

Abstract
BACKGROUND:
Noninvasive, precision monitoring of hepatocellular carcinoma (HCC) treatment efficacy would greatly facilitate personalized therapy and improve patient outcomes. We hypothesize that quantifying methylated circulating tumor DNA (ctDNA) can be used to effectively monitor HCC burden without the need for biopsy.
STUDY DESIGN:
Blood samples were collected from 25 patients, 21 with HCC and 4 with benign liver masses, at various timepoints throughout the course of treatment at a high-volume academic medical center. Quantification of methylated ctDNA molecules assessed CpG sites on more than 550 preselected cancer-specific amplicons. The tumor methylation score (TMS) was calculated by measuring the difference between the amount of methylation in the plasma and buffy coat with a normal cutoff value of 120 or less.
RESULTS:
Among 10 patients with surgical HCC (5 surgical resections and 5 liver transplants), TMS revealed a statistically significant, rapid postoperative decline in 9. One patient who had a persistently elevated TMS on postoperative day 1 was subsequently found to have had metastatic disease. Patients in the negative control cohort all had normal-range pre- and postoperative TMS. Preoperative TMS correlated moderately with tumor burden on pathology (Spearman r = 0.54) of surgical specimens. From 11 subjects undergoing systemic therapy or Y90 radioembolization, analysis of 16 time periods demonstrated that the change in TMS (ΔTMS) was better associated with tumor progression than the change in Δalpha-fetoprotein (area under the curve 0.800 and 0.783, respectively). A composite score combining ΔTMS and Δalpha-fetoprotein further improved performance for detecting tumor progression with an area under the curve of 0.892.
CONCLUSIONS:
These findings indicate that ctDNA methylation scores can effectively evaluate changes in tumor burden without the need for tumor biopsy.
With hepatocellular carcinoma (HCC) already being the third highest cause of cancer mortality worldwide, the impact of this disease is projected to increase an additional 50% for the next 20 years.1,2 Early diagnosis significantly improves prognosis, especially as early-stage HCC can usually be treated surgically, providing the best rates of survival.1,3,4 In cases where surgical management is not possible, alternative options such as systemic therapies or locoregional treatments, including ablation or transarterial chemo- or radioembolization, may be employed as a primary treatment or as a downstaging strategy.1,3 Among these patients, frequent and real-time monitoring holds the potential to greatly enhance prognosis by enabling physicians to assess treatment response and make individualized, timely decisions.
Currently, alpha-fetoprotein (AFP) is the most commonly used biomarker for HCC monitoring,2,5 but this method is limited by its low sensitivity, missing up to 40% of cases.6,7 Although other markers have been explored, none have been validated in a large enough cohort to become routinely used.2 In recent years, circulating tumor DNA (ctDNA) quantification has emerged as a promising method for “liquid biopsies,” allowing minimally invasive detection of tumors through blood samples.8 Some studies even suggest that ctDNA can detect disease progression or recurrence more reliably than AFP.9 ctDNA consists of extracellular DNA fragments that are shed into the systemic circulation by tumor cells.8 The noninvasive nature of liquid biopsies permits frequent, dynamic monitoring throughout treatment, decreasing risks and discomfort of an invasive procedure and reducing reliance on potentially inaccurate tumor biopsies, which may yield inaccurate or nonrepresentative results in cases of heterogeneous tumors depending on the biopsy sampling location.8
Distinction of ctDNA from normal circulating DNA can be done through various methods.10 Mutation-based ctDNA quantification is limited by a low sensitivity of 65%,11 primarily due to the extensive variability of mutations. Even the most common mutations are not consistently present. For instance, telomerase reverse transcriptase promoter mutations, considered the most common in HCC, are estimated to be present in only up to 59% of cases.12 Given the inconsistency of common mutations across patients, mutation-based ctDNA quantification typically requires analysis of tumor tissue specimens to identify the mutations to be looked for within the circulating cell-free DNA.
DNA methylation is an epigenetic mechanism that involves the addition of a methyl group to a cytosine component in CpG sites, a process that typically leads to the silencing of gene expression.13,14 Because the methylation of tumor suppressor genes is involved in the development of malignancies, tumors typically exhibit distinctive methylation patterns, which can be used to quantify ctDNA.13 Methylation patterns identified in ctDNA display a significantly higher level of consistency when compared with mutations, which exhibit considerable variability and low sensitivity.8,13 Therefore, we hypothesize that quantifying methylation patterns in a panel of preselected cancer-specific amplicons will allow for an efficient way to monitor HCC burden while sparing the need for invasive tumor biopsies.
METHODS
Study cohort
A total of 25 patients were included in this study. Informed consent was obtained from all recipients before enrollment. This study was approved by the IRB at the University of Florida. All research was conducted in concordance with the Declaration of Helsinki. Detailed demographic data, including age, sex, race, ethnicity, BMI, diagnosis, and treatment regimens, were collected (Table 1). The study was structured into 2 steps with specific objectives, and patients were enrolled accordingly.
Step 1: The primary aim of this step was to assess the method’s capacity to differentiate patients with HCC from those without and to detect substantial changes in tumor burden. To this end, in step 1, we included patients undergoing surgical treatment for HCC or for benign hepatic cysts.
Step 2: On confirmation of the method’s feasibility and ability to distinguish the presence and absence of HCC, the study progressed to the second step, which aimed to investigate the method’s ability to detect more subtle changes in tumor burden. In step 2, we enrolled eleven patients undergoing nonsurgical treatment.
Table 1.
Patient Demographics and Medical History
| Variable | Hepatocellular carcinoma patients | Control patients |
|---|---|---|
| Age, y, median (IQR) | 63 (10) | 61 (45.5) |
| Sex, n (%) | ||
| Male | 17 (81.0) | 1 (25) |
| Female | 4 (19.0) | 3 (75) |
| BMI, kg/m2, median (IQR) | 24.8 (8.7) | 31.0 (15.3) |
| Race, n (%) | ||
| White | 16 (76.2) | 4 (100) |
| Black | 4 (19.0) | 0 (0) |
| Other | 1 (4.8) | 0 (0) |
| Ethnicity, n (%) | ||
| Non-Hispanic or Latino | 19 (90.5) | 3 (75) |
| Hispanic or Latino | 2 (9.5) | 0 (0) |
| American or Alaska Native | 0 (0) | 1 (25) |
| Underlying disease, n (%) | ||
| Alcoholic cirrhosis | 4 (19.0) | N/A |
| Nonalcoholic steatohepatitis cirrhosis | 5 (23.8) | N/A |
| Hepatitis C virus cirrhosis | 10 (47.6) | N/A |
| Other | 2 (9.6) | N/A |
| Treatment type, n (%) | ||
| Surgical resection | 5 (23.8) | 4 (100) |
| Liver transplantation | 5 (23.8) | 0 (0) |
| Systemic therapy | 9 (42.8) | 0 (0) |
| Radioembolization | 2 (9.6) | 0 (0) |
Sample collection and processing
Blood samples were collected at various timepoints throughout the course of treatment at a high-volume academic medical center. From surgical patients, 1 sample was collected preoperatively and up to 2 samples were collected postoperatively, at timepoint 1 (1 to 60 days after the operation) and timepoint 2 (61 to 230 days after the operation). Samples were collected into anticoagulant EDTA K2 tubes and centrifuged at 4° C at 1,000g for 15 minutes to separate the components into buffy coat and plasma fractions. The resulting fractions were stored at −80° C for subsequent ctDNA quantification.
