

Abstract
Systemic lupus erythematosus (SLE) and its renal manifestation Lupus nephritis (LN) are characterized by a dysregulated immune system, autoantibodies, and injury to the renal parenchyma. Iron accumulation and ferroptosis in the immune effectors and renal tubules are recently identified pathological features in SLE and LN. Ferroptosis is an iron dependent non-apoptotic form of regulated cell death and ferroptosis inhibitors have improved disease outcomes in murine models of SLE, identifying it as a novel druggable target. In this review, we discuss novel mechanisms by which iron accumulation and ferroptosis perpetuate immune cell mediated pathology in SLE/LN. We highlight intra-renal dysregulation of iron metabolism and ferroptosis as an underlying pathogenic mechanism of renal tubular injury. The basic concepts of iron biology and ferroptosis are also discussed to expose the links between iron, cell metabolism and ferroptosis, that identify intracellular proferroptotic enzymes and their protein conjugates as potential targets to improve SLE/LN outcomes.
Keywords: SLE, Lupus nephritis, Iron, Ferroptosis
1. Introduction
Systemic lupus erythematosus (SLE) is the most prevalent form of lupus, an autoimmune disease characterized by the loss of tolerance to self-DNA, histones, and ribonucleoproteins [1,2]. The adaptive immune cells of individuals with SLE have limited ability to discern between self and non-self-antigens [3], which results in the production of autoantibodies that form circulating and in-situ immune complexes (IC) that induce multiple organ inflammation and pathology [2,4]. While the etiology of SLE is incompletely understood, a combination of genetic susceptibility, hormonal changes, and exogenous factors (e.g., allergens, infection, heavy metals) can trigger the onset [5]. Circulating and in situ IC disproportionately affect the highly vascular kidneys, resulting in lupus nephritis (LN). LN is the most severe end-organ complication of SLE that leads to end-stage kidney disease (ESKD) [6,7]. Since IC deposits are the dominant feature in LN patients, glomeruli are considered the initiating site of renal pathology. However, tubulointerstitial inflammation and pathology strongly correlate with renal dysfunction independent of the extent of glomerular damage and predict worse outcomes [8-11].
Our understanding of SLE has come a long way since the discovery of the disease by Cazenave and Biett in 1833 and Hebra in 1846 [12]. However, novel pathogenic mechanisms of human SLE and LN are continuously uncovered [13,14]. While these new challenges influence disease outcomes, they also provide novel opportunities for intervention [15,16]. A recent addition to this list is iron (Fe) metabolism [17,18] and ferroptosis, an iron-dependent form of regulated cell death [19,20].
In this review, we briefly introduce the fundamentals of iron metabolism and ferroptosis and discuss novel iron-centric mechanisms of immune effectors and parenchymal cells contributing to the pathogenesis of SLE/LN.
2. Fundamentals of iron biology and ferroptosis
Iron is essential for physiology, cell survival, and proliferation, and reducing intracellular iron induces cell cycle arrest and apoptosis [21]. Without iron, cells cannot proceed from the G1 to the S phase of the cell cycle [22]. Fe-containing proteins catalyze critical reactions involved in oxygen sensing [23,24], energy metabolism [25], and DNA synthesis (e.g., ribonucleotide reductase) [22]. Bioactive or labile Fe (Fe2+) participates in the Fenton reaction and catalyzes the generation of reactive oxygen species (ROS), which induce oxidative stress and lipid oxidation [26,27]. Hence systemic and cellular iron metabolism is dynamic, tightly regulated, and a well-orchestrated symphony with evolutionary conserved pathways that control cellular iron levels [28]. The hepcidin-ferroportin axis primarily regulates systemic iron metabolism [29]. At a mechanistic level, the flow of iron out of the cells is controlled by hepcidin through two known mechanisms: occlusion of the open-outward conformation of ferroportin by hepcidin [30] and hepcidin-induced endocytosis and degradation of ferroportin [31]. In contrast, cellular iron metabolism is a complex and coordinated symphony involving multiple iron importers, chaperons, storage molecules, and iron export [32,33].
The role of iron regulation in immunometabolism and immune-related disease is documented [34] and in SLE, the pathological consequences of abnormal iron metabolism and consequent mitochondrial and cellular dysfunctions are reported [18,35,36]. We have previously reviewed the details of renal iron metabolism in context of LN [17]. Iron accumulation, oxidative stress, and lipid peroxidation, the major biochemical characteristics of ferroptosis [37], are observed in SLE/LN [38-40]. There is a recent spurt in the literature reporting the occurrence of ferroptosis in SLE/LN by multiple independent groups [40-45].
