

Abstract
Background
Annotation of eukaryotic genomes is a complex endeavor that requires the integration of evidence from multiple, often contradictory, sources. With the ever-increasing amount of genome sequence data now available, methods for accurate identification of large numbers of genes have become urgently needed. In an effort to create a set of very high-quality gene models, we used the sequence of 5,000 full-length gene transcripts from Arabidopsis to re-annotate its genome. We have mapped these transcripts to their exact chromosomal locations and, using alignment programs, have created gene models that provide a reference set for this organism.
Results
Approximately 35% of the transcripts indicated that previously annotated genes needed modification, and 5% of the transcripts represented newly discovered genes. We also discovered that multiple transcription initiation sites appear to be much more common than previously known, and we report numerous cases of alternative mRNA splicing. We include a comparison of different alignment software and an analysis of how the transcript data improved the previously published annotation.
Conclusions
Our results demonstrate that sequencing of large numbers of full-length transcripts followed by computational mapping greatly improves identification of the complete exon structures of eukaryotic genes. In addition, we are able to find numerous introns in the untranslated regions of the genes.
Background
The scientific community has recently witnessed the publication of several large eukaryotic genomes in various stages of completion, including the human genome [1,2], the nematode Caenorhabditis elegans [3], the fruit fly Drosophila melanogaster [4], and the model plant Arabidopsis thaliana [5,6]. Each of these genomes contains over 10,000 genes, and as scientists attempt to study these genes more closely, the need for accurate gene models becomes increasingly apparent. For high-throughput genome sequencing projects, equally rapid high-throughput genome annotation is necessary, and bioinformaticists use a variety of computational methods to generate this annotation. Despite many improvements in recent years, these computational methods still fall short of producing correct models for every gene. In order to improve the annotation and facilitate further research, it is essential that we develop methods to identify genes correctly.
Annotation of a gene model should include a precise description of where the genomic DNA sequence is transcribed into messenger RNA, the positions in the mRNA of any and all introns, and the translated protein sequence of the gene. If alternative splice variants are present, these should be annotated as well. Computational methods for genome annotation have several shortcomings that result in the following errors in annotation.
Gene prediction programs predict exon boundaries correctly only about 80% of the time, even for the most intensively studied organisms [7,8,9]. Thus a gene with five exons will be completely correct only 0.85 = 33% of the time, and genes with more exons will be even less likely to be correct. Gene prediction programs also tend to merge and split gene models. Thus two real genes may be merged into one predicted transcript, or vice versa. In addition, programs to align genomic DNA to protein sequences often miss small exons, especially when the homologous proteins are not well conserved. Annotation protocols also tend to miss short genes. For example, recent work has shown that at least one large family of Arabidopsis genes encodes a short (80-120 amino acid) protein similar to a secreted polypeptide ligand for a receptor-like kinase that functions in meristems [10]. Most of these were missed in the original, automated annotation of the Arabidopsis genome. Alignment programs also make mistakes when genes occur in tandemly repeated copies. Finally, alignment of protein sequence to genomic DNA cannot predict untranslated regions (UTRs), and the leading ab initio gene prediction programs (Genscan [11], GlimmerM [12], Genemark.HMM [13]) have great difficulty predicting UTRs; most of them predict only the coding portion of a transcript.
The solution to many of these problems is to identify the complete sequence of the transcribed portions of the genome. Sequencing the mature transcripts (spliced mRNA) solves three major problems: first, it permits accurate identification of the 5' and 3' UTRs. Second, in conjunction with complete genomic sequence, it enables alignment software to identify the precise locations of all introns. Third, it aids in the discovery of new genes.
Results and discussion
We used sequences from 5,000 full-length transcripts sequenced by Ceres, Inc. and released to the public in March 2001 ([14], and at GenBank as accession numbers AY084215-AY089214). We merged this data with a small set of full-length transcripts created in a pilot study, yielding 5,016 complementary DNA (cDNA) sequences in total. As described in detail below, after comparing the cDNA alignments to the previous annotation, we found that 62% of the matching gene models correctly predicted the exon-intron structure for the gene, 33% needed to be modified, and 5% represented previously undiscovered genes.
We mapped and aligned the cDNA sequences to the five chromosomes of A. thaliana. This step is not entirely straightforward, in part because several different programs are available for aligning cDNA to genomic sequence (see Materials and methods). Although the programs are similar, they are not identical, and we conducted a detailed comparison in order to determine which one would produce the best results for these data. Note that the conclusions from this comparison might change for alignments between cDNAs and genomic sequences from different species - for example, alignments between human cDNAs and the mouse genome - where the genome and the cDNAs match less closely than in this study. Because the cDNA sequences are derived from two A. thaliana ecotypes (Wassilewskija and Landsberg erecta; see Materials and methods) that differ from the genome's ecotype (Columbia), we expected to find some polymorphisms when we compared the cDNAs to the genome; on average, the three ecotypes are more than 99% identical.
The four programs used for the alignment comparison were sim4 [15], dds/gap2 [16], est_genome [17] and GeneSeqer [18]. Three of these are general-purpose alignment algorithms designed to map expressed sequence tag (EST) or cDNA sequences to a genome. One program, GeneSeqer, is more specialized, in that it has particular subroutines designed to recognize splice sites in A. thaliana, and it is able to find AT-AC introns. The other three programs require only that an intron begin with the dinucleotide GT and end with AG. Note that none of these programs is tuned to find AT-AC introns [19] or any other introns with non-consensus splice sites.
The first question we asked in the comparison was how often the four programs produced identical alignments between the cDNA sequences and the genome. Because the programs differed in how they treated the first and last nucleotides of the cDNA, we ignored those nucleotides in deciding if two alignments were identical. The number of identical alignments is shown in Table 1. The table shows that the two programs in greatest agreement with each another were gap2 and GeneSeqer, which agreed on 4,839 out of 5,016 alignments. The total number of cDNAs for which all four programs agreed was 4,124. This initial comparison does not provide a true picture of the extent of agreement, though, because most of the differences were in the alignment at the 5' or 3' ends of the cDNAs.
Table 1.
