCs have unique metabolic hubs that provide opportunities for the development of LSC targeting strategies.
A notable metabolic difference between enriched LSCs and HSPCs was in arginine metabolism, specifically in elevated abundance of spermidine. Arginine has been widely studied in cancer and even evaluated as a therapeutic target in clinical trials for patients with AML (30). To date, arginine-targeting strategies have failed clinically because of the ability of cancer cells to up-regulate arginine biosynthesis. Because of the lower SAT1 abundance observed in AML cells compared with NBM, we examined a polyamine export strategy to reduce polyamine abundance. We chose to use DENSpm as a polyamine-depleting agent because it has been previously shown to be safe in clinical trials in metastatic breast cancer, lung cancer, and hepatocellular carcinoma (46–48). However, polyamine export is not the only strategy to target polyamines in cancer. Polyamine abundance is often increased in MYC-driven malignancies because of up-regulated transcriptional expression of MYC-target ODC1, the enzyme catalyzing the first step of polyamine biosynthesis (49). Pharmacologic inhibition of ODC1 using DFMO has shown promising results in preclinical cancer models (23, 50) but, in clinical trials, has shown little to no cytotoxic effects (21, 34, 35). Data suggest that the lack of efficacy is due to the ability of cancer cells to take up polyamines from the microenvironment, thereby compensating for the loss of polyamine biosynthesis (17). Inhibition of polyamine uptake in combination with DMFO treatment has shown promising results in preclinical models of diffuse intrinsic pontine glioma (17) and is currently being evaluated in a phase 1B/2A trial (NCT05500508). Our work suggests that the combination of DFMO + the polyamine transporter inhibitor AMXT 1501 may also be a promising approach for the treatment of AML.
We show that polyamine depletion decreases LSC function by promoting myeloid cell differentiation in primary human AML specimens. Several strategies that result in AML cell death through the promotion of myeloid cell differentiation have been explored pre-clinically and clinically (51). This study shows differentiation, specifically of enriched LSCs and not HSPCs upon polyamine depletion, providing an additional clinically relevant approach to induce differentiation in LSCs. Elevating spermidine abundance has been previously reported to enhance the self-renewal capacity of HSCs (52), suggesting that polyamines do biologically contribute to HSC biology, but our work shows that enriched HSPCs tolerate decreases in polyamine abundance to a greater extent than LSCs. In vivo administration of DENSpm decreased human leukemic burden in most AML specimens examined, indicating that polyamine depletion may be a promising strategy to treat patients with AML. Understanding why a subset of AML patient specimens may not respond to polyamine depletion will be an important area of further investigation. AML is a metabolically heterogeneous disease (12), and several common AML mutations are known to regulate metabolism and influence response to metabolism-targeting therapies (9–12, 14). Future studies should evaluate whether response to polyamine depletion is related to the mutational status of the AML specimens. Some AML specimens evaluated in vivo in our study were derived from patients with AML who had relapsed on conventional cytotoxic chemotherapy (data file S1). Further, several specimens shown to be responsive to polyamine depletion in our study are resistant to the BCL2 inhibitor venetoclax. The inclusion of specimens representing these patient populations is important because they have particularly poor outcomes and limited therapeutic options and would likely be the first patient populations to be included in clinical trials evaluating polyamine depletion strategies in patients with AML.
Mechanistically, we show that polyamine depletion treatment decreases protein synthesis by reducing eIF5A hypusination, resulting in decreased expression of proteins important for maintaining stemness in LSCs. Therapies targeting global protein synthesis in AML have shown promise in preclinical models but have had limiting toxicities in some clinical trials (53). eIF5A regulates the translation of only a subset of mRNAs, specifically mRNAs that encode for regions rich in amino acid sequences that are less efficient at forming peptide bonds, such as proline-rich regions (39, 40). It is likely that targeting the synthesis of a subset of proteins as opposed to global protein synthesis may result in less toxicity and still maintain a high efficacy whether LSC essential protein abundance is reduced. One of the proteins with reduced expression upon polyamine depletion and inhibition of eIF5AH was KAT7. KAT7 has been previously examined in AML and LSCs and has been shown to be essential for maintaining LSC function and stemness through its role in regulating expression of myeloid differentiation genes (43, 44). Rescue experiments revealed that KAT7 is a critical part of the mechanisms by which polyamine depletion targets the clonogenic potential of AML—albeit not the full mechanism because KAT7 overexpression only partially rescues DENSpm-induced cell death. Future studies evaluating KAT7 and other proteins differentially expressed upon polyamine depletion or potentially other functions of spermidine in LSCs are warranted.