2.1. Ferroptosis
Ferroptosis was identified as a distinct iron-dependent, regulated form of cell death driven by excessive lipid peroxidation [37]. It is independent of other metals and is morphologically, biochemically, and genetically distinct from apoptosis and necrosis [37]. Ferroptosis is now appreciated as a widespread and evolutionary conserved form of cell death across species since ferroptosis-like cell death in plants, protozoa, and fungi has also been observed [46,47].
Ferroptosis-driven lipid peroxidation disrupts the thickness, permeability, and structure of membrane bilayers, which is lethal to the cells [48,49]. Lipid peroxidation can occur via the selective enzymatic or random non-enzymatic free radical chain reaction [50,51]. Both enzymatic and non-enzymatic lipid peroxidation involve abstraction of the bis-allylic hydrogen, rearrangement of the resonance radical structure, addition of the molecular oxygen that generates peroxyl radical to form hydroperoxy lipid [52]. Reaction rates of non-enzymatic peroxidation of poly-unsaturated fatty acids (PUFA) are proportional to the number of bis-allylic hydrogens [53]. In contrast to this random profile of free radical peroxidation, enzymatic lipid peroxidation by di-oxygenases (e.g., cyclooxygenases [COXs] and LOXs) is highly substrate-selective and product-specific [54].
The identification of the specific lipids and sites that drive ferroptosis and the enzymes that promote their generation and incorporation into cell membranes has been a critical discovery in ferroptosis research [55,56]. Ferroptosis is now defined as an organized oxidation of only one class of phospholipids, phosphatidylethanolamines (PEs), with a specificity toward two fatty acyls-arachidonoyl (AA) and adrenoyl (AdA) [55]. PUFA, a common component of phospholipids, have enhanced susceptibility to ferroptosis. The presence of multiple double bonds in PUFAs increases the propensity to form highly reactive peroxyl radicals that cause irreparable membrane damage [57]. Additionally, lipid peroxidation or ferroptosis breakdown products, including 4-hydroxynonenal (HNE) and malondialdehyde (MDA), are damaging to cellular processes because they form adducts with proteins and DNA [58,59]. Ferroptosis is inhibited by the canonical system xc−-/GHS/GPX4 pathway and promoted by enzymes such as long-chain acyl-co-enzyme A (CoA) synthase 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3), lipoxygenase (LOX), and cytochrome P450s (CYPs), or non-enzymatically by Fe-dependent free radical-induced peroxidation [60-62].
The canonical control of ferroptosis entails four key steps (Fig. 1): 1) uptake of cystine via the cystine-glutamate antiporter, system xc−; 2) reduction of cystine to cysteine; 3) synthesis and reduction of glutathione to reduced glutathione (GSH); and 4) GSH-dependent activation of the enzyme glutathione peroxidase 4 (GPX4), a selenoprotein that reduces toxic phospholipid hydroperoxides (PE-OOH) to corresponding non-toxic alcohols (PE-OH) [63-65].