Circulating tumor DNA quantification
For quantification of methylated ctDNA molecules, CpG sites were assessed on more than 550 preselected cancer-specific amplicons as previously described.15 The tumor methylation score (TMS), representing the number of methylated tumor molecules per 1,000 input genomic equivalents, was calculated by measuring the difference between the number of methylated molecules per 1,000 genomic equivalents in the plasma and buffy coat. A cut-off value of 120 has been established for differentiating cancer-positive and cancer-negative samples with the use of healthy volunteers.15
Statistical analysis
Wilcoxon paired tests or Kruskal–Wallis and Dunn’s multiple comparisons tests were conducted to determine significant differences in TMS and AFP. Correlation analysis was performed between TMS and tumor burden. The Youden index was used to choose the cut point for dichotomization. Receiver operating characteristic (ROC) curves and Fisher’s exact tests were performed to evaluate test performance. A p value of <0.05 was considered statistically significant. All statistical analyses were performed using the GraphPad Prism version 10.0.2 for Windows (Boston, MA) or JMP Pro 17.0.0 (Cary, NC).
RESULTS
Distinguishing major tumor burden changes
Fourteen patients, consisting of 5 undergoing surgical resection for HCC, 5 receiving liver transplants for HCC, and 4 undergoing surgical resections of benign hepatic masses, serve as a negative control group.
Comparative analysis of the preoperative vs postoperative TMS scores from the HCC surgical patients revealed that the preoperative TMS was significantly higher than the first (p = 0.008) and second (p = 0.031) postoperative TMS measurements, revealing a consistent postoperative decline in 9 of 10 patients with HCC (Fig. 1A). Comparative analysis of change in AFP (ΔAFP) throughout postoperative course showed no statistically significant differences between preoperative and postoperative values (Fig. 2). One patient who underwent resection exhibited a persistently elevated TMS on postoperative day 1. This patient is not included in the quantitative analysis as the patient had a very early recurrence indicating metastatic disease at the time of resection and persistent tumor burden. On postoperative day 14, abdominal CT showed a metastatic subxiphoid lesion. The patient was subsequently diagnosed with widespread metastasis to the brain, bones, and lung. This patient’s preoperative score was 26,700, and the postoperative score was 39,400. The postoperative TMS from this patient was collected on postoperative day 1. As the half-life of ctDNA is estimated to be up to 2 hours,15 it is unlikely that the increase in TMS was related to the primary tumor. A possible explanation is that the surgical stress prompted the metastatic tumor to increase shedding of ctDNA.
Figure 1.
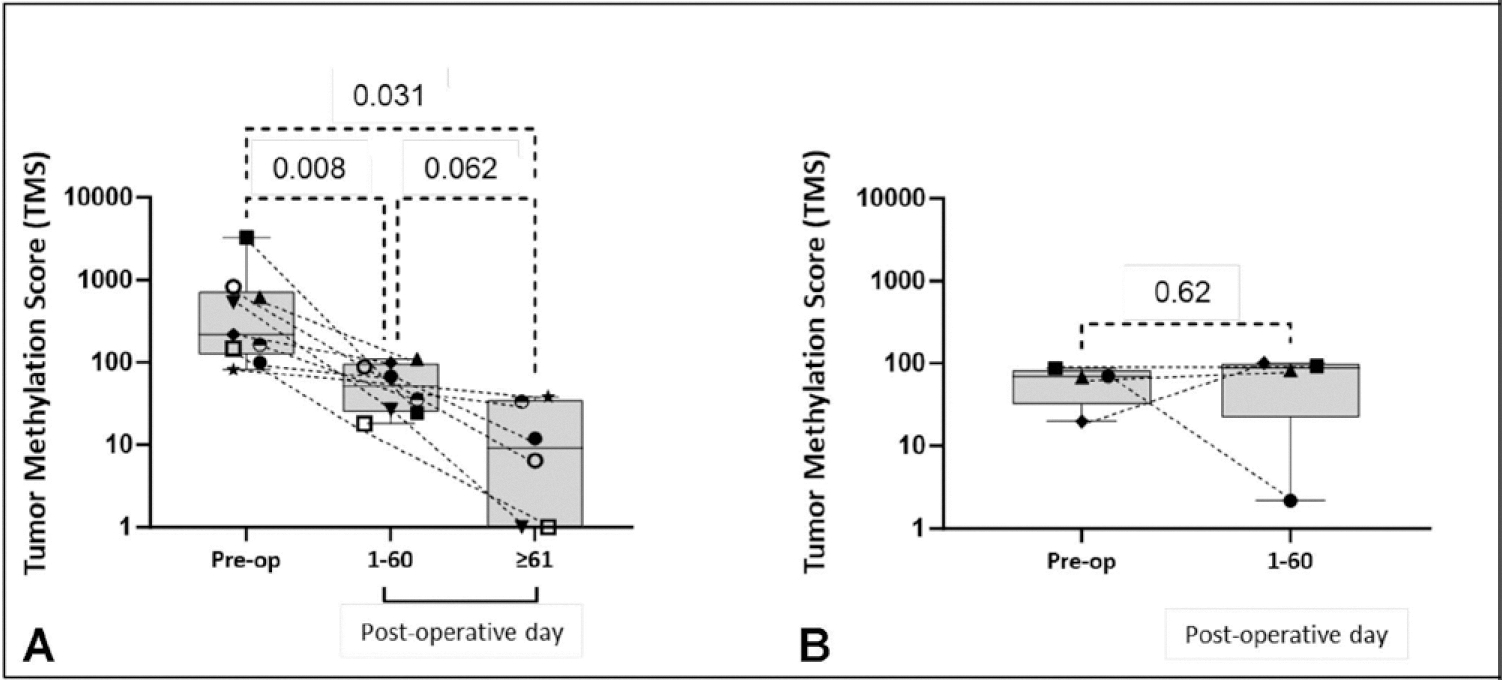
Change in tumor methylation score (TMS) after resection or transplantation for hepatocellular carcinoma (HCC). (A) HCC resection and transplant patients; and (B) control. Dashed lines connect TMS of each patient in their respective postoperative courses.
