

ABSTRACT
Hereditary fructose intolerance (HFI) is characterized by liver damage and a secondary defect in N‐linked glycosylation due to impairment of mannose phosphate isomerase (MPI). Mannose treatment has been shown to be an effective treatment in a primary defect in MPI (i.e., MPI‐CDG), which is also characterized by liver damage. Therefore, the aims of this study were to determine: (1) hepatic nucleotide sugar levels, and (2), the effect of mannose supplementation on hepatic nucleotide sugar levels and liver fat, in a mouse model for HFI. Aldolase B deficient mice (Aldob −/− ) were treated for four weeks with 5% mannose via the drinking water and compared to Aldob −/− mice and wildtype mice treated with regular drinking water. We found that hepatic GDP‐mannose and hepatic GDP‐fucose were lower in water‐treated Aldob −/− mice when compared to water‐treated wildtype mice (p = 0.002 and p = 0.002, respectively), consistent with impaired N‐linked glycosylation. Of interest, multiple other hepatic nucleotide sugars not involved in N‐linked glycosylation, such as hepatic UDP‐glucuronic acid, UDP‐xylose, CMP‐N‐acetyl‐beta‐neuraminic acid, and CDP‐ribitol (p = 0.002, p = 0.003, p = 0.002, p = 0.002), were found to have altered levels as well. However, mannose treatment did not correct the reduction in hepatic GDP‐mannose levels, nor was liver fat affected. Aldob −/− mice are characterized by hepatic nucleotide sugar abnormalities, but these were not abrogated by mannose treatment. Future studies are needed to identify the underlying mechanisms responsible for the abnormal hepatic nucleotide sugar pattern and intrahepatic lipid accumulation in HFI.
Trial Registration: PCT ID: PCTE0000340, this animal experiment is registered at (https://preclinicaltrials.eu/).
Keywords: aldolase B, congenital disorders of glycosylation, fructose intolerance, genetic diseases, glycosylation, nucleotide sugars
Abbreviations
- Aldob −/−
aldolase B knockout
- CDG
congenital disorder of glycosylation
- F1P
fructose 1‐phosphate
- HFI
hereditary fructose intolerance
- MPI
mannose phosphate isomerase
1. Introduction
Hereditary fructose intolerance (HFI; OMIM #229600) is an autosomal recessive inborn error of metabolism [1, 2], with an estimated incidence of 1:18000–20 000 in live births [3, 4]. HFI is caused by mutations in the gene encoding aldolase B (ALDOB). Aldolase B (EC 4.1.2.13), which is predominantly expressed in the liver, kidney, and small intestine [5], catalyses the cleavage of fructose 1,6‐bisphosphate and fructose 1‐phosphate (F1P) into triose molecules [6]. In patients with HFI, ingestion of fructose results in the accumulation of F1P and concomitant depletion of ATP, which are believed to cause symptoms such as nausea, vomiting, hypoglycaemia, and liver and kidney failure [7, 8, 9, 10].
Treatment of HFI consists of a lifelong fructose‐restricted diet [11, 12, 13]. We and others recently showed that, despite this treatment, HFI patients are characterized by intrahepatic lipid accumulation when compared to healthy controls [14, 15, 16]. Some HFI patients even displayed signs of hepatic inflammation and fibrosis [14, 17]. The underlying mechanism responsible for the liver derangements in HFI remains to be elucidated [18]. We recently demonstrated that glucokinase regulatory protein and carbohydrate response element binding protein mediate F1P‐stimulated de novo lipogenesis in a mouse model for HFI, but these factors did not affect intrahepatic lipid accumulation [19].
HFI patients are also characterized by a secondary defect in N‐linked glycosylation likely caused by F1P‐mediated impairment of mannose phosphate isomerase (MPI) (Figure 1) [20]. We and others recently showed that the abnormal transferrin glycosylation patterns can be used as a diagnostic biomarker to identify dietary‐treated HFI patients [21, 22].
FIGURE 1.