Fig. 1.
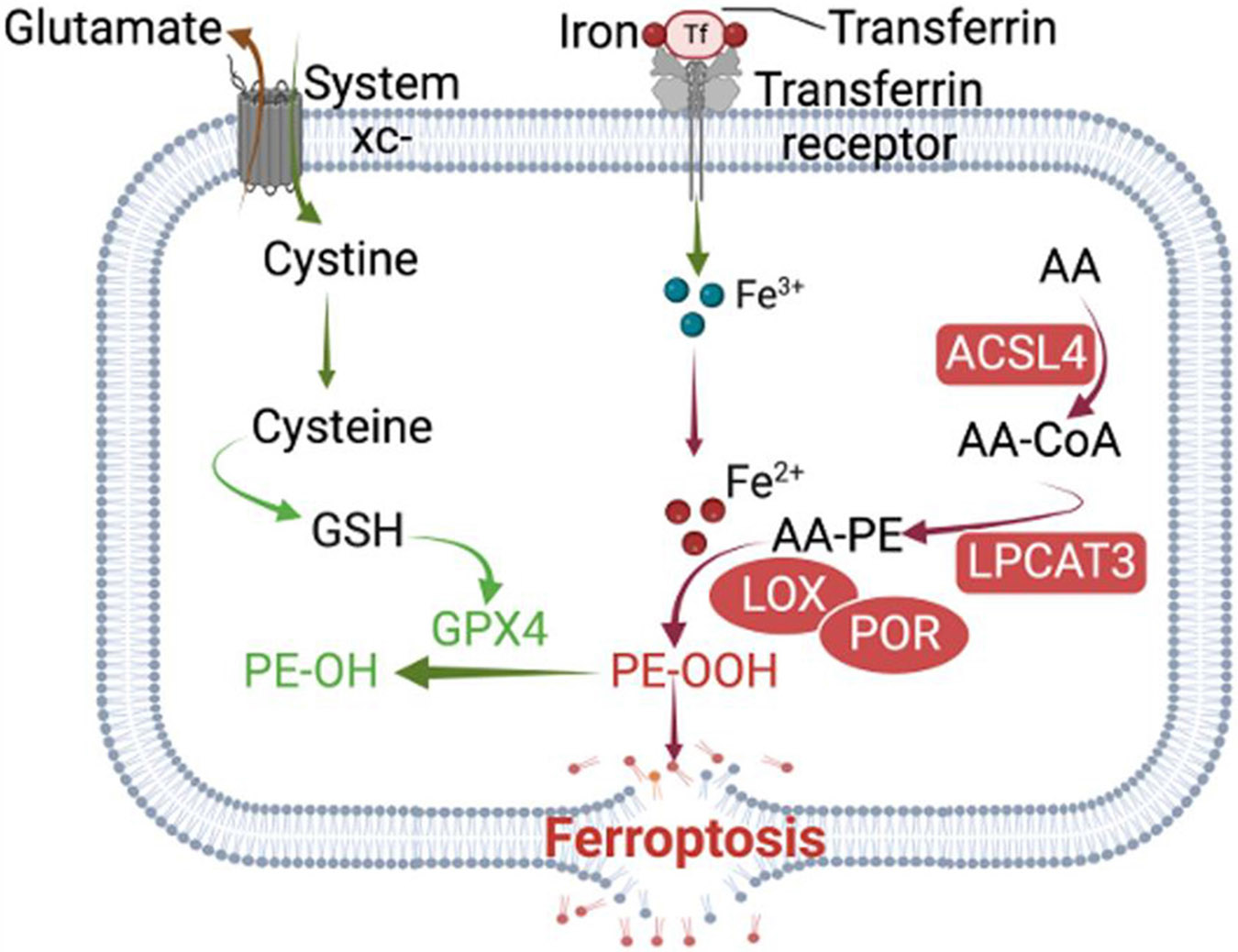
Adequate availability of cysteine via the cystine-glutamate antiporter, system xc – prevents the dynamic lipid-induced toxicity under physiological conditions and in pathology. Cysteine is essential for GSH synthesis, which in turn is required for the activity of GPX4 to convert toxic lipid hydroperoxides (PE-OOH) into inert lipid alcohols (PE-OH). The long-chain acyl-coenzyme A (CoA) synthase 4 (ACSL4) in conjunction with lysophosphatidylcholine acyltransferase 3 (LPCAT3) converts poly unsaturated fatty acid (PUFA) like Arachidonic acid (AA) into AA-CoA intermediate that is ultimately esterified into phosphatidylethanolamine (PE). Then, acid-15-lipoxygenase (ALOX15) catalyzes the oxidation of these esterified-PE and excessive AA-CoA to its PE-OOH, thus promoting ferroptosis. Intracellular Fe2+ promotes lipid peroxidation and ferroptosis by increasing the catalytic activity of ALOX15 to exacerbate ROS. Additionally, Fe2+ promotes the non-enzymatic arm of ferroptosis by participating, in the Fenton reaction to catalyze the generation of reactive free radicals, which target unsaturated phospholipids and convert them to toxic phospholipids hydroperoxides.
System xc−, GPX4: Attenuate ferroptosis; Iron, ACSL4, LPCAT3; Promote ferroptosis.
The long-chain acyl-coenzyme A (CoA) synthase 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) were the first proferroptotic identified gene products [55]. To initiate lipid peroxidation on cell membranes (Fig. 1), ACSL4 first catalyzes the conversion of arachidonic acid (AA) into AA-CoA, followed by the esterification of AA-CoA into phosphatidylethanolamine (PE) to form AA-PE with the assistance of LPCAT3. Then, acid-15-lipoxygenase (ALOX15) catalyzes the oxidation of AA-PE to its toxic hydroperoxy form (PE-OOH) [55,66,67]. In addition, lipoxygenases (LOXs) catalyze the oxidation of excessive PUFA-CoA and contribute to ferroptosis [68].
In a non-enzymatic manner, intracellular Fe2+ can promote lipid peroxidation and initiate ferroptosis via the Fenton reaction or by increasing the catalytic activity of ALOX15, which has an iron-binding site and produces ROS [69] (Fig. 1). By participating in the Fenton reaction, Fe2+ can react with hydrogen peroxide to produce highly reactive free radicals such as OH·, OH−. Then, OH·, in the presence of oxygen, attacks the C═C double bonds of PUFAs and produces PE-OOH [70]. The oxidation state of iron is vital for its ability to contribute to ferroptosis: Fe2+ promotes ferroptosis, whereas Fe3+ is generally inert and stored in ferritin, except in the active site of lipoxygenases, where Fe (III) is the active form of the enzyme.