Figure 2.
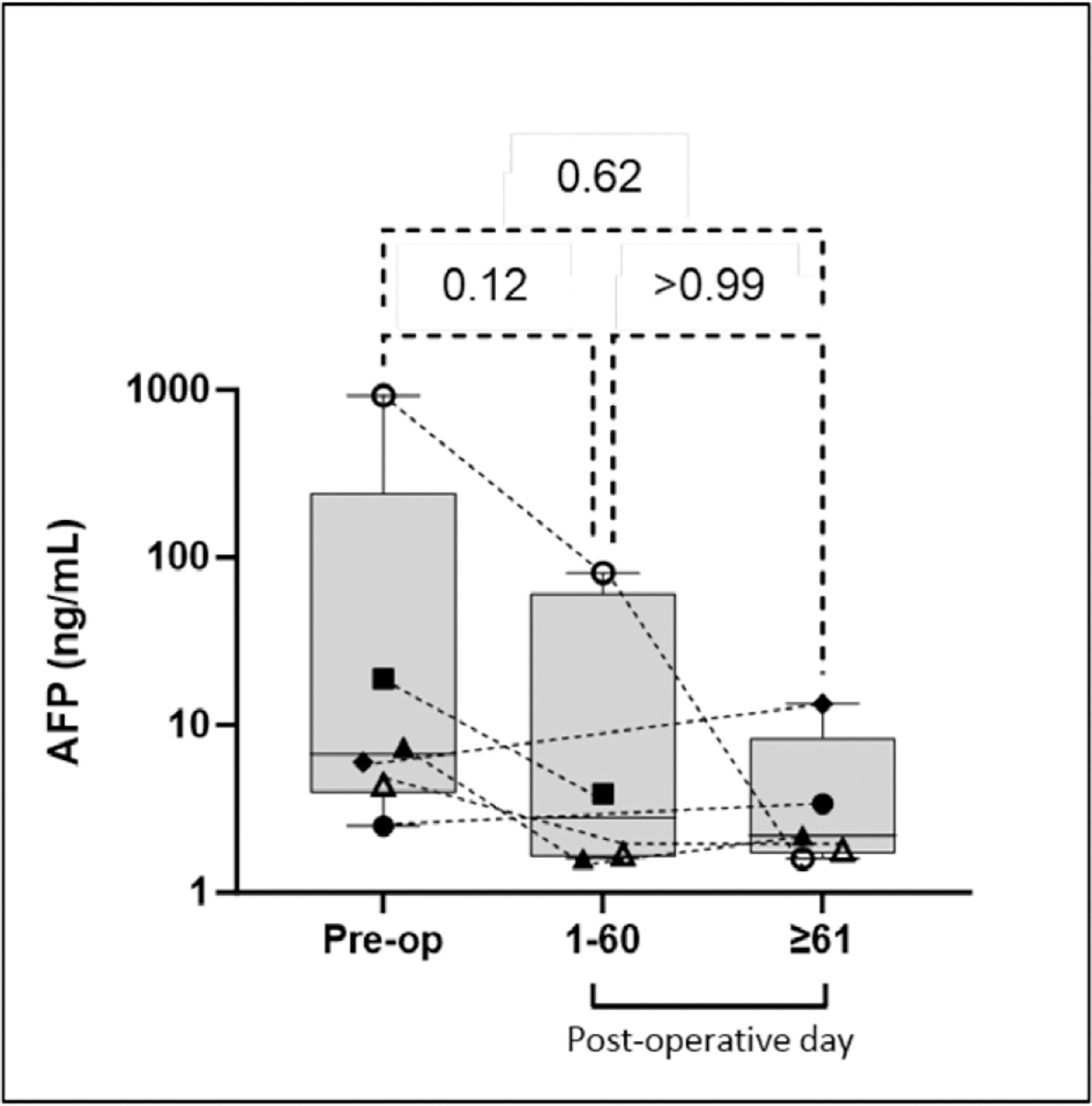
Change in alpha-fetoprotein (AFP) after resection or transplantation for hepatocellular carcinoma. The dashed lines connect the AFP of each patient in their respective postoperative courses.
Among this cohort, preoperative TMS was higher than the cutoff in 8 of 10 patients, with a sensitivity of 80% in detecting tumor presence, compared with AFP with a sensitivity of 40% (Table 2). Of note, the 2 instances in which TMS was negative coincided with negative AFP. In the first instance, the patient had very low-grade tumor, classified as well-differentiated by the pathologist. The second patient had undergone Y90 therapy 46 days before transplantation and, although the pathologist observed 30% of remaining viable tumor, the tumor injury and necrosis may have impacted the ctDNA production. Analysis of the negative control cohort revealed that all subjects had normal-range TMS in both pre- and postoperative samples with no significant change (Fig. 1B), determining a specificity of 100%. Notably, AFP was not routinely measured in the negative control patients, so its specificity could not be adequately determined.
Table 2.
Comparison between Tumor Methylation Score and Alpha-Fetoprotein in Detecting Tumor Presence among Surgical Patients
| Variable | Tumor methylation score, % | Alpha-fetoprotein, % |
|---|---|---|
| Sensitivity | 80 | 40 |
| Specificity | 100 | N/A |
N/A, not applicable.
To assess whether TMS was directly proportional to tumor burden, we performed a correlation analysis between preoperative TMS and tumor burden on pathology among surgical patients. Tumor burden was defined as the largest diameter of viable tumor, as described by pathology. The analysis demonstrated a moderate positive association, with a Spearman r value of 0.54 (Fig. 3).
Figure 3.