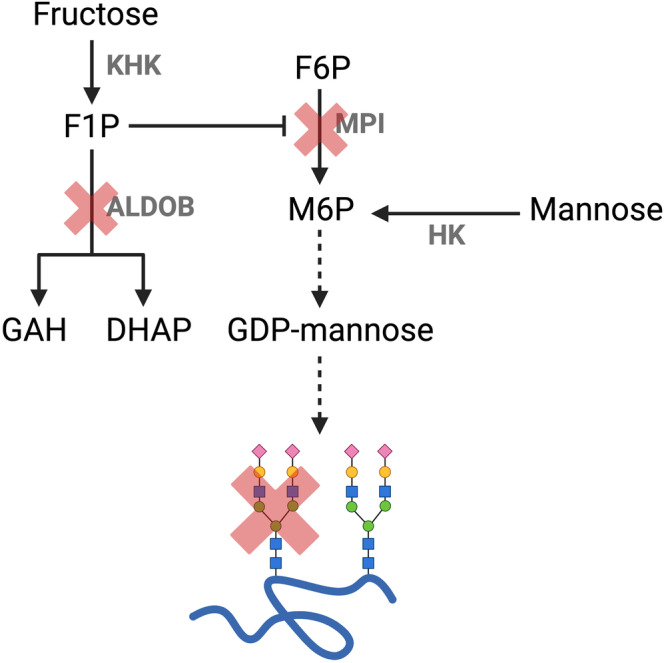
Fructose 1‐phosphate mediated impairment of mannose phosphate isomerase and N‐linked glycosylation in aldolase B deficiency. Abbreviations: ALDOB, aldolase B; DHAP, dihydroxyacetone phosphate; F1P, fructose 1‐phosphate; F6P, fructose 6‐phosphate; GAH, glyceraldehyde; GDP‐mannose, guanosine diphosphate mannose; HK, hexokinase; KHK, ketohexokinase; M6P, mannose 6‐phosphate; MPI, mannose phosphate isomerase. Figure created with (Biorender.com).
Of interest, patients with a congenital disorder of glycosylation caused by a primary defect in MPI (MPI‐CDG) [23], are also characterized by hepatic fat accumulation and hepatic fibrosis [24, 25] (but are otherwise clinically different to HFI). Since treatment of MPI‐CDG consists of mannose supplementation (which bypasses the enzymatic defect in MPI) [23], it might also be an alternative or add‐on treatment for HFI [18].
Therefore, the aims of this present study were to determine: (1) hepatic nucleotide levels in a mouse model for HFI, and (2), the effect of mannose supplementation on hepatic nucleotide sugar levels and the fatty liver phenotype in HFI.
2. Materials and Methods
2.1. Animals
All experimental procedures were approved by the Animal Experiments Committee of Maastricht University (Maastricht, The Netherlands; AVD1070020187086) and in compliance with the relevant guidelines from the Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
We used aldolase B knockout (Aldob −/−) mice since they exhibit similar metabolic features including hepatic F1P accumulation and the fatty liver phenotype as seen in HFI patients [26, 27]. Aldob −/− mice in the C57BL/6NTac background were generated as previously described (Figure S1 and Table S1) [26]. All mice were maintained in temperature‐ and humidity‐controlled specific pathogen–free conditions on a 12‐h‐dark and 12‐h‐light cycle (lights on from 7:00 am to 7:00 pm) and allowed ad libitum access to a fructose‐free diet (Bioserv, catalog F6700).
(FIGURE S2) depicts a flow diagram of the experimental design. Female mice were caged according to genotype (with at least one litter mate; researcher non‐blinded) and underwent a 2‐week acclimatisation period to the experimental location prior to the start of the experiment. After the 2‐week acclimatisation period, water treatment (control group) was non‐randomly allocated to female wildtype mice. Water treatment (control group) and mannose treatment (experimental group) were randomly allocated per cage of female Aldob −/− mice.
Next, eight‐week‐old water‐treated wildtype mice (n = 7) and water‐treated Aldob −/− mice (n = 7) were followed‐up for 4 weeks. In parallel, eight‐week‐old Aldob −/− mice (n = 6) were treated for 4 weeks with 5% mannose (D‐[+]‐Mannose, Sigma‐Aldrich, CAS No.: 3458‐28‐4) via the drinking water (~200 mg/day, as done before to treat MPI‐deficient mice [28]).
After the 4‐week experiment, all animals were sacrificed by CO2/O2 inhalation (8 AM). After sacrifice, livers were immediately collected, snap‐frozen in liquid nitrogen, and stored at −80°C.
2.2. Analysis of Hepatic Nucleotide Sugars by Triple‐Quad Mass Spectrometry
Levels of hepatic nucleotide sugars were measured by triple‐quad mass spectrometry according to published methods [29, 30]. For quantification of hepatic GDP‐mannose levels, quantified as a proxy and a downstream product of MPI activity (Figure 1), we made use of an external GDP‐mannose calibration curve. Commercial GDP‐mannose (Sigma, G‐5131, CAS 103301–73‐1) was diluted in MilliQ water resulting in concentration series of 0.025, 0.05, 0.1, 0.2, 0.4, 0.8, 1.0, and 2.0 μM. Protein concentrations of the liver homogenates were determined by a Lowry protein assay. Calibration curves were measured in the same series, and intensities of GDP‐mannose in the samples was correlated with the standard curve to calculate concentrations per protein. Relative hepatic nucleotide sugars were normalized based on the sum of GDP‐mannose, GDP‐fucose, UDP‐galactose, UDP‐glucose, UDP‐N‐hexosamine (i.e., the combined peaks of UDP‐N‐acetylgalactosamine and UDP‐N‐acetylglucosamine), UDP‐glucuronic acid, UDP‐xylose, CMP‐N‐acetyl‐beta‐neuraminic acid, and CDP‐ribitol. Data was processed in Skyline 20.2. The levels of hepatic nucleotide sugars are shown as the relative abundance of each nucleotide sugar.