Integrating research involving metabolism, iron regulation, and ROS biology has yielded insights into distinct aspects of ferroptosis. For example, metabolism explains how the critical substrates of ferroptosis are generated and remodeled; iron regulation indicates how the availability of Fe2+ is controlled and compartmentalized; and ROS biology shows how endogenous defenses against lipid peroxidation function. The dysregulation of these three pillars of ferroptosis is also a salient feature in SLE/LN [17,71-74]. For example, in SLE, metabolic reprogramming alters the CD4+ T and B cell responses and their effector functions [75-78]. Iron accumulation in immune cells drives their cytokine production and differentiation into pathogenic effectors in SLE [79-81]. Additionally, improper iron sequestration and accumulation in the renal tubules promotes ferroptosis in LN [42]. Finally, ROS-induced pathology in different organelles as well as the role of lipid mediators in SLE and LN have also been documented [40,82,83]. These observations support an inherent role of ferroptosis in SLE and LN.
Below, we focus on literature that has identified dysregulated iron metabolism and the occurrence of ferroptosis in neutrophils, T cells, B cells, and the proximal tubular epithelial cells (PTEC) of the kidneys, the critical effectors in the pathogenesis of SLE/LN.
2.2. Neutrophils, iron and ferroptosis
Neutrophils are the most numerous white blood cells in the body and are primarily studied in settings of innate responses following acute injury. Neutrophils secrete lipocalin-2 to limit bacteria from scavenging host iron from the bloodstream [84,85]. They also secrete myeloperoxidase (MPO), a hemoprotein whose Fe3+/Fe2+ redox states are critical to its anti-microbial effects [86]. In addition, neutrophils produce large amounts of lactoferrin to scavenge iron, inhibiting bacterial proliferation [87]. By binding its cognate receptor, lactoferrin promotes the maturation, migration, and cell proliferation of macrophages and monocytes [88]. As such, neutrophils present an active iron metabolism as a part of their normal immunological response. The role of neutrophils in SLE/LN is increasingly appreciated [89-91]. LN kidneys have an elevated α-defensin gene transcript, a neutrophil product associated with neutrophil extracellular traps (NETS) and co-relates positively with local IFN-I expression [92]. In SLE patients, neutrophils exhibit an increased tendency to undergo apoptosis, which positively correlates with anti-dsDNA antibody titers and disease activity [93,94]. Increased MPO (iron-containing hemoprotein)-DNA complexes in SLE patients are observed [95]. Neutrophils of SLE patients also demonstrate an increased propensity to undergo netosis and release neutrophil extracellular traps (NETs), the fibrous strands composed of self-antigens including dsDNA, histones, chromatin, granule proteins, and mitochondrial DNA (mtDNA) [96,97]. Dysfunctional or reduced DNase due to genetic factors leads to impaired degradation of NETS and is associated with worse outcomes of LN [98,99]. This mechanism is now considered a key source of self-antigens in SLE.
The pathogenic role of neutrophil ferroptosis in SLE was first reported by a collaborative effort between P. Lipsky, G. Tsokos, and X. Zhang groups [45]. Using sera and neutrophils from SLE patients as well as murine models, they demonstrated that autoantibodies and IFN-α contained in SLE serum induce neutrophil ferroptosis [45]. Mechanistically, autoantibodies and type I IFNs cooperatively promote the nuclear translocation of cAMP response element modulator (CREMα) and its binding to the Gpx4 promoter in neutrophils, suppressing neutrophil GPX4 expression, promoting their ferroptosis and worsening disease outcomes. The authors found a conserved cAMP response element (CRE) upstream of the human Gpx4 promoter. SLE serum, or normal serum supplemented with SLE IgG or IFN-α recapitulated this effect in healthy donor neutrophils. The importance of GPX4 in neutrophil health was demonstrated by neutrophil specific Gpx4 haploinsufficiency, which led to lipid peroxidation and ferroptosis, and recapitulated key clinical features of human SLE in mice. Finally, Liproxstatin-1, a ferroptosis inhibitor that traps lipid-derived free radicals to inhibit the propagation of nonenzymatic lipid peroxidation [100], ameliorated disease outcomes in MRL/lpr lupus-prone mice. Thus, these studies provided a novel proof of concept that neutrophil ferroptosis is a source of self-antigens that may be presented to the T cells to perpetuate SLE.