Number of identical cDNA alignments produced by four different programs on a set of 5,016 cDNA sequences
| Program | gap2 | est_genome | GeneSeqer |
| sim4 | 4,819 | 4,342 | 4,784 |
| gap2 | 4,274 | 4,839 | |
| est_genome | 4,257 |
To determine how well the programs agreed at the ends of the alignments, we compared the lengths of the alignments. Because the cDNAs derive from a different ecotype of Arabidopsis than the genomic sequence, there are a few polymorphisms in the UTRs, but even these regions are usually > 98% identical. Differences between ecotypes sometimes interrupt an open reading frame (ORF). The result is that a gene in one ecotype may be a pseudogene, or a severely truncated gene, in the other [20]. We addressed this problem by first aligning cDNAs to the genome, and then always using the genomic sequence to create the predicted protein. Therefore the alignments should span the entire length of the cDNA. Our analysis indicates that gap2 and sim4 performed the best at matching the entire cDNA to the genome, as shown in Table 2.
Table 2.
Comparison of lengths of cDNA alignments for the 5,016 cDNA sequences
| gap2 | est_genome | GeneSeqer | |
| sim4 | 32/34 | 6/652 | 31/67 |
| gap2 | 2/708 | 3/50 | |
| est_genome | 706/708 | 706/733 |
Entries show the number of sequences for which the program listed along the top produced a longer alignment than the program listed on the left; for example, the entry 32/34 indicates that gap2 produced a longer alignment in 32 cases out of the total 34 for which sim4 and gap2 had alignments of different lengths.
Two immediate observations from Table 2 are that gap2 produced longer alignments than any of the other programs, and that est_genome produced shorter alignments than all the others. Sim4 and gap2 are closest to each other, disagreeing on alignment length in only 34 cases, with gap2 generating the longer alignment on 32 of those. GeneSeqer has a few more differences than gap2, but is clearly much closer to gap2 than est_genome. Even though est_genome often produces shorter alignments, the unaligned sequence is almost always restricted to a small number of terminal nucleotides.
A more critical question is how many times the programs agreed on the precise locations of all the splice sites for a particular transcript. This question is answered for all pairwise comparisons between the programs in Table 3. Overall, there were 4,918 cDNAs that yielded identical splice sites (and therefore identical introns) regardless of which program was used to generate the alignment. As Table 3 shows, the programs disagreed on fewer than 100 alignments, less then 2% of the total. This still leaves open the question of whether one program is clearly superior on these problematic alignments. We therefore evaluated the 98 cDNAs (5,016 - 4,918) for which there was disagreement among the programs to determine the cause of the differences. In 64 of these cases, three programs agree and only one disagrees, while in the remaining 32 cases, there was no alignment shared by a majority of the programs. We looked individually at all 64 cases in which a majority agreed to evaluate whether or not a 'majority wins' rule would always produce the correct result, and came to the following conclusions.
Table 3.
Number of cDNA alignments, out of 5,016 total, for which all splice sites are identical
| Program | gap2 | est_genome | GeneSeqer |
| sim4 | 4,946 | 4,965 | 4,960 |
| gap2 | 4,954 | 4,955 | |
| est_genome | 4,961 |
Originally, there were 140 cDNAs for which the programs did not all agree on the locations of the introns. Of these, 106 were cases in which only one program disagreed with the three others. These differences were communicated to the authors of the GeneSeqer program (V. Brendel) and the sim4 program (L. Florea). Both authors identified bugs in their systems, fixed the bugs, and issued new releases, which were then re-run on all the data used in this study. The result was that these two programs are in much closer agreement with gap2 and with each other than previously.
Gap2 disagreed with the other three programs in 24 alignments. The most common reason for the disagreement was an erroneous alignment to non-consensus splice sites (other than GT and AG). Gap2's alignment appeared to be incorrect for all 24 cases. Sim4 disagreed with the other three programs 16 times. Like gap2, in most cases these were due to erroneous non-consensus splice sites. However, in one case, sim4 seems to have found a correct alignment missed by the other three programs. GeneSeqer disagreed with the majority 14 times, sometimes as the result of a tendency to create additional short exons. On the other hand, GeneSeqer is excellent at identifying potential short exons, especially at the termini: in four cases out of 14, GeneSeqer's alignment contained an additional exon that results in a greater percent identity in the overall alignment to the genome. Est_genome disagreed with the other three programs on 10 alignments; in all 10 cases the majority was correct. The mistake in eight of these alignments was that a short exon was missed.
Overall, it does indeed matter which program is used to align cDNA sequences to genome sequences. Three of the four programs do an excellent job of extending the alignment to cover the full length of the cDNA; only est_genome consistently failed to extend the alignments. When the programs disagree, this sometimes indicates that an exon was missed by one or more programs, and manual inspection is necessary to determine the best alignment. Finally, there is a substantial difference in computational speed. Sim4 is more than 200 times faster than any of the other programs, which can have a significant impact in efforts such as this to align large numbers of sequences. For the searches in this study, the average computation time per cDNA (on an 850 MHz Pentium III computer) for sim4 was 0.026 second; for gap2, 6.4 seconds; for est_genome, 9.2 seconds; and for Gene-Seqer, 12.2 seconds. GeneSeqer's speed per search increases dramatically, approximating the speed of sim4, if the cDNAs are submitted as a batch rather than one at a time. In addition, memory requirements make it impossible to run some programs on a standard desktop (dds/gap2 runs out of memory if one attempts to align a cDNA to a whole chromosome, for example).
Re-annotation of the Arabidopsis genome
The alignments generated from the cDNA sequences were used to create new gene models for the corresponding genes in the A. thaliana genome. Many of the genes have been manually curated, but many others were created by automated scripts [5,6]. Manual curation is still ongoing.
We used the cDNA alignments to create new gene models automatically according to the following criteria. As described above, there were 4,918 cDNAs for which all alignment programs agreed on the positions of all introns. Using a majority voting scheme for the remaining 98 cDNAs did not always give a correct answer, as discussed above, therefore we used these only after manual inspection. We assume the protein-coding region is the longest ORF on the forward strand, and required it to span at least 40% of the cDNA length. This allowed us to create 4,809 gene models automatically, leaving 109 cDNAs that were inspected manually to determine if they represent RNA genes, pseudogenes or other types of sequence. In one case, cDNA Ceres: 104289, the protein-coding region was actually on the opposite strand, corresponding to expressed protein At2g23670, and Ceres: 20125 matched the correct strand, supporting the gene annotation. (This could be explained in several ways: as an example of antisense-mediated translational control, as two separate proteins on opposite strands, perhaps expressed in different cell types, or simply erroneous data.) In most of the other cases, the problematic cDNA is either an RNA gene or a likely pseudogene.