Our study has some limitations. This work relies on enrichment of LSCs using cell surface markers and relative ROS abundance. No perfect markers exist to isolate a pure population of LSCs; therefore, the best approach we have is to combine validated LSC enrichment methods with analysis of LSC function. Another limitation of the study is that the ability of KAT7 to rescue LSC function was only assessed using colony-formation assays as opposed to engraftment analysis, which was not feasible because of technical limitations of these experiments. Because of this limitation, we were only able to conclude that KAT7 is important for AML clonogenic potential and not LSC function. Last, the number of primary human AML specimens used in this study is limited and precludes the understanding of the mechanisms that drive heterogeneity in response to polyamine-depleting therapies, which will be an important for future studies to evaluate.
In this study, we show that polyamine depletion is an effective therapy in targeting LSCs in preclinical models. DENSpm has a favorable toxicity profile in phase 1 and 2 clinical trials in non–small lung, hepatocellular carcinoma, and metastatic breast cancer (46–48). However, these trials also demonstrated a lack of efficacy for DENSpm as a single agent (46–48). It is possible that polyamine depletion will only be effective in a subset of cancers that are dependent on eIF5A-regulated genes, which are likely to vary on the basis of cancer type–specific gene expression. DFMO + AMXT 1501 are currently being evaluated in clinical trials. If they are determined to be safe and efficacious in advanced solid tumors (NCT03536728 and NCT05500508), this combination strategy may be a promising approach for rapid clinical translation into evaluation in AML.
MATERIALS AND METHODS
Study design
The first objective of this study was to identify metabolite differences in enriched LSCs compared with normal human HSPCs. The second objective was to evaluate the therapeutic potential of targeting a metabolic pathway up-regulated in LSCs compared with HSPCs. Last, the third objective was to understand why the identified pathway represents a vulnerability in LSCs but not HSPCs. To identify metabolic differences between LSCs and HSPCs, we performed metabolomics analysis using mass spectrometry. Using the power calculator in metaboanalyst 5.0, we determined that specimens from 18 patients with AML were required for 70% power to detect differences in metabolites with an FDR < 0.05. Complementary mass spectrometry analysis led to the identification of spermidine metabolism as up-regulated in LSCs. Spermidine metabolism was targeted using several inhibitors, including DENSpm and DFMO + AMXT-1501. The efficacy and toxicity of these agents were determined by measuring polyamine abundance using mass spectrometry, colony-forming assays, cell viability assays, and xenograft models. The differential dependency on spermidine between LSCs and HSPCs was interrogated using transcriptomic analysis, proteomic analysis, Western blotting, flow cytometry, and colony-formation assays. All experiments were performed in AML cell lines and samples from patients with AML. Cryopreserved specimens isolated from deidentified patients with AML were used for the study without age or gender preferences. Investigators and data analyzers were blinded for the evaluation of polyamine-depleting agents administered in vivo. Mouse experiments were designed to provide 70 to 80% power for a target effect size of 1.2 to 1.5. All mice were randomly allocated into experimental groups. Mice receiving vehicle and experimental agents were included in the same cages to account for this potential confounder. All experiments were performed with at least three independent biological replicates. No experimental data points were removed from the studies.
Human specimens
Patient AML cells were obtained from donors who gave informed consent for sample procurement under the Princess Margaret Leukemia Tissue Bank protocol or under the University of Colorado tissue procurement protocol. Experiments were performed in accordance with UHN’s Research Ethics Board, under protocol 20–5031. Human bone marrow mononuclear cells and human bone marrow CD34+ progenitor cells were purchased from Lonza. See data file S1 for additional details on the human AML specimens.
