

Abstract
Extracellular vesicles (EVs), submicron-sized membranous structures released by cells, serve as vehicles of tissue-specific proteins and nucleic acids, facilitating intercellular communication and playing roles in pathophysiological processes. Leveraging their unique characteristics, EVs have emerged as promising drug delivery nanocarriers. Electroporation (EP) and ultrasonication (US) are among the prevalent techniques used for loading exogenous drugs into EVs owing to their simplicity and efficiency. However, the effectiveness of the two methods in depleting initial EV cargo has been overlooked. But this information is indispensable, as the bioactive residuals of EVs, notably derived from tumor or stem donor cells, may impact downstream therapeutic effects. Bridging this knowledge gap, therefore, can guide the selection of optimal drugs and loading methods tailored to therapeutic objectives. Here, we used high-throughput sequencing to investigate the protein and small RNA cargo of EVs treated with EP and US, respectively. We found that US exhibits higher efficacy in depleting EV cargo compared to EP, while US may also deplete essential endogenous molecules for combination therapy. Neither method demonstrated significant selectivity in cargo depletion, but they might preferentially retain few specific molecules. Additionally, membrane proteins are more prone to loss during US and EP treatments than cytoplasmic proteins.
Keywords: Extracellular vesicles, Ultrasonication, Electroporation, Drug delivery
1. Introduction
Extracellular vesicles (EVs) are submicron, cell-derived, lipid-bilayer-enclosed vesicles released by cells. Based on size and biogenesis, EVs are classified into exosomes, microvesicles, and apoptotic bodies.1–3 EVs act as intracellular messenger in both physiological and pathological states.4 They can selectively encapsulate proteins and nucleic acids from their parent cell and deliver them to recipient cells, ultimately regulating cellular functions.5–8 Alternatively, EVs can directly modulate recipient cells by interacting with their membrane receptors.9 Indeed, their role in facilitating intercellular communication is upheld by EV properties. Being naturally occurring, EVs are biocompatible and can evade phagocytosis,10 especially autologous ones.11 The unique composition of membrane proteins, lipids, and polysaccharides shields EVs, allowing them to circulate for extended periods in the body, maximizing their therapeutic potential.12 The small size aids in their ability to traverse physical barriers, augmenting their deep tissue penetration.13 Leveraging these appealing characteristics, EVs are being explored as nanocarriers for drug delivery in translational medicine.
Current methods for loading exogeneous drugs into EVs can be categorized into pre-loading and post-loading.13–16 Pre-loading entails loading drugs into donor cells, which subsequently produces EVs wrapping the drug.16,17 Post-loading involves directly loading the drug into EVs using techniques, including co-incubation, electroporation (EP), ultrasonication (US), extrusion, and freeze-thaw cycles.18 In addition, hybrid vesicles for drug delivery can be prepared through membrane fusion between EVs and drug preloaded liposomes.19 Pre-loading may alter the phenotype of donor cell due to the presence of the drug, affecting EV production, composition, and safety. Moreover, pre-loading may restrict the encapsulation of certain drugs, e.g., vulnerable proteins or nucleic acids, within donor cells. While hybrid vesicle could enhance product yield to some extent, achieving efficient and controlled fusion is highly challenging. The fusion process may also compromise the distinctive properties of EVs. Therefore, post-loading is favored due to their simplicity, versatile, and cost-effectiveness. More specifically, among the five post-loading methods, EP and US are the most widely used. Co-incubation and freeze-thaw methods typically result in low loading efficiency,20,21 especially for loading macromolecules. Extended loading time and significant temperature changes also contribute to EV degradation, membrane protein denaturation, and EV aggregation/agglomeration. Extrusion, though efficient for drug loading, suffers from frequent filter membrane clogging, hindering its practicality.15 In comparison, EP and US generally overcome these mentioned drawbacks.
An ideal loading method should also efficiently deplete the initial cargo of EVs. It ensures maximum space for exogenous drug to be loaded. Meanwhile, it minimizes the potential impact of residual cargo, which could influence therapeutic effects or induce unforeseen biological effects.22 For example, tumor derived EVs are commonly used as drug delivery nanocarriers because of their immune evasion, tumor homing effect, and scalable production. However, the residual cargo may foster tumor growth and metastasis,23–25 raising safety concerns about their use.26 While stem-cell derived EVs demonstrated their safety,27 the residual cargo may exhibit tumor-suppressive or tumor-promoting properties, depending on the type of stem cells.28 In brief, understanding the relationship between the loading method, EV integrity, and residual cargo would aid in selecting the appropriate method for specific therapeutic applications.29,30 However, a comparative study between EP and US has not yet been conducted. Here, leveraging high-throughput nucleic acid and protein sequencing for high accuracy, sensitivity, and repeatability,31–34 we compare the alterations in EV composition induced by EP and US. Our findings indicate that US outperforms EP for EV cargo depletion.
1.1. Theory
Both US and EP can deplete EV initial cargo. We hypothesize that US is more effective than EP in depleting initial cargo under commonly employed experimental conditions due to US’s more intense mechanical disruption.
2. Materials and methods
Cell culture
MDA-MB-231 (HTB-26) cells were obtained from ATCC and underwent a fluorescence test for mycoplasma contamination. The cells were cultured in DMEM medium at 37 °C, supplemented with 10% FBS, 100 units/mL penicillin, 100 μg/mL streptomycin, and 1 × non-essential amino acids, in a humidified atmosphere containing 5% CO2. During the entire experiment, all cells did not exceed 30 passages.
EV harvest and purification
Cells were incubated in T-225 flasks until cellular confluence reached ~50%. The culture medium was replaced with FBS-free DMEM after gently rinsed with PBS. The cells were further incubated for 48h (final cellular confluence ~70%). Subsequently, the cell culture supernatant was centrifuged at 12,000g at 4 °C for 15 min to dispose cell debris. Afterwards, the cell culture supernatant was ultracentrifuged at 160,000g, 4 °C for 4 h. The EV pellets were resuspended with 200 μL of PBS and were stored at −80 °C. Before use, the frozen EV samples were thawed and were filtered by 100 kD reverse osmosis (RO) membrane with centrifuge at 5,000g for 15 min to remove free protein and nucleic acids. The purified EVs were resuspend with pure water.