2.3. Analysis of Hepatic Fat by Biochemical Assay
Livers were homogenized in 250 μL SET buffer (250 mM sucrose, 2 mM EDTA, and 10 mM Tris) using a Mini‐bead beater homogenizer (Biospec). Hepatic triglycerides were measured using a colorimetric test (Triglycerides FS 5'ecoline, Diagnostic System GmbH, Holzheim, Germany) as described previously [31]. Hepatic triglyceride levels were corrected for protein content by using a BCA assay (BCA kit, Sigma‐Aldrich, Germany) according to the manufacturer's instructions.
2.4. Statistical Analyses
Data in figures are presented as box‐and‐whisker plots indicating the sample minimum, median, and sample maximum. Data graphics were performed using GraphPad Prism 5.01 (La Jolla California, USA). Statistical analyses were performed with the use of the Statistical Package for Social Sciences (Version 25.0; IBM, Chicago, IL). Data were analysed with Mann–Whitney U tests and Hochberg‐adjusted to correct for multiple testing. A value of p < 0.05 was regarded as statistically significant. No animals, experimental units, or data points were excluded.
3. Results
Water‐treated wildtype mice had a higher body weight when compared to water‐treated Aldob −/− mice and mannose‐treated Aldob −/− mice (p = 0.006 and p = 0.028, respectively, Figure 2). Noteworthy, mannose‐treated Aldob −/− mice had a higher body weight in comparison to water‐treated Aldob −/− mice (p = 0.035, Figure 2), which was already evident at the start of the experiment (p = 0.022 at baseline, data not shown).
FIGURE 2.
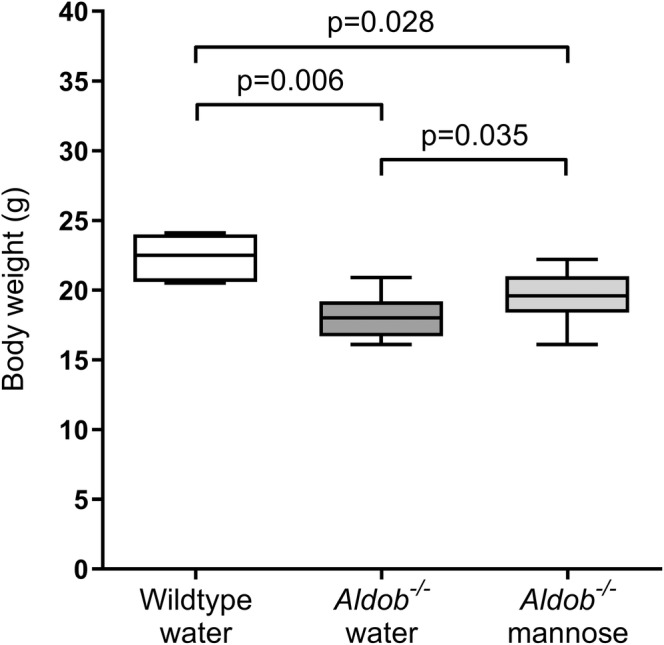
Effects of aldolase B deficiency and mannose supplementation on body weight. Body weight (g) in female water‐treated wildtype (Aldob +/+ ) (n = 7), water‐treated Aldob −/− (n = 7), and mannose‐treated Aldob −/− mice (n = 6). Data are presented as sample minimum, median, and sample maximum. Analysed with Mann–Whitney U tests with Hochberg‐adjustment.
Four out of the 6 mannose‐treated Aldob −/− mice developed severe morphological eye defects (i.e., cloudy eyes and smaller eyecups), within 1 week after mannose supplementation initiation. Of interest, none of the mice with eye defects seemed to be affected in terms of welfare, weight gain, and eating and drinking behavior.
3.1. Effects of Aldolase B Deficiency and Mannose Supplementation on Hepatic Nucleotide Sugars
We measured hepatic GDP‐mannose and GDP‐fucose (formed from GDP‐mannose) levels as a proxy and a downstream product of MPI activity. Absolute hepatic GDP‐mannose was lower in water‐treated Aldob −/− mice when compared to water‐treated wildtype mice (p = 0.002, Figure 3A).
FIGURE 3.