Animal studies
Animal studies were performed in accordance with UHN’s Animal Resource Center, under animal use protocol 6366 and in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care–accredited animal facility of Cincinnati Children’s Hospital Medical Center (IACUC2023-1032). NSG-SGM3 (RRID:IMSR_JAX:032474) mice 8 to 12 weeks of age were conditioned with busulfan (Sigma-Aldrich; B2635) at 20 to 25 mg/kg intraperitoneally [10% dimethyl sulfoxide and 90% phosphate-buffered saline (PBS)] as a single injection 24 hours before engraftment. For ex vivo experiments, primary cells (AML or healthy bone marrow mononuclear cells) were plated as described previously and treated with DENSpm (10 μM), venetoclax (50 nM), or vehicle 72 hours before engraftment. For eIF5A knockdown experiments, primary cells were transfected with siRNA targeting eIF5A or a nontargeting scrambled control using the methods described below 24 hours before engraftment. About 15 min before transplantation, cells were treated with OKT3 (Bio X Cell, BE0001-2) at 1 μg per 1 million cells in PBS to deplete CD3+ immune cells and to reduce the occurrence of graft-versus-host disease. Filtered cells were injected by tail vein at a concentration of 0.25 to 1 million cells per mouse. After engraftment was established (8 to 10 weeks), mice were sacrificed, and both femurs were dissected out. The condyle was removed, and cells were harvested by centrifugation or femur crushing. Cells were then treated with red blood cell lysis buffer (Sigma-Aldrich, 11814389001) to remove red blood cell fraction, and the remaining cells were evaluated by flow cytometry as described below. For in vivo experiments, freshly thawed primary cells (AML or healthy bone marrow mononuclear cells) were treated with OKT3, filtered, and injected into NSG-SGM3 mice as previously described. Once leukemia burden reached 10% in the bone marrow, mice were injected with DENSpm (60 mg/kg) 5 days/week for 2 weeks or with venetoclax (100 mg/kg) 5 days/week for 1 week. Both femurs were dissected out 2 hours after the last injection and processed as previously described. For secondary engraftment, equal numbers of human leukemic cells from vehicle- and DENSpm-treated mice were transplanted into NSG-SGM3 mice at a concentration of 0.5 million cells per mouse.
Statistical analysis
Raw, individual-level data are presented in data file S2. The numbers of animals, patient specimens, and experimental or biological replicates are indicated in the figure legends. Each graph represents the mean, and error bars represent SD. Differences among multiple groups were determined by one-way or two-way analysis of variance (ANOVA). Normality was determined using Shapiro-Walk test. Differences between two groups was determined by two-tailed Student’s t test with paired or unpaired analysis when data passed normality test or by unpaired Mann-Whitney test when data did not pass normality test. FDR was calculated to account for multiple testing when appropriate. The specific analysis used is indicated in each figure legend. P values less than 0.05 were considered significant. P values are indicated with * on each graph (*P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001) and are shown in figures. Unless otherwise indicated, graphs and analysis were generated using GraphPad Prism software.
Supplementary Material
Acknowledgments:
We thank the Princess Margaret Cancer Center Leukemia Tissue Bank and the University of Colorado Hematology Program for providing primary AML samples; J. Flewelling (Princess Margaret Cancer Center) and J. Fellers and J. Widner (Cincinnati Children’s Hospital Medical Center) for administrative assistance; A. Tajik, E. Tsao, P. Joshi, S. Adeel, S. Moreira, T. Chen, and Z. Balde (Princess Margaret Cancer Center) for the help in animal sacrifice and dissection; and D. Starczynowski and L. Miles for the thoughtful feedback on the manuscript.
Funding:
This work was supported in part by the Leukemia and Lymphoma Society Special Fellow and Career Achievement Award 3398-20 (to C.L.J.), the Leukemia and Lymphoma Society Discovery grant 8035-23 (to C.L.J.), the Canadian Cancer Society Emerging Scholars 707144 (to C.L.J.), Cincinnati Children’s Hospital Research Foundation (to C.L.J.), and the Rally Foundation (to C.L.J.). The Princess Margaret Cancer Center Foundation and the Ontario Ministry of Health supported C.L.J., K.J.H., A.N.T., M.D.M., A.A., and B.R. This work was supported by the NIH through the University of Colorado Cancer Center Support Grant P30CA046934 (to A.D.), 1F31CA250361-01 (to R.C.-H.), and R50 CA211404 (to M.W.). This work was supported by NIDDK U54 DK126108 at Cincinnati Children’s Hospital Medical Center and their Flow Cytometry and Comprehensive Mouse Cores. This work was supported by the American Hematology Graduate Student Scholarship (to T.L.).
Footnotes
Supplementary Materials
The PDF file includes:
Other Supplementary Material for this manuscript includes the following:
Data files S1 to S3
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. All materials are commercially available. RNA-seq data are available at Gene Expression Omnibus (GSE274658). Proteomics data are available through MassIVE (MSV000093543).
