

Abstract
The aim of this single‐centre retrospective observational study was to evaluate the safety, tolerability, and efficacy of an in‐class combination therapy switch from bosentan plus sildenafil to ambrisentan plus tadalafil in children with pulmonary arterial hypertension. Children aged over 5 years who were established on sildenafil plus bosentan were offered to undergo a therapy switch from May 2014 to May 2021 and, if remaining in the service, followed up to May 2024. Children with Eisenmenger syndrome, open intra or extra‐cardiac shunt, or with pulmonary hypertension‐associated lung disease were excluded. As part of a structured clinical program children were assessed via walk test, echocardiography, cardiac magnetic resonance imaging (CMRI), cardiopulmonary exercise testing, and serum biomarkers. Fifty‐two children were included, 33 in the switch group and 19 in the control group. Clinical characteristics at diagnosis and baseline assessments did not differ between groups. All children tolerated the medication switch. Over a median 13.0 [12.0,13.7] week follow‐up in the switch group there was a significant improvement in World Health Organization functional class (WHO FC, p < 0.001); reduction in estimated right ventricular systolic pressure by echocardiography of 7 mmHg (p = 0.03) and a 2% increase (p = 0.03) in right ventricular ejection fraction on CMRI. There was a sustained improvement in WHO FC (p < 0.01) in the switch group at medium‐term follow‐up of 40.9 [35.2,49.3] weeks. Long‐term outcome of transplant‐ or Potts shunt‐free survival was comparable between the two groups.
Keywords: pulmonary artery hypertension, pulmonary hypertension, rare pediatric lung disease, treatment
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a rare, progressive, and heterogenous disease in children for which there is no cure. Outcomes for children in the modern drug era remain poor. Transplant‐free survival from recent international registries estimates survival of 74%–80% at 5 years. 1 , 2 Clinical trials assessing the efficacy of novel PAH therapies in children are limited by the ability to recruit sufficiently large cohorts to power studies and lack of consensus on trial endpoints for pediatric populations. Prospective studies involving children have instead primarily focused on the pharmacokinetics and safety profile of new therapies in PAH, 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 with only a single randomized control trial (RCT) in children. 9 Instead, the use of PAH therapies in children has been established in clinical practice through the extrapolation of evidence from adult trials and evidence from real‐world observational studies in children to inform international consensus guidance. 11 , 12 , 13 Superiority of up‐front combination therapy with ambrisentan plus tadalafil compared to monotherapy with either ambrisentan or tadalafil in treatment‐naive adults was demonstrated by the multi‐centre RCT AMBITION. 14 Subsequent real‐world prospective and retrospective studies in both adults and children have also shown up‐front and sequential combination therapy with alternative combinations of drugs in PAH to have additional benefit. 15 , 16 , 17 , 18 , 19 , 20 , 21 Increased adoption in clinical practice of combination therapy use in children has been demonstrated in recent UK and REVEAL registries 1 , 22 and is also reflected in recent clinical guidelines. 11 , 12 , 23 However, superiority of different PAH therapy combinations in children has not been demonstrated. Efficacy of an in‐class PAH therapy switch has not been studied widely, with only a single open‐label adult RCT showing this to be a potential option for treatment escalation. 24 Children with PAH in the UK who meet recommendations for dual oral therapy have historically been first established on combination therapy with bosentan plus sildenafil. Once established on bosentan plus sildenafil children and their families have been offered the option to switch to combination therapy with ambrisentan plus tadalafil. Safety and pharmacokinetics for both ambrisentan and tadalafil as monotherapy 3 , 4 , 25 , 26 , 27 , 28 , 29 and in combination 30 have already been established in small cohorts of children. The European Medicines Agency (EMA) only recently approved ambrisentan for use in children above the age of 8 years in 2021 and Tadalafil for the use in children above the age of 2 years in 2023. 31 However, neither are currently approved for use in children by the United States (US) Food and Drug Administration (FDA). In this retrospective analysis we consider the safety, tolerability, and efficacy of switching children established on dual combination therapy with bosentan plus sildenafil to ambrisentan plus tadalafil within a structured clinical care program.
METHODS
Study setting and design
This is a single‐centre retrospective cohort study of children with a diagnosis of PAH. A change in clinical practice guidelines at our institution from May 1, 2014, permitted offering children with a primary diagnosis of PAH the possibility to switch oral combination therapy from bosentan plus sildenafil to ambrisentan plus tadalafil. Children were eligible if aged over of 5 years during the study period ending May 1, 2021, with the lower age cut‐off based on available safety data at the time. 3 , 4 Children with Eisenmenger syndrome, prevalent intra‐ and extra‐cardiac systemic‐to‐pulmonary shunt, (including those with a palliative Potts shunt), and children with early postoperative pulmonary hypertension were excluded. Children with comorbid lung disease that was contributory to their PAH were also excluded. Medication switch was offered if, in the view of the treating clinician, the child was not achieving anticipated clinical targets or was demonstrating worsening disease trajectory despite being established on bosentan plus sildenafil (with or without the addition of a prostanoid). 32 Dosing for ambrisentan and tadalafil was based on pediatric retrospective cohorts and observational studies. 3 , 4 In clinical practice, dose banding for age and weight was used for both ambrisentan and tadalafil, shown in Supporting Information S1: Table 1. Children were included in the analysis if established on combination therapy with bosentan plus sildenafil, with or without additional prostanoid therapy. Two groups of children were identified: (1) switch group, that is, those who completed the combined medication switch and (2) control group, that consisted of children who met all other inclusion criteria but were not considered for a therapeutic switch or to whom the switch was offered but declined (see Figure 1). Children were followed within a structured clinical program with 3 to 6 monthly multi‐parametric, noninvasive assessments 33 from May 1, 2014 to May1, 2024.
FIGURE 1.
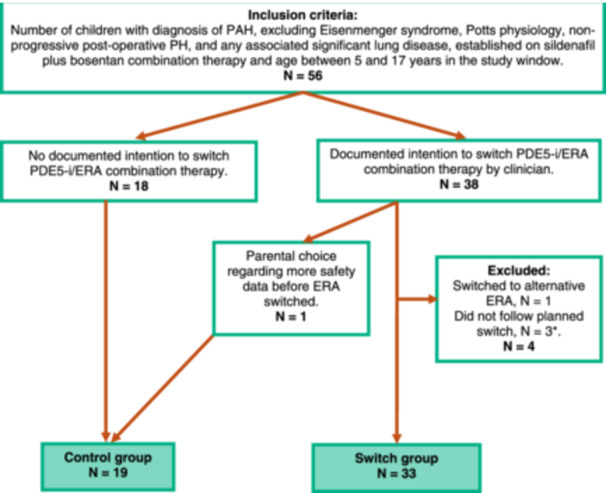
Inclusion criteria for the switch and control groups. Key: CHD, congenital heart disease; ERA, endothelin receptor antagonist; PAH, pulmonary arterial hypertension; PDE5‐i, phosphodiesterase type‐5 inhibitor; review window = May 1, 2014 to May 1, 2021. * Patients excluded as the planned switch did not follow local practice: switch done at the same time without interval assessment in view of compliance issues (1); unable to undergo assessments (1); and prolonged switch period of 18 months (1).
This retrospective analysis utilizes anonymized data collected for routine clinical care covered under ethical approval 17/LO/0008, allowing individual consent to be waived.
Data collection and assessment structure
Data were collected on a standard minimum set of clinical assessments as detailed in Figure 2: anthropometry, World Health Organization functional class (WHO FC), 6‐minute walk distance (6MWD), and echocardiography. Height and weight were expressed as z‐scores for normal British children, including the use of Down syndrome‐specific growth charts. 34 , 35 Echocardiographic indices associated with outcomes in children with PAH included in the analysis were: right atrial area (RAA); estimated right ventricular systolic pressure (RVSP); tricuspid annular plane systolic excursion (TAPSE); and left ventricular systolic and diastolic eccentricity indices (LVEIs and LVEId). 36 At study entry and at follow‐up after medication‐switch children underwent cardiac magnetic resonance imaging (CMRI). Image processing and analysis of CMRI was performed as previously detailed. 37 , 38 Parameters included in the analysis were: right ventricular ejection fraction (RVEF); right ventricular end‐diastolic volume indexed to body surface area (RVEDVi); and estimate of right ventricular afterload (est. mPAP). For children undergoing therapeutic change, it is local practice for additional multi‐parameter assessments before and following any medication change to assess for clinical response. Therefore, those in the switch group also underwent cardiopulmonary exercise testing (CPET) and/or N‐terminal pro B‐type natriuretic peptide (NT‐proBNP). Parameters assessed on CPET for children with PAH in routine clinical practice have been described previously and those included in the analysis were peak oxygen consumption (VO2) and ventilatory efficiency slope (VE/VCO2). 39 Liver function tests, including aspartate aminotransferase (AST), alanine aminotransferase (ALT), and bilirubin, in addition to hemoglobin concentration and platelet count, were monitored monthly.