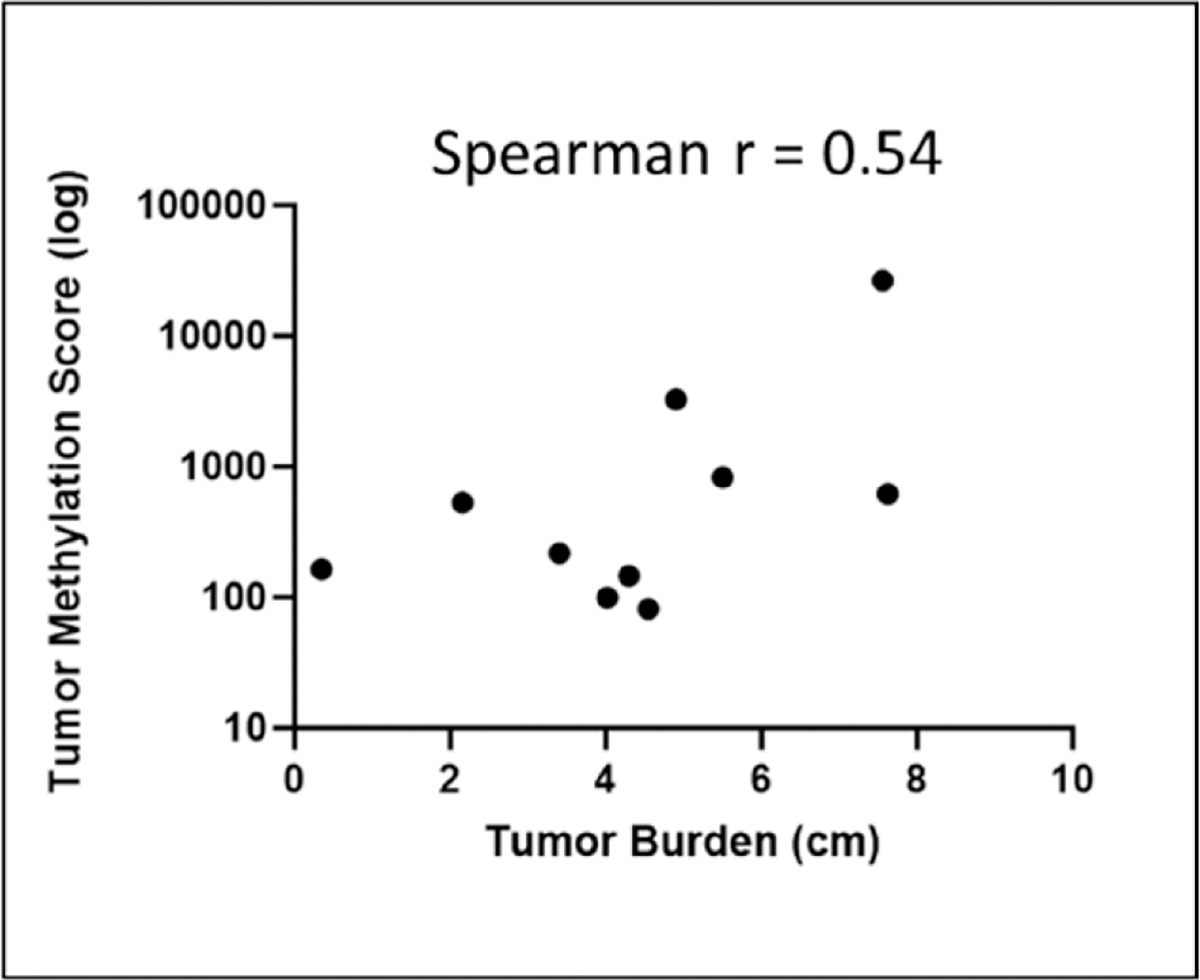
Correlation analysis between tumor burden (longest dimension of viable tumor) and tumor methylation score showed a moderate positive correlation.
These findings suggest that methylation-based ctDNA can accurately differentiate the presence and absence of HCC, prompting the continuation to the next step.
Distinguishing minor tumor burden changes
To assess the efficacy of methylated ctDNA quantification in detecting more subtle changes in tumor burden, we analyzed 16 time periods from 9 different patients receiving systemic therapy and 2 patients undergoing Y90 radioembolization. Systemic therapy regimens varied among patients, including nivolumab, lenvatinib, or atezolizumab plus bevacizumab. Time periods were defined as the time between pairs of blood samples and were categorized into “progression,” “stable,” or “response” groups based on modified Response Evaluation Criteria in Solid Tumors. The change in TMS (ΔTMS) and change in AFP (ΔAFP) during each time period were calculated. Comparative analysis revealed that ΔTMS distinguished periods of response (complete or partial response) from periods of disease progression (Fig. 4). Conversely, ΔAFP did not distinguish the groups in this analysis. These results suggest that ΔTMS is more effective at detecting tumor variations than ΔAFP (Fig. 3).
Figure 4.
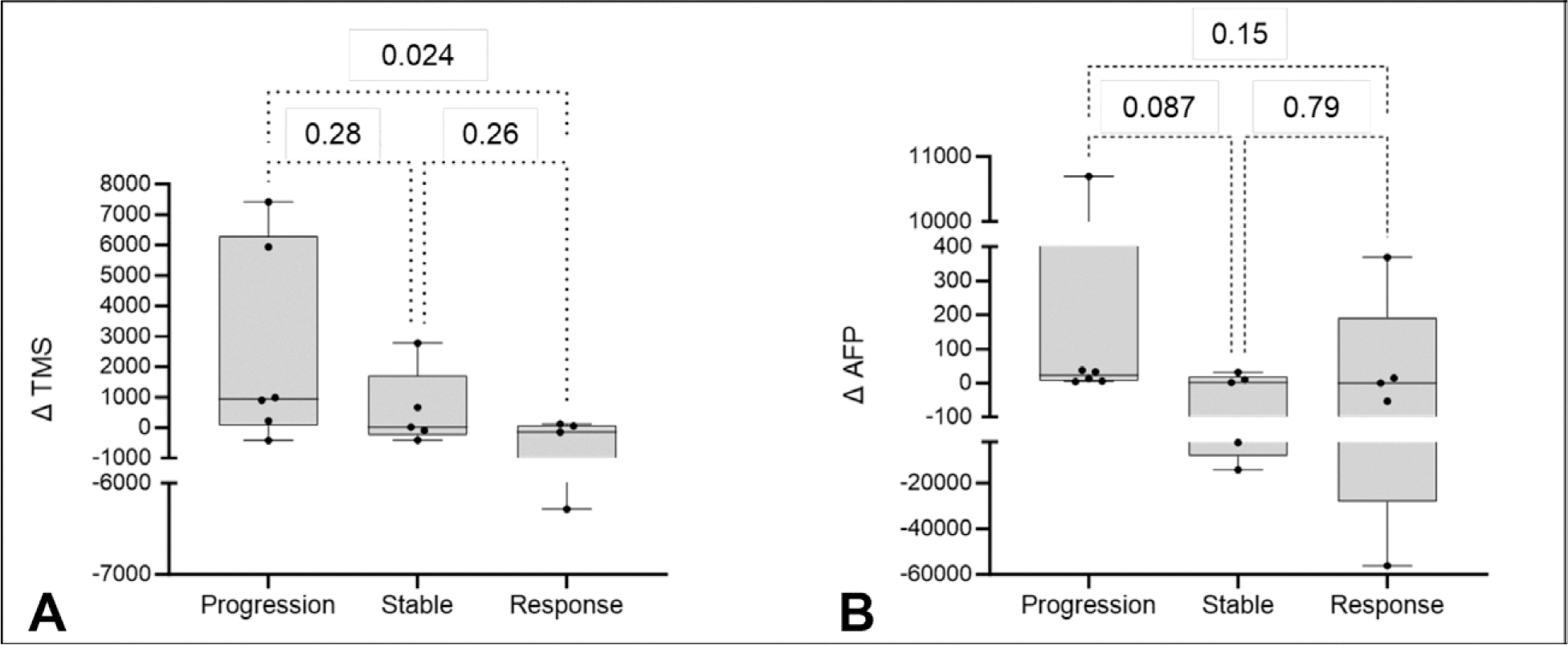
Among systemic therapy and radioembolization patients, (A) Δtumor methylation score was better associated with tumor progression and response than (B) Δalpha-fetoprotein.