REFERENCES AND NOTES
- 1.DiNardo CD, Erba HP, Freeman SD, Wei AH, Acute myeloid leukaemia. Lancet 401, 2073–2086 (2023). [DOI] [PubMed] [Google Scholar]
- 2.Shlush LI, Mitchell A, Heisler L, Abelson S, Ng SWK, Trotman-Grant A, Medeiros JJF, Rao-Bhatia A, Jaciw-Zurakowsky I, Marke R, McLeod JL, Doedens M, Bader G, Voisin V, Xu C, McPherson JD, Hudson TJ, Wang JCY, Minden MD, Dick JE, Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 547, 104–108 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Jones CL, Inguva A, Jordan CT, Targeting energy metabolism in cancer stem cells: Progress and challenges in leukemia and solid tumors. Cell Stem Cell 28, 378–393 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanahan D, Hallmarks of cancer: New dimensions. Cancer Discov. 12, 31–46 (2022). [DOI] [PubMed] [Google Scholar]
- 5.DeVilbiss AW, Zhao Z, Martin-Sandoval MS, Ubellacker JM, Tasdogan A, Agathocleous M, Mathews TP, Morrison SJ, Metabolomic profiling of rare cell populations isolated by flow cytometry from tissues. eLife 10, e61980 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nemkov T, Reisz JA, Gehrke S, Hansen KC, D’Alessandro A, High-throughput metabolomics: Isocratic and gradient mass spectrometry-based methods. Methods Mol. Biol 1978, 13–26 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, Pei S, Khan N, Adane B, Ye H, Krug A, Reinhold D, Smith C, DeGregori J, Pollyea DA, Jordan CT, Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell 34, 724–740.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raffel S, Falcone M, Kneisel N, Hansson J, Wang W, Lutz C, Bullinger L, Poschet G, Nonnenmacher Y, Barnert A, Bahr C, Zeisberger P, Przybylla A, Sohn M, Tönjes M, Erez A, Adler L, Jensen P, Scholl C, Fröhling S, Cocciardi S, Wuchter P, Thiede C, Flörcken A, Westermann J, Ehninger G, Lichter P, Hiller K, Hell R, Herrmann C, Ho AD, Krijgsveld J, Radlwimmer B, Trumpp A, BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 551, 384–388 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Stevens BM, Jones CL, Pollyea DA, Culp-Hill R, D’Alessandro A, Winters A, Krug A, Abbott D, Goosman M, Pei S, Ye H, Gillen AE, Becker MW, Savona MR, Smith C, Jordan CT, Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat. Cancer 1, 1176–1187 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas D, Wu M, Nakauchi Y, Zheng M, Thompson-Peach CAL, Lim K, Landberg N, Köhnke T, Robinson N, Kaur S, Kutyna M, Stafford M, Hiwase D, Reinisch A, Peltz G, Majeti R, Dysregulated lipid synthesis by oncogenic IDH1 mutation is a targetable synthetic lethal vulnerability. Cancer Discov. 13, 496–515 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sabatier M, Birsen R, Lauture L, Mouche S, Angelino P, Dehairs J, Goupille L, Boussaid I, Heiblig M, Boet E, Sahal A, Saland E, Santos JC, Armengol M, Fernández-Serrano M, Farge T, Cognet G, Simonetta F, Pignon C, Graffeuil A, Mazzotti C, Avet-Loiseau H, Delos O, Bertrand-Michel J, Chedru A, Dembitz V, Gallipoli P, Anstee NS, Loo S, Wei AH, Carroll M, Goubard A, Castellano R, Collette Y, Vergez F, Mas VM-D, Bertoli S, Tavitian S, Picard M, Récher C, Bourges-Abella N, Granat F, Kosmider O, Sujobert P, Colsch B, Joffre C, Stuani L, Swinnen JV, Guillou H, Roué G, Hakim N, Dejean AS, Tsantoulis P, Larrue C, Bouscary D, Tamburini J, Sarry JE, C/EBPα confers dependence to fatty acid anabolic pathways and vulnerability to lipid oxidative stress-induced ferroptosis in FLT3-mutant leukemia. Cancer Discov. 13, 1720–1747 (2023). [DOI] [PubMed] [Google Scholar]