Using the alignments from the 4,809 gene models, we updated the annotation of the genome, and evaluated how this had changed the previous annotation. For the vast majority of genes, 5' and 3' UTRs had not been annotated previously, and these were added with the incorporaton of the cDNA data. More interesting is how the protein-coding regions changed. Of the gene models, 2,978 contained identical protein-coding regions to what had already been annotated and required only UTR refinements, but 1,591 were adjusted, yielding more accurate protein sequences. Some of these contain very short 'micro-exons' that are usually missed by ab initio gene prediction programs. Perhaps most significant was the addition of 240 completely novel genes not previously included in the Arabidopsis genome annotation. Of the 240 novel genes, 92 have significant homology to known proteins, and the rest do not match any previously described proteins. In summary, we found that 62% of the matching gene models further validated the existing exon-intron structure for the gene, 33% needed to be corrected, and 5% represented previously undiscovered genes.
Micro-exons
We also used the cDNA alignments to detect 'micro-exons', very short exons that are typically missed by both gene-finding programs and alignment algorithms. Using new software protocols we developed, we found 47 micro-exons, ranging from 3 to 25 base pairs (bp) in length, distributed evenly across all five chromosomes.
To find micro-exons, we analyzed the results of sim4 alignments using all 5,016 Ceres cDNAs. Sim4 identified 36 cDNAs encoding exons of 25 bp or less. In an effort to identify additional micro-exons, sim4 alignments containing imperfect intron-exon boundaries were examined. We selected only those cases with near-perfect alignments, requiring that all but one or two exons have 100% identity. We then checked to see if the 1-2 exons with slightly lower identity were misaligned as the result of the presence of a small, undetected, exon. We used the 5 bp segments at the boundaries of the exon as probes. If these 5 bp probes mismatched in the original alignment, we searched the adjacent intron (that is, the intron identified by the initial alignment) for short exons that would produce a perfect match with the cDNA. We also required that any new exon would generate introns with a standard GT-AG consensus on either end. This procedure therefore yielded valid exon-intron structures that always improved the identity of the alignment between cDNA and genomic DNA. Figure 1 shows an example of the cDNA alignments before and after inserting a micro-exon.
Figure 1.
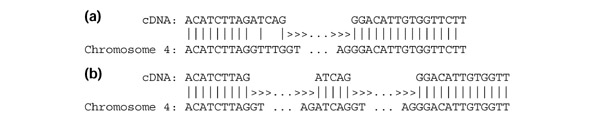
An example showing how a micro-exon improves a cDNA alignment. (a) Alignment showing the boundaries of the fourth and fifth exons from the sim4 alignment of cDNA Ceres:20761 to chromosome 4. (b) The improvement resulting from insertion of a micro-exon; all three exons now align with 100% identity to the cDNA sequence. Intron positions are shown by '>' in the alignment.
Using this method, we were able to identify 11 additional micro-exons, all shorter than 12 bp. An extraordinarily short exon of only 3 bp was identified, corresponding to exon 2 of disease-resistance gene RAR1 (At5g51700). A listing of these micro-exons from all chromosomes is shown in Table 4. Note that in some cases the length of the micro-exons is not a multiple of three; for these, one of the preceding or following exons had its boundary realigned to maintain the reading frame. In comparison to the other alignment programs examined, GeneSeqer proved to be highly competent in identifying micro-exons; 46 of the 47 micro-exons were identified by GeneSeqer using the default settings. After lowering the minimum exon length cut-off to 1 bp, all 47 were identified.
Table 4.
Micro-exons from each of the five chromosomes, listed in order of increasing length
| Locus | Gene name | cDNA accession | Exon number | Exon length (nucleotides) | Micro-exon sequence |
| At5g51700 | RAR1 | Ceres:99615 | 2 of 6 | 3 | AG<GGA>GT |
| At1g63290 | D-ribulose-5-phosphate-3-epimerase | Ceres:37843 | 2 of 8 | 5 | AG<GACGG>GT |
| At3g01850 | D-ribulose-5-phosphate-3-epimerase | Ceres:2398 | 2 of 9 | 5 | AG<GACGG>GT |
| At4g01610 | Cysteine protease | Ceres:20761 | 5 of 11 | 5 | AG<ATCAG>GT |
| At5g14030 | Expressed protein | Ceres:16313. | 5 of 6 | 6 | AG<GCCAAG>GT |
| At2g38880 | Putative CCAAT-binding transcription factor subunit | Ceres:7805. | 4 of 7 | 6 | AG<TTGGAG>GT |
| At2g07340 | Expressed protein | Ceres:34060. | 4 of 6 | 7 | AG<GAAGAAC>GT |