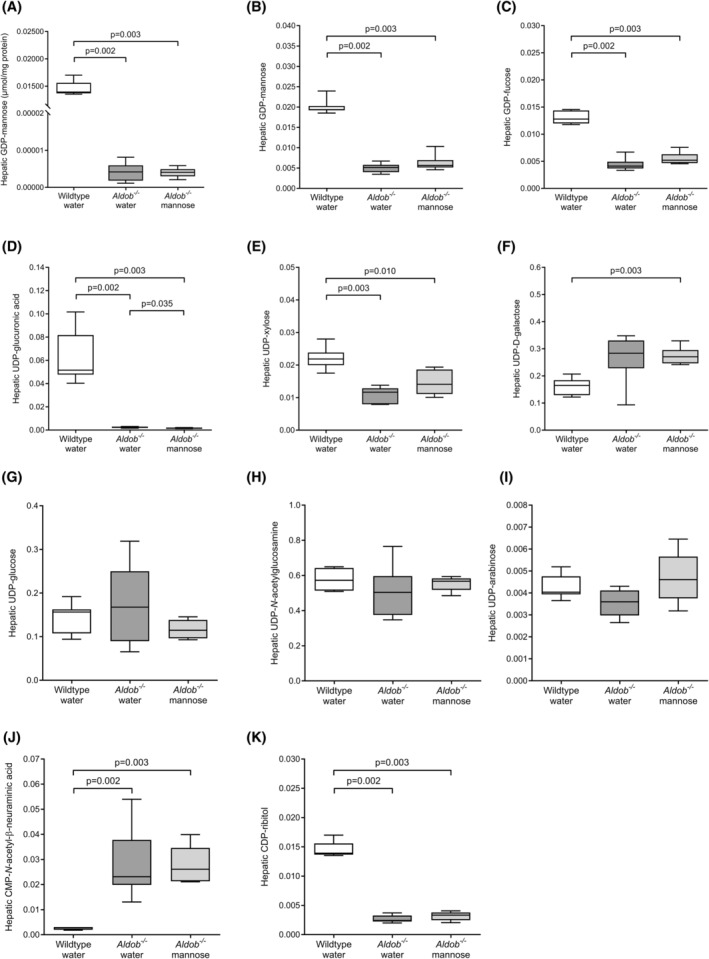
Effects of aldolase B deficiency and mannose supplementation on hepatic nucleotide sugars. (A) Absolute hepatic GDP‐mannose (μmol/mg protein) in female water‐treated wildtype (Aldob +/+ ) (n = 7), water‐treated Aldob −/− (n = 7), and mannose‐treated Aldob −/− mice (n = 6). (B–K) Relative levels of hepatic nucleotide sugars (relative abundance of each nucleotide sugar) in the same groups as panel A. Data are presented as sample minimum, median, and sample maximum. Analysed with Mann–Whitney U tests with Hochberg‐adjustment.
Next, we analysed the relative levels of hepatic GDP‐mannose and GDP‐fucose. Consistent with the absolute levels, relative hepatic GDP‐mannose was lower in water‐treated Aldob −/− mice when compared to water‐treated wildtype mice (p = 0.002, Figure 3B). Hepatic GDP‐fucose was also lower in water‐treated Aldob −/− mice when compared to water‐treated wildtype mice (p = 0.002, Figure 3C).
We also analysed the relative levels of other hepatic nucleotide sugars that are not directly related to MPI activity. Both hepatic UDP‐glucuronic acid and hepatic UDP‐xylose were lower in water‐treated Aldob −/− mice compared to water‐treated wildtype mice (p = 0.002 and p = 0.003, respectively, Figure 3D,E). Hepatic UDP‐galactose, UDP‐glucose, hepatic UDP‐N‐acetylglucosamine, and hepatic UDP‐arabinose were not different between the groups (Figure 3F–I). In addition, hepatic CMP‐N‐acetyl‐beta‐neuraminic acid was higher (p = 0.002, Figure 3J), while hepatic CDP‐ribitol was lower in water‐treated Aldob −/− mice compared to water‐treated wildtype mice (p = 0.002, Figure 3K).
Last, we studied the effects of 4‐week mannose supplementation on hepatic nucleotide sugars in Aldob −/− mice. Absolute hepatic GDP‐mannose was not different between water‐treated Aldob −/− mice and mannose‐treated Aldob −/− mice (Figure 3A). Furthermore, apart from hepatic UDP‐glucuronic acid (p = 0.035, Figure 3D), mannose treatment did not affect the relative levels of hepatic sugar nucleotides (Figure 3B,C,E–K).
3.2. Effects of Aldolase B Deficiency and Mannose Supplementation on Liver Fat
Water‐treated Aldob −/− mice had a higher intrahepatic lipid content when compared to wildtype mice (p = 0.05, Figure 4), consistent with previous studies demonstrating a fatty liver phenotype in Aldob −/− mice [19, 26, 27, 32].
FIGURE 4.