- 12.Erdem A, Marin S, Pereira-Martins DA, Cortés R, Cunningham A, Pruis MG, de Boer B, van den Heuvel FAJ, Geugien M, Wierenga ATJ, Brouwers-Vos AZ, Rego EM, Huls G, Cascante M, Schuringa JJ, The glycolytic gatekeeper PDK1 defines different metabolic states between genetically distinct subtypes of human acute myeloid leukemia. Nat. Commun 13, 1105 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones CL, Stevens BM, Pollyea DA, Culp-Hill R, Reisz JA, Nemkov T, Gehrke S, Gamboni F, Krug A, Winters A, Pei S, Gustafson A, Ye H, Inguva A, Amaya M, Minhajuddin M, Abbott D, Becker MW, DeGregori J, Smith CA, D’Alessandro A, Jordan CT, Nicotinamide metabolism mediates resistance to venetoclax in relapsed acute myeloid leukemia stem cells. Cell Stem Cell 27, 748–764.e4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nechiporuk T, Kurtz SE, Nikolova O, Liu T, Jones CL, D’Alessandro A, Culp-Hill R, d’Almeida A, Joshi SK, Rosenberg M, Tognon CE, Danilov AV, Druker BJ, Chang BH, McWeeney SK, Tyner JW, The TP53 apoptotic network is a primary mediator of resistance to BCL2 Inhibition in AML cells. Cancer Discov. 9, 910–925 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pegg AE, Functions of polyamines in mammals. J. Biol. Chem 291, 14904–14912 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y, Kramer DL, Diegelman P, Vujcic S, Porter CW, Apoptotic signaling in polyamine analogue-treated SK-MEL-28 human melanoma cells. Cancer Res. 61, 6437–6444 (2001). [PubMed] [Google Scholar]
- 17.Khan A, Gamble LD, Upton DH, Ung C, Yu DMT, Ehteda A, Pandher R, Mayoh C, Hébert S, Jabado N, Kleinman CL, Burns MR, Norris MD, Haber M, Tsoli M, Ziegler DS, Dual targeting of polyamine synthesis and uptake in diffuse intrinsic pontine gliomas. Nat. Commun 12, 971 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kramer DL, Fogel-Petrovic M, Diegelman P, Cooley JM, Bernacki RJ, McManis JS, Bergeron RJ, Porter CW, Effects of novel spermine analogues on cell cycle progression and apoptosis in MALME-3M human melanoma cells. Cancer Res. 57, 5521–5527 (1997). [PubMed] [Google Scholar]
- 19.Nakanishi S, Cleveland JL, Targeting the polyamine-hypusine circuit for the prevention and treatment of cancer. Amino Acids 48, 2353–2362 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakanishi S, Li J, Berglund AE, Kim Y, Zhang Y, Zhang L, Yang C, Song J, Mirmira RG, Cleveland JL, The polyamine-hypusine circuit controls an oncogenic translational program essential for malignant conversion in MYC-driven lymphoma. Blood Cancer Discov. 4, 294–317 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Shaughnessy JA, Demers LM, Jones SE, Arseneau J, Khandelwal P, George T, Gersh R, Mauger D, Manni A, Alpha-difluoromethylornithine as treatment for metastatic breast cancer patients. Clin. Cancer Res 5, 3438–3444 (1999). [PubMed] [Google Scholar]
- 22.Schipper RG, Deli G, Deloyer P, Lange WP, Schalken JA, Verhofstad AA, Antitumor activity of the polyamine analog N1, N11-diethylnorspermine against human prostate carcinoma cells. Prostate 44, 313–321 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Schultz CR, Geerts D, Mooney M, El-Khawaja R, Koster J, Bachmann AS, Synergistic drug combination GC7/DFMO suppresses hypusine/spermidine-dependent eIF5A activation and induces apoptotic cell death in neuroblastoma. Biochem. J 475, 531–545 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Wallick CJ, Gamper I, Thorne M, Feith DJ, Takasaki KY, Wilson SM, Seki JA, Pegg AE, Byus CV, Bachmann AS, Key role for p27Kip1, retinoblastoma protein Rb, and MYCN in polyamine inhibitor-induced G1 cell cycle arrest in MYCN-amplified human neuroblastoma cells. Oncogene 24, 5606–5618 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Wang C, Ruan P, Zhao Y, Li X, Wang J, Wu X, Liu T, Wang S, Hou J, Li W, Li Q, Li J, Dai F, Fang D, Wang C, Xie S, Spermidine/spermine N1-acetyltransferase regulates cell growth and metastasis via AKT/β-catenin signaling pathways