Additionally, ROC curves were performed comparing periods with progression vs periods with tumor response or stability. In this cohort, ΔTMS exhibited an area under the curve (AUC) of 0.800, superior to ΔAFP with an AUC of 0.783 (Fig. 5). Using the Youden index to derive cutoffs of 222 for ΔTMS and 4 ng/mL for ΔAFP, we compared the performance of each test with their respective cutoffs in distinguishing tumor progression from stability or response and found that both ΔTMS and ΔAFP were significantly different between the groups (p = 0.035 and 0.034, respectively). Using these cutoffs, ΔTMS had a sensitivity of 83% and specificity of 80%, whereas ΔAFP had a sensitivity of 100%, but a specificity of 60%.
Figure 5.
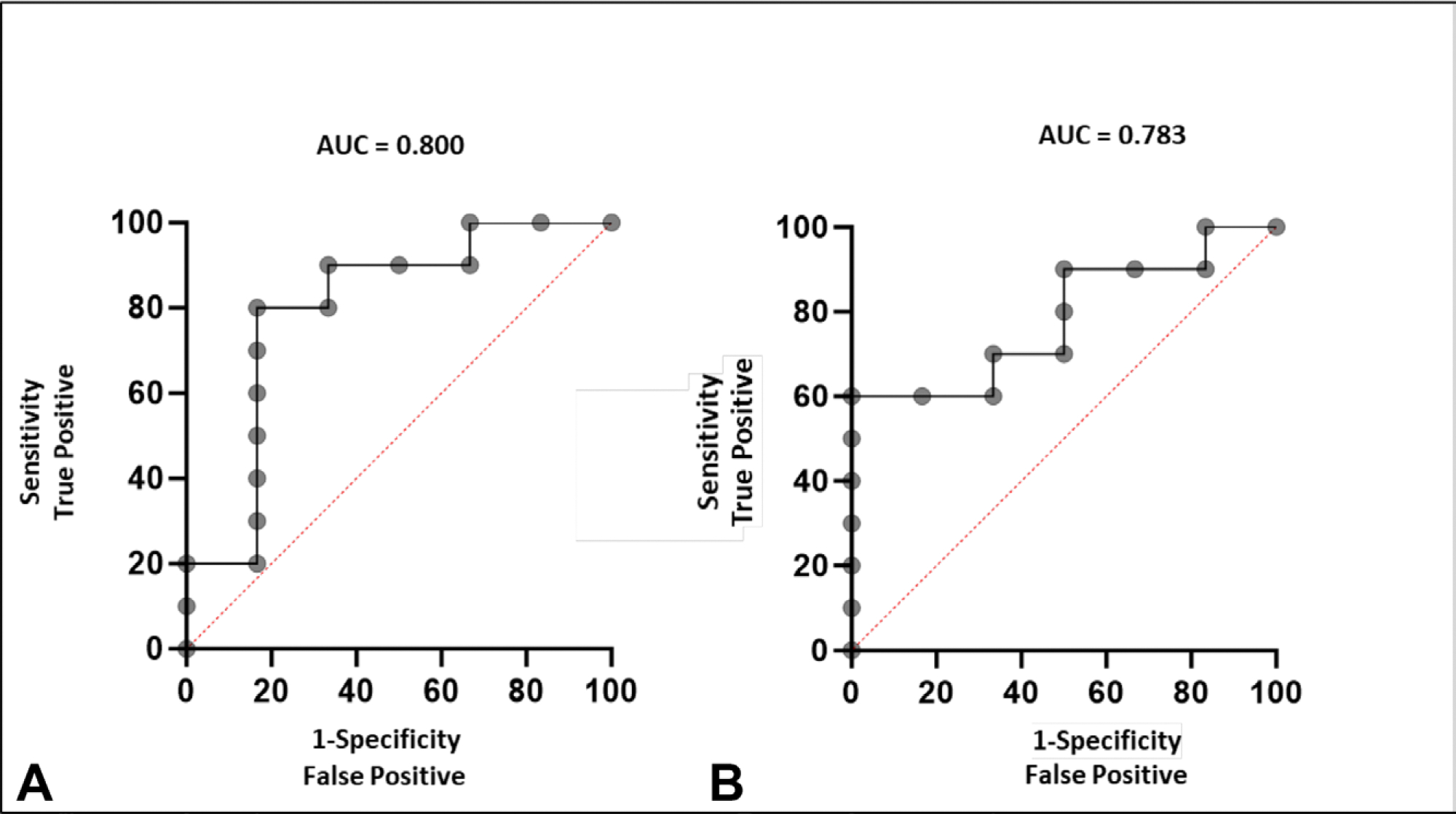
Receiver operating characteristic curves comparing the performance of (A) Δtumor methylation score and (B) Δalpha-fetoprotein in distinguishing periods of tumor progression from periods of tumor response or stability.
A composite score was formulated using a combination of ΔTMS and ΔAFP. The description of the scoring system is described in Table 3. The respective cutoff values were previously defined using the Youden index. ROC curve analysis revealed an improved performance with an AUC of 0.892 (Fig. 6). Employing a cutoff of 4 points, determined by Youden index analysis, the composite score exhibited a sensitivity of 83% and a specificity of 90%. Additionally, Fisher’s exact test was performed and revealed that the cutoff of 4 points could significantly distinguish tumor progression from tumor response or stability (p = 0.008).
Table 3.
Composite Score Combining ΔTumor Methylation Score and ΔAlpha-Fetoprotein
| Variable | 0 point | 1 point | 2 points |
|---|---|---|---|
| ΔTumor methylation score | ≤0 | 1–222 | >222 |
| ΔAlpha-fetoprotein | ≤0 | 1–4 | >4 |
Figure 6.