EV Characterization
The morphology of EVs was characterized using transmission electron microscopy (TEM). Briefly, 10 μL of each sample was loaded onto a 400-mesh Formvar-coated copper grid and incubated for 3 min at room temperature (RT). The samples were then drained using filter paper and stained with 1% filtered uranyl acetate solution for 1 min. The prepared samples were imaged with a Hitachi TEM at an acceleration voltage of 100 kV. The concentration and the size distribution of EVs were accessed by NanoSight NS300 system. EVs were diluted 100 × in particle free water (Mili-Q) to fit with the detection range. Ten 60-s videos were recorded and analyzed. Report was given by the NanoSight build-in software. Classic EV protein biomarkers (CD63 and TSG101) were analyzed via Western blot. In brief, 200 μL of EV lysates were prepared by adding 50 μL of RIPA lysis buffer on ice, followed by 5 min incubation. Samples were then mixed with 5 × loading buffer and heated at 95 °C for 20 min. The gels were run at 60 V for stacking and 100 V for separating. Proteins were then semi-drily transferred to a PVDF membrane (Bio-Rad mini blotting system) at 25 V for 7 min. The membranes were blocked for 1 h in 5% skimmed milk dissolved in TBS. The proteins were detected by incubation with primary antibodies conjugated with HRP (CD63: sc-5275 and TSG101: sc-7964). The membranes were washed 3 × 10 min before imaging.
Electroporation
GenePulser Xcell electroporator system (Bio-Rad) was used for electroporation. In brief, 200 μL of purified EVs were ultrafiltered using a 100 kDa RO membrane at 5,000g for 15 min and were resuspended in electroporation buffer at 4 °C. After electroporation at 350 V and 150 mF in 0.4 cm electroporation cuvetes using the GenePulser Xcell electroporator system, the samples were incubated at 37 °C for 30 min to ensure the membrane of the EVs recovered. Recovery was assessed by TEM assay as stated above. EVs were then washed with pure water twice by ultrafiltered using a 100 kD RO membrane at 5,000g for 15 min to remove leaked EV cargo. The EP-EVs collected were then analyzed by Nanosight NS300 system, strictly following protocol described above. RNase inhibitor (Thermo Fisher Scientific, N9090119)/protease inhibitor (Sigma-Aldrich, P8340) was added before storage.
Ultra-sonication
The 200 μL of purified EVs were sonicated (2 kHz, 20% power, 6 cycles by 4 s pulse/2 s pause), in ice bath for 2 min. The sonication process was repeated twice. After sonification the samples were incubated at 37 °C for 30 min to allow EV membrane recover. The downstream washing and characterization were followed by the protocol described above. RNase inhibitor (Thermo Fisher Scientific, N9090119)/protease inhibitor (Sigma-Aldrich, P8340) was added before storage.
smRNA sequencing
The RNA cargo of EVs were extracted using the miRNeasy Mini Kit (Qiagen), with five biological replicates prepared for each group. RNA sequencing was conducted at the SUNY Upstate University. Quality and adapter trimming of Illumina reads were performed using Trimmomatic. All reads were then aligned to the human reference genome hg38 using STAR. COMPSRA was employed to annotate and count smRNA species. Reads with at least 1 aligned read were counted as annotated items. Basepair was used to analyze the similarity levels among the samples in our dataset, generating the principal component analysis (PCA) plot, expression heatmap. MATLAB was used to generate venn diagram and MA plot. All downstream statistical analyses and data visualizations were conducted online.
Mass spectroscopy
Protein concentrations of negative control EVs (NC-EV), electroporation EVs (EP-EV), and ultrasonication EVs (US-EV) were quantified using Sypro Ruby staining on SDS-PAGE (SYPRO™ Protein Gel Stains, Thermo Fisher Scientific) against a 2 μg standard of E. coli protein. Three biological replicates of NC, EP-EV, and US-EV protein samples (9 samples in total, 16 μg each) were analyzed via stain-free SDS-PAGE, electrophoresed at 100 V for 15 min. From each triplicate EV sample, one gel slice (fraction) was excised, while four gel slices (fractions) from each of the three replicate EV samples were cut into 1 mm cubes for in-gel digestion and extraction of tryptic peptides. The gel pieces underwent sequential washing with 200 μL of deionized water, a 1:1 mixture of 100 mM ammonium bicarbonate (Ambic)/acetonitrile (ACN), and ACN. Reduction was performed with 70 μL of 10 mM dithiothreitol (DTT) in 100 mM Ambic for 1 h at 56 °C, followed by alkylation with 100 μL of 55 mM iodoacetamide in 100 mM Ambic at room temperature in the dark for 45 min. After alkylation, the gel slices were dried and rehydrated with 50 μL of trypsin in 50 mM Ambic and 10% ACN (20 ng/μL) at 37 °C for 16 h. The digested peptides were extracted twice with 70 μL of 50% ACN containing 5% formic acid (FA) and once with 70 μL of 90% ACN containing 5% FA. The three extracts from each sample were combined, filtered using a 0.22-μm spinning unit (Corning, USA), and dried using a Speedvac SC110. The in-gel tryptic digests of EV proteins were reconstituted in 40 μL of 2% ACN containing 0.5% FA, spiked with 50 fmol of tryptic digests of yeast enolase as an internal standard. Peptide samples were analyzed by nanoLC-tandem mass spectrometry (MS/MS) using an Orbitrap Fusion™ Tribrid™ mass spectrometer equipped with a nanospray Flex Ion Source and coupled with a Dionex UltiMate 3000 RSLCnano system. The gel-extracted peptide samples (9 μL for NC-EV proteins and 15 μL for each fraction of EP-EV or US-EV proteins) were injected onto a PepMap C-18 RP nano trapping column (75 μm i.d. × 20 mm) at a flow rate of 20 μL/min for rapid sample loading and then separated on a PepMap C-18 RP nano column (75 μm i.d. × 25 cm) at 35 °C. The tryptic peptides were eluted in a 120-min gradient of 5%–33% ACN in 0.1% FA at 300 nL/min, followed by a 7-min ramp to 90% ACN-0.1% FA and an 8-min hold at 90% ACN-0.1% FA. The column was re-equilibrated with 0.1% FA for 25 min prior to the next run. The Orbitrap Fusion was operated in positive ion mode with a spray voltage of 1.6 kV and a source temperature of 275 °C. External calibration for FT, IT, and quadrupole mass analyzers was performed. In data-dependent acquisition (DDA) analysis, the instrument operated using an FT mass analyzer in MS scan mode to select precursor ions followed by 3-s “Top Speed” data-dependent CID ion trap MS/MS scans at 1.6 m/z quadrupole isolation for precursor peptides with multiple charged ions above a threshold ion count of 10,000 and normalized collision energy of 30%. MS survey scans were conducted at a resolving power of 120,000 (fwhm at m/z 200) for the mass range of m/z 375–1575. Dynamic exclusion parameters were set at 35 s of exclusion duration with a 10-ppm exclusion mass width. All data were acquired using Xcalibur 4.3 operation software. The DDA raw files for CID MS/MS were processed using Proteome Discoverer (PD) 2.5 software with the Sequest HT algorithm for database searches. The PD 2.5 processing workflow included an additional Minora Feature Detector node for precursor ion-based quantification, facilitating protein identification and relative quantitation analysis within triplicate samples. Database searches were conducted against a Homo sapiens database containing 81,725 sequences from NCBI and a human EVs database with 60,444 sequences from Vesiclepedia. Up to two missed trypsin cleavage sites were allowed. The peptide precursor tolerance was set to 10 ppm, and the fragment ion tolerance was set to 0.6 Da. Variable modifications included methionine oxidation, deamidation of asparagine/glutamine, carboxylation on tryptophan, acetylation on the protein N-terminal, and a fixed modification of cysteine carbamidomethylation. Identified peptides were further filtered for a maximum 1% FDR using the Percolator algorithm in PD 2.5, with additional peptide confidence set to high and peptide mass accuracy ≤5 ppm. Relative quantitation of identified proteins among three replicates in each group was assessed using the label-free quantitation (LFQ) workflow in PD 2.5. Precursor abundance intensity for each peptide identified by MS/MS was automatically determined, and the unique peptides for each protein in each sample were summed and used to calculate protein abundance. Normalization was performed against the spiked yeast enolase protein.