in hepatocellular and colorectal carcinoma cells. Oncotarget 8, 1092–1109 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Zabala-Letona A, Arruabarrena-Aristorena A, Martín-Martín N, Fernandez-Ruiz S, Sutherland JD, Clasquin M, Tomas-Cortazar J, Jimenez J, Torres I, Quang P, Ximenez-Embun P, Bago R, Ugalde-Olano A, Loizaga-Iriarte A, Lacasa-Viscasillas I, Unda M, Torrano V, Cabrera D, van Liempd SM, Cendon Y, Castro E, Murray S, Revandkar A, Alimonti A, Zhang Y, Barnett A, Lein G, Pirman D, Cortazar AR, Arreal L, Prudkin L, Astobiza I, Valcarcel-Jimenez L, Zuñiga-García P, Fernandez-Dominguez I, Piva M, Caro-Maldonado A, Sánchez-Mosquera P, Castillo-Martín M, Serra V, Beraza N, Gentilella A, Thomas G, Azkargorta M, Elortza F, Farràs R, Olmos D, Efeyan A, Anguita J, Muñoz J, Falcón-Pérez JM, Barrio R, Macarulla T, Mato JM, Martinez-Chantar ML, Cordon-Cardo C, Aransay AM, Marks K, Baselga J, Tabernero J, Nuciforo P, Manning BD, Marjon K, Carracedo A, mTORC1-dependent AMD1 regulation sustains polyamine metabolism in prostate cancer. Nature 547, 109–113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stevens BM, O’Brien C, Jordan CT, Jones CL, Enriching for human acute myeloid leukemia stem cells using reactive oxygen species-based cell sorting. STAR Protoc. 2, 100248 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pei S, Pollyea DA, Gustafson A, Stevens BM, Minhajuddin M, Fu R, Riemondy KA, Gillen AE, Sheridan RM, Kim J, Costello JC, Amaya ML, Inguva A, Winters A, Ye H, Krug A, Jones CL, Adane B, Khan N, Ponder J, Schowinsky J, Abbott D, Hammes A, Myers JR, Ashton JM, Nemkov T, D’Alessandro A, Gutman JA, Ramsey HE, Savona MR, Smith CA, Jordan CT, Monocytic subclones confer resistance to venetoclax-based therapy in patients with acute myeloid leukemia. Cancer Discov. 10, 536–551 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pei S, Shelton IT, Gillen AE, Stevens BM, Gasparetto M, Wang Y, Liu L, Liu J, Brunetti TM, Engel K, Staggs S, Showers W, Sheth AI, Amaya ML, Minhajuddin M, Winters A, Patel SB, Tolison H, Krug AE, Young TN, Schowinsky J, McMahon CM, Smith CA, Pollyea DA, Jordan CT, A novel type of monocytic leukemia stem cell revealed by the clinical use of venetoclax-based therapy. Cancer Discov. 13, 2032–2049 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tabe Y, Lorenzi PL, Konopleva M, Amino acid metabolism in hematologic malignancies and the era of targeted therapy. Blood 134, 1014–1023 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsai HJ, Jiang SS, Hung WC, Borthakur G, Lin SF, Pemmaraju N, Jabbour E, Bomalaski JS, Chen YP, Hsiao HH, Wang MC, Kuo CY, Chang H, Yeh SP, Cortes J, Chen LT, Chen TY, A phase II study of arginine deiminase (ADI-PEG20) in relapsed/refractory or poor-risk acute myeloid leukemia patients. Sci. Rep 7, 11253 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen CL, Hsu SC, Ann DK, Yen Y, Kung HJ, Arginine signaling and cancer metabolism. Cancers 13, 3451 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pegg AE, Spermidine/spermine-N1-acetyltransferase: A key metabolic regulator. Am. J. Physiol. Endocrinol. Metabol 294, E995–E1010 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Sinicrope FA, Velamala PR, Song L, Viggiano TR, Bruining DH, Rajan E, Gostout CJ, Kraichely RE, Buttar NS, Schroeder KW, Kisiel JB, Larson MV, Sweetser SR, Sedlack RR, Sinicrope SN, Richmond E, Umar A, Della’Zanna G, Noaeill JS, Meyers JP, Foster NR, Efficacy of difluoromethylornithine and aspirin for treatment of adenomas and aberrant crypt foci in patients with prior advanced colorectal neoplasms. Cancer Prev. Res 12, 821–830 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Messing E, Kim KM, Sharkey F, Schultz M, Parnes H, Kim D, Saltzstein