| At2g41710 | AP2 domain transcription factor | Ceres:41462 | 3 of 9 | 9 | AG<TTTATCTAG>GT |
| At2g36190 | Beta-fructofuranosidase | Ceres:118038 | 2 of 6 | 9 | AG<ATCCAAATG>GT |
| At4g13720 | Auxin-regulated protein | Ceres:8361 | 4 of 8 | 10 | AG<GGCCATACAT>GT |
| At4g29510 | Arginine methyltransferase (pam1) | Ceres:38601 | 2 of 9 | 11 | AG<GAATCCATGAA>GT |
| At3g55260 | Beta-N-acetylhexosaminidase | Ceres:118286 | 7 of 15 | 17 | AG<GTTTGCCAAAATGAGAG>GT |
| At1g80380 | Auxin-regulated protein | Ceres:117698 | 3 of 7 | 18 | AG<GTACCTAGGTACAATAAG>GT |
| At3g55630 | Tetrahydrofolylpolyglutamate synthase | Ceres:230791. | 6 of 15 | 18 | AG<GAGAAAACCAGCAATGAG>GT |
| At5g61530 | Auxin-regulated protein | Ceres:152557 | 5 of 10 | 19 | AG<GGAGTTGCCAGCTCAGATG>GT |
| At5g03880 | Auxin-regulated protein | Ceres:37668 | 4 of 12 | 19 | AG<CTGTCCCTTCTGCCGGAAG>GT |
| *At4g23470 | Expressed protein | Ceres:25694. | 7 of 8 | 19 | AG<GGTTTGCGCATGTATGCAG>GT |
| At1g67320 | Expressed protein | Ceres:116252. | 7 of 18 | 20 | AG<TTGAAAACATTTACTACAAG>GT |
| At3g50210 | Flavonol synthase | Ceres:25787. | 8 of 12 | 20 | AG<TGGAGCTCACACTGACTATG>GT |
| At4g37680 | Expressed protein | Ceres: 262351. | 2 of 5 | 21 | AG<GGTTTGTCTTTCGAAATTCAG>GT |
| At3g60340 | Palmitoyl-protein thioesterase | Ceres:38539. | 5 of 12 | 21 | AG<ACATCAGTTGTTTGTGAGAAG>GT |
| At3g23600 | Expressed protein | Ceres:11339. | 2 of 7 | 22 | AG<GTTTTGAAGCTCCAAACTTAAG>GT |
| At5g46030 | Expressed protein | Ceres:15222. | 4 of 5 | 22 | AG<CTTTGACTGAAGCTATAGACAT>GT |
| At4g33925 | Expressed protein | Ceres:24360. | 2 of 5 | 22 | AG<TAACCGAAGAACAGCTCTCAAT>GT |
| *At4g23470 | Expressed protein | Ceres:25694. | 2 of 8 | 22 | AG<ATTGTTGCTTCGCGTTGTGGTG>GT |
| At3g13860 | Chaperonin, putative | Ceres:38045. | 2 of 17 | 22 | AG<CTCGTCTACTTCCAGGAAACTG>GT |
| At1g73180 | Expressed protein | Ceres:108165. | 13 of 14 | 23 | AG<TTACTTGGAATAAGCACAACAGG>GT |
| At5g09830 | Expressed protein | Ceres:37422. | 2 of 3 | 23 | AG<GAAGTCATTGACATATCTGGAGG>GT |
| At2g23930 | Small nuclear ribonucleoprotein E | Ceres:4850. | 2 of 4 | 23 | AG<GTACATGGATAAGAAGCTCCAAA>GT |
| At1g66940 | Expressed protein | Ceres:110066. | 3 of 5 | 24 | AG<AATCTAATATTAGATGGATAATAG>GT |
| At5g51100 | Expressed protein | Ceres:126592. | 8 of 9 | 24 | AG<CACGCTTACTATCTGGATTTTGAG>GT |
| At1g05070 | Expressed protein | Ceres:13725. | 2 of 3 | 24 | AG<AGCTCAGTAATGCTTCTTTTGCTG>GT |
| At2g32580 | Expressed protein | Ceres:16625. | 2 of 3 | 24 | AG<GACTCAGCAATGGTTCATTCACTG>GT |
| At2g29960 | Cyclophilin | Ceres:19211. | 4 of 6 | 24 | AG<AAAACTTCAGAGCTTTGTGCACAG>GT |
| At1g65220 | Expressed protein | Ceres:21223. | 2 of 8 | 24 | AG<CTCAAAGGAGAAGCCCACTCTCGG>GT |
| At5g23310 | Iron superoxide dismutase 3 | Ceres:26637. | 7 of 8 | 24 | AG<CACTCTTATTATCTGGACTACAAG>GT |
| At4g25100 | Superoxide dismutase | Ceres:32935. | 6 of 7 | 24 | AG<CATGCTTACTACCTTGACTTCCAG>GT |
| At3g55920 | Cyclophilin-like protein | Ceres:94608. | 5 of 8 | 24 | AG<AGAACTTTCGGTCACTTTGCACGG>GT |
| At1g77060 | Carboxyphosphonoenol-pyruvate mutase, putative | Ceres:12293. | 4 of 6 | 25 | AG<GACCAAGCATGGCCAAAGAAGTGTG>GT |
| At4g15900 | PRL1 protein | Ceres:123113. | 2 of 17 | 25 | AG<CAAGCAGATTCGTCTCAGCCATAAG>GT |
| At2g47640 | Putative small nuclear ribonucleoprotein D2 | Ceres:26123. | 3 of 6 | 25 | AG<CAAGCCAATGGAAGAGGATACCAAT>GT |
| At2g41630 | Transcription factor IIB (TFIIB) | Ceres:2657. | 2 of 7 | 25 | AG<GTTGGGACTTGTTGCAACTATCAAG>GT |
| At3g62840 | Small nuclear ribonucleoprotein | Ceres:32457. | 3 of 5 | 25 | AG<TAAACCAATGGAAGAGGATACCAAC>GT |
| At2g21270 | Putative ubiquitin fusion-degradation protein | Ceres:34470. | 4 of 10 | 25 | AG<CCACAACTTGAAAGTGGTGACAAGA>GT |
| At3g10330 | Transcription initiation factor IIB (TFIIB) | Ceres:38950. | 2 of 7 | 25 | AG<GTTGGGACTTGTTGCGACCATCAAG>GT |
| At1g42480 | Expressed protein | Ceres:42677. | 7 of 9 | 25 | AG<ATTGCTGGAGGAAACTGAAGATGAG>GT |
| At2g23985 | Expressed protein | Ceres:252843. | 2 of 4 | 25 | AG<TGTCTTGTTCAGGTGAACAAAAAAG>GT |
*At4g23470 contains two micro-exons.
One indication that these micro-exons are correct (in addition to the identity with the cDNA) is that many of them are homologous to exons in other Arabidopsis genes. For example, a search of GenBank in late 2001 revealed that the micro-exon of Ceres: 118038 is homologous to exons from five different cDNAs (accession numbers gi: 15028118, gi:6683111, gi:14517549, gi:15027838, and gi:16974574). The consensus sequence of these exons, ATCCTAA(T/C)G, has been previously characterized as a micro-exon in the potato invertase gene [21].