FIGURE 2.
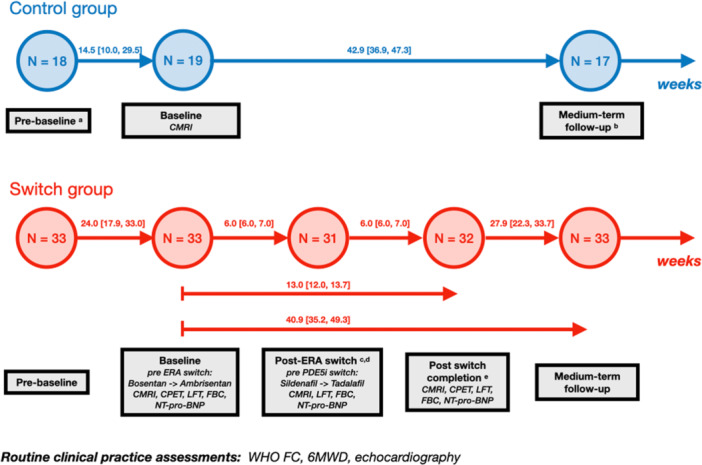
Flow diagram demonstrating routine and structured assessments for the switch group and routine assessment for the control group. Duration between assessments is given as a median and inter‐quartile range ([]) in weeks. Routine assessments included WHO FC, 6MWD, and echocardiogram. For the switch group, children underwent additional assessments including CPET, CMRI and NT‐proBNP, LFT, and FBC. The control group underwent CMRI at study initiation. Two children underwent adaptation to remote assessments due to the COVID‐19 pandemic, and therefore, only WHO FC was obtained: one child in control group at pre‐baseline assessment and one child in switch group at both post‐switch completion and medium‐term follow‐up assessments. Additional reasons for missed assessments were: a One child had not entered the service before their study initiation visit. b Two children underwent lung transplantation before medium‐term follow‐up. c One child had a deterioration in their disease requiring an unplanned admission after baseline assessment and before post‐ERA switch assessment, therefore the timing between switching of the two medication classes was truncated and their post‐ERA switch assessment was dropped. d No assessment due to the COVID‐19 pandemic, medication switched. e Acute, non‐PAH related illness. Key: FC, functional class; 6MWD, 6‐min walk test; CPET, cardio‐pulmonary exercise testing; CMRI, cardiac magnetic resonance imaging; NT‐proBNP, N‐terminal pro B‐type natriuretic peptide; LFT, liver function test; FBC, full blood count.
Timepoints
Timepoints for assessments are summarized in Figure 2. Children in the switch group therefore underwent a baseline assessment before medication switch and at approximately 6 weeks following each medication switch (post‐ERA switch assessment and post‐switch completion assessment). Children in both groups underwent a medium‐term follow‐up assessment at approximately 6–12 months after the baseline visit. Long‐term outcome was assessed at the first occurrence of Potts shunt, lung transplant, death, transition to adult care or the end of the review window.
Statistical analysis
Continuous variables are presented as median and inter‐quartile range [IQR] or mean and standard deviation (SD) as appropriate. Categorical variables are presented as a number and percentage (%). Missing data was not imputed. Comparisons were made for the medication switch‐group between baseline assessment before ERA switch and at each of the following time points: pre‐baseline, post‐ERA switch, post‐switch completion and medium‐term follow‐up assessments and additionally for post‐ERA switch to post switch completion assessments. For children who did not undergo medication switch in the control group comparisons were made between baseline and medium‐term follow‐up assessments. Comparison between groups were made using the paired t‐test or Wilcoxon signed rank test for continuous variables and Chi‐squared or Fisher's exact test for nominal categorical variables as appropriate. Comparisons of geometric mean for NT‐proBNP were made to assess any change across assessments. Kaplan–Meier curves were constructed in addition to univariate Cox regression survival analysis for the determination of the composite outcome of all‐cause mortality, lung transplantation, or Potts shunt (referred to as transplant‐ or Potts shunt‐free survival). Transplant‐ or Potts shunt‐free survival time was taken from baseline assessment to either the time of death, transplantation, Potts shunt, or the first censoring event. Children were censored at transition to adult services or at the end of the review window.
RESULTS
Patient groups and assessment completion
Fifty‐six children met the inclusion criteria (see Figure 1). Thirty‐eight children intended to undergo a combined switch to ambrisentan plus tadalafil, with four of these children being excluded from our analysis and 1 child entering the control group (details presented in Figure 1). The reasons for undergoing the medication switch were (a) disease progression as assessed by treating clinician (n = 15); (b) stable disease but no satisfactory change in symptom burden with current medication (n = 16); and (c) persistent mildly elevated transaminases on bosentan (n = 2). Nineteen children who continued with combination therapy with bosentan plus sildenafil and did not undergo a switch in medication formed the control group. Attendance at structured clinical follow‐up assessments, median duration between assessments and reasons for missed in‐person assessments in the switch group are shown in Figure 2. In the control group, one child did not have a pre‐baseline assessment due to becoming eligible on their first review at our institution, taken as their baseline assessment. Children who had an adapted remote assessment due to the COVID‐19 pandemic were assigned a WHO FC and underwent routine blood monitoring only. One child in the switch group was unable to perform a 6‐minute walk test, CPET or CMRI due to co‐morbidities but tolerated echocardiography and blood testing for NT‐proBNP. Hence, they were included in the analysis.
Characteristics of switch and control groups at diagnosis and baseline assessment
The demographic and clinical characteristics of both the switch and control groups are described in Table 1. Half of the children had PAH associated with repaired congenital heart disease (CHD, 56%), followed by idiopathic or heritable PAH (27%), coincidental cardiac shunt (13%), and portal hypertension (4%). Children had moderate to severe haemodynamic disease at diagnosis with mean pulmonary artery pressure (mPAP) of 45.7 ± 12.1 mmHg and indexed pulmonary vascular resistance (PVRi) of 13.5 ± 8.6 WU.m2. There was no difference in age at the commencement of dual agent combination therapy with sildenafil plus bosentan between the switch and control groups (7.0 ± 3.8 vs. 7.1 ± 4.2 years, p = 0.94). At baseline assessment, both the switch and control groups were similar in terms of age, PAH etiology and most measures of disease severity. However, children in the switch group had significantly greater 6 MWD (401 ± 92 m vs. 300 ± 103 m, p < 0.01) and had been treated for longer, although this was not statistically significant (49.2 ± 35.5 vs. 29.7 ± 33.7 months, p = 0.06). Over a third (37%) of children were on triple therapy at baseline assessment, and 8% were also treated with a calcium channel blocker, there was no significant difference in additional therapy between the two groups.
TABLE 1.