D, Wilding G, Randomized prospective phase III trial of difluoromethylornithine vs placebo in preventing recurrence of completely resected low risk superficial bladder cancer. J. Urol 176, 500–504 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Wang L, Liu Y, Qi C, Shen L, Wang J, Liu X, Zhang N, Bing T, Shangguan D, Oxidative degradation of polyamines by serum supplement causes cytotoxicity on cultured cells. Sci. Rep 8, 10384 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE, A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367, 645–648 (1994). [DOI] [PubMed] [Google Scholar]
- 38.Kang HA, Hershey JW, Effect of initiation factor eIF-5A depletion on protein synthesis and proliferation of Saccharomyces cerevisiae. J. Biol. Chem 269, 3934–3940 (1994). [PubMed] [Google Scholar]
- 39.Park MH, Nishimura K, Zanelli CF, Valentini SR, Functional significance of eIF5A and its hypusine modification in eukaryotes. Amino Acids 38, 491–500 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henderson A, Hershey JW, Eukaryotic translation initiation factor (eIF) 5A stimulates protein synthesis in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A 108, 6415–6419 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang E, Zhou H, Nadorp B, Cayanan G, Chen X, Yeaton AH, Nomikou S, Witkowski MT, Narang S, Kloetgen A, Thandapani P, Ravn-Boess N, Tsirigos A, Aifantis I, Surface antigen-guided CRISPR screens identify regulators of myeloid leukemia differentiation. Cell Stem Cell 28, 718–731.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saksouk N, Avvakumov N, Champagne KS, Hung T, Doyon Y, Cayrou C, Paquet E, Ullah M, Landry AJ, Côté V, Yang XJ, Gozani O, Kutateladze TG, Côté J, HBO1 HAT complexes target chromatin throughout gene coding regions via multiple PHD finger interactions with histone H3 tail. Mol. Cell 33, 257–265 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MacPherson L, Anokye J, Yeung MM, Lam EYN, Chan YC, Weng CF, Yeh P, Knezevic K, Butler MS, Hoegl A, Chan KL, Burr ML, Gearing LJ, Willson T, Liu J, Choi J, Yang Y, Bilardi RA, Falk H, Nguyen N, Stupple PA, Peat TS, Zhang M, de Silva M, Carrasco-Pozo C, Avery VM, Khoo PS, Dolezal O, Dennis ML, Nuttall S, Surjadi R, Newman J, Ren B, Leaver DJ, Sun Y, Baell JB, Dovey O, Vassiliou GS, Grebien F, Dawson SJ, Street IP, Monahan BJ, Burns CJ, Choudhary C, Blewitt ME, Voss AK, Thomas T, Dawson MA, HBO1 is required for the maintenance of leukaemia stem cells. Nature 577, 266–270 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Au YZ, Gu M, De Braekeleer E, Gozdecka M, Aspris D, Tarumoto Y, Cooper J, Yu J, Ong SH, Chen X, Tzelepis K, Huntly BJP, Vassiliou G, Yusa K, KAT7 is a genetic vulnerability of acute myeloid leukemias driven by MLL rearrangements. Leukemia 35, 1012–1022 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baell JB, Leaver DJ, Hermans SJ, Kelly GL, Brennan MS, Downer NL, Nguyen N, Wichmann J, McRae HM, Yang Y, Cleary B, Lagiakos HR, Mieruszynski S, Pacini G, Vanyai HK, Bergamasco MI, May RE, Davey BK, Morgan KJ, Sealey AJ, Wang B, Zamudio N, Wilcox S, Garnham AL, Sheikh BN, Aubrey BJ, Doggett K, Chung MC, de Silva M, Bentley J, Pilling P, Hattarki M, Dolezal O, Dennis ML, Falk H, Ren B, Charman SA, White KL, Rautela J, Newbold A, Hawkins ED, Johnstone RW, Huntington ND, Peat TS, Heath JK, Strasser A, Parker MW, Smyth GK, Street IP, Monahan BJ, Voss AK, Thomas T, Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature 560, 253–257 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Goyal L, Supko JG, Berlin J, Blaszkowsky LS, Carpenter A, Heuman DM, Hilderbrand SL, Stuart KE, Cotler S, Senzer NN, Chan E, Berg CL, Clark JW, Hezel AF, Ryan DP, Zhu AX, Phase 1 study of N1,N-diethylnorspermine (DENSPM) in patients with advanced hepatocellular carcinoma. Cancer Chemother. Pharmacol 72, 1305–1314 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Wolff AC, Armstrong DK, Fetting JH, Carducci MK, Riley CD, Bender JF, Casero RA Jr., Davidson NE, A phase II study of the polyamine analog N1,N11-diethylnorspermine (DENSpm) daily for five days every 21 days in patients with previously treated metastatic breast cancer. Clin. Cancer Res 9, 5922–5928 (2003). [PubMed] [Google Scholar]