Data Analysis
Data analyses were carried out using SPSS 23, excel, and MATLAB software. The statistical significance was determined by Student’s t-test and ANOVA test. All tests were two-sided, and p-value <0.05 were considered statistically significant.
3. Results
EV Characterization
To circumvent significant EV loss, mitigate physical damage and cargo leakage, and alleviate relevant batch-to-batch variation, we refrained from manually filtering cell culture supernatant using 0.22-μm filters. Therefore, without pre-filtration the majority of harvested EVs fell within a diameter range of 30–600 nm (Fig. 1A), and EV average concentration was 3.90 × 1011 particles/mL. RO filtration was used to remove free proteins and nucleic acids in harvested EV samples. It did not alter the size distribution of the washed out EVs (Fig. 1B), but it led to a mild increase in average concentration to 4.03 × 1011 particles/mL because of trace liquid retention in the RO membrane device. The subsequent EP and US treatment altered the size distribution of EVs, resulting in multiple peaks. Both induced the formation of micro-scale vesicles due to either membrane fusion among EVs or EV aggregation/agglomeration (Fig. 1C and D). Accordingly, EV average concentration following EP and US treatment decreased to 3.05 × 1011 and 2.66 × 1011 particles/mL, respectively. TEM analysis revealed typical saucer-shaped EV morphology, with additional presence of EV aggregates in the EP and US groups. Western blot analysis readily detected the membrane protein CD63 and the cytosolic protein TSG101 in the control group (Fig. 1E). In contrast, TSG101 was significantly depleted in the EP and US groups. While CD63 remained detectable in the EP and US groups, its signal intensity also decreased. The blots reflected the loss of EV integrity and proteins during EP or US treatment.
Fig. 1. Characterization of EVs.
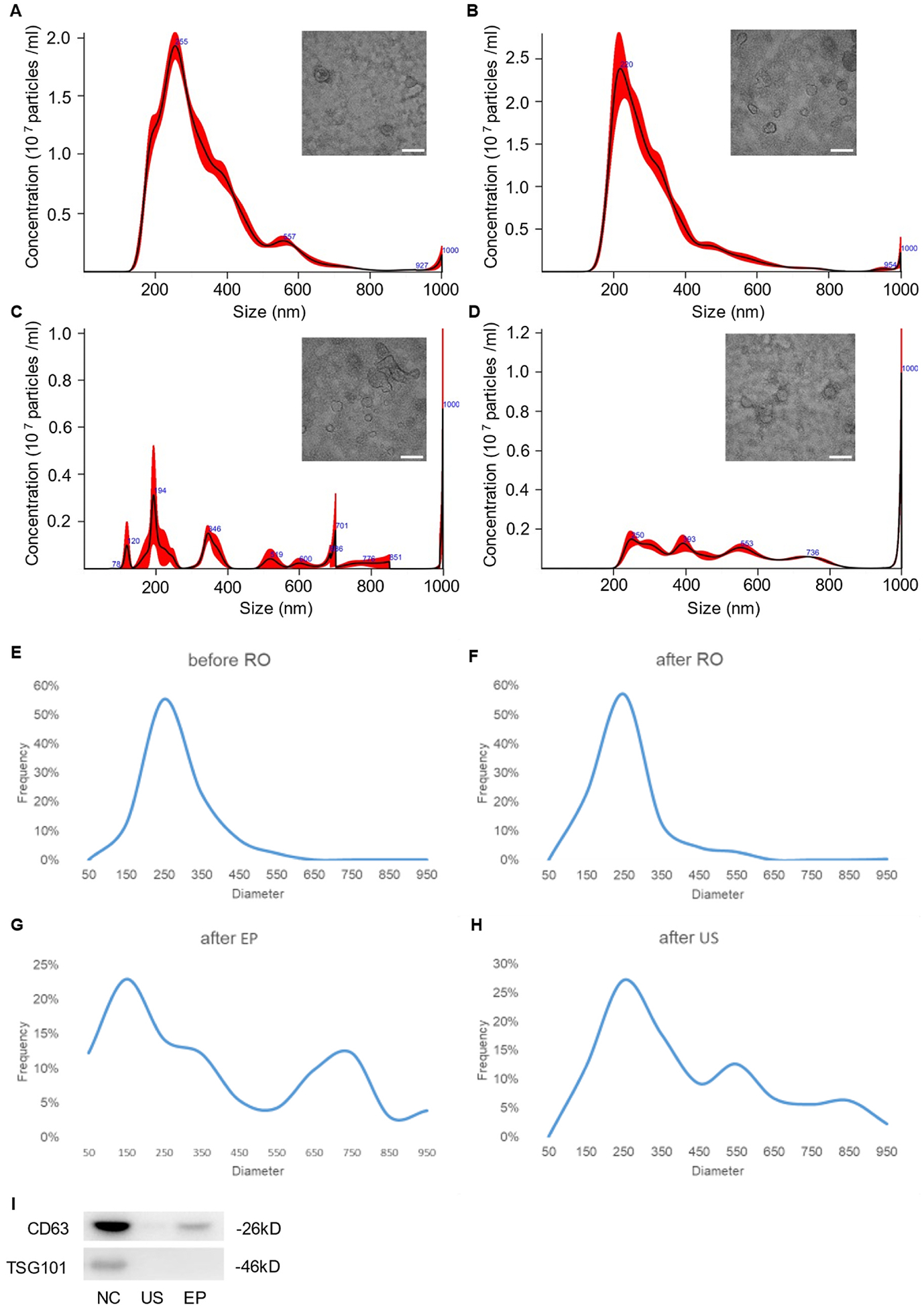
(A) Size distribution and TEM image of EVs before RO. (B) Size distribution and TEM image of EVs after RO. (C) Size distribution and TEM image of EVs after EP. (D) Size distribution and TEM image of EVs after US. Scale bars in A-C is 100 nm. (E) Size distribution based on TEM before RO. (F) Size distribution based on TEM after RO. (G) Size distribution based on TEM after EP. (H) Size distribution based on TEM after US. (I) Western blot analysis of CD63 and TSG101 from NC, US, and EP groups, respectively.