Demographic and clinical characteristics of switch and control groups at diagnosis and baseline assessments.
| Characteristics | All | Switch | Control | p | |||
|---|---|---|---|---|---|---|---|
| N | 52 | 33 | 19 | ‐ | |||
| At diagnosis | |||||||
| Female | 37 (72) | 25 (76) | 12 (63) | 0.56 | |||
| Age (years) | 5.5 ± 3.9 | 5.4 ± 3.6 | 5.6 ± 4.4 | 0.85 | |||
| WSPH classificationb | |||||||
| 1.1: Idiopathic | 12 (23) | 8 (24) | 4 (21) | 0.97 | |||
| 1.2: Heritable | 2 (4) | 1 (3) | 1 (5) | ||||
| 1.4.3: a/w Portal hypertension | 2 (4) | 1 (3) | 1 (5) | ||||
| 1.4.4.3: a/w Co‐incidental shunt | 7 (13) | 4 (12) | 3 (16) | ||||
| 1.4.4.4: a/w Postoperative CHD | 29 (56) | 19 (58) | 10 (53) | ||||
| Co‐morbidities | |||||||
| Down syndrome | 8 (15) | 4 (12) | 4 (19) | 0.44 | |||
| Other chromosomal abnormality/associated gene variant | 4 (8) | 2 (6) | 2 (11) | 0.55 | |||
| Haemodynamicsb | n | n | n | ||||
| mPAP (mmHg) | 49 | 45.7 ± 12.1 | 30 | 44.1 ± 11.6 | 19 | 48.2 ± 12.8 | 0.27 |
| PVRi (WU.m2) | 48 | 13.5 ± 8.6 | 30 | 11.6 ± 5.9 | 18 | 16.5 ± 11.4 | 0.11 |
| At study initiation | |||||||
| Age (years) | 10.5 ± 3.6 | 11.1 ± 3.3 | 9.5 ± 4.1 | 0.17 | |||
| Sildenafil plus bosentan therapy | |||||||
| Age established (years) | 7.0 ± 3.9 | 7.0 ± 3.8 | 7.1 ± 4.2 | 0.94 | |||
| Duration of therapy (months) | 42.1 ± 35.8 | 49.2 ± 35.5 | 29.7 ± 33.7 | 0.06 | |||
| Other therapy | |||||||
| Prostanoid | 19 (37) | 11 (33) | 8 (42) | 0.77 | |||
| CCB | 4 (8) | 3 (9) | 1 (5) | 1.00 | |||
| Anthropometry | |||||||
| Weight z‐score | −0.37 ± 1.38 | −0.29 ± 1.46 | −0.51 ± 1.26 | 0.57 | |||
| Height z‐score | −0.5 ± 2.15 | −0.06 ± 1.47 | −0.65 ± 1.32 | 0.18 | |||
| WHO functional class | |||||||
| I or II | 29 (56) | 17 (52) | 12 (63) | 0.24 | |||
| III or IV | 21 (40) | 16 (48) | 5 (26) | ||||
| 6MWT | n | n | n | ||||
| Walk distance (m) | 40 | 378 ± 102 | 30 | 401 ± 92* | 10 | 308 ± 103* | <0.01 |
| Echocardiography | n | n | n | ||||
| RAA (cm2) | 30 | 13.3 ± 3.6 | 20 | 14.1 ± 3.3 | 10 | 11.5 ± 3.7 | 0.08 |
| RVSP (mmHg) | 48 | 68.9 ± 27.0 | 31 | 67.0 ± 24.5 | 17 | 72.3 ± 31.7 | 0.55 |
| TAPSE (mm) | 43 | 16.7 ± 4.7 | 30 | 15.9 ± 3.9 | 13 | 18.7 ± 5.8 | 0.13 |
| LVEI diastole | 43 | 1.33 ± 0.25 | 28 | 1.29 ± 0.21 | 15 | 1.40 ± 0.31 | 0.22 |
| LVEI systole | 44 | 1.75 ± 0.50 | 29 | 1.69 ± 0.51 | 15 | 1.88 ± 0.47 | 0.23 |
| Cardiac MRI | n | n | n | ||||
| RVEDVI (mL/m2) | 42 | 101.2 ± 36.7 | 32 | 98.9 ± 39.1 | 10 | 108.4 ± 28.1 | 0.41 |
| RVEF (%) | 42 | 51.7 ± 11.4 | 32 | 53.3 ± 8.9 | 10 | 46.5 ± 16.7 | 0.24 |
| Estimated mPAP (mmHg) | 40 | 56.0 ± 15.5 | 30 | 54.7 ± 14.7 | 10 | 59.8 ± 17.8 | 0.43 |
Note: Data represented as a number, n, percentage (%), and mean ± s.d. Statistically significant p values (<0.05) are shown in bold font.
Key: a/w., associated with; CHD, congenital heart disease; IV, intravenous; LVEI, left ventricular eccentricity index; mPAP, mean pulmonary artery pressure; MRI, magnetic resonance imaging; PVRi, pulmonary vascular resistance indexed to body surface area; RAA, right atrial area; RVEDVi, right ventricular end‐diastolic volume indexed to body surface area; RVEF, right ventricular ejection fraction; RVSP, estimate of right ventricular systolic pressure on echocardiography; s/c, sub‐cutaneous; TAPSE, tricuspid annular plane systolic excursion; WHO, World Health Organization; WSPH, World Symposium on Pulmonary Hypertension; 6MWT, 6‐min walk test.
a6th WSPH classification used. 40
First haemodynamic assessment at or after diagnosis.
Assessments during medication switch
Changes between assessments during structured follow‐up in the medication switch group are shown in Table 2. Eleven children were on triple therapy with a prostanoid; changes in assessments during structured follow‐up are shown in Supporting Information S1: Table 2.
TABLE 2.
Clinical, biometric, echocardiographic, and cardiac MRI assessed parameters across assessments in the switch group summarized as mean ± standard deviation.
| Primary assessment: Baseline assessment to switch completion assessment | Switch phase 1: Baseline assessment to post ERA switch assessment | Switch phase 2: post ERA switch assessment to switch completion assessment | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Bosentan + sildenafil switched to ambrisentan + tadalafil | Bosentan + sildenafil switched to ambrisentan + sildenafil | Ambrisentan + sildenafil switched to ambrisentan + tadalafil | |||||||
| n | Δ | p | n | Δ | p | n | Δ | p | |
| WHO FCa | |||||||||
| III/IV (%) | ‐ | 48 vs. 13a | <0.01 | ‐ | 48 vs. 16 | <0.01 | ‐ | 16 vs. 13 | 0.73 |
| Exercise assessment | |||||||||
| 6MWD (m) | 23 | 7 [−21, 35] | 0.62 | 27 | −8 [−32, 16] | 0.51 | 23 | 21 [5,37] | 0.01 |
| VE/VCO2 slope | 25 | −3.5 [−7.1, 0.1] | 0.06 | ||||||
| VE/VCO2 slope % changeb | 25 | −7 [−14.4, 0.3] | 0.06 | ||||||
| Peak VO2 (L/min/m2) | 25 | 0.6 [−1.7, 2.9] | 0.60 | ||||||
| Peak VO2% changeb | 25 | 3.4 [−7.6, 14.4] | 0.53 | ||||||
| Biochemical measure | |||||||||
| NT‐proBNP ratioc | 26 | 0.96 [0.83, 1.10] | 0.55 | 25 | 1.00 [0.89, 1.13] | 0.97 | 26 | 0.93 [0.85, 1.01] | 0.08 |
| Echocardiographic parameters | |||||||||
| RAA (cm2) | 17 | 0.2 [−1.1, 1.6] | 0.72 | 16 | 0.1 [−1.1, 1.4] | 0.84 | 19 | 0.7 [−0.2, 1.7] | 0.13 |
| RVSP (mmHg) | 27 | −7 [−13, −1] | 0.03 | 29 | ‐8 [−14, −3] | <0.01 | 26 | 3 [−3, 9.0] | 0.3 |
| Eccentricity index diastole | 27 | −0.007 [−0.09, 0.08] | 0.87 | 26 | 0.07 [−0.05, 0.19] | 0.22 | 26 | −0.11 [−0.25, 0.03] | 0.13 |
| Eccentricity index systole | 28 | 0.01 [−0.18, 0.16] | 0.90 | 26 | −0.01 [−0.20, 0.18] | 0.90 | 26 | 0.02 [−0.25, 0.29] | 0.89 |
| TAPSE (mm) | 28 | 0.02 [−1.4, 1.5] | 0.98 | 29 | 0.16 [−0.8, 1.2] | 0.74 | 29 | 0.1 [−1.6, 1.7] | 0.95 |
| Cardiac MRI parameters | |||||||||
| RVEDVi (mL/m2) | 30 | 5.7 [0.8, 10.6] | 0.02 | 29 | 3.3 [0.6, 6.1] | 0.02 | 27 | 2.9 [−1.9, 7.6] | 0.23 |
| RVEF (%) | 30 | 1.8 [0.2, 3.5] | 0.03 | 29 | 1.1 [−0.3, 2.5] | 0.11 | 27 | 0.3 [−1.5, 2.0] | 0.76 |
| Estimated mPAP (mmHg) | 28 | 0.7 [−4.7, 3.4] | 0.74 | 26 | 0.3 [−3.5, 4.1] | 0.88 | 25 | −0.6 [−4.3, 3.1] | 0.73 |
Note: Comparison of parameters between assessments summarized as difference, Δ, confidence interval, [], number of paired pre/post data, n, and p value, p. Except for: ** comparison between % of children with the same or improved WHO FC and those with a worse WHO FC. Statistically significant p values (<0.05) are shown in bold font.