Splicing anomalies
Analysis of cDNA sequences can help to estimate the frequency of alternative splicing in a species. Alternative splicing appears to be relatively common in animals [22,23]; in plants this phenomenon has been less frequently observed, possibly as a result of the smaller collections of ESTs compared with mammalian systems. Recently, some reports have appeared documenting a small number of cases [24,25]. We examined the alignments of cDNAs to the genome, looking for examples where more than one cDNA aligned to overlapping locations on the same chromosome in such a way as to predict a different splicing pattern. The working hypothesis was that if two cDNAs mapped to the same locus, but presented distinct sets of exons, this would constitute evidence of alternative splicing, or possibly another type of splicing anomaly. We broadened the search for splicing anomalies by including in this protocol all the complete cDNAs available from GenBank, including the Institute for Physical and Chemical Research (RIKEN) collection described below. A total of 1,515 Ceres transcripts overlapped another transcript, of which 1,129 overlapped a sequence from the RIKEN set.
This protocol identified 158 genes with apparent splicing anomalies, each of which was inspected manually. They fall into many different classes, representing different genetic events, as follows: 27 alignments indicate an alternative 3' acceptor site for an intron; 17 alignments indicate an alternative 5' donor site for an intron; 23 alignments indicate that one or more introns remained unspliced. In some cases more than one intron was unspliced; for example, in one interesting case only one intron was spliced in the RIKEN transcript (gi: 15146259), whereas four introns were spliced from the corresponding Ceres transcript (Ceres:3992, corresponding to gene At2g35520). These unspliced transcripts may arise from nuclear rather than mature cytoplasmic mRNA sequences. Six alignments indicate that an internal exon is missing in one isoform; presumably the adjacent introns are spliced as a single intron containing the exon sequence. Fifty-seven alignments suggest possible alternative transcription initiation sites. For 17 of these transcripts, the putative initiation site was shifted far enough in the 3' direction to move past the first donor site, making it impossible to splice out the first intron, producing an additional 5' exon in one of the transcripts. Many of the other transcripts contained one or more additional 5' exons as a result of alternative initiation sites. Thirteen alignments suggest alternative 3' polyadenylation (poly(A)) sites that affect splicing. The prediction of poly(A) sites can be confounded by misannealing of the oligo(dT) primers used for reverse transcription; for example, the presence of multiple adenines within the 3' UTR can be mistaken for a poly(A) site. Misannealing cannot explain the presence of unspliced intronic sequence found at the terminus of 12 of these 13 transcripts, suggesting that these putative poly(A) sites are genuine and have an impact on splicing. We have found similar evidence for the occurrence of multiple poly(A) sites in RACE-PCR experiments directed at cloning cDNAs from hypothetical genes. Finally, 15 alignments display multiple splicing anomalies, falling into more than one of the categories above.
Table 5 lists many of these alternatively spliced genes; the complete list, with graphical and textual alignment data, is available on-line [26] and is also provided as Additional data with this paper online. Figure 2 highlights several interesting examples. In Figure 2a, the alternative 3' splice site on the second intron leads to a shift in the reading frame, producing a different protein sequence. In Figure 2b, alignments of several cDNAs indicate that the last intron is unspliced. Figure 2c shows that different 5' ends lead to differing 5' introns and exons, while not changing the protein sequence in this particular example. Figure 2d shows a centrally located exon that is spliced out along with the surrounding introns. Figure 2e contains three different 5' transcription start sites, three different 3' termination sites, and two unspliced introns in the middle transcript. The unspliced introns occur within exon 2 of GI:14335057, which corresponds to three exons and two introns in both the other transcripts. Note that some of the alternative splicing events occur within the same ecotype.
Table 5.
Alternative acceptor and donor splice sites, alternative 5'exons, and exon skipping examples based on cDNA alignments
| Locus | Gene name | cDNA accessions |
| Alternative acceptor splice sites | ||
| At3g58710 | WRKY DNA-binding protein | Ceres:100465, gi:15991735 |
| At5g35680 | Expressed protein | Ceres:11304, gi:14596002 |
| At2g38860 | Expressed protein | Ceres:114031, gi:13122287 |
| At4g31550 | DNA-binding protein | Ceres:11953, gi:15384214 |
| At1g22700 | Expressed protein | Ceres:120133, gi:15294175 |
| At4g30480 | Expressed protein | Ceres:12573, gi:14423435 |
| At1g52870 | Expressed protein | Ceres:126586, gi:14326544 |
| At5g41810 | Expressed protein | Ceres:126660, gi:14532565 |
| At1g63970 | Expressed protein | Ceres:15758, gi:11386014 |
| At2g33830 | Auxin-regulated protein | Ceres:1711, gi:11127600 |
| At5g20040 | IPP transferase | Ceres:19250, gi:14279069 |
| At3g55330 | Oxygen-evloving complex subunit | Ceres:21674, Ceres:3747 |
| At1g60850 | RNA polymerase subunit | Ceres:21961, gi:514321 |
| At1g76405 | Expressed protein | Ceres:23773, gi:13358245 |
| At1g22630 | Expressed protein | Ceres:37537, gi:15010607 |
| At1g02500 | S-adenosylmethionine synthase | Ceres:37800, gi:15450420 |
| At4g20380 | Zinc finger protein Lsd1 | Ceres:38456, gi:1872520 |
| At3g54380 | Expressed protein | Ceres:38778, gi:14423485 |
| At3g04830 | Expressed protein | Ceres:38917, gi:15293268 |
| At1g11840 | Lactoylglutathione lyase | Ceres:39107, gi:11094298 |