Similarity of protein cargo
EP and US treatments led to a substantial decrease in EV proteins. On average, the EP group retained only 40.9%, and the US group retained only 24.3% of their original cargo protein mass (NC: 4.40 μg; EP: 1.80 μg; US: 1.07 μg). These values were semi-quantified by SDS-PAGE and compared against an E. coli protein standard. To ensure high-confidence protein identification in subsequent mass spectrometry (MS) analysis, only proteins with at least two detected peptides were included for downstream analysis. MS identified 2217 proteins in total, with 2,165, 1,713, and 1205 proteins found in the NC, EP, and US groups, respectively. The similarity of protein composition between EP and NC was 75.9%, between US and NC was 53.4%, and between EP and US was 64.9% (Fig. 2A). The relatively low similarity between EP and US was mainly attributed to the depletion of additional 544 (511 + 33) proteins by US. After MS data normalization (i.e., individual protein abundance relative to total protein abundance), we plotted proteins detected across all three replicates. The volcano plot revealed that the majority exhibited no significant change in relative abundance compared to the average loss after EP or US treatment. Only ~3% of the proteins showed a significant difference after EP or US treatment (Fig. 2B and C). The heatmap also corroborated the trend of protein loss and revealed that a limited number of proteins were up- or down-regulated (Fig. 2D). Taken together, we deduced that EP and US treatment did not exhibit significantly selective depletion or retention of most proteins. We observed 30 and 23 upregulated proteins in EP and US groups, respectively. Despite the absolute decrease in the quantity of these proteins following EP or US treatment, their relative abundance somehow increased. Gene ontology analysis indicated that these proteins are primarily associated with functions related to the ribosome, lysosome, proteasome, and cancer pathways.
Fig. 2. Similarity of protein cargo.
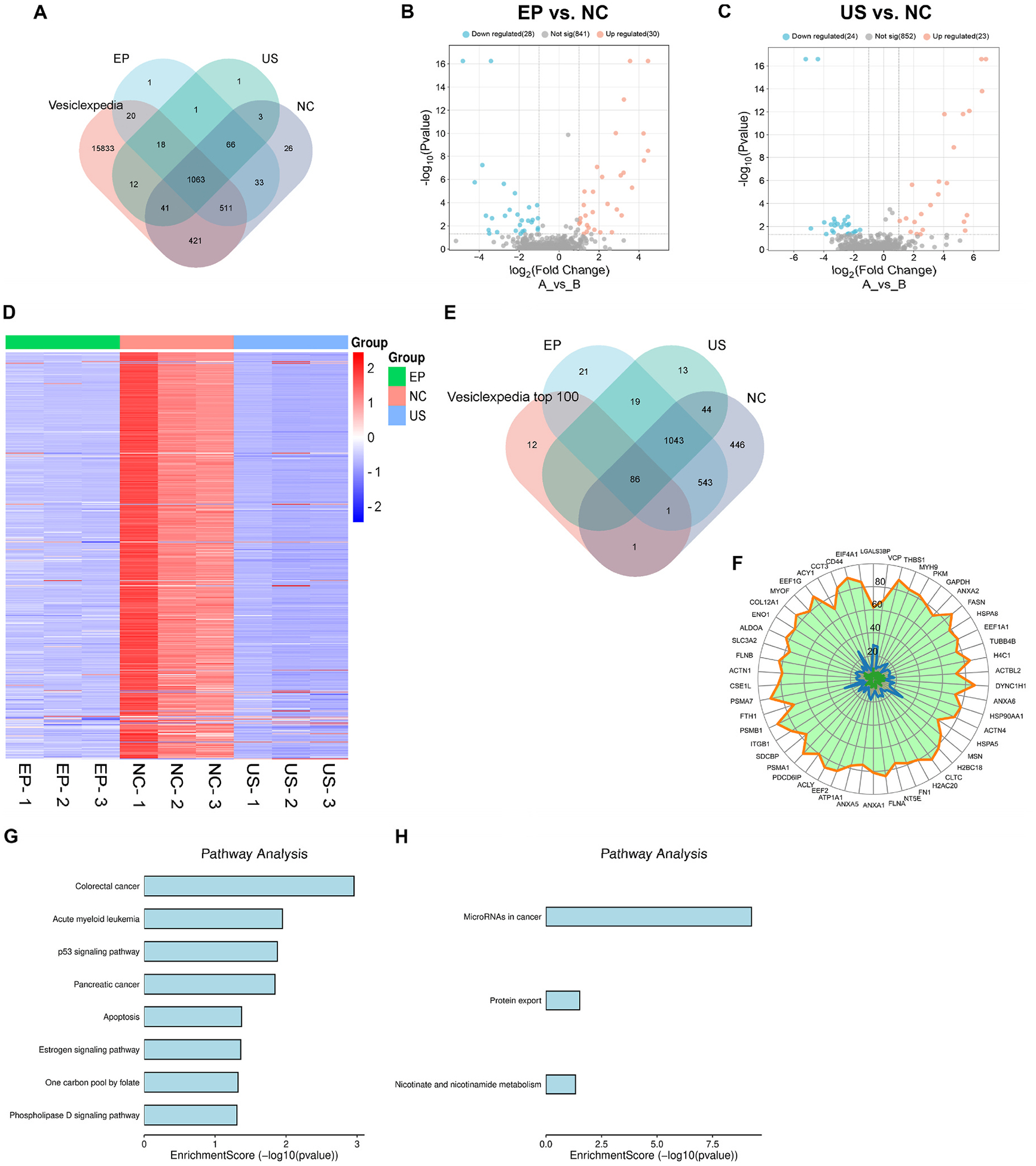
(A) Venn diagram of proteins detected in each group comparing with Vesiclepedia. (B) Volcano plot showing the differential protein expression profiles between EP and NC group. (C) Volcano plot showing the differential protein expression profiles between US and NC group. (D) Heatmap showing the differential protein expression profiles across three groups. (E) Venn diagram of proteins detected comparing with top 100 high-abundance proteins listed in Vesiclepedia. (F) Radar chart of top 50 high-abundance proteins in NC (orange boundary), EP (blue boundary), and US (green boundary) groups, respectively. (G) GO analysis of most likely to be lost proteins in EP group. (H) GO analysis of most likely to be lost proteins in US group.