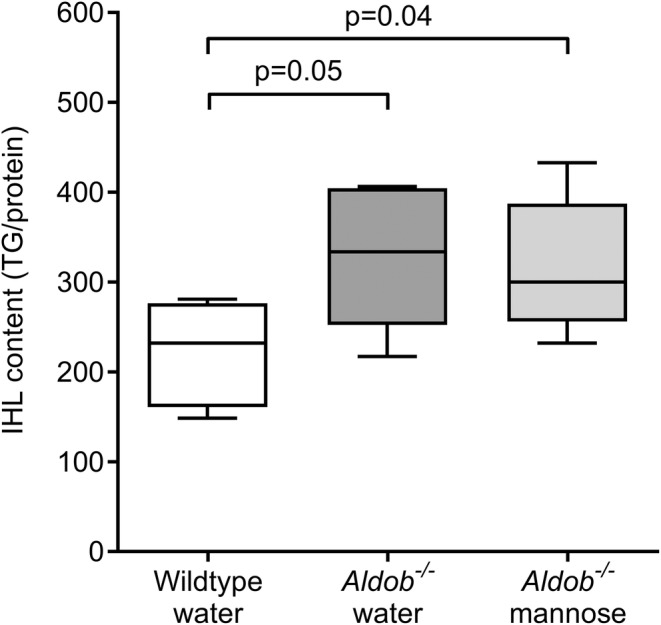
Effects of aldolase B deficiency and mannose supplementation on liver fat. Intrahepatic lipid (IHL) content (triglycerides/protein) in female water‐treated wildtype (Aldob +/+ ) (n = 7), water‐treated Aldob −/− (n = 7), and mannose‐treated Aldob −/− mice (n = 6). Data are presented as sample minimum, median, and sample maximum. Analysed with Mann–Whitney U tests with Hochberg‐adjustment.
Next, we studied the effects of 4‐week mannose supplementation on liver fat in Aldob −/− mice. In contrast to our hypothesis, intrahepatic lipid content did not differ between water‐treated Aldob −/− mice and mannose‐treated Aldob −/− mice (p = 0.731, Figure 4).
4. Discussion
We found abnormal levels of hepatic nucleotide sugars in a mouse model for HFI, but mannose supplementation did not reverse these nucleotide sugars abnormalities nor the fatty liver phenotype.
Glycosylation, the addition of a carbohydrate chain to proteins, is one of the most common post‐translational modifications and affects many aspects of protein function (e.g., protein folding, enzyme activity, and cell‐to‐cell and cell‐to‐extracellular matrix interactions) [33]. MPI‐CDG is a primary defect in glycosylation caused by mutations in MPI and is characterized by, amongst others, hypoglycaemia, hepatic steatosis, and fibrosis [23]. Treatment with oral mannose has been shown to improve most symptoms of the disease, as it can serve as a substrate for mannose 6‐phosphate, independent from MPI (Figure 1) [23, 34]. Of interest, HFI shows some clinical overlap with MPI‐CDG. Patients with HFI also present with hepatic steatosis and, in some cases, hepatic fibrosis [14, 15, 16, 17]. Furthermore, previous studies have shown that HFI patients are characterized by a secondary impairment of glycosylation due to F1P‐mediated inhibition of MPI (Figure 1) [14, 35, 36]. In agreement, to the best of our knowledge, we are the first to demonstrate hepatic nucleotide sugar abnormalities in a mouse model that phenocopies HFI.
Besides lower hepatic GDP‐mannose and hepatic GDP‐fucose levels, in line with F1P‐mediated impairment of MPI (Figure 1), we also found other relative hepatic nucleotide sugars abnormalities in Aldob −/− mice. These findings could either be spurious (relative differences due to normalization) or have a true biological explanation. Previous animal studies have demonstrated a higher glucose uptake in Aldob −/− mice in comparison to wildtype mice [27, 32]. Consequent glucose phosphorylation results in increased glucose 6‐phosphate levels which could, amongst others, facilitate the conversion into glucose 1‐phoshate. The latter can be converted into UDP‐galactose and UDP‐glucose (via the Leloir pathway) [37, 38], and could explain the suggestively higher levels of hepatic UDP‐galactose in Aldob −/− mice (p = 0.052, Figure 3F).