| At1g02090 | COP9 complex subunit | Ceres:40042, gi:3288822 |
| At1g79650 | DNA repair protein RAD23 | Ceres:40579, gi:14334441 |
| At2g25625 | Expressed protein | Ceres:465, gi:14334615 |
| At4g02640 | Expressed protein | Ceres:6568, gi:10954094 |
| At3g11930 | Ethylene-responsive protein | Ceres:7474, gi:13926249 |
| At2g20820 | Expressed protein | Ceres:91872, gi:14190456 |
| At3g09150 | Expressed protein | Ceres:98026, gi:13359272 |
| Alternative donor splice sites | ||
| At1g16460 | Mercaptopyruvate sulfurtransferase | Ceres:111646, gi:6009982 |
| At5g16540 | Zinc finger protein 3 | Ceres:113763, gi:4689375 |
| At2g41070 | bZIP family transcription factor | Ceres:114632, gi:13346156 |
| At2g36000 | Expressed protein | Ceres:123727, gi:14532493 |
| At3g21175 | Expressed protein | Ceres:12996, gi:14596058 |
| At5g61880 | Expressed protein | Ceres:146274, gi:14517497 |
| At3g14230 | RAP2 family protein | Ceres:158240, gi:15450917 |
| At3g03890 | Expressed protein | Ceres:18355, gi:14190432 |
| At1g67700 | Expressed protein | Ceres:19973, gi:15215605 |
| At3g55630 | Tetrahydrofolylpolyglutamate synthase | Ceres:230791, gi:15292866 |
| At2g21620 | Expressed protein | Ceres:31655, gi:15320407 |
| At4g10100 | Expressed protein | Ceres:35962, gi:6635742 |
| At1g23950 | Expressed protein | Ceres:41387, gi:15146261 |
| At1g24260 | Floral homeotic protein | Ceres:5055, gi:2345157 |
| At2g39730 | Expressed protein | Ceres:7114, gi:15450670 |
| At3g07760 | Expressed protein | Ceres:7246, gi:15451021 |
| At3g06720 | Importin alpha | Ceres:9351, gi:4191743 |
| Alternative 5' exons | ||
| At3g57810 | Expressed protein | Ceres:101256, Ceres:29384 |
| At3g03780 | Methionine synthase | Ceres:111720, gi:14532771 |
| At5g59890 | Actin depolymerizing factor 4 | Ceres:11691, gi:15215858 |
| At3g49010 | 60S ribosomal protein L13 | Ceres:12182, gi:15292840 |
| At5g52210 | GTP-binding protein | Ceres:16621, gi:1184980 |
| At1g01100 | Acidic ribosomal protein | Ceres:24367, gi:15293082 |
| At2g41430 | ERD15 | Ceres:31388, gi:13926319 |
| At3g08580 | Adenylate translocator | Ceres:36818, gi:1433 |
| At5g05000 | GTP-binding protein | Ceres:6734, gi:1151243 |
| At3g48880 | Expressed protein | Ceres:99337, gi:15028346 |
| Exon skipping | ||
| At5g54940 | Translation initiation factor | Ceres:103464, Ceres:32071 |
| At2g46800 | Zinc transporter | Ceres:207558, gi:4206639 |
| At5g53860 | Expressed protein | Ceres:22860, gi:15215799 |
| At5g27840 | TOPP8 Ser/Thr protein phosphatase type-1 | Ceres:38656, gi:14596132 |
| At3g23280 | Expressed protein | Ceres:41648, gi:15010671 |
| At1g77080 | Expressed protein | Ceres:92459, gi:11545544, gi:11545546, gi:13649968 |
A full list with illustrations and supporting alignment data is available at [26].
Figure 2.

Alternative splice variants discovered by cDNA alignments. Red bars indicate the protein-coding portion of each exon. Black bars indicate noncoding exons and the UTR portions of the initial and terminal exons. Exon boundaries that line up exactly between two or more cDNAs are highlighted in blue. Thin lines connecting the exons represent introns. The genes involved are: (a) auxin-regulated protein, At2g20820, chromosome (chr) 2; (b) SKP1-interacting partner 5 (SKIP5), At3g54480, chr 3; (c) acidic ribosomal protein, At1g01100, chr 1; (d) auxin-regulated protein, At5g53860, chr 5; (e) unknown expressed protein, At2g45740, chr 2.
Neither collection of cDNAs can be considered a random sample of transcripts, and therefore the number of examples of alternative splicing discovered in this data (approximately 10% of the overlapping transcripts) should not be used to extrapolate to the entire genome. The discovery of transcripts with different introns spliced out raises the question of whether the different spliced products are translated and whether the splicing differences reflect programmed developmental variation or simply splicing errors. It is not possible to answer these questions now, but incomplete splicing and consequential variants in plants have been noted previously to be associated with gene silencing and were postulated to reflect the regulated production of aberrant RNA products not destined to be translated [27]. One clear conclusion is that alternative splicing can be discovered via analysis of cDNAs and genomic sequence, and that a fuller collection of cDNAs will provide a valuable resource for more discoveries about splicing and gene regulation.
Are the sequences full-length?
An independent project to sequence complete Arabidopsis cDNAs is ongoing by the SPP consortium [28], using clones created by K. Shinozaki at RIKEN in Japan. These sequences are publicly available from GenBank (search for "RIKEN cDNA Arabidopsis"). These data provided the opportunity to compare the two sets of cDNAs and measure independently how many of them appear to cover the entire length of the predicted mRNA transcript. The sequencing of the RIKEN cDNAs generated 2,996 sequences as of October 2001; we compared these to the 5,016 cDNAs from Ceres and found 1,129 sequences that are contained in both data sets. Of the 1,129 sequences, 941 alignments yield the same exon-intron structure for the underlying gene. We then asked, for each of the sequences containing identical introns, do the 5' and 3' ends match, and if not, how large is the difference? The results are illustrated in Figure 3.
Figure 3.
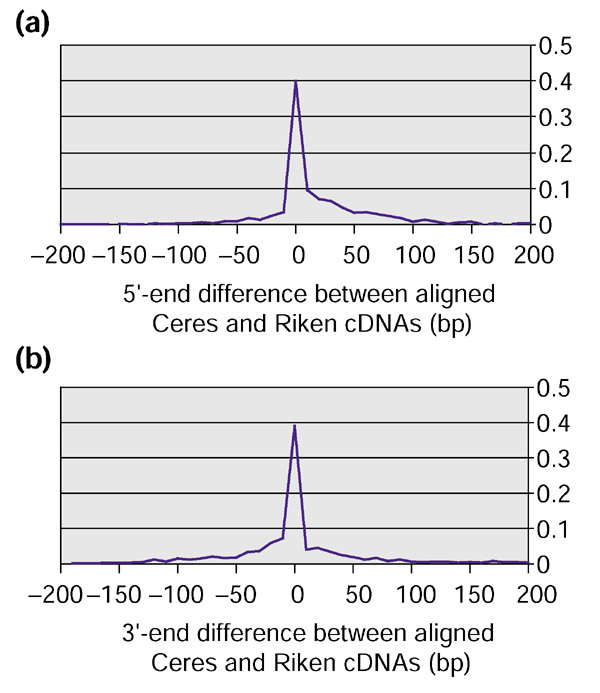
Comparison of the lengths of the 941 cDNAs from the clones that are contained in both the Ceres and RIKEN collections. (a) Comparison of the 5'-end difference between Ceres and RIKEN clones; (b) comparison of the 3'-end difference between Ceres and RIKEN clones. Peak height indicates the percentage of sequences with a length difference as indicated along the horizontal axis. Positive values on the horizontal axis correspond to longer Ceres clones, while negative values correspond to longer RIKEN clones.