Key: Echocardiographic parameters: LVEI, left ventricular eccentricity index; RAA, right atrial area; RVSP, estimate of right ventricular systolic pressure on echocardiography; TAPSE, tricuspid annular plane systolic excursion. Cardiac magnetic resonance imaging (MRI) parameters: est. mPAP, estimated mean pulmonary artery pressure; RVEDVi, right ventricular end‐diastolic volume indexed to body surface area; RVEF, right ventricular ejection fraction. Cardio‐pulmonary exercise parameters: VO2, oxygen consumption; VE/VCO2, ventilatory efficiency slope. NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide. WHO FC, World Health Organization functional class.
% of children in FC III or IV in those assigned.
VE/VCO2 slope % change and VO2% change are represented as means.
NT‐proBNP is represented as the difference in geometric means and is expressed as a ratio.
Primary assessment, baseline assessment to post‐switch completion assessment
In the 13.0 [12.0,13.7] weeks between baseline (before medication switch) and post‐switch completion assessments, there was a significant improvement in WHO FC (Figure 3a, p < 0.001) with all children either maintaining or improving their WHO FC. Echocardiography‐derived estimated RVSP fell (−7 mmHg CI = [−13, −1] mmHg, p = 0.03), and CMRI‐derived RVEF increased (1.8%, CI = [0.2, 3.5]%, p = 0.03). However, cardiac MRI assessed RVEDVi increased (5.7 mL/m2, CI = [0.8, 10.6] mL/m2, p = 0.02). There was an improvement in the VE/VCO2 slope on CPET (−3.5, CI = [−7.1, −0.1], p = 0.07), although this change was not significant. Neither 6MWD, NT‐proBNP, additional echocardiographic nor CMRI parameters changed significantly. For children on triple therapy with a prostanoid, 27% were in WHO FC III/IV at baseline assessment compared to no children at post‐switch completion assessment at 12.2 [12.0, 13.5] weeks, although this did not reach statistical significance. There was no significant change in any of the other assessed parameters in children on triple therapy with a prostanoid.
FIGURE 3.
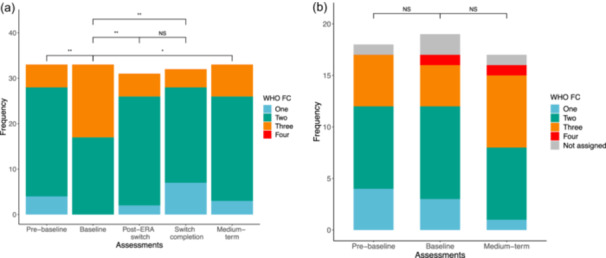
World Health Organization functional class (WHO FC) assigned to (a) the switch group and (b) control group across assessments. NS = nonsignificant, p values: * p < 0.01, **p < 0.001.
Switch phase 1, baseline assessment to post‐ERA switch assessment
In the 6.0 [6.0, 7.0] weeks following ERA switch, there was a significant improvement in WHO FC (Figure 3a, p < 0.001), with only one child (3%) experiencing a worsening in their FC. There was a reduction in echocardiography‐derived estimate of RVSP (−8 mmHg, 95%CI = [−14, −3] mmHg, p = 0.005). Cardiac MRI assessed RVEDVi increased (3.3 mL/m2, 95%CI = [0.6, 6.1] mL/m2, p = 0.02). Neither 6MWD, NT‐proBNP, additional echocardiographic nor CMRI parameters changed significantly.
Switch phase 2, post‐ERA switch assessment to post‐switch completion assessment
In the 6.0 [6.0, 7.0] weeks following PDE5i switch there was a significant increase in 6MWD (21 m, 95%CI = [5,37] m, p = 0.01). There was no significant change in WHO FC (Figure 3a). There was no significant change in NT‐proBNP, echocardiographic and CMRI parameters.
Comparison between switch and control groups
Change in assessment parameters between pre‐baseline to baseline assessments and baseline to medium‐term assessments is summarized in Table 3 and Supporting Information S1: Table 2.
TABLE 3.
Clinical, biometric, and echocardiographic assessed parameters across pre‐baseline, baseline and medium‐term follow‐up assessments in the switch and control groups.
| Pre‐baseline assessment to baseline assessment | Baseline assessment to medium‐term assessment | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Switch | Control | Switch | Control | |||||||||
| Bosentan + sildenafil | Bosentan + sildenafil | Bosentan + sildenafil switched to ambrisentan + tadalafil | Bosentan + sildenafil | |||||||||
| n | Δ | p | n | Δ | p | n | Δ | p | n | Δ | p | |
| WHO FC** | ||||||||||||
| III/IV (%) | ‐ | 15 vs. 48* | <0.01 | 29 vs. 29 | 1.0 | ‐ | 48 vs. 21* | 0.04 | ‐ | 29 vs. 50 | 0.30 | |
| Exercise assessment | ||||||||||||
| 6MWD (m) | 24 | 1 [−33, 36] | 0.94 | 8 | −37 [−71, −3]* | 0.04 | 26 | 8 [−14, 20] | 0.47 | 5 | 26 [−16, 68] | 0.16 |
| Echocardiographic parameters | ||||||||||||
| RAA (cm2) | 10 | 1.8 [0.2, 3.5]* | 0.03 | 6 | 0.4 [−1.7, 2.5] | 0.66 | 13 | 0.5 [−0.8, 1.9] | 0.42 | 8 | 2.3 [−1.8, 6.3] | 0.23 |
| RVSP (mmHg) | 27 | 2 [−6, 10] | 0.57 | 13 | 1.9 [−16.2, 19.9] | 0.82 | 27 | −3 [−11, 4] | 0.39 | 13 | 9.4 [−2.6, 21.5] | 0.11 |
| Eccentricity index diastole | 22 | −0.05 [−0.17, 0.08] | 0.45 | 10 | −0.12 [−0.33, 0.08] | 0.20 | 23 | 0.08 [−0.03, 0.18] | 0.16 | 11 | 0.12 [−0.16, 0.40] | 0.36 |
| Eccentricity index systole | 22 | 0.00 [−0.19, 0.18] | 0.95 | 10 | −0.10 [−0.64, 0.44] | 0.69 | 24 | 0.63 [−0.50, 1.76] | 0.26 | 11 | 0.37 [−0.17, 0.91] | 0.16 |
| TAPSE (mm) | 21 | 1.1 [−0.5, 2.7] | 0.17 | 9 | 1.7 [−1.3, 4.7] | 0.23 | 24 | 0.8 [−0.6, 2.2] | 0.22 | 10 | 2.6 [−6.8, 1.6] | 0.19 |
Note: Data summarized as mean ± standard deviation. Comparison of parameters between assessments summarized as difference, Δ, confidence interval, [], number of paired pre/post data, n, and p‐value, p.
Key: Echocardiographic parameters: LVEI, left ventricular eccentricity index; RAA, right atrial area; RVSP, estimate of right ventricular systolic pressure on echocardiography; TAPSE, tricuspid annular plane systolic excursion. WHO FC, World Health Organization functional class.
=p < 0.05.
% of children in FC III or IV in those assigned.
Pre‐baseline assessment to baseline assessment
There was no significant difference in the duration from pre‐baseline to baseline assessment between the switch and control groups (24.0 [17.9, 33.0] weeks vs. 14.5 [10.0, 29.5] weeks, p = 0.36) (Figure 1). There was a significant worsening in WHO FC in the switch group, with an increase in the proportion of children in WHO FC III or IV between pre‐baseline to baseline assessment (15% vs. 48%, p < 0.01, Figure 3a). There was no significant change in WHO FC in the control group (Figure 3b). Children in the control group walked on average 37 m less between pre‐baseline and baseline assessments (95%CI = [−71, −3] m, p = 0.04). Children in the switch group had a mean increase in RAA (1.8 cm2, 95%CI = [0.2, 3.5] cm2, p = 0.03). There was no significant change in any other assessed parameters in either group.