2.3. T cells, iron, and ferroptosis
Pioneering work by the Tsokos group has shown that the aberrant and heightened T cell receptor (TCR) signaling in SLE T cells is due to the substitution of CD3ζ protein in the TCR–CD3 complex by the Fc receptor common gamma subunit chain (FcRγ) [101,102]. FcRγ recruits Syk instead of ZAP-70, resulting in the higher calcium influx and activation of T cells [103]. Auto-reactive CD4+ T cells are expanded in active SLE, produce effector cytokines, and invade the kidneys [104]. These intrarenal CD4+ T cells are oligoclonal, indicating local accumulation of antigen-specific T cells in inflamed kidneys [105].
Proliferation and effector functions of immune cells are energy-expensive processes. T cells require iron for many metabolic and redox reactions as well as heme- and Fe-S-containing enzymes essential for cell division, metabolism, and cytokine production [106,107]. Human studies showed that iron deficiency reduced the number of circulating T lymphocytes and their blastogenic response to mitogens [108,109]. CD71 (TfR1: transferrin receptor-1) mediated uptake of transferrin-bound iron and albumin-heme complex is the principal mechanism of cellular iron import [110], and its expression on T lymphocytes increases within the first few minutes of TCR/CD3 engagement [111]. In line with this observation, downregulation of TfR1 is associated with the induction of T cell anergy, a process whereby an activated T cell becomes tolerant and functionally inactive [112]. Furthermore, hematopoietic-specific deletion of the ferritin heavy chain gene Fth1, which encodes for the intracellular iron sequestering protein FtH, reduced the numbers of lymphocytes while other immune cell types such as granulocytes and monocytes are unaffected [113]. The proposed mechanism entails increased free intracellular Fe2+ levels, leading to oxidative stress and cell death [113]. However, upon stimulation, FtH-deficient CD4+ T cells proliferated more before ultimately dying, highlighting an obligatory function of iron in cell proliferation. Given that proliferating CD4+ T cells import high levels of iron, it is not surprising that GPX4 is essential for their survival and expansion, highlighting the importance of preventing lipid peroxidation and iron dysregulation in proliferating CD4+ T cells [114].
Iron contributes to activation-induced T-cell expansion by positively regulating IL-2R signaling and mitochondrial function [115]. Iron deposits have been observed in the brains of patients with multiple sclerosis, an autoimmune, CD4+ T cell-driven disease, and iron-deficient mice fail to develop autoimmune encephalomyelitis [116]. At a mechanistic level, iron promoted GM-CSF and IL-2 production in CD4+ T cells by stabilizing the RNA-binding protein PCBP1, and iron deficiency in T cells improved outcomes [117].
In the context of SLE, Zhao et al. demonstrated for the first time a pathological consequence of iron accumulation in the CD4+ T cells from patients [79]. This novel study identified a reduced expression of 3-hydroxy butyrate dehydrogenase 2 (BDH2), a modulator of intracellular iron homeostasis in the CD4+ T cells of SLE patients. BDH2 is a member of the short-chain dehydrogenase family of reductases, and it catalyzes a rate-limiting step in the biogenesis of the mammalian siderophore [118]. Loss of BDH2 impaired intracellular iron sequestration and increased the availability of Fe2+. Excess iron elevated global DNA hydroxymethylation levels, corresponding to a reduced DNA methylation levels in SLE CD4+ T cells as compared to healthy controls [79], leading to overexpression of immune-related genes. The same group has recently identified BDH2 deficiency-driven labile iron accumulation in SLE CD4+ T cells as promoting the demethylation and hence upregulation of the BCL6 gene, and thereby promoting T follicular helper (Tfh) cell differentiation [80,81]. Tfh cells are a key effector subset in SLE with a frequency that correlates with disease activity [119]. This work identified a functional role for iron in CD4+ T cell biology and the development into pathogenic effectors in SLE. Lipid peroxidation leading to ferroptosis in Tfh cells is not surprising, given that these cells have excess labile iron. Tfh cells show intensified lipid peroxidation and altered mitochondrial morphology, resembling the features of ferroptosis that is kept in check by the seleno-enzyme GPX4, which is necessary for Tfh cell survival [120]. The deletion of GPX4 in T cells selectively abrogated Tfh cells and germinal center responses in immunized mice, and selenium supplementation enhanced GPX4 expression in T cells, increased Tfh cell numbers and promoted antibody responses [120].
Collectively, these studies highlight that activated CD4+ T cells immediately turn on their iron import machinery for proliferative and metabolic requirements. In the context of autoimmunity, iron can act as a chaperon to stabilize cytokine mRNA or induce epigenetic modifications to polarize into specialized effectors. Hence, CD4+ T effector cells have defense mechanisms to counter iron-mediated pathology like ferroptosis.