MS detected 87, 86, and 88 proteins in EP, US, and NC groups, respectively, out of the top 100 high-abundance EV proteins listed in Vesiclepedia (Fig. 2E). The high alignment rate of ~90% indicated the reliability of our MS data. Out of the 88 most abundant proteins, 13 were identified as membrane proteins present in all groups. An additional 13 were cytosolic proteins with close ties to plasma membrane. The remaining 62 were cytosolic proteins residing within the interior space of EVs. These 87 and 86 proteins remained detectable following the EP or US treatment due to their high initial abundance. This finding also suggested that the depletion of high-abundance proteins induced by EP or US treatment might occur primarily through a random process. Otherwise, there would not be a difference of only one protein composition between two groups. Notably, the loss of the housekeeping protein, GAPDH, was nearly equivalent to the average protein loss rate. Using GAPDH as an internal reference, we found that most of the 13 high-abundance membrane proteins, including CD9, CD63, and CD81, exhibited a significantly higher loss rate after EP treatment than cytosolic proteins. This tendency was even more pronounced in the US group. We speculated that the loss of membrane proteins after EP or US treatment could be attributed to the disruption of the protein-lipid association. Upon dissociation from lipids, these membrane proteins, particularly multipass membrane proteins, may be unable to reassemble properly and effectively, leading to their loss. The radar chart of the top 50 proteins further illustrated that the protein retention level after EP and US treatment is positively correlated with its initial abundance (Fig. 2F). But we acknowledged that EP and US treatments may influence the abundance of few individual proteins. EP may preferentially retain cytosolic proteins, such as FTH1 (~21 kDa), ACY1 (~50 kDa), LGALS3BP (~90 kDa), VCP (~97 kDa), and HSPA5 (~78 kDa), while US may show preferential retention of LGALS3BP and VCP. It is unclear why EP and US treatments preferentially retain these proteins. This phenomenon might be attributed to random error, considering that only three biological replicates were analyzed. Certain proteins closely associated with cancer were more prone to loss after EP (Fig. 2G). Additionally, proteins linked to miRNA in cancer were more likely to be lost after US (Fig. 2H).
Similarity of smRNA cargo
The average yield of total RNA extracted from the NC, EP, US groups was 240.27, 29.43, and 5.30 ng respectively. Approximately only 12.24% and 2.21% of the RNA cargo were retained after EP and US treatment, respectively. Due to the low RNA quantity in the US group, the three biological replicates were pooled for sequencing. On average, 77.95% of reads mapped to human total RNA for NC sample, 84.85% for EP-treated samples, and 83.81% for US-treated samples respectively. The mapping data suggested that the samples were of good quality. The annotation result of COMPSRA indicated that there were no significant alterations in smRNA composition among the three groups (Fig. 3A). Differential expression was observed between NC, EP, and US groups (Fig. 3B). Top 1000 differential gene expressions of smRNAs were further compared (Fig. 3C). The similarity between EP and NC was 65.0%, between US and NC was 50.2 %, and between EP and US was 58.9%. Volcano plots and MA plots were used to visualize both the magnitude and statistical significance of differential expression between two groups (Fig. 3D and E). Overall, while a number of smRNAs showed relatively increased expression, more smRNAs were downregulated following EP and US treatment. We compared 50 of the most depleted and retained smRNAs in both EP and US group (Fig. 4). Gene ontology analysis showed that smRNAs related to EV function were more likely to be depleted, while those related to EV generation and cell survival were less likely to be lost. Moreover, EP-treated EVs exhibited enriched smRNAs associated with membrane protein function, localization, EV biogenesis, receptor activity, and organ development. Conversely, US treatment primarily affect signaling pathway-related smRNAs. Nevertheless, these observations suggest that EP and US may partially preserve some biofunctions of EVs. Furthermore, EP treatment depleted smRNAs associated with immune system suppression and miRNA function, potentially weakening the tumor-promoting activity of MDA-MB-231 EVs. Similarly, US treatment primarily depleted smRNAs involved in angiogenesis and translation inhibition, potentially achieving the same anti-tumor effect. These contrasting findings, which include both anti-tumor and pro-tumor effects, also highlight the multifaceted nature of EVs.
Fig. 3. Similarity of smRNA cargo.