Although Aldob −/− mice presented with reduced hepatic GDP‐mannose (and GDP‐fucose) levels, mannose supplementation did not correct the hepatic nucleotide sugars abnormalities nor liver fat. Aldob −/− mice were treated with 5% mannose via the drinking water for 4 weeks (~200 mg/day). It could be speculated that the mannose dose was too low and/or mannose was not properly taken up by the liver. Unfortunately, we did not measure serum mannose levels in this study. Of note, we based the dose of mannose on a previous study by He and colleagues in which MPI‐deficient mice were treated with 5% mannose in the drinking water for 7 days (providing ∼200 mg mannose/day), which increased serum mannose levels by 20%–40% [28]. The authors found that 5% mannose supplementation restored ICAM‐1 expression in the vasculature of MPI‐deficient mice [28], demonstrating that the dose of mannose was efficacious to improve the inflammatory response. Furthermore, another study reported severe morphological eye defects in 45% of mannose‐treated MPI‐deficient mice [39]. These defects were clearly evident at 2–8 weeks after birth but none of the MPI‐deficient mice had eye defects when mannose was added to the drinking water at 6–8 weeks of age (i.e., 2–4 weeks after full eye development) [39]. In the present study, 67% of mannose‐treated Aldob −/− mice developed eye defects, including cloudy eyes and smaller eyecups. The Aldob −/− mice were 8 weeks of age prior to the start of mannose treatment and yet developed eye defects, which shows that mannose treatment induces eye toxicity after 8 weeks of age in Aldob −/− mice. Taken together, these data collectively suggest that 5% mannose supplementation via the drinking water must have been efficacious. However, it cannot be excluded that a higher dose of mannose is needed to reverse the hepatic nucleotide sugars abnormalities and the fatty liver phenotype in Aldob −/− mice. Noteworthy, other secondary defects in glycosylation, like classical galactosemia, are also not associated with chronic fatty liver disease.
In conclusion, we found abnormal levels of hepatic nucleotide sugars in a mouse model for HFI, as seen in patients in HFI. Furthermore, mannose supplementation (as used for the treatment of MPI‐CDG) does not appear to be a suitable alternative or add‐on treatment for HFI. Future studies are needed to identify the underlying mechanisms responsible for the abnormal hepatic nucleotide sugar pattern and intrahepatic lipid accumulation in HFI.
Ethics Statement
All institutional and national guidelines for the care and use of laboratory animals were followed.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Figure S1. PCR‐genotype analysis of Aldob‐targeted mice. The results of representative PCR‐genotyping for Aldob in wildtype (Aldob +/+ , 316 bp), heterozygous (Aldob +/− , 316 bp and 436 bp), and homozygous (Aldob −/−, 436 bp) mice. The reference is a 100 bp DNA ladder. The primers are listed in table 1 below.
Figure S2. Flow diagram of experimental design. Figure created with Biorender.com.
Table S1. Primer set for Aldob genotyping.
Data S1.
Acknowledgements
Support by Petra M.G. Niessen, Margee Teunissen, Vicky M.M.J. Vermeulen, and Marjo P.H. van de Waarenburg for the conduct of the experiments is gratefully acknowledged.
Communicating Editor: Jerry Vockley
Funding: This study was supported by the Dutch Diabetes Research Foundation (personal grant #2017.82.004 to MCGJB) and a Catalyst Grant from United for Metabolic Diseases (UMD‐CG‐2022‐004), which is financially supported by Metakids. Publication of this article was made possible by a ZonMw grant from the program More Knowledge with Fewer Animals (grant number 114024071).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Chambers R. and Pratt R., “Idiosyncrasy to Fructose,” Lancet 18, no. 271 (1956): 340. [DOI] [PubMed] [Google Scholar]
- 2. Herrs H. and Joassin G., “Anomalie de l'aldolase hepatique dans l'intolerance au fructose,” Enzymologia Biologica et Clinica 1 (1961): 4–14. [PubMed] [Google Scholar]
- 3. James C. L., Rellos P., Ali M., Heeley A. F., and Cox T. M., “Neonatal Screening for Hereditary Fructose Intolerance: Frequency of the Most Common Mutant Aldolase B Allele (A149P) in the British Population,” Journal of Medical Genetics 33, no. 10 (1996): 837–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gitzelmann R. and Baerlocher K., “Vorteile und Nachteile der Fructose in der Nahrung,” Paediatr Fortbildungsk Prax 37 (1973): 40. [Google Scholar]