2.4. B cells, iron, and ferroptosis
We have a limited understanding of whether iron and ferroptosis in B cells play a role in SLE. In lupus, the aberrant activation of TLR7 by self-nucleic acids leads to a break in tolerance and perpetuation of the autoimmune feed-forward loop [121]. Additionally, RNA-associated autoantigens activate B cells by combining B cell antigen receptor and TLR7 engagement [122]. Since B cell intrinsic TLR7 signaling plays a critical role in viral infections [123-125] and iron plays an essential role in B cell anti-viral and immunization responses [125], we may draw parallels to gain insights on the role of iron in B cell effector function during the evolution of SLE.
B cells that receive the appropriate activation signals in the presence or absence of T cells initiate an energy-intensive differentiation program to develop into highly proliferative, antibody-secreting plasma cells [126]. During this process, B cells utilize iron in different forms and cellular compartments. Patients with a Tyr20His substitution in the TFRC gene encoding for TfR1, the principal receptor for uptake of transferrin-bound iron, have significantly reduced numbers of memory B cells, impaired B cell proliferation, and impaired IgE production [127]. Iron is essential for B cell proliferation as iron deficiency suppresses cyclin E1 induction and S phase entry in activated B cells [128]. Iron-dependent histone 3 lysine 9 demethylation controls B cell proliferation, and iron-deficient individuals and mice exhibit a significantly reduced antibody response to the measles vaccine compared to iron-normal control [128]. Activated B cells also utilize iron-sulfur (Fe─S) clusters as cofactors for the activity of DNA polymerases, helicases, and glycosylases [128]. These Fe─S cluster proteins are synthesized in the mitochondria, and the Fe-S-glutathione intermediate is exported to the cytoplasm for maturation by the mitochondrial transporter ABCB7 [129,130]. Conditional deletion of ABCB7 in B cells blocked bone marrow B cell development at the pro-B cell stage. Surprisingly though, the loss of ABCB7 in pro-B cells increased intracellular iron and replication-induced DNA damage but did not increase cellular or mitochondrial ROS, ferroptosis, or apoptosis. Why ABCB7-mediated export of Fe─S intermediates are required for bone marrow B cell development, proliferation, and class switch recombination but dispensable for peripheral B cell homeostasis in mice remains unanswered [128]. The importance of interaction between secreted iron binding proteins and B cell function was showcased by a study that showed that the HIV-1 accessory protein Nef induces the production and secretion of ferritin, an iron-sequestration protein, in infected macrophages in a NF-κB-dependent manner [131], which then induces B-cell hyperactivation and exhaustion. This study identified soluble ferritin heavy and light chains as the essential soluble factor secreted by infected macrophages responsible for B-cell proliferation. B cells express T cell immunoglobulin-domain and mucin-domain 2 (TIM2) [132], and TfR1 [133]; the receptors for soluble ferritin. In vitro exposure to the supernatant from cultures of Nef-expressing or HIV-1-infected macrophages led to B cells proliferation and differentiation into memory B cells and plasma cells. Exogenous ferritin induced B-cell proliferation and the expression of the activation markers, which were suppressed when ferritin was immunodepleted from the supernatants, demonstrating a causal effect of ferritin on B cells.
Ferritin is uptaken via the TIM-2 receptor-mediated endocytosis and, eventually, transits through the lysosomal compartment, distinguishing it from the transferrin-based mechanism, the classical vehicle for cellular iron delivery [134]. Secreted serum ferritin levels mirror the degree of acute and chronic inflammation, independent of iron status [135,136]. Serum ferritin levels are increased in SLE/LN patients and correlate with disease activity [137,138]. Hepcidin, present at high levels in SLE [139,140] can degrade ferroportin and attenuate systemic iron availability [29]. Whether secreted ferritin acts as a non-classical source of iron to support B cell function remains to be evaluated.
Within B cell subsets, B1 and marginal zone (MZ) B cells display increased sensitivity to lipid peroxidation and ferroptosis compared to follicular B cells [141]. Increased metabolic requirements and differences in lipid metabolism between B1/MZ and follicular B cells dictates the requirement of GPX4 to scavenge lipid ROS and inhibit ferroptosis. Fat uptake and active breakdown of lipid droplets fuel the mitochondrial Krebs cycle to generate ATP, which is critical for B1 cells to sustain mitochondrial oxidative phosphorylation [142]. ACSL-mediated activation of fatty acids is required for lipid-droplet formation [143]. Mechanistically, ACSL4 promotes the intracellular lipogenesis and lipid droplets accumulation to enhance fatty acid oxidation (FAO) and adenosine triphosphate production by upregulating the FAO rate-limiting enzyme CPT1A (carnitine palmitoyltransferase 1 isoform A) [144]. However, ACSL4 also enriches cellular membranes with long polyunsaturated omega-6 fatty acids and is essential for ferroptosis execution. Thus, activation of ACSL4 during lipid-droplet formation and lipolysis may collaterally facilitate ferroptosis in Gpx4-deficient B1 and MZ B cells. Moreover, since high intracellular free fatty acids levels also cause oxidative stress [145], B1/MZ B cells may require GPX4 to prevent accentuated ROS and ferroptosis. Consequently, GPX4 is dispensable for the development and maintenance of follicular B cells, antibody responses, and germinal center reactions, whereas B1 and MZ B cells die by ferroptosis in the absence of GPX4 [141]. These data shed light on the complex interplay between subset-dependent-B cell metabolism, their redox biology and ferroptosis.