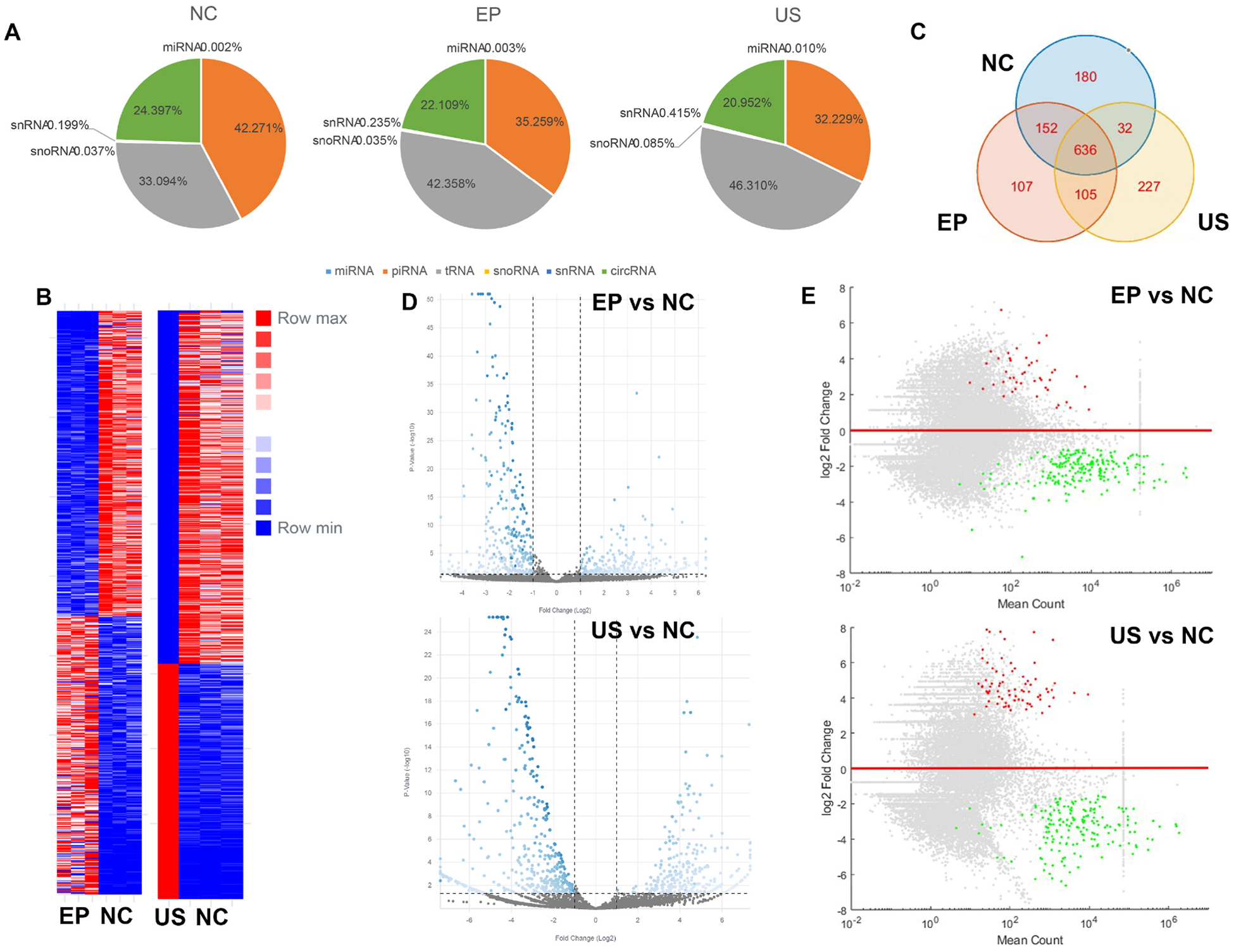
(A) Annotation of aligned smRNA in each group. (B) Heatmap showing the differential smRNA level between EP, US, and NC. (C) Venn diagram of top 1000 smRNAs in each group. (D) Volcano plot showing the differential smRNA level between EP, US, and NC. (E) MA plot showing the differential smRNA level between EP, US, and NC.
Fig. 4. GO enrichment of smRNA analysis.

(A) The related bioprocess of the most retained RNAs in EP group. (B) The related bioprocess of the most retained RNAs in US group. (C) The related bioprocess of the most depleted RNAs in EP group. (D) The related bioprocess of the most depleted RNAs in US group.
4. Discussion
During EP, high-voltage pulses create temporary pores in EV membranes. Pore size and number depend on pulse strength and duration. These pores allow drugs to passively diffuse or be driven electrophoretically into the EV interior space.35 After the pulse, pores can be spontaneously and rapidly sealed. Loading lasts only a few milliseconds, followed by EVs resting for 0.5–1 h. The primary advantage of EP is its efficient delivery of various therapeutic agents, including chemical compounds, nucleic acids, and proteins, into EVs. Notably, EP is a relatively efficient, with loading efficiencies ranging from 2% to 60%.36–39 Small, ionized drugs tend to achieve higher efficiency, while un-ionized macromolecules may exhibit low efficiency. US works by creating cavitation, a phenomenon caused by the propagation of ultrasound waves through a medium. These waves generate cycles of compression and rarefaction, leading to the formation and violent collapse of microscopic microbubbles. The rapid collapse of these microbubbles within microseconds creates intense shear forces. These forces are powerful enough to disrupt EV membranes. Subsequently, the disrupted membrane lipids can spontaneously reassemble. Surrounding drugs can become encapsulated within these newly formed vesicles.17 US takes a few minutes, followed by EV resting. Various drugs can also be used via US. To prevent denaturation of EV proteins or therapeutic proteins caused by the heat generated during US, samples are normally immersed in ice-cold water. In addition, a pause of ~30s during US allows the sample temperature to quickly cool down. Studies have reported that loading efficiency ranging from 20% to 70%, influenced by factors such as the type of drugs and experimental conditions.36,38,40,41 Overall, US tends to be more efficient than EP in drug loading.42 A few reasons may result in the difference. EP might not always achieve optimal size and number of pores for drug entering. The uniformity of electric field strength may affect loading efficiency. In contrast, US disrupts EV membranes in a non-specific manner. US creates large openings, enabling a wide range of molecules to enter EVs.
On the other hands, the initial cargo of EVs can be depleted into the surrounding environment through damaged membranes caused by EP or US. Because US can create large openings, EV cargo may be depleted more efficiently compared to EP. Moreover, we found RNA cargo can be depleted more efficiently compared to protein cargo. Smaller size and simpler structure allow smRNAs to be more readily depleted via EP and US compared to large proteins, which struggle with steric hindrance during passage through transient openings. In EV-based drug delivery, US could be a favorable option. This is because US can more efficiently clear the EV’s native cargo, creating space for the desired therapeutic payload. In addition, considering that EVs derived from tumor cells or stem cells might harbor bioactive molecules with potential to influence physiological or pathological processes, US could be used to efficiently remove these molecules, mitigating unforeseen downstream effects. Specifically, almost all RNA cargo could be removed by the US. Furthermore, US is a viable option if the intended therapeutic payload risks interacting or binding with EV’s initial cargo, potentially weakening their overall therapeutic effect. But in the scenario of combination therapy, while certain initial microRNAs and proteins of EVs have therapeutic effects and need to be preserved, while simultaneously requiring the loading of exogenous drugs, such as adjuvants, EP could be used for drug loading. However, US treatment might not be ideal for EVs where their biocompatible “outer shell” (composed of outer membrane proteins and polysaccharides) is crucial. This is because US treatment can efficiently deplete proteins.