- 48.Hahm HA, Ettinger DS, Bowling K, Hoker B, Chen TL, Zabelina Y, Casero RA Jr., Phase I study of N1,N11-diethylnorspermine in patients with non-small cell lung cancer. Clin. Cancer Res 8, 684–690 (2002). [PubMed] [Google Scholar]
- 49.Bello-Fernandez C, Packham G, Cleveland JL, The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc. Natl. Acad. Sci. U.S.A 90, 7804–7808 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.LoGiudice N, Le L, Abuan I, Leizorek Y, Roberts SC, Alpha-difluoromethylornithine, an irreversible inhibitor of polyamine biosynthesis, as a therapeutic strategy against hyperproliferative and infectious diseases. Med. Sci 6, 12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stubbins RJ, Karsan A, Differentiation therapy for myeloid malignancies: Beyond cytotoxicity. Blood Cancer J. 11, 193 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luo Y, Shao L, Chang J, Feng W, Liu YL, Cottler-Fox MH, Emanuel PD, Hauer-Jensen M, Bernstein ID, Liu L, Chen X, Zhou J, Murray PJ, Zhou D, M1 and M2 macrophages differentially regulate hematopoietic stem cell self-renewal and ex vivo expansion. Blood Adv. 2, 859–870 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kantarjian HM, O’Brien S, Cortes J, Homoharringtonine/omacetaxine mepesuccinate: The long and winding road to food and drug administration approval. Clin. Lymphoma Myeloma Leuk 13, 530–533 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Brien C, Ling T, Berman JM, Culp-Hill R, Reisz JA, Rondeau V, Jahangiri S, St-Germain J, Macwan V, Astori A, Zeng A, Hong JY, Li M, Yang M, Jana S, Gamboni F, Tsao E, Liu W, Dick JE, Lin H, Melnick A, Tikhonova A, Arruda A, Minden MD, Raught B, D’Alessandro A, Jones CL, Simultaneous inhibition of Sirtuin 3 and cholesterol homeostasis targets acute myeloid leukemia stem cells by perturbing fatty acid β-oxidation and inducing lipotoxicity. Haematologica 108, 2343–2357 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, Barnes I, Berry A, Bignell A, Sala SC, Chrast J, Cunningham F, Di Domenico T, Donaldson S, Fiddes IT, Girón CG, Gonzalez JM, Grego T, Hardy M, Hourlier T, Hunt T, Izuogu OG, Lagarde J, Martin FJ, Martínez L, Mohanan S, Muir P, Navarro FCP, Parker A, Pei B, Pozo F, Ruffier M, Schmitt BM, Stapleton E, Suner MM, Sycheva I, Uszczynska-Ratajczak B, Xu J, Yates A, Zerbino D, Zhang Y, Aken B, Choudhary JS, Gerstein M, Guigó R, Hubbard TJP, Kellis M, Paten B, Reymond A, Tress ML, Flicek P, GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liao Y, Smyth GK, Shi W, featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 58.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, Sergushichev A, Fast gene set enrichment analysis. bioRxiv 060012 [Preprint] (2021); 10.1101/060012. [DOI] [Google Scholar]
- 61.Merico D, Isserlin R, Stueker O, Emili A, Bader GD, Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLOS ONE 5, e13984 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Benjamini Y, Hochberg Y, Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat 57, 289–300 (1995). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. All materials are commercially available. RNA-seq data are available at Gene Expression Omnibus (GSE274658). Proteomics data are available through MassIVE (MSV000093543).