B cell ferroptosis was very recently reported in human and murine SLE [44]. Single-cell RNA sequencing and gene set enrichment analysis of B cell subsets revealed a significant enrichment of genes associated with pathways related to cell death, iron metabolism, iron ion binding, and response to oxidative stress in SLE patients. The expression of GPX4was reduced in both naive and memory B cells from SLE patients, and ultrastructural analysis of B cells from SLE patients showed mitochondrial condensation, swelling, with reduced or absent crista, features associated with ferroptosis. These features were recapitulated in B cells isolated from the MRL/lpr mice, and Liproxstatin-1 reversed lipid peroxidation in purified human B cells exposed to SLE serum, thus confirming B cell ferroptosis across species in settings of SLE/LN. We had earlier reported anomalous iron deposits in the splenic white pulp of nephritic MRL/lpr mice [38], and by identifying the occurrence of B cell ferroptosis in aged MRL/lpr mice, Chen et al. [44] mechanistically furthered our observations.
2.5. Renal iron handling: a potential clue to increased susceptibility of renal tubules to iron mediated pathology
Iron accumulation in renal tubular epithelial cells of LN patients and lupus-prone mice has been documented [38,146]. In the kidneys, the proximal tubular epithelial cells (PTECs) are the most metabolically active, and as such, they contain more mitochondria than any other cells in the kidney [147]. Mitochondria, the primary source of ROS [148,149], produce highly reactive and toxic hydroxyl radicals (OH) via metal-dependent breakdown using cellular transition metals, most notably iron [148,150]. Little is known about iron handling by the glomerular cells, and most of our understanding of renal iron handling is based on studies on the tubular compartment [17,151]. The distal renal tubules express proteins associated with iron import. The expression of light and heavy chain ferritin mediated iron storage is low, and the iron exporter ferroportin is not detected in distal tubules [146]. The lack of expression of ferroportin and lower expression of ferritin might render the distal tubules susceptible to iron-mediated damage. However, majority of literature dwelling on iron-induced pathology in renal tubules reports PTEC pathology and we know little about iron mediated injury in distal tubules [152-155]. This may be because, unlike the distal renal tubules, PTECs express receptors for iron import, high levels of light and heavy chain ferritin, as well as ferroportin [146]. Hence, PTECs, but not the distal renal tubules, are more likely to participate in iron recycling [146]. Under physiological conditions, a fraction of transferrin-bound Iron (TBI) is filtered by the glomerulus into the renal tubular lumen and almost entirely reabsorbed by renal tubular epithelial cells [156,157]. TBI is imported from the apical surface of the PTEC via TfR1 and megalin-cubulin endocytic complex [158,159], whereas non-transferrin bound iron (NTBI) is imported by ZIP8 and/or ZIP14 [160], such that iron loss in the urine is minimal. We have previously discussed the details of PTEC iron transport in health and LN [17].
The injury to the glomerular structure increases the permeability of all proteins, including transferrin and albumin. Unlike TfR1, the receptor for transferrin, which is post-transcriptionally downregulated in cells that accumulate excess iron [161], the expression of the megalin-cubulin endocytic complex, ZIP8, and ZIP14 is not regulated by the iron content of the cells [162]. Furthermore, PTECs also reabsorb albumin [163,164], which is a heme-and iron-carrier protein [165-167], whose excessive uptake can increase the iron content of the tubular cells. Consequently, following the breakdown of the glomerular filtration barrier, iron can be taken up by PTECs in a disproportionate manner. Thus, the combination of excessive filtered iron uptake due to a compromised glomerular function in LN and a high mitochondrial content render the PTECs susceptible to iron-catalyzed, ROS-mediated injury and ferroptosis.