A few concerns should be noted. Both EP and US involve temporary disruption of EV membranes, impacting EV size and quality. They often result in EV aggregation/agglomeration and fusion. Aggregation/agglomeration could result from the translocation of positively charged lipids from the inner to the outer membrane during membrane disruption.43 The fusion event could be ascribed to the intrinsic self-assembly behavior of lipids, fostering non-selective EV-EV membrane fusion.44 Accordingly, the additional filtration is compulsory to re-collect vesicles with a size less than 300 nm for proper drug delivery. However, the additional purification will lead to significant EV loss. Industrial manufacturers may find it unappealing due to the low yield and high production cost. If subsequent purification fails to eliminate EVs with positively charged lipids on their outer membranes, macrophages and dendritic cell might readily recognize and clear these EVs from circulation, potentially lowering therapeutic effects.45 For the clinical translation of exogenous drug-loaded EVs, regulatory considerations are also crucial aspects that demand attention. Key concerns, such as the depletion of the EV’s initial cargo, the overall purity of the drug-loaded EVs within the population, the capacity and efficiency for loading exogenous drugs, potential variability between production batches, and other uncertainties, may also cause regulator bodies to hesitate. Lastly, we did not investigate depletion efficiency using various parameters, e. g., voltage and pulse duration in EP, as well as amplitude, cycles, and duration time in US. Indeed, drawing from our established protocols and consulting published studies, we compiled a commonly used parameters for EP and US.20,41,46–49 While we acknowledge that optimizing parameters can impact outcomes, in our opinion, the effect size may be limited. Therefore, we directly compare the depletion effect of the two methods using respective representative settings. Nevertheless, we acknowledge that a more powerful EP setup could deplete cargo as efficiently as US. However, the potential adverse effects of the high-energy EP on EVs remain unexplored.
5. Conclusions
In summary, our findings suggest that US may be more efficient than EP for depleting original cargo from EVs. The depletion, generally speaking, is random and non-selective. smRNA cargo can be more efficiently depleted compared to protein cargo. Membrane proteins are more prone to loss compared to cytosolic proteins during EP or US treatment. Both methods can damage EVs, reduce their biocompatibility, and limit the yield of drug-loaded EVs. The optimal choice between EP and US for drug loading depends on the specific treatment strategy, drug properties, and characteristics of the EVs themselves. Given cargo cannot be completely depleted, the residual molecules within the EVs could retain diverse functionalities, which warrant attention in treatment. In individual studies, the operational parameters of EP and US should be judiciously optimized based on specific requirement and objectives.
Acknowledgment
We thank Proteomics Facility at Cornell University and Cancer Center at Cold Spring Harbor Laboratory for their sequencing services.
Funding sources
The work was partially supported by National Cancer Institute R01CA230339 and R37CA255948.
Footnotes
Conflicts of interest
All authors declare that they have no conflicts of interest and no competing interest.
CRediT authorship contribution statement
Yundi Chen: Writing – original draft, Visualization, Formal analysis, Data curation, Conceptualization. Le Wang: Visualization, Data curation. Xu Yu: Writing – review & editing, Supervision. Wenjun Mao: Software, Resources. Yuan Wan: Writing – review & editing, Supervision, Resources, Project administration, Funding acquisition, Conceptualization.
References
- 1.Ashoub MH, Salavatipour MS, Kasgari FH, Valandani HM, Khalilabadi RM. Extracellular Microvesicles: Biologic Properties, Biogenesis, and Applications in Leukemia. vol. 479. 2024:419–430. [DOI] [PubMed] [Google Scholar]
- 2.Omrani M, Beyrampour-Basmenj H, Jahanban-Esfahlan R, et al. Global Trend in Exosome Isolation and Application: An Update Concept in Management of Diseases. vol. 479. 2024:679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang AJII. Taylor & Francis. 2024:1–4. [Google Scholar]
- 4.Wen Y, Chen Y, Wang G, et al. Factors Influencing the Measurement of the Secretion Rate of Extracellular Vesicles. vol. 145. 2020:5870–5877. [DOI] [PubMed] [Google Scholar]
- 5.Yáñez-Mó M, Siljander P, Andreu Z, et al. Biological properties of extracellular vesicles and their physiological functions. 2015;4, 27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song C, Sun Y, Chen Y, et al. Differential Diagnosis of Pulmonary Nodules and Prediction of Invasive Adenocarcinoma Using Extracellular Vesicle DNA. vol. 14. 2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng Q, Ding H, Wang L, et al. Circulating Exosomal miR-96 as a Novel Biomarker for Radioresistant Non-small-cell Lung Cancer. 2021. 2021, 5893981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, Xia Y, Smollar J, Mao W, Wan Y. The roles of small extracellular vesicles in lung cancer: molecular pathology, mechanisms. diagnostics, and therapeutics. 2021;1876, 188539. [DOI] [PubMed] [Google Scholar]
- 9.Jabalee J, Towle R, Garnis CJC. The role of extracellular vesicles in cancer: cargo, function, and therapeutic implications. 2018;7:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng G, Li W, Ha L, et al. Self-assembly of Extracellular Vesicle-like Metal–Organic Framework Nanoparticles for Protection and Intracellular Delivery of Biofunctional Proteins. vol. 140. 2018:7282–7291. [DOI] [PubMed] [Google Scholar]
- 11.Vader P, Breakefield XO, Wood M. Extracellular Vesicles: Emerging Targets for Cancer Therapy. vol. 20. 2014:385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abhange K, Makler A, Wen Y, et al. Small extracellular vesicles. cancer. 2021;6:3705–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herrmann IK, Wood MJA, Fuhrmann GJ Nn. Extracellular Vesicles as a Next-Generation Drug Delivery Platform. vol. 16. 2021:748–759. [DOI] [PubMed] [Google Scholar]
- 14.Han L, Zhao Z, He C, et al. Removing the Stumbling Block of Exosome Applications in Clinical and Translational Medicine: Expand Production and Improve Accuracy. vol. 14. 2023:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kenari AN, Cheng L, Hill AFJM. Methods for Loading Therapeutics into Extracellular Vesicles and Generating Extracellular Vesicles Mimetic-Nanovesicles. vol. 177. 2020:103–113. [DOI] [PubMed] [Google Scholar]