Baseline assessment to medium‐term assessment
Two children in the control group underwent lung transplantation before medium‐term follow‐up. Therefore, 50 children were assessed at medium‐term follow‐up. There was no difference in the duration from baseline to medium‐term follow‐up assessment between the switch and control groups (40.9 [35.2, 49.3] weeks vs. 42.9 [36.9, 47.3] weeks, p = 0.38). The proportion of children with WHO FC III or IV in the switch group reduced from baseline to medium‐term assessment (48% vs. 21%, p = 0.04) and there was no significant change in the control group. Overall, there was a sustained improvement in WHO FC at medium‐term assessment in the switch group (Figure 3a, p < 0.01) and none in the control group (Figure 3b). There was no significant change in other assessed parameters in either group. In the 11 children on triple therapy in the switch group there was a sustained improvement in WHO FC from baseline assessment to medium‐term assessment at 23.0 [17.8, 27.0] weeks, although this did not meet statistical significance (proportion in WHO FC III/IV: 27% vs. 9%, p = 0.58). There was no significant change in any of the other assessed parameters in the children on triple therapy with a prostanoid. Escalation to triple therapy with the addition of a prostanoid by medium‐term assessment in prostanoid‐naive children was the same in both groups (2 (9%) in switch group compared to 1 (9%) in control group).
Longer‐term clinical outcomes
There was no significant difference in duration of follow‐up from baseline assessment between the switch and control groups (52.6 [32.8, 66.8] months vs. 41.8 [16.4, 69.1] months, p = 0.22). During long‐term follow‐up in the switch group, 2 (6%) children underwent a Potts shunt and 2 (6%) underwent insertion of an atrial‐flow regulator. In the control group, 1 (5%) child underwent atrial septostomy. Five children (15%) underwent lung transplant in the switch group and 5 (26%) in the control group. Four children (12%) in the switch group died, compared to none in the control group. Those in the switch group had transplant‐ or Potts shunt‐free survival of 97%, 97%, and 84% at 1, 2, and 3 years respectively, compared to 85%, 73%, and 68% in the control group (Figure 4). However, the difference in outcome only met statistical significance at 2 years (hazard ratio at 1 year, 0.47, 95%CI [0.14–1.62], p = 0.2; 2 years, 0.13, 95%CI [0.01–1.13], p = 0.03; and 3 years, 0.47, 95%CI [0.14–1.64], p = 0.2).
FIGURE 4.
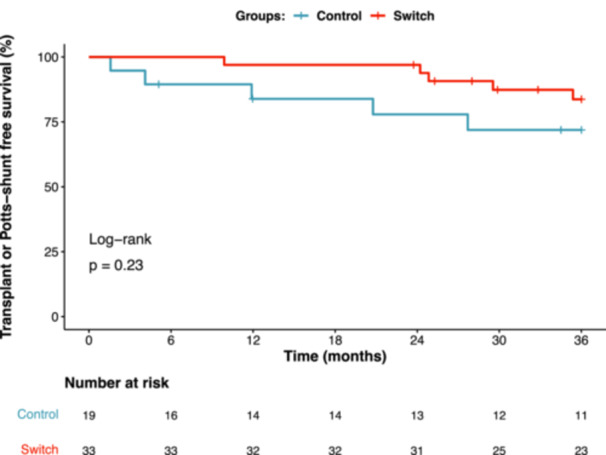
Kaplan–Meier analysis for transplant‐ or Potts shunt‐free survival comparing the switch and control groups.
Safety and tolerability
At medium‐term follow‐up, no children became anemic or experienced a significant elevation in liver transaminases that would require dose adjustment or discontinuation of ambrisentan after switching from bosentan. A total of seven children (21%) experienced adverse events (AE) that were considered minor, self‐limiting and did not persist beyond post‐switch completion assessment: headache (n = 3), pedal edema (n = 2), dizziness (n = 1) and transient elevation in serum sodium (n = 1). One further child was temporarily switched back to sildenafil plus bosentan at 6 months post‐switch completion as they developed urticaria and angioedema. They were subsequently diagnosed with idiopathic urticaria and angioedema and were successfully switched back to tadalafil plus ambrisentan without recurrence of reported symptoms.
DISCUSSION
In this study, we provide a robust assessment of the effects of an in‐class medication switch in children with PAH using routine data obtained within a structured clinical program with multiparametric assessment. We have shown that a medication switch from bosentan plus sildenafil to ambrisentan plus tadalafil in children with PAH was safe, well tolerated, and associated with both short‐ and medium‐term improvement in functional status.
To date only one adult study has assessed the effect of an in‐class medication switch in patients with PAH24 with our study being the first to do so in children. Moreover, in comparison to adult cohorts, there is a paucity of evidence for drug efficacy in children with PAH due to a lack of RCTs and prospective studies. Studies involving children are often designed primarily to assess safety and dosing rather than efficacy due to ethical considerations. As a result, there is frequently a significant time lag or absence in the licensing of new drugs for children with PAH. As an alternative to RCTs and prospective studies, real‐world retrospective observational studies in children, such as this study, offer a unique opportunity to study longer‐term outcomes of therapeutic changes and provide unique insights into the practicalities of medication switches in children. While not directly comparable, our results align with the safety, tolerability, and efficacy seen in adult RCTs and pediatric prospective studies of PAH therapies.
To assess drug efficacy, we used a range of measures across the domains of functional status, exercise and cardiovascular assessment. A subset of these measures have been considered as exploratory noninvasive endpoints in a handful of prospective studies in children with PAH, including time to clinical worsening, 27 , 41 , 42 change in WHO FC from baseline, 7 , 26 , 27 , 29 , 41 change in 6MWD, 7 , 26 , 27 , 29 change in CPET parameters, 9 , 43 echocardiographic parameters 7 , 26 , 27 , 44 and health surveys. 27 , 41 We found that WHO FC improved from baseline following ERA switch, following medication switch completion, and at medium‐term follow‐up (40.9 [35.2, 49.3] weeks). This result is consistent with previous prospective studies of both ambrisentan and tadalafil demonstrating improved or maintained WHO FC at 24 weeks in children with PAH. 26 , 27 In pediatric PAH WHO FC has been shown to be a surrogate for survival and is recommended as a treatment goal. 11 Changes in exercise tolerance were assessed using 6‐minute walk test and CPET, which both have limitations of feasibility in young children, limiting their use in trials. To date only two trials in children have considered change in 6MWD as an outcome, both of which were underpowered to draw strong conclusions. 7 , 27 Despite including children less than the accepted age of 7 years in assessment of 6MWD we observed good data completeness, with 75% of children performing a walk test at baseline. In our cohort, following a switch of sildenafil to tadalafil, children walked 21 m farther. This is a similar result to the 58 m increase in 6MWD seen in the open‐label extension study of children treated with ambrisentan, although this was over a median 3.5‐year follow‐up period. 29 Only a single pediatric trial has used CPET parameters as an endpoint, assessing efficacy of sildenafil monotherapy in treatment naïve children. 9 , 43 We observed a 7.1% reduction in VE/VCO2 and a 3.4% increase in peak VO2 in the 13 weeks from baseline, although neither result was statistically significant. This finding is comparable to the pediatric STARTS‐2 study that showed 9.7% reduction in VE/VCO2 and 7.7% increase in peak VO2 at 16 weeks of treatment with sildenafil. 43 Measures of right ventricular function and estimates of pulmonary artery pressure were assessed using echocardiographic and CMRI parameters in addition to surrogate marker NT‐proBNP for ventricular strain. Echocardiographic parameters were included in a recent post‐hoc analysis of 64 children treated with ambrisentan that showed a high degree of variability with small changes in only three parameters over the 24‐weeks of treatment. 44 Nevertheless, we included a selection of echocardiographic parameters that showed a small but significant change in RVSP and RAA in the medication switch group. As an alternative to echocardiography, CMRI has been shown to provide robust and reproducible assessment in children. 38 , 45 It can also provide can provide an estimate of mean pulmonary artery pressure. 37 Moreover, measures of RV volume and function have been shown to correlate well with 6MWD, WHO FC, tricuspid doppler‐derived estimates of systolic pulmonary artery pressure, and invasive measurement of mPAP and to predict outcomes in children with PAH. 38 More recently, two adult trials have used CMRI as an endpoint. 24 , 46 Hence, our analysis is the first to demonstrate the potential use of CMRI as an exploratory endpoint for pediatric studies assessing therapeutic efficacy in PAH. In children with PAH NT‐proBNP has been demonstrated as a prognostic marker 47 and hence its use in routine surveillance in our institution. However, there was significant data missingness due adoption on NT‐proBNP testing occurring later in the review window. Therefore, NT‐proBNP did not demonstrate utility in our comparison. Likewise, recent trials that have incorporated NT‐proBNP as a secondary or exploratory endpoint in children with PAH have not been able to demonstrate a clinically meaningful change due to studies being underpowered. 26 , 27 , 29
The duration of follow‐up was longer than most RCTs assessing efficacy of PAH therapy. Median duration of follow‐up was 50.5 28 [68] months, allowing for long‐term assessment of medication switch efficacy. At longer‐term follow‐up escalation in therapy with a prostanoid and occurrence of either all‐cause mortality, lung transplantation, or Potts shunt were high in both the control and switch groups. However, composite outcomes for children who underwent medication switch were comparable with those in the control group.