- 5. Fagerberg L., Hallstrom B. M., Oksvold P., et al., “Analysis of the Human Tissue‐Specific Expression by Genome‐Wide Integration of Transcriptomics and Antibody‐Based Proteomics,” Molecular & Cellular Proteomics 13, no. 2 (2014): 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hers H. and Kusaka T., “Le metabolisme du fructose‐1‐phosphate dans le foie,” Biochimica et Biophysica Acta 11 (1953): 427–437. [DOI] [PubMed] [Google Scholar]
- 7. Odièvre M., Gentil C., Gautier M., and Alagille D., “Hereditary Fructose Intolerance in Childhood: Diagnosis, Management, and Course in 55 Patients,” American Journal of Diseases of Children 132, no. 6 (1978): 605–608. [DOI] [PubMed] [Google Scholar]
- 8. Mock D. M., Perman J. A., Thaler M. M., and R. C. Morris, Jr. , “Chronic Fructose Intoxication After Infancy in Children With Hereditary Fructose Intolerance: A Cause of Growth Retardation,” New England Journal of Medicine 309, no. 13 (1983): 764–770. [DOI] [PubMed] [Google Scholar]
- 9. Von Ruecker A., Endres W., Shin Y., Butenandt I., Steinmann B., and Gitzelmann R., “A Case of Fatal Hereditary Fructose Intolerance. Misleading Information of Formula Composition,” Helvetica Paediatrica Acta 36, no. 6 (1981): 599–600. [PubMed] [Google Scholar]
- 10. Levin B., Snodgrass G., Oberholzer V., Burgess E. A., and Dobbs R., “Fructosaemia: Observations on Seven Cases,” American Journal of Medicine 45, no. 6 (1968): 826–838. [DOI] [PubMed] [Google Scholar]
- 11. Ali M., Rellos P., and Cox T. M., “Hereditary Fructose Intolerance,” Journal of Medical Genetics 35, no. 5 (1998): 353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cox T. M., “Aldolase B and Fructose Intolerance,” FASEB Journal 8, no. 1 (1994): 62–71. [DOI] [PubMed] [Google Scholar]
- 13. Li H., Byers H. M., Diaz‐Kuan A., et al., “Acute Liver Failure in Neonates With Undiagnosed Hereditary Fructose Intolerance due to Exposure From Widely Available Infant Formulas,” Molecular Genetics and Metabolism 123, no. 4 (2018): 428–432. [DOI] [PubMed] [Google Scholar]
- 14. Simons N., Debray F. G., Schaper N. C., et al., “Patients With Aldolase B Deficiency Are Characterized by an Increased Intrahepatic Triglyceride Content,” Journal of Clinical Endocrinology and Metabolism 104 (2019): 5056–5064. [DOI] [PubMed] [Google Scholar]
- 15. Di Dato F., Spadarella S., Puoti M. G., et al., “Daily Fructose Traces Intake and Liver Injury in Children With Hereditary Fructose Intolerance,” Nutrients 11, no. 10 (2019): 2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aldámiz‐Echevarría L., de Las H. J., Couce M. L., et al., “Non‐alcoholic Fatty Liver in Hereditary Fructose Intolerance,” Clinical Nutrition 39, no. 2 (2020): 455–459. [DOI] [PubMed] [Google Scholar]
- 17. Zheng M., Huang D. Q., Konkwo C., et al., “Genomic Analysis of Lean Individuals With NAFLD Identifies Monogenic Disorders in a Prospective Cohort Study,” JHEP Reports 5, no. 4 (2023): 100692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Buziau A. M., Schalkwijk C. G., Stehouwer C. D. A., Tolan D. R., and Brouwers M., “Recent Advances in the Pathogenesis of Hereditary Fructose Intolerance: Implications for Its Treatment and the Understanding of Fructose‐Induced Non‐alcoholic Fatty Liver Disease,” Cellular and Molecular Life Sciences 77, no. 9 (2020): 1709–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buziau A. M., Oosterveer M. H., Wouters K., et al., “Hepatic Glucokinase Regulatory Protein and Carbohydrate Response Element Binding Protein Attenuation Reduce de Novo Lipogenesis but Do Not Mitigate Intrahepatic Triglyceride Accumulation in Aldob Deficiency,” Molecular Metabolism 87 (2024): 101984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jaeken J., Pirard M., Adamowicz M., Pronicka E., and van Schaftingen E., “Inhibition of Phosphomannose Isomerase by Fructose 1‐Phosphate: An Explanation for Defective N‐Glycosylation in Hereditary Fructose Intolerance,” Pediatric Research 40, no. 5 (1996): 764–766. [DOI] [PubMed] [Google Scholar]
- 21. Panis B., Janssen L. E. F., Lefeber D. J., Simons N., Rubio‐Gozalbo M. E., and Brouwers M., “Development of Tools to Facilitate the Diagnosis of Hereditary Fructose Intolerance,” JIMD Reports 64, no. 5 (2023): 353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cano A., Alcalde C., Belanger‐Quintana A., et al., “Transferrin Isoforms, Old but New Biomarkers in Hereditary Fructose Intolerance,” Journal of Clinical Medicine 10, no. 13 (2021): 2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saudubray J.‐M. B. M., García‐Cazorla A., and Walter J. H., Inborn Metabolic Diseases: Diagnosis and Treatment (Berlin Heidelberg: Springer, 2022). [Google Scholar]