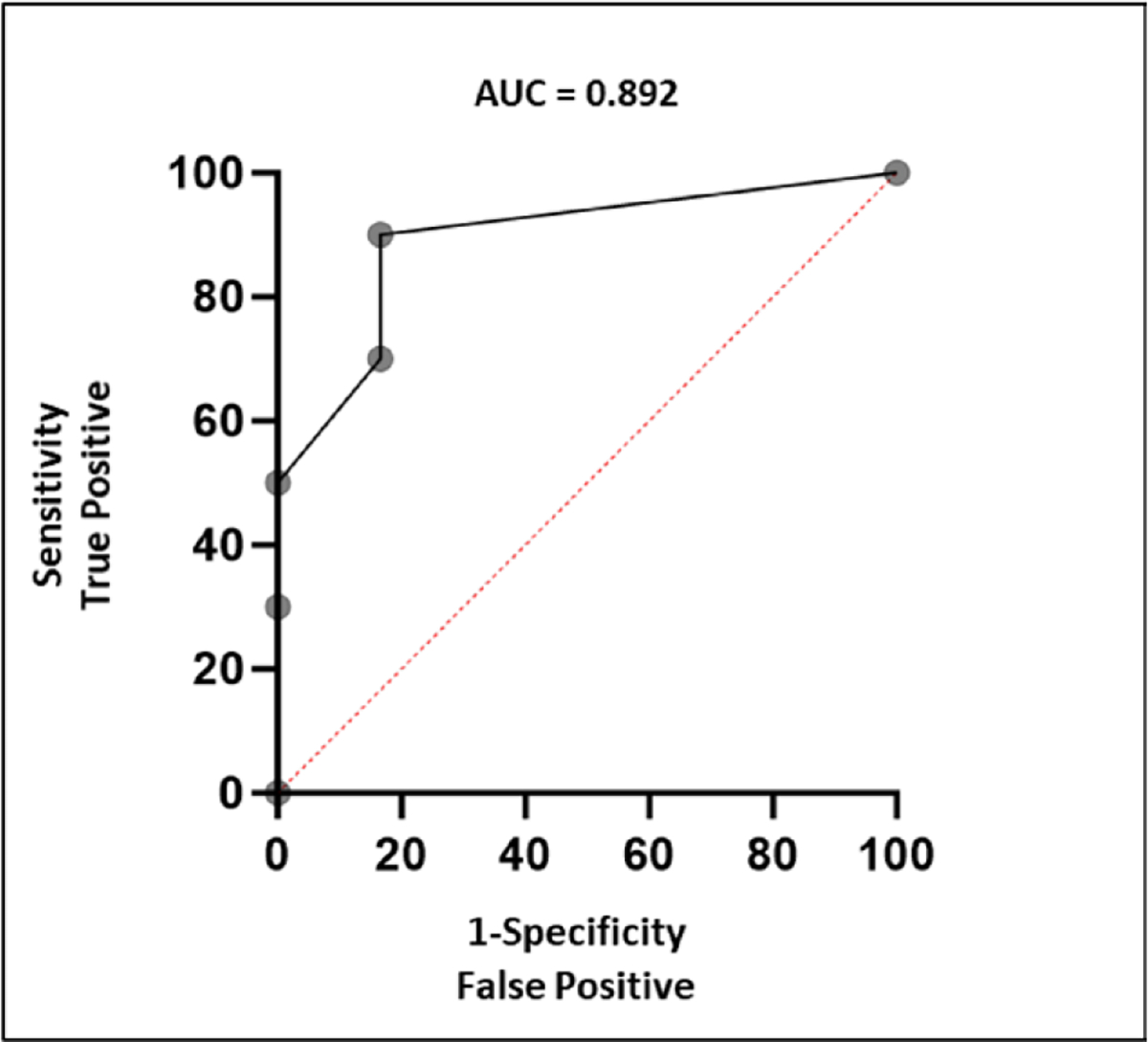
Receiver operating characteristic curve showing the performance of the composite score combining Δtumor methylation score and Δalpha-fetoprotein in detecting periods of tumor progression. AUC, area under the curve.
Overall diagnostic performance of tumor methylation score and alpha-fetoprotein
In total, TMS was positive in 17 of 21 patients with HCC, with a sensitivity of 80.9% (Table 4). Notably, there were no false-positive results in the negative control cohort, indicating a specificity of 100%. Conversely, AFP, using a cutoff value of 20 ng/mL in the same patients and at the same timepoints, exhibited a markedly lower sensitivity of 51.7%, with positive AFP values recorded in only 12 of 21 patients with HCC. AFP values were not consistently measured in the negative control patients as there was no suspicion for malignancy.
Table 4.
Comparison Between Tumor Methylation Score and Alpha-Fetoprotein in Overall Cohort
| Variable | Tumor methylation score, % | Alpha-fetoprotein, % |
|---|---|---|
| Sensitivity | 80.9 | 51.7 |
| Specificity | 100 | N/A |
N/A, not applicable.
DISCUSSION
With a high mortality rate globally,1,2 it is extremely important to find a reliable blood-based biomarker for diagnosis and monitoring of HCC. AFP, the current standard-of-care blood test for HCC monitoring, is hampered by its low sensitivity.6,7 In recent years, liquid biopsies have emerged as alternative methods for HCC diagnosis and monitoring,10 although none have yet achieved routine clinical implementation. In this study, we introduce a novel methylated ctDNA quantification method, which assesses a preselected panel of more than CpG sites to quantify TMS and evaluate its performance in HCC monitoring.
Our data indicate that TMS effectively detected the presence of HCC and distinguished it from noncancerous patients with benign hepatic lesions. We also found that TMS was a sensitive method for detecting changes in tumor burden, both in cases with substantial changes, such as after surgical removal of HCC via hepatic resection or transplantation and in more subtle tumor burden fluctuations observed among patients undergoing systemic therapy or radioembolization—treatments associated with progressive, slower tumor burden changes. We also encountered a patient who exhibited an increased TMS score after surgery, who notably was later determined to have metastatic disease.
Comparison with alpha-fetoprotein
In comparison to AFP, TMS demonstrated superior performance across various aspects. In our study, the overall sensitivity of AFP in detecting the presence of HCC was of 51.7%, whereas TMS identified HCC in 80.9% of patients. Focusing on the surgical cohort alone, the only 2 instances in which TMS was negative coincided with negative AFP. Of those 2 patients, 1 had undergone Y90 therapy 46 days before the operation that was correlated with the blood sample and TMS measurement, and the other had a tumor classified as very low grade. Regarding the assessment of major tumor burden changes, comparison of TMS before and after surgery revealed a statistically significant decrease postoperatively, whereas the same did not apply to AFP (Fig. 1).