Adverse events were minimal and self‐limiting in children switched to ambrisentan plus tadalafil, with headache and pedal edema being the most common. These AEs were also the most commonly reported in adults in the results of the AMBITION study. However, we did not observe AEs of nasal congestion, anemia, or syncope in our pediatric cohort. 14 The AE profiles in our study are in‐keeping with pediatric cohorts of patients treated with either ambrisentan or tadalafil, which found that headache was the most commonly occurring treatment‐related AE. 3 , 4 , 26 , 28 , 29 Lower AE rates in our cohort of children were likely attributed to prior established combination therapy with bosentan plus sildenafil. This has previously been shown in studies of children for those who were treatment naïve experiencing higher rates of AEs, than those undergoing an in‐class medication switch. 3 , 4 , 26 , 27 , 28 Combination therapy with ambrisentan and tadalafil has been shown to be safe and tolerable in a small cohort of children in the retrospective study by Issapour et al. 30 However, only three children in this study were switched from bosentan plus sildenafil, whereas our study, with a larger number of children, confirms safety and tolerability of this in‐class medication switch.
This study is limited by its retrospective design as compared to a prospective randomized controlled study, which is the gold‐standard for assessing treatment efficacy and superiority. However, this study utilizes children who did not undergo medication switch as a control group, therefore allowing treatment effects to be compared. Although the sample size for each group was small, it was reasonable in comparison to retrospective pediatric studies in rare diseases. Data for some children was incomplete due to the study's real‐world setting and retrospective design, with some assessments not being available or performed. Data missingness was further compounded by the study window encompassing the COVID‐19 pandemic, which was declared in March 2020 in the UK. There was a temporary halt to in‐person consultations during this time. Therefore, the number of children included in the analyses differed for each variable, depending on corresponding follow‐up data.
The US FDA has highlighted the need in PAH trials to identify outcome measures that reflect how a patient “feels, functions or survives.” 48 We have assessed the effect of a therapeutic change on “function” (WHO FC, exercise testing, and right ventricular function) and “survival,” but due to the retrospective design of this review we were unable to assess how a patient “feels” through patient‐reported outcome measures.
CONCLUSION
A switch in combination therapy from bosentan plus sildenafil to ambrisentan plus tadalafil was well tolerated in children with PAH. The therapeutic switch was associated with improvements across a range of noninvasive parameters assessed during routine reviews within a structured clinical care program. At long‐term follow‐up transplant‐ or Potts shunt‐free survival in children who underwent combination oral medication switch appeared to be comparable with those who were not switched.
AUTHOR CONTRIBUTIONS
Shahin Moledina, Nikmah Idris, and Kathy Elterefi conceptualized the study. Cara Morgan, Nikmah Idris, Kathy Elterefi, Luca Di Ienno, Andrew Constantine, and Sadia Quyam performed data curation. Cara Morgan performed formal analysis and data visualization. Supervision was performed by Shahin Moledina. Cara Morgan and Shahin Moledina wrote the original draft. The final manuscript was reviewed by Cara Morgan, Kathy Elterefi, Roberta Bini, Andrew Constantine, Sadia Quyam, and Shahin Moledina.
CONFLICT OF INTEREST STATEMENT
AC has received an educational grant, payment for lectures/educational events, and nonfinancial support from Janssen‐Cilag Ltd. SM has acted as a consultant for Janssen‐Cilag Ltd and GSK. CM, NI, KE, LDI, SQ and RB declare no conflicts of interest.
ETHICS STATEMENT
This retrospective analysis utilizes anonymized data collected for routine clinical care covered under ethical approval 17/LO/0008, allowing individual consent to be waived.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
It is with sadness that we inform the reader that Dr Nikmah Idris has passed away. Her vision, dedication and contribution were central in this work. We sincerely thank all clinicians associated with the UK Pulmonary Hypertension Service for Children, who share the care for these complex patients. Without their cooperation and collaborative work, this study would not have been possible. The authors thank the Dinosaur Trust and Great Ormond Street Children's Charity for supporting research capacity within the Paediatric Pulmonary Hypertension Unit. All research at Great Ormond Street Hospital NHS Foundation Trust and UCL Great Ormond Street Institue of Child Health is made possible by the NIHR Great Ormond Street Hospital Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. This research received no specific grant from any funding agency in the public, commercial, or not‐for‐profit sectors.
Morgan C, Idris N, Elterefi K, Di enno L, Constantine A, Quyam S, Bini R, Moledina S. Safety, tolerability, and efficacy of an in‐class combination therapy switch from bosentan plus sildenafil to ambrisentan plus tadalafil in children with pulmonary arterial hypertension. Pulm Circ. 2024;14:e70011. 10.1002/pul2.70011
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Constantine A, Dimopoulos K, Haworth SG, Muthurangu V, Moledina S. Twenty‐Year experience and outcomes in a national pediatric pulmonary hypertension service. Am J Respir Crit Care Med. 2022;206:758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marín MJC, Rotés AS, Ogando AR, Soto AM, Jiménez MQ, Camacho JLG, Sonnenfeld IR, Bonora AM, Brotons DCA, Galdó AM. Assessing pulmonary hypertensive vascular disease in childhood. data from the Spanish registry. Am J Respir Crit Care Med. 2014;190:1421–1429. 10.1164/rccm.201406-1052OC [DOI] [PubMed] [Google Scholar]
- 3. Takatsuki S, Calderbank M, Ivy DD. Initial experience with tadalafil in pediatric pulmonary arterial hypertension. Pediatr Cardiol. 2012;33:683–688. 10.1007/s00246-012-0180-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Takatsuki S, Rosenzweig EB, Zuckerman W, Brady D, Calderbank M, Ivy DD. Clinical safety, pharmacokinetics, and efficacy of ambrisentan therapy in children with pulmonary arterial hypertension. Pediatr Pulmonol. 2013;48:27–34. 10.1002/ppul.22555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barst RJ, Maislin G, Fishman AP. Vasodilator therapy for primary pulmonary hypertension in children. Circulation. 1999;99:1197–1208. [DOI] [PubMed] [Google Scholar]
- 6. Lammers AE, Hislop AA, Flynn Y, Haworth SG. Epoprostenol treatment in children with severe pulmonary hypertension. Heart. 2007;93:739–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hansmann G, Meinel K, Bukova M, Chouvarine P, Wåhlander H, Koestenberger M. Selexipag for the treatment of children with pulmonary arterial hypertension: first multicenter experience in drug safety and efficacy. J Heart Lung Transplant. 2020;39:695–706. [DOI] [PubMed] [Google Scholar]