- 16.Blomme EA, Will Y. Toxicology Strategies for Drug Discovery: Present and Future. vol. 29. 2016:473–504. [DOI] [PubMed] [Google Scholar]
- 17.Chen C, Sun M, Wang J, Su L, Lin J, Yan X. Active Cargo Loading into Extracellular Vesicles: Highlights the Heterogeneous Encapsulation Behaviour. vol. 10. 2021, e12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Abreu RC, Ramos CV, Becher C, et al. Exogenous Loading of miRNAs into Small Extracellular Vesicles. vol. 10. 2021, e12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sato YT, Umezaki K, Sawada S, et al. Engineering Hybrid Exosomes by Membrane Fusion with Liposomes. vol. 6. 2016, 21933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haney MJ, Klyachko NL, Zhao Y, et al. Exosomes as Drug Delivery Vehicles for Parkinson’s Disease Therapy. vol. 207. 2015:18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pomatto MAC, Bussolati B, D’Antico S, et al. Improved Loading of Plasma-Derived Extracellular Vesicles to Encapsulate Antitumor miRNAs. vol. 13. 2019:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wen Y, Fu Q, Soliwoda A, et al. Cell-derived Nanovesicles Prepared by Membrane Extrusion Are Good Substitutes for Natural Extracellular Vesicles. vol. 1. 2022, 100004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y, Xue F, Russo A, Wan YJ Tp j. Proteomic Analysis of Extracellular Vesicles Derived from MDA-MB-231 Cells in Microgravity. vol. 40. 2021:108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wan Y, Xia Y-Q, Zheng S. Extruded Small Extracellular Vesicles: Splinters of Circulating Tumour Cells May Promote Cancer Metastasis?. vol. 127. 2022:1180–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, Xu Y, Wang J, et al. Statins Lower Lipid Synthesis but Promote Secretion of Cholesterol-Enriched Extracellular Vesicles and Particles. vol. 12. 2022, 853063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taghikhani A, Hassan ZM, Ebrahimi M, Moazzeni S. microRNA Modified Tumor-derived Exosomes as Novel Tools for Maturation of Dendritic Cells. vol. 234. 2019:9417–9427. [DOI] [PubMed] [Google Scholar]
- 27.Wang L, Abhange K, Wen Y, et al. Preparation of Engineered Extracellular Vesicles Derived from Human Umbilical Cord Mesenchymal Stem Cells with Ultrasonication for Skin Rejuvenation. vol. 4. 2019:22638–22645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie Y, Wu J, Xu A, et al. Heterologous Human/rat HER2-specific Exosome-Targeted T Cell Vaccine Stimulates Potent Humoral and CTL Responses Leading to Enhanced Circumvention of HER2 Tolerance in Double Transgenic HLA-A2/her2 Mice. vol. 36. 2018:1414–1422. [DOI] [PubMed] [Google Scholar]
- 29.Chen Y, Wang L, Zheng M, et al. Engineered Extracellular Vesicles for Concurrent Anti-PDL1 Immunotherapy and Chemotherapy. vol. 9. 2022:251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quinn Z, Mao W, Xia Y, John R, Wan YJ Bm. Conferring Receptors on Recipient Cells with Extracellular Vesicles for Targeted Drug Delivery. vol. 6. 2021:749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu X, Zhou L, Shi X, Xu G. New Advances in Analytical Methods for Mass Spectrometry-Based Large-Scale Metabolomics Study. vol. 121. 2019, 115665. [Google Scholar]
- 32.Kim DK, Kang B, Kim OY, et al. EVpedia: An Integrated Database of High-throughput Data for Systemic Analyses of Extracellular Vesicles. vol. 2. 2013, 20384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hartjes T, Slotman J, Vredenbregt MS, et al. EVQuant; High-Throughput Quantification and Characterization of Extracellular Vesicle (Sub) Populations, 2020.2010.2021.348375. 2020. [Google Scholar]
- 34.Liu T, Zhang Q, Zhang J, et al. EVmiRNA: A Database of miRNA Profiling in Extracellular Vesicles. vol. 47. 2019:D89–D93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beebe SJ. Ultrashort Electric Pulse Effects in Biology and Medicine. vols 33–75. Springer; 2021. [Google Scholar]
- 36.Kim MS, Haney MJ, Zhao Y, et al. Development of Exosome-Encapsulated Paclitaxel to Overcome MDR in Cancer Cells. vol. 12. 2016:655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Q, Wu D, Wang Y, Chen ZJM. Exosomes as novel delivery systems for application in traditional. Chin Med. 2022;27:7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang L, Wang G, Mao W, et al. Bioinspired Engineering of Fusogen and Targeting Moiety Equipped Nanovesicles. vol. 14. 2023:3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodríguez-Morales B, Antunes-Ricardo M, González-Valdez JJP. Exosome-mediated Insulin Delivery for the Potential Treatment of Diabetes Mellitus. vol. 13. 2021:1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wan Y, Wang L, Zhu C, et al. Aptamer-conjugated Extracellular Nanovesicles for Targeted Drug Delivery. vol. 78. 2018:798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJA. Delivery of siRNA to the Mouse Brain by Systemic Injection of Targeted Exosomes. vol. 29. 2011:341–345. [DOI] [PubMed] [Google Scholar]
- 42.Lee J, Lee J-H, Chakraborty K, Hwang J, Lee Y-KJ R a. Exosome-based Drug Delivery Systems and Their Therapeutic Applications. vol. 12. 2022:18475–18492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Slusky JS, Dunbrack R. Charge Asymmetry in the Proteins of the Outer Membrane. vol. 104. 2013:539a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Staufer O, Dietrich F, Rimal R, et al. Bottom-up assembly of biomedical relevant fully synthetic extracellular vesicles. 2021;7, eabg6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cocco RE, Ucker D. Distinct Modes of Macrophage Recognition for Apoptotic and Necrotic Cells Are Not Specified Exclusively by Phosphatidylserine Exposure. vol. 12. 2001:919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lennaárd AJ, Mamand DR, Wiklander RJ, El Andaloussi S, Wiklander OPJP. Optimised Electroporation for Loading of Extracellular Vesicles with Doxorubicin. vol. 14. 2021:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian Y, Li S, Song J, et al. A Doxorubicin Delivery Platform Using Engineered Natural Membrane Vesicle Exosomes for Targeted Tumor Therapy. vol. 35. 2014:2383–2390. [DOI] [PubMed] [Google Scholar]
- 48.Li Y-J, Wu JY, Wang JM, Hu XB, Cai JX, Xiang DX. Gemcitabine Loaded Autologous Exosomes for Effective and Safe Chemotherapy of Pancreatic Cancer. vol. 101. 2020:519–530. [DOI] [PubMed] [Google Scholar]
- 49.Sancho-Albero M, Encabo-Berzosa MDM, Beltrán-Visiedo M, et al. Efficient Encapsulation of Theranostic Nanoparticles in Cell-Derived Exosomes: Leveraging the Exosomal Biogenesis Pathway to Obtain Hollow Gold Nanoparticle-Hybrids. vol. 11. 2019:18825–18836. [DOI] [PubMed] [Google Scholar]