Several observations can be made about these results. First, it is important to note that the Ceres clones were selected for full-length sequencing from among a large number of clustered 5' sequences (see Materials and methods), whereas the RIKEN clones were sequenced on the 3' end followed by clustering and selection of a clone for sequencing [29]. The methods for creating the full-length cDNA sequences at both centers involve multiple sequencing runs, followed by assembly of the overlapping sequences. Second, we observed that in the Ceres data, many mRNAs appeared to have two or more putative alternative transcription start sites. This became apparent when different cDNA assemblies were found to overlap exactly except for an extension on the 5' end on one or more clones. It is interesting to note that when the RIKEN clones were longer or shorter on the 5' end, clones of the equivalent length could often be found in the Ceres collection. Multiple clones with the same 5' end provided strong validation that these were truly representative of alternative transcription initiation sites or repeatable artifacts of the cloning process. Overall, there were 397 Ceres clones that were > 10 bp longer on the 5' end, and 136 RIKEN clones that were longer on the 5' end. If alternative transcription initiation is the correct explanation, then it is relatively common. It is worth noting that in almost all cases, both alternative cDNAs contain complete ORFs.
On the 3' end, the Ceres and RIKEN databases each contained 316 sequences that were >10 bp longer than their match from the other set. If these represent alternative polyadenylation sites or stabilized ends of RNA that get polyadenylated, then these are quite common. Further investigation will be necessary to determine if the 3' end of transcripts truly varies at such a high frequency.
In summary, work described in this study on Arabidopsis illustrates the utility of full-length cDNAs for finding alternative splice variants, short exons, UTRs, short genes and alternative transcription start sites. The annotation of eukaryotic genomes is currently an inexact and developing science, and the results described here demonstrate the power of full-length cDNA sequences for improving the quality of multiple aspects of genome annotation.
Materials and methods
Preparation and sequencing of cDNA
Starting material for cDNA synthesis was polysomal RNA isolated from the top-most inflorescence tissues (ecotype Wassilewskija) and from roots (ecotype Landsberg erecta). RNA from roots of Landsberg erecta was used to construct the libaries because of the availability of high-quality RNA. Nine parts inflorescence to one part root, as measured by wet mass, was used to make three size-fractionated libraries. Because the ecotypes were mixed before library construction, we cannot determine the source ecotype for any individual cDNA. Polysomal RNA was isolated from a detergent-generated supernatant on a 2 M sucrose cushion. To capture full-length cDNAs, an oligonucleotide is first attached to intact 5' ends, taking advantage of the cap. After first- and second-strand synthesis, the full-length cDNAs were selected, size fractionated and cloned into pBluescript. The ligation mixture was transformed into bacteria, selected on appropriate antibiotics and picked into 384-well microtiter plates. In repeated rounds of sequencing, several tens of thousands of clones from the three libraries were sequenced from the 5' end, the sequences clustered, and the clone with the longest 5' end in each cluster selected for complete sequencing.
The number of clones sequenced in each round depended on the percentage of new full-length clones that could be obtained from each of the size-fractionated libraries. As the clones reported in this study came from non-normalized libraries, only three rounds of 5' sequencing were employed; 42,000 in the first round, 59,000 in the second round and 22,000 in the final round. Following each round of 5' sequencing, all sequences were clustered using a clustering algorithm that forms separate clusters if there are more than 6 nucleotide differences in any 30-nucleotide window of the match. In this way, clones would not fall into separate clusters simply because of ecotypic differences, different putative transcription start sites or sequencing errors. However, they would cluster separately if alternative splicing occurred in the first approximately 500 nucleotides and involved more than 6 nucleotides. Following clustering, the clone that was longest on the 5' end was selected for full-length sequencing. If clones were of comparable length on the 5' end, the clone to be sequenced was selected from the library with the highest percentage of full-length clones. Sequencing of 5' ends was performed on capillary sequencers (Molecular Dynamics); full-length sequencing was done on ABI377 sequencers using primer walking. The 5,016 clones analyzed in this study were selected from all full-length clones based on length (> 400 nucleotides), non-redundancy (eliminating alternatively spliced clones), and length of the putative ORF relative to overall clone length.
Alignment of cDNA sequences to the A. thaliana genome
Four programs were used to align all 5,016 Ceres cDNA sequences to the A. thaliana genome as follows. First, each program was used to align each cDNA sequence to the genome. Some programs cannot efficiently handle a search comparing a cDNA to a 30 + Mb eukaryotic chromosome, and to compensate for those programs, we created a modified procedure that first used BLASTN to identify and extract a region of 20,000 bp surrounding the gene. Each cDNA was aligned to the corresponding 20 kb genome sequence segment using all four programs with default parameter settings. The resulting alignments were then compared automatically to generate the comparison data appearing in the main text. The programs are sim4, available from [30]; dds/gap2, available from [31]; GeneSeqer, available from [32]; and est_genome, available from [33].
Gene models were constructed by first recreating the cDNA sequence using the Arabidopsis genome sequence, employing the longest alignment for which all programs predicted identical splice sites. The longest ORF was identified along the forward strand of the cDNA followed by a division of the ORF into protein-coding exon segments and untranslated regions of exons. These constructed gene models were then compared to the existing gene annotation at the mapped genomic region. Previously annotated gene structures that disagreed with the cDNAs were replaced by the cDNA alignment-based gene models, and new gene models were created where pre-existing gene annotations were lacking.
Additional data files
Additional data corresponding to anomalous splicing, including png image files and text-formatted multiple alignments, is available.
Supplementary Material
Additional data corresponding to anomalous splicing
Acknowledgments
Acknowledgements
We thank Stephen Mount for helpful comments and discussion. S.L.S. and N.V. were supported in part by NSF grants IIS-9902923 and KDI-9980088, and by NIH grant R01-LM06845. B.J.H., C.D.T. and O.W. were supported in part by NSF cooperative agreement DBI-9813586.