Assessment of each method’s performance in detecting minor changes in tumor burden revealed that TMS had an overall performance superior to AFP. Quantitative analysis showed statistically significant differences in ΔTMS between periods of progression and response, whereas ΔAFP did not show such distinctions. After establishing cutoffs of 222 for ΔTMS and 4 ng/mL for ΔAFP, analysis showed that both tests could adequately and similarly distinguish tumor progression from stability or response. However, TMS had a slightly superior performance in detecting tumor progression with an AUC of 0.800, superseding AFPs AUC of 0.783. The shorter half-life of ctDNA of up to 2 hours,16 compared with AFPs half-life of 5 to 7 days,17 could explain the better performance of TMS. One factor that may have overestimated AFPs performance in step 2 was that within this cohort, 9 of 11 patients (82%) had AFP-producing HCC, whereas in the overall population, this number only reaches up to 60%.6,7
As both methods revealed similar performance in detecting tumor progression, we theorized that their synergy could achieve an enhanced performance. For this aim, we devised a composite score combining ΔTMS and ΔAFP, which revealed an impressive enhancement in performance, with an AUC of 0.892, sensitivity of 83% and specificity of 90%. This composite score shows promising results and could improve the clinical utility of both markers.
Comparison with other “liquid biopsies”
The term “liquid biopsy” encompasses methods intended to assess solid tumors through minimally invasive blood samples. Several methods are currently under study, including the assessment of circulating tumor cells, circulating miRNA, and ctDNA. In this context, ctDNA has stood out as one of the most promising.10 There are various methods that aim to distinguish ctDNA from nontumoral cell-free DNA. Gene integrity analysis focuses on the size of ctDNA molecule, noting that tumor DNA molecules in plasma are typically shorter. Somatic mutations are also widely explored as a potential method, which is faded to be limited by a low sensitivity due to the high variability and low frequency of these mutations.10 The most common mutations include telomerase reverse transcriptase, detected in 59% to 63% of patients, followed by TP53 in 48% and CTNNB1 in 37% of patients.18 A study reported a sensitivity of 68% in HCC stages I to III.19 Because it is hardly feasible to target a multitude of possible mutations, frequently mutation-based ctDNA quantification is often accompanied by tumor biopsy to identify target mutations, enabling personalized approaches.19,20 In contrast, methylated ctDNA is much more consistently present, not requiring a biopsy.
Methylation of tumor suppressor genes represents one of the first events that occur in tumor development,13 suggesting that methylated ctDNA could serve as a sensitive and early biomarker. Currently, methylation-based ctDNA quantification seems to be the most promising field and is the most intensely investigated ctDNA quantification method.9 It can be done by quantifying methylation sites, methylation site expression, or by 5-hydroxymethylcytosine detection.10,21 The current study focuses on quantifying methylated molecules at preselected sites. Many of earlier studies have tested this method by assessing the differential methylation in a limited number of markers.22,23 Huang and colleagues24 examined 7 CpG sites in the promoter region of INK4A and reached a sensitivity of 74.2%, Xu and colleagues8 tested a larger panel of 10 methylation sites across different genes, significantly increasing the sensitivity to 83%. This current study tested a panel of 550 cancer-specific, but not HCC-specific, amplicons, and had a sensitivity of 80.9% when identifying HCC in this cohort. Earlier studies on methylated ctDNA have primarily tested the method’s performance in diagnosing, or generally detecting the presence of HCC. However, a distinctive contribution of our study is that we have longitudinally followed up with patients undergoing systemic therapy, to investigate the method’s performance in assessing subtle changes in between treatments, and showed promising results as described previously.
Limitations
A recognized limitation of our study is the relatively small cohort. However, it is noteworthy that this limitation is not unique to our investigation and is the case of most studies on the topic to date.10,21 Another potential limitation is the effect of heterogeneity in the timing and handling of sample collections on the TMS values. The variations in sampling the timepoints are a direct reflection of the practical clinical environment, with patients coming to follow-up visits or for treatments at different times. Although recognizing these limitations, this preliminary study shows promising data that warrant further investigation in a larger and more diverse cohort.
CONCLUSIONS
In summary, the results of this study indicate that methylated ctDNA quantification can effectively evaluate changes in tumor burden while bypassing the requirement for invasive tumor biopsies. This method could be applied in the postoperative setting. Our results show that TMS consistently declined in patients postoperatively, and the only patient who had an increased TMS was subsequently found to have metastatic disease, therefore with persistent disease at the time of the sample. It also has the potential of being applied in personalized medicine in the selection of systemic therapies, as it can help detect whether a tumor is responding to a therapeutic regimen. It could further have the potential of improving patient prognosis by early detection of tumor unresponsiveness to treatment, allowing for timely changes to therapy and individualized therapeutic decisions. The results of this study represent a promising first step toward a reliable noninvasive HCC tumor burden monitoring method and encourage further studies with prospective data collection.
Acknowledgment:
We thank Patrick Ye and Sydne Langpap for providing extensive technical support and assistance in the interpretation of the TMS scores, as well as critical feedback for the writing of the manuscript.
Support:
This study was supported in part by the National Institutes of Health/National Center for Advancing Translational Sciences (grant no. 1UH2TR002087).
Abbreviations and Acronyms
- AFP
alpha-fetoprotein
- ctDNA
circulating tumor DNA
- HCC
hepatocellular carcinoma
- ROC
receiver operating characteristic
- TMS
tumor methylation score
Biographies
DR REBEKAH WHITE (La Jolla, CA): I want to emphasize how novel this work is. We have been using circulating tumor DNA (ctDNA) for several years, but this has been based almost exclusively on tumor mutation. So, the use of the methylation patterns is highly novel, and indeed liberating, for tumors such as hepatocellular carcinoma (HCC), in which we often do not know what mutations we are looking for and would rather not have to biopsy the tumor.
As you mentioned, the panel was derived as a pan-cancer marker. So, it is not specific for HCC. Did you look in parallel at patients undergoing liver resection for other malignancy, such as colorectal liver metastases?
Regarding the normalization process for the buffy coat layer, I presume this improved the performance of the assay in the preliminary studies, but it is known that surgery can affect DNA methylation patterns due to stress. That is 1 of the ways we regulate expression of immune genes.
So, do you or the company have those data on separating the plasma from the buffy coat layer? Did you see changes in the methylation patterns in the buffy coat layer in patients undergoing operation?
Regarding the sensitivity of 80%, it is not terrible, but it is also not great. Could you tell us a little more about the 2 patients who had a negative Tumor Methylation Score (TMS)? Were these small tumors? Did these patients perhaps not have cirrhosis?
Finally, how are we going to improve this assay? This is obviously an early version of the assay, so is the answer going to be to look at more or different markers, or are there further improvements in the technology to improve that sensitivity?
DR CHRISTOPHER WOLFGANG (New York, NY): For nearly a decade, we have heard a great deal of press about potential liquid biopsy in the management of cancer, yet in most of surgical oncology, liquid biopsy has not lived up to its potential or has not been integrated into our practice.