- 8. Schweintzger S, Koestenberger M, Schlagenhauf A, Grangl G, Burmas A, Kurath‐Koller S, Pocivalnik M, Sallmon H, Baumgartner D, Hansmann G, Gamillscheg A. Safety and efficacy of the endothelin receptor antagonist macitentan in pediatric pulmonary hypertension. Cardiovasc Diagn Ther. 2020;10:1675–1685. 10.21037/cdt.2020.04.01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barst RJ, Ivy DD, Gaitan G, Szatmari A, Rudzinski A, Garcia AE, Sastry BKS, Pulido T, Layton GR, Serdarevic‐Pehar M, Wessel DL. A randomized, double‐blind, placebo‐controlled, dose‐ranging study of oral sildenafil citrate in treatment‐naive children with pulmonary arterial hypertension. Circulation. 2012;125:324–334. 10.1161/CIRCULATIONAHA.110.016667 [DOI] [PubMed] [Google Scholar]
- 10. Beghetti M, Haworth SG, Bonnet D, Barst RJ, Acar P, Fraisse A, Ivy DD, Jais X, Schulze‐Neick I, Galiè N, Morganti A, Dingemanse J, Kusic‐Pajic A, Berger RMF. Pharmacokinetic and clinical profile of a novel formulation of bosentan in children with pulmonary arterial hypertension: the FUTURE‐1 study. Br J Clin Pharmacol. 2009;68:948–955. 10.1111/j.1365-2125.2009.03532.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rosenzweig EB, Abman SH, Adatia I, Beghetti M, Bonnet D, Haworth S, Ivy DD, Berger RMF. Paediatric pulmonary arterial hypertension: updates on definition, classification, diagnostics and management. Eur Respir J. 2019;53:1801916. 10.1183/13993003.01916-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, Carlsen J, Coats AJS, Escribano‐Subias P, Ferrari P, Ferreira DS, Ghofrani HA, Giannakoulas G, Kiely DG, Mayer E, Meszaros G, Nagavci B, Olsson KM, Pepke‐Zaba J, Quint JK, Rådegran G, Simonneau G, Sitbon O, Tonia T, Toshner M, Vachiery JL, Vonk Noordegraaf A, Delcroix M, Rosenkranz S, Schwerzmann M, Dinh‐Xuan AT, Bush A, Abdelhamid M, Aboyans V, Arbustini E, Asteggiano R, Barberà JA, Beghetti M, Čelutkienė J, Cikes M, Condliffe R, de Man F, Falk V, Fauchier L, Gaine S, Galié N, Gin‐Sing W, Granton J, Grünig E, Hassoun PM, Hellemons M, Jaarsma T, Kjellström B, Klok FA, Konradi A, Koskinas KC, Kotecha D, Lang I, Lewis BS, Linhart A, Lip GYH, Løchen ML, Mathioudakis AG, Mindham R, Moledina S, Naeije R, Nielsen JC, Olschewski H, Opitz I, Petersen SE, Prescott E, Rakisheva A, Reis A, Ristić AD, Roche N, Rodrigues R, Selton‐Suty C, Souza R, Swift AJ, Touyz RM, Ulrich S, Wilkins MR, Wort SJ. 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;43:3618–3731. [DOI] [PubMed] [Google Scholar]
- 13. Avitabile CM, Vorhies EE, Ivy DD. Drug treatment of pulmonary hypertension in children. Pediatric Drugs. 2020;22:123–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Galiè N, Barberà JA, Frost AE, Ghofrani HA, Hoeper MM, McLaughlin VV, Peacock AJ, Simonneau G, Vachiery JL, Grünig E, Oudiz RJ, Vonk‐Noordegraaf A, White RJ, Blair C, Gillies H, Miller KL, Harris JHN, Langley J, Rubin LJ. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015;373:834–844. 10.1056/NEJMoa1413687 [DOI] [PubMed] [Google Scholar]
- 15. Sitbon O, Sattler C, Bertoletti L, Savale L, Cottin V, Jaïs X, De Groote P, Chaouat A, Chabannes C, Bergot E, Bouvaist H, Dauphin C, Bourdin A, Bauer F, Montani D, Humbert M, Simonneau G. Initial dual oral combination therapy in pulmonary arterial hypertension. Eur Respir J. 2016;47:1727–1736. 10.1183/13993003.02043-2015 [DOI] [PubMed] [Google Scholar]
- 16. Escribano Subías P, Aurtenetxe Pérez A, Pérez Olivares C, Gómez Climent L, Diago Cabezudo JI, Perelló MF. Recent advances in the management of pulmonary arterial hypertension: lessons from the upfront combination of ambrisentan and tadalafil. Expert Rev Respir Med. 2021;15:493–504. 10.1080/17476348.2021.1878027 [DOI] [PubMed] [Google Scholar]
- 17. Kataoka M, Satoh T, Matsubara H, Yamamoto K, Inada T, Umezawa K, Takahashi T, Nakano A, Fukuda K. Safety and efficacy of ambrisentan–phosphodiesterase type 5 (PDE5) inhibitor combination therapy for Japanese pulmonary arterial hypertension patients in real‐world clinical practice. Circ Rep. 2019;1:268–275. 10.1253/circrep.CR-19-0029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boucly A, Savale L, Jaïs X, Bauer F, Bergot E, Bertoletti L, Beurnier A, Bourdin A, Bouvaist H, Bulifon S, Chabanne C, Chaouat A, Cottin V, Dauphin C, Degano B, De Groote P, Favrolt N, Feng Y, Horeau‐Langlard D, Jevnikar M, Jutant EM, Liang Z, Magro P, Mauran P, Moceri P, Mornex JF, Palat S, Parent F, Picard F, Pichon J, Poubeau P, Prévot G, Renard S, Reynaud‐Gaubert M, Riou M, Roblot P, Sanchez O, Seferian A, Tromeur C, Weatherald J, Simonneau G, Montani D, Humbert M, Sitbon O. Association between initial treatment strategy and long‐term survival in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2021;204:842–854. 10.1164/rccm.202009-3698OC [DOI] [PubMed] [Google Scholar]
- 19. Sitbon O, Cottin V, Canuet M, Clerson P, Gressin V, Perchenet L, Bertoletti L, Bouvaist H, Picard F, Prévot G, Bergot E, Simonneau G. Initial combination therapy of macitentan and tadalafil in pulmonary arterial hypertension. Eur Respir J. 2020;56:2000673. 10.1183/13993003.00673-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fu W, He W, Li Y, Chen Y, Liang J, Lei H, Fu L, Chen Y, Ren N, Jiang Q, Shen Y, Ma R, Wang T, Wang X, Zhang N, Xiao D, Liu C. Efficacy and safety of novel‐targeted drugs in the treatment of pulmonary arterial hypertension: a Bayesian network meta‐analysis. Drug Delivery. 2021;28:1007–1019. 10.1080/10717544.2021.1927243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grünig E, Jansa P, Fan F, Hauser JA, Pannaux M, Morganti A, Rofael H, Chin KM. Randomized trial of macitentan/tadalafil single‐tablet combination therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2024;83:473–484. [DOI] [PubMed] [Google Scholar]
- 22. Barst RJ, McGoon MD, Elliott CG, Foreman AJ, Miller DP, Ivy DD. Survival in childhood pulmonary arterial hypertension. Circulation. 2012;125:113–122. [DOI] [PubMed] [Google Scholar]
- 23. Hansmann G, Koestenberger M, Alastalo T‐P, Apitz C, Austin ED, Bonnet D, Budts W, D'Alto M, Gatzoulis MA, Hasan BS, Kozlik‐Feldmann R, Kumar RK, Lammers AE, Latus H, Michel‐Behnke I, Miera O, Morrell NW, Pieles G, Quandt D, Sallmon H, Schranz D, Tran‐Lundmark K, Tulloh RMR, Warnecke G, Wåhlander H, Weber SC, Zartner P. 2019 updated consensus statement on the diagnosis and treatment of pediatric pulmonary hypertension: the european pediatric pulmonary vascular disease network (EPPVDN), endorsed by AEPC, ESPR and ISHLT. J Heart Lung Transplant. 2019;38:879–901. [DOI] [PubMed] [Google Scholar]
- 24. Hoeper MM, Al‐Hiti H, Benza RL, Chang SA, Corris PA, Gibbs JSR, Grünig E, Jansa P, Klinger JR, Langleben D, McLaughlin VV, Meyer GMB, Ota‐Arakaki J, Peacock AJ, Pulido T, Rosenkranz S, Vizza CD, Vonk‐Noordegraaf A, White RJ, Chang M, Kleinjung F, Meier C, Paraschin K, Ghofrani HA, Simonneau G, Olschewski H, Delcroix M, Andrade‐Lima M, de Amorim Corrêac R, Figueiredo Campos F, Ota Arakaki J, Meyer G, De Souza R, Langleben D, Al‐Hiti H, Jansa P, Mellemkjær S, Bauer F, Montani D, Simonneau G, Drömann D, Ghofrani HA, Grünig E, Halank M, Held M, Hoeper M, Klose H, Kneidinger N, Leuchte H, Opitz C, Rosenkranz S, Wilkens H, Wirtz H, Karvounis H, Pitsiou G, Orfanos S, D'Alto M, Ghio S, Vizza C, Vitulo P, Nakayama T, Maki H, Tatebe S, de los Rios Ibarra M, Pulido T, Van Dijk A, Vonk‐Noordegraaf A, Roleder T, Castro G, Loureiro M, Robalo‐Martins S, Barberá J, Lázaro M, Perez‐Penate G, Román A, Cheng CC, Hsu CH, Hsu HH, Atahan E, Mogulkoc Bishop N, Okumus N, Onen Z, Chang HJ, Chang SA, Lee JS, Kim HK, Coghlan J, Corris P, Church A, Condliffe R, Gibbs J, Peacock A, Wort S, Allen R, Allen S, Awdish R, Benza R, DeSouza S, Feldman J, Johri S, Klinger J, Layish D, McConnell J, McLaughlin V, Migliore C, Rahaghi F, Rischard F, Robbins I, Satterwhite L, Shah T, Sulica R, White R. Switching to riociguat versus maintenance therapy with phosphodiesterase‐5 inhibitors in patients with pulmonary arterial hypertension (REPLACE): a multicentre, open‐label, randomised controlled trial. Lancet Respir Med. 2021;9:573–584. [DOI] [PubMed] [Google Scholar]