References
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Waterston R, Sulston J. The genome of Caenorhabditis elegans. Proc Natl Acad Sci USA. 1995;92:10836–10840. doi: 10.1073/pnas.92.24.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Bevan M, Mayer K, White O, Eisen JA, Preuss D, Bureau T, Salzberg SL, Mewes HW. Sequence and analysis of the Arabidopsis genome. Curr Opin Plant Biol. 2001;4:105–110. doi: 10.1016/s1369-5266(00)00144-8. [DOI] [PubMed] [Google Scholar]
- Burge CB, Karlin S. Finding the genes in genomic DNA. Curr Opin Struct Biol. 1998;8:346–354. doi: 10.1016/s0959-440x(98)80069-9. [DOI] [PubMed] [Google Scholar]
- Lin X, Kaul S, Rounsley S, Shea TP, Benito MI, Town CD, Fujii CY, Mason T, Bowman CL, Barnstead M, et al. Sequence and analysis of chromosome 2 of the plant Arabidopsis thaliana. Nature. 1999;402:761–768. doi: 10.1038/45471. [DOI] [PubMed] [Google Scholar]
- Pavy N, Rombauts S, Dehais P, Mathe C, Ramana DV, Leroy P, Rouze P. Evaluation of gene prediction software using a genomic data set: application to Arabidopsis thaliana sequences. Bioinformatics. 1999;15:887–899. doi: 10.1093/bioinformatics/15.11.887. [DOI] [PubMed] [Google Scholar]
- Cock JM, McCormick S. A large family of genes that share homology with CLAVATA3. Plant Physiol. 2001;126:939–942. doi: 10.1104/pp.126.3.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge CB, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Salzberg SL, Pertea M, Delcher AL, Gardner MJ, Tettelin H. Interpolated Markov models for eukaryotic gene finding. Genomics. 1999;59:24–31. doi: 10.1006/geno.1999.5854. [DOI] [PubMed] [Google Scholar]
- Lukashin AV, Borodovsky M. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 1998;26:1107–1115. doi: 10.1093/nar/26.4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceres data at TIGR ftp://ftp.tigr.org/pub/data/a_thaliana/ceres
- Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 1998;8:967–974. doi: 10.1101/gr.8.9.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Adams MD, Zhou H, Kerlavage AR. A tool for analyzing and annotating genomic sequences. Genomics. 1997;46:37–45. doi: 10.1006/geno.1997.4984. [DOI] [PubMed] [Google Scholar]
- Mott R. EST_GENOME: a program to align spliced DNA sequences to unspliced genomic DNA. Comput Appl Biosci. 1997;13:477–478. doi: 10.1093/bioinformatics/13.4.477. [DOI] [PubMed] [Google Scholar]
- Usuka J, Zhu W, Brendel V. Optimal spliced alignment of homologous cDNA to a genomic DNA template. Bioinformatics. 2000;16:203–211. doi: 10.1093/bioinformatics/16.3.203. [DOI] [PubMed] [Google Scholar]
- Tarn WY, Steitz JA. A novel spliceosome containing U11, U12, and U5 snRNPs excises a minor class (AT-AC) intron in vitro. Cell. 1996;84:801–811. doi: 10.1016/s0092-8674(00)81057-0. [DOI] [PubMed] [Google Scholar]
- Kliebenstein DJ, Lambrix VM, Reichelt M, Gershenzon J, Mitchell-Olds T. Gene duplication in the diversification of secondary metabolism: tandem 2-oxoglutarate-dependent dioxygenases control glucosinolate biosynthesis in Arabidopsis. Plant Cell. 2001;13:681–693. doi: 10.1105/tpc.13.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson CG, Hedley PE, Watters JA, Clark GP, McQuade C, Machray GC, Brown JW. Requirements for mini-exon inclusion in potato invertase mRNAs provides evidence for exon-scanning interactions in plants. RNA. 2000;6:422–433. doi: 10.1017/s1355838200992173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstrohm AC, Greenleaf AL, Garcia-Blanco MA. Co-transcriptional splicing of pre-messenger RNAs: considerations for the mechanism of alternative splicing. Gene. 2001;277:31–47. doi: 10.1016/s0378-1119(01)00695-3. [DOI] [PubMed] [Google Scholar]
- Grabowski PJ, Black DL. Alternative RNA splicing in the nervous system. Prog Neurobiol. 2001;65:289–308. doi: 10.1016/s0301-0082(01)00007-7. [DOI] [PubMed] [Google Scholar]
- Lazar G, Goodman HM. The Arabidopsis splicing factor SR1 is regulated by alternative splicing. Plant Mol Biol. 2000;42:571–581. doi: 10.1023/a:1006394207479. [DOI] [PubMed] [Google Scholar]
- Sakai H, Hua J, Chen QG, Chang C, Medrano LJ, Bleecker AB, Meyerowitz EM. ETR2 is an ETR1-like gene involved in ethylene signaling in Arabidopsis. Proc Natl Acad Sci USA. 1998;95:5812–5817. doi: 10.1073/pnas.95.10.5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anomalous mRNA splicing in Arabidopsis http://www.tigr.org/tdb/e2k1/ath1/splicing_anomalies.html
- Metzlaff M, O'Dell M, Hellens R, Flavell RB. Developmentally and transgene regulated nuclear processing of primary transcripts of chalcone synthase A in petunia. Plant J. 2000;23:63–72. doi: 10.1046/j.1365-313x.2000.00793.x. [DOI] [PubMed] [Google Scholar]
- Salk Institute Genomic Analysis Laboratory: cDNA sequencing status http://signal.salk.edu/cdnastatus.html
- Salk Institute Genomic Analysis Laboratory: cDNA isolation and sequencing http://signal.salk.edu/cprotocol.html
- sim4 http://globin.cse.psu.edu/
- dds/gap2 http://genome.cs.mtu.edu/aat/aat.html
- GeneSeqer http://bioinformatics.iastate.edu/cgi-bin/gs.cgi
- est_genome http://www.hgmp.mrc.ac.uk/Registered/Option/est_genome.html
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional data corresponding to anomalous splicing