Liquid biopsy is a general term for the measurement of biomarkers within bodily fluid. It is not really a single test but can include circulating cell-free DNA, circulating tumor cells, extracellular vesicles, and proteins, among others. The excitement about liquid biopsy is that it has the potential to provide a molecular assessment of the primary tumor in the absence of an invasive biopsy. Just as importantly, it has the potential to provide real-time feedback and assessment of treatment response on a day-to-day basis, almost like a cancer complete blood count.
The test that was performed herein is a variation of a circulating cell-free DNA, which I thought was very innovative
Footnotes
Disclosure Information: Nothing to disclose.
Disclosures outside the scope of this work: Dr Hughes receives book royalties from Wolter’s Kluwer.
Disclaimer: Circulating tumor DNA testing was performed by BillionToOne, Inc.
Presented at the Southern Surgical Association 135th Annual Meeting, Hot Springs, VA, December 2023.
Contributor Information
Isabella Angeli-Pahim, Department of Surgery, University of Florida College of Medicine, Gainesville, FL.
Anastasia Chambers, Department of Surgery, University of Florida College of Medicine, Gainesville, FL.
Sergio Duarte, Department of Surgery, University of Florida College of Medicine, Gainesville, FL.
Daiki Soma, Department of Surgery, University of Florida College of Medicine, Gainesville, FL.
Thiago Beduschi, Department of Surgery, University of Florida College of Medicine, Gainesville, FL.
Ilyas Sahin, Department of Medicine, University of Florida College of Medicine, Gainesville, FL.
Steven Hughes, Department of Surgery, University of Florida College of Medicine, Gainesville, FL.
Ali Zarrinpar, Department of Surgery, University of Florida College of Medicine, Gainesville, FL.
REFERENCES
- 1.Rumgay H, Arnold M, Ferlay J, et al. Global burden of primary liver cancer in 2020 and predictions to 2040. J Hepatol 2022;77:1598–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marrero JA, Kulik LM, Sirlin CB, et al. Diagnosis, staging, and management of hepatocellular carcinoma: 2018 practice guidance by the American Association for the Study of Liver Diseases. Hepatology 2018;68:723–750. [DOI] [PubMed] [Google Scholar]
- 3.European Association for the Study of the Liver. easloffice@easlofficeeu, European Association for the study of the liver EASL clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol 2018;69:182–236. [DOI] [PubMed] [Google Scholar]
- 4.Ferrante ND, Pillai A, Singal AG. Update on the diagnosis and treatment of hepatocellular carcinoma. Gastroenterol Hepatol (N Y) 2020;16:506–516. [PMC free article] [PubMed] [Google Scholar]
- 5.Piñero F, Dirchwolf M, Pessôa MG. Biomarkers in hepatocellular carcinoma: diagnosis, prognosis and treatment response assessment. Cells 2020;9:1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lok AS, Sterling RK, Everhart JE, et al. Des-gamma-carboxy prothrombin and alpha-fetoprotein as biomarkers for the early detection of hepatocellular carcinoma. Gastroenterology 2010;138:493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stefaniuk P, Cianciara J, Wiercinska-Drapalo A. Present and future possibilities for early diagnosis of hepatocellular carcinoma. World J Gastroenterol 2010;16:418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu R-H, Wei W, Krawczyk M, et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater 2017;16:1155–1161. [DOI] [PubMed] [Google Scholar]
- 9.Chen VL, Xu D, Wicha MS, et al. Utility of liquid biopsy analysis in detection of hepatocellular carcinoma, determination of prognosis, and disease monitoring: a systematic review. Clin Gastroenterol Hepatol 2020;18:2879–2902.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu X, Li J, Gassa A, et al. Circulating tumor DNA as an emerging liquid biopsy biomarker for early diagnosis and therapeutic monitoring in hepatocellular carcinoma. Int J Biol Sci 2020;16:1551–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiong Y, Xie C-R, Zhang S, et al. Detection of a novel panel of somatic mutations in plasma cell-free DNA and its diagnostic value in hepatocellular carcinoma. Cancer Manag Res 2019;11:5745–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nault JC, Mallet M, Pilati C, et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun 2013;4:2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papanicolau-Sengos A, Aldape K. DNA methylation profiling: an emerging paradigm for cancer diagnosis. Annu Rev Pathol 2022;17:295–321. [DOI] [PubMed] [Google Scholar]
- 14.Curradi M, Izzo A, Badaracco G, Landsberger N. Molecular mechanisms of gene silencing mediated by DNA methylation. Mol Cell Biol 2002;22:3157–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye PP, Viens RA, Shelburne KE, et al. Molecular counting enables accurate and precise quantification of methylated ctDNA for tumor-naive cancer therapy response monitoring. medRxiv 2023. [Google Scholar]
- 16.Kustanovich A, Schwartz R, Peretz T, Grinshpun A. Life and death of circulating cell-free DNA. Cancer Biol Ther 2019;20:1057–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DelaCourt A, Mehta A. Beyond glyco-proteomics-Understanding the role of genetics in cancer biomarkers. Adv Cancer Res 2023;157:57–81. [DOI] [PubMed] [Google Scholar]
- 18.Lim HY, Merle P, Weiss KH, et al. Phase II studies with refametinib or refametinib plus sorafenib in patients with RAS-mutated hepatocellular carcinoma. Clin Cancer Res 2018;24:4650–4661. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Yao Y, Xu Y, et al. Pan-cancer circulating tumor DNA detection in over 10,000 Chinese patients. Nat Commun 2021;12:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McDonald BR, Contente-Cuomo T, Sammut S-J, et al. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer. Sci Transl Med 2019;11:eaax7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kopystecka A, Patryn R, Leśniewska M, et al. The use of ctDNA in the diagnosis and monitoring of hepatocellular carcinoma-literature review. Int J Mol Sci 2023;24:9342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu N, Fan X-P, Fan Y-C, et al. Hypomethylated ubiquitin-conjugating Enzyme2 Q1 (UBE2Q1) gene promoter in the serum is a promising biomarker for hepatitis B virus-associated hepatocellular carcinoma. Tohoku J Exp Med 2017;242:93–100. [DOI] [PubMed] [Google Scholar]
- 23.Pasha HF, Mohamed RH, Radwan MI. RASSF1A and SOCS1 genes methylation status as a noninvasive marker for hepatocellular carcinoma. Cancer Biomark 2019;24:241–247. [DOI] [PubMed] [Google Scholar]
- 24.Huang G, Krocker JD, Kirk JL, et al. Evaluation of INK4A promoter methylation using pyrosequencing and circulating cell-free DNA from patients with hepatocellular carcinoma. Clin Chem Lab Med 2014;52:899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]