- 25. Shiva A, Shiran M, Rafati M, Zamani H, Babazadeh K, Saeedi M, Ala S. Oral tadalafil in children with pulmonary arterial hypertension. Drug Res. 2015;66:7–10. 10.1055/s-0034-1395510 [DOI] [PubMed] [Google Scholar]
- 26. Ivy D, Beghetti M, Juaneda‐Simian E, Miller D, Lukas MA, Ioannou C, Okour M, Narita J, Berger RMF. A randomized study of safety and efficacy of two doses of ambrisentan to treat pulmonary arterial hypertension in pediatric patients aged 8 years up to 18 years. J Pediatr: X. 2020;5:100055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ivy D, Bonnet D, Berger R, Meyer GMB, Baygani S, Li B. Efficacy and safety of tadalafil in a pediatric population with pulmonary arterial hypertension: phase 3 randomized, double‐blind placebo‐controlled study. Pulm Circ. 2021;11:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Youssef DE, Handler SS, Richards SM, Sheppard CA, Smith J, Tillman K, Pietrosanu M, Kirkpatrick E, Bates A. Multicenter review of a tadalafil suspension formulation for infants and children with pulmonary hypertension: a North American experience. Front Pediatr. 2023;11:1055131. 10.3389/fped.2023.1055131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ivy D, Beghetti M, Juaneda‐Simian E, Ravindranath R, Lukas MA, Machlitt‐Northen S, Scott N, Narita J, Berger RMF. Long‐term safety and tolerability of ambrisentan treatment for pediatric patients with pulmonary arterial hypertension: an open‐label extension study. Eur J Pediatr. 2024;183:2141–2153. 10.1007/s00431-024-05446-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Issapour A, Frank B, Crook S, Hite MD, Dorn ML, Rosenzweig EB, Ivy DD, Krishnan US. Safety and tolerability of combination therapy with ambrisentan and tadalafil for the treatment of pulmonary arterial hypertension in children: real‐world experience. Pediatr Pulmonol. 2022;57:724–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. European Medicines Agency . Assessment report—Volibris. https://www.ema.europa.eu/en/documents/variation-report/volibris-h-c-839-x-0061-g-epar-assessment-report-variation_en.pdf. 2021.
- 32. British and Irish Legal Information Institute. Gillick v West Norfolk and Wisbech AHA; 1985. [Google Scholar]
- 33. Moledina S. Risk stratification in paediatric pulmonary arterial hypertension. In: Molecular mechanism of congenital heart disease and pulmonary hypertension. Singapore: Springer Singapore; 2020. p. 177–182. [Google Scholar]
- 34. Wright CM, Williams AF, Elliman D, Bedford H, Birks E, Butler G, Sachs M, Moy RJ, Cole TJ. Using the new UK‐WHO growth charts. BMJ. 2010;340:c1140. [DOI] [PubMed] [Google Scholar]
- 35. Styles ME. New cross sectional stature, weight, and head circumference references for down's syndrome in the UK and Republic of Ireland. Arch Dis Child. 2002;87:104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lammers AE, Marek J, Diller GP, Haworth SG, Moledina S. Prognostic value of transthoracic echocardiography in children with pulmonary arterial hypertension. J Am Heart Assoc. 2023;12:e023118. 10.1161/JAHA.121.023118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pandya B, Quail MA, Steeden JA, McKee A, Odille F, Taylor AM, Schulze‐Neick I, Derrick G, Moledina S, Muthurangu V. Real‐time magnetic resonance assessment of septal curvature accurately tracks acute hemodynamic changes in pediatric pulmonary hypertension. Circ: Cardiovasc Imaging. 2014;7:706–713. [DOI] [PubMed] [Google Scholar]
- 38. Moledina S, Pandya B, Bartsota M, Mortensen KH, McMillan M, Quyam S, Taylor AM, Haworth SG, Schulze‐Neick I, Muthurangu V. Prognostic significance of cardiac magnetic resonance imaging in children with pulmonary hypertension. Circ: Cardiovasc Imaging. 2013;6:407–414. 10.1161/CIRCIMAGING.112.000082 [DOI] [PubMed] [Google Scholar]
- 39. Abumehdi MR, Wardle AJ, Nazzal R, Charalampopoulos A, Schulze‐Neick I, Derrick G, Moledina S, Giardini A. Feasibility and safety of cardiopulmonary exercise testing in children with pulmonary hypertension. Cardiol Young. 2016;26:1144–1150. [DOI] [PubMed] [Google Scholar]
- 40. Galiè N, McLaughlin VV, Rubin LJ, Simonneau G. An overview of the 6th world symposium on pulmonary hypertension. Eur Respir J. 2019;53:1802148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Berger RMF, Haworth SG, Bonnet D, Dulac Y, Fraisse A, Galiè N, Ivy DD, Jaïs X, Miera O, Rosenzweig EB, Efficace M, Kusic‐Pajic A, Beghetti M. FUTURE‐2: results from an open‐label, long‐term safety and tolerability extension study using the pediatric FormUlation of bosenTan in pulmonary arterial hypeRtEnsion. Int J Cardiol. 2016;202:52–58. 10.1016/j.ijcard.2015.08.080 [DOI] [PubMed] [Google Scholar]
- 42. Berger RMF, Gehin M, Beghetti M, Ivy D, Kusic‐Pajic A, Cornelisse P, Grill S, Bonnet D. A bosentan pharmacokinetic study to investigate dosing regimens in paediatric patients with pulmonary arterial hypertension: FUTURE‐3. Br J Clin Pharmacol. 2017;83:1734–1744. 10.1111/bcp.13267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Russell S, Beghetti M, Oudiz R, Balagtas C, Zhang M, Ivy D. Effects of oral sildenafil on exercise capacity in children with pulmonary arterial hypertension: a randomised trial. Open Heart. 2019;6:001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Beghetti M, Berger RMF, Bonnet D, Grill S, Lesage C, Lemarie JC, Ivy DD. Echocardiographic changes and Long‐Term clinical outcomes in pediatric patients with pulmonary arterial hypertension treated with bosentan for 72 weeks: A post‐hoc analysis from the FUTURE 3 study. Front Pediatr. 2021;9:681538. 10.3389/fped.2021.681538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Muthurangu V, Lurz P, Critchely JD, Deanfield JE, Taylor AM, Hansen MS. Real‐time assessment of right and left ventricular volumes and function in patients with congenital heart disease by using high spatiotemporal resolution radial k‐t SENSE. Radiology. 2008;248:782–791. [DOI] [PubMed] [Google Scholar]
- 46. Vonk Noordegraaf A, Channick R, Cottreel E, Kiely DG, Marcus JT, Martin N, Moiseeva O, Peacock A, Swift AJ, Tawakol A, Torbicki A, Rosenkranz S, Galiè N. The REPAIR study. JACC: Cardiovasc Imaging. 2022;15:240–253. [DOI] [PubMed] [Google Scholar]
- 47. Ploegstra M‐J, Zijlstra WMH, Douwes JM, Hillege HL, Berger RMF. Prognostic factors in pediatric pulmonary arterial hypertension: a systematic review and meta‐analysis. Int J Cardiol. 2015;184:198–207. [DOI] [PubMed] [Google Scholar]
- 48. Sitbon O, Gomberg‐Maitland M, Granton J, Lewis MI, Mathai SC, Rainisio M, Stockbridge NL, Wilkins MR, Zamanian RT, Rubin LJ. Clinical trial design and new therapies for pulmonary arterial hypertension. Eur Respir J. 2019;53:1801908. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.