- 24. Mention K., Lacaille F., Valayannopoulos V., et al., “Development of Liver Disease Despite Mannose Treatment in Two Patients With CDG‐Ib,” Molecular Genetics and Metabolism 93, no. 1 (2008): 40–43. [DOI] [PubMed] [Google Scholar]
- 25. Marques‐da‐Silva D., Dos Reis F. V., Monticelli M., et al., “Liver Involvement in Congenital Disorders of Glycosylation (CDG). A Systematic Review of the Literature,” Journal of Inherited Metabolic Disease 40, no. 2 (2017): 195–207. [DOI] [PubMed] [Google Scholar]
- 26. Oppelt S. A., Sennott E. M., and Tolan D. R., “Aldolase‐B Knockout in Mice Phenocopies Hereditary Fructose Intolerance in Humans,” Molecular Genetics and Metabolism 114, no. 3 (2015): 445–450. [DOI] [PubMed] [Google Scholar]
- 27. Lanaspa M. A., Andres‐Hernando A., Orlicky D. J., et al., “Ketohexokinase C Blockade Ameliorates Fructose‐Induced Metabolic Dysfunction in Fructose‐Sensitive Mice,” Journal of Clinical Investigation 128, no. 6 (2018): 2226–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. He P., Srikrishna G., and Freeze H. H., “N‐Glycosylation Deficiency Reduces ICAM‐1 Induction and Impairs Inflammatory Response,” Glycobiology 24, no. 4 (2014): 392–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van Scherpenzeel M., Conte F., Büll C., et al., “Dynamic Analysis of Sugar Metabolism Reveals the Mechanisms of Action of Synthetic Sugar Analogs,” Glycobiology 32, no. 3 (2022): 239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haskovic M., Coelho A. I., Lindhout M., et al., “Nucleotide Sugar Profiles Throughout Development in Wildtype and Galt Knockout Zebrafish,” Journal of Inherited Metabolic Disease 43, no. 5 (2020): 994–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shiri‐Sverdlov R., Wouters K., van Gorp P. J., et al., “Early Diet‐Induced Non‐alcoholic Steatohepatitis in APOE2 Knock‐In Mice and Its Prevention by Fibrates,” Journal of Hepatology 44, no. 4 (2006): 732–741. [DOI] [PubMed] [Google Scholar]
- 32. Liu G., Wang N., Zhang C., et al., “Fructose‐1,6‐Bisphosphate Aldolase B Depletion Promotes Hepatocellular Carcinogenesis Through Activating Insulin Receptor Signaling and Lipogenesis,” Hepatology 74, no. 6 (2021): 3037–3055. [DOI] [PubMed] [Google Scholar]
- 33. Lefeber D. J., Freeze H. H., Steet R., and Kinoshita T., “Congenital Disorders of Glycosylation,” in Essentials of Glycobiology (Internet), 4th ed., eds. Varki A., Cummings R. D., Esko J. D., et al. (Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press, 2022), 599–614. [PubMed] [Google Scholar]
- 34. Sharma V., Ichikawa M., and Freeze H. H., “Mannose Metabolism: More Than Meets the Eye,” Biochemical and Biophysical Research Communications 453, no. 2 (2014): 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Quintana E., Sturiale L., Montero R., et al., “Secondary Disorders of Glycosylation in Inborn Errors of Fructose Metabolism,” Journal of Inherited Metabolic Disease 32, no. Suppl 1 (2009): S273–S278. [DOI] [PubMed] [Google Scholar]
- 36. Adamowicz M., Ploski R., Rokicki D., et al., “Transferrin Hypoglycosylation in Hereditary Fructose Intolerance: Using the Clues and Avoiding the Pitfalls,” Journal of Inherited Metabolic Disease 30, no. 3 (2007): 407. [DOI] [PubMed] [Google Scholar]
- 37. Holden H. M., Rayment I., and Thoden J. B., “Structure and Function of Enzymes of the Leloir Pathway for Galactose Metabolism,” Journal of Biological Chemistry 278, no. 45 (2003): 43885–43888. [DOI] [PubMed] [Google Scholar]
- 38. Demirbas D., Coelho A. I., Rubio‐Gozalbo M. E., and Berry G. T., “Hereditary Galactosemia,” Metabolism 83 (2018): 188–196. [DOI] [PubMed] [Google Scholar]
- 39. Sharma V., Nayak J., DeRossi C., et al., “Mannose Supplements Induce Embryonic Lethality and Blindness in Phosphomannose Isomerase Hypomorphic Mice,” FASEB Journal 28, no. 4 (2014): 1854–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. PCR‐genotype analysis of Aldob‐targeted mice. The results of representative PCR‐genotyping for Aldob in wildtype (Aldob +/+ , 316 bp), heterozygous (Aldob +/− , 316 bp and 436 bp), and homozygous (Aldob −/−, 436 bp) mice. The reference is a 100 bp DNA ladder. The primers are listed in table 1 below.
Figure S2. Flow diagram of experimental design. Figure created with Biorender.com.
Table S1. Primer set for Aldob genotyping.
Data S1.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.