2.6. Renal tubules, iron, and ferroptosis
Reports of the beneficial effects of modulating iron metabolism in SLE/LN are limited. Non-heme iron levels in the kidneys of (New Zealand Black X New Zealand White) F1 (NZB/W) mice are increased compared with healthy New Zealand White (NZW) mice in an age- and strain-dependent manner [36]. The expression of TfR1 is attenuated in tubules from NZB/W compared to NZW mice, and ferritin expression increased, consistent with increased iron accumulation and compensatory downregulation of uptake pathways. Treating these mice with deferiprone, an FDA-approved iron chelator, delayed the onset of albuminuria even though anti-dsDNA IgG levels were comparable to the vehicle-treated group. We have shown the beneficial effect of exogenous hepcidin in reducing renal iron accumulation, labile iron content, and glomerular and tubular injury in MRL/lpr mice [38]. As in the NZB/W mice, hepcidin treatment did not reduce renal IC deposits and serum autoantibodies, but it mitigated intrarenal cytokine production, immune cell infiltration, and tubular injury without worsening lupus-associated anemia. Hepcidin was protective even when administered to mice with existing proteinuria, highlighting its therapeutic potential. Our study challenges the existing paradigm that suggests that inhibition of ferroportin-induced iron export should worsen iron-mediated injury [168]. We observed that intermittent administration of exogenous hepcidin more than doubled the expression of renal FtH, a cytoprotective molecule [38]. The protective role of FtH in a rodent model of thymocyte antigen-1-induced glomerulonephritis was previously highlighted by Cheng et al. who showed that reduced expression of FtH accelerated mesangial cell death [169]. Furthermore, forced expression of wild-type FtH made the cells more resistant to ROS-mediated injury, and this salutary effect was not observed in FtH mutants that lost the capacity for iron storage and ferroxidase activity [169].
A common theme in the reno-protective effect of iron chelator or hepcidin was the lack of difference in circulating autoantibodies and glomerular IC deposits between vehicle and drug-treated animals, suggesting a direct effect on renal parenchyma. Thus, these data support the ongoing hypothesis that approaches to increase renal parenchymal cell resistance may mitigate progression to renal disease in SLE.
Since most iron resorption and accumulation occurs in the tubular compartment of the kidneys, it is not surprising that ferroptosis predominantly occurs in this segment of the nephron. Ferroptosis-related differentially expressed genes were identified in the glomeruli and tubulointerstitium in kidney biopsies of healthy controls and LN patients [20,170]. These studies identified genes associated with antioxidant system inhibitors and ferroptosis suppressors significantly altered in LN. Expression of some of these genes was positively correlated with immune cell infiltration in glomeruli [20,170]. However, most of the data in these reports is based on transcriptomics, which has not been validated by animal studies that can open avenues for mechanistic and therapeutic queries.
We have recently described the occurrence of intra-renal ferroptosis in LN patients and in female MRL/lpr and male (NZW X BXSB) F1 mice, two spontaneous models of murine LN using protein, gene, and lipidomic analyses [42]. Our study, for the first time, showed that the protein expression of 4-hydroxynonenal (4-HNE), a marker for lipid peroxidation [171], and ACSL4 are increased in the iron accumulating tubular segments of the nephron in both human and murine LN kidneys. In line with an active ferroptosis program, we reported that SLC7A11 (cystine importer), the glutathione synthesis pathway, and GPX4 was attenuated in the nephritic kidneys. GPX4 is a glutathione-dependent enzyme, and glutathione synthesis requires adequate cystine supply. As pièce de resistance, we observed a significant increase in the esterification of the sn-2 chain of PE with adrenic acid (C22:4) (P-18:0/22:4), the preferred substrate for lipid peroxidation [172] in the nephritic kidneys [42]. To highlight the importance of labile iron Fe2+ sequestration in the tubular compartment during the evolution of LN, we showed that FtH deficiency selectively in PTECs exacerbated tubular injury and ferroptosis in a model of nephrotoxic serum-induced immune complex glomerulonephritis [42]. This observation further validated our previous study [38], which showed that hepcidin-induced increase in intrarenal FtH is associated with amelioration of LN. LN patients' serum induced ferroptosis in human PTECs, which was reversed by Liproxstatin-2, a novel next-generation ferroptosis inhibitor, in a prophylactic and therapeutic approach, identifying ferroptosis as a druggable target to mitigate tubular injury in LN.
2.7. Conclusion
The studies highlighted in this review suggest that a finely tuned iron metabolism supports cellular bioenergetic, but when dysregulated, can cause ferroptosis, contributing to immune and tubular cell pathology in SLE and LN. We have highlighted articles that focus on the dysregulation of iron metabolism in immune and parenchymal cells involved in the pathogenesis of SLE/LN. During the evolution of SLE and its progression to LN, innate and adaptive immune cells use iron for varying functions. Novel approaches have revealed the Janus face of iron in immune cells, wherein it transcends its obligatory physiological functions to drive pathology and, consequently, the outcomes of SLE/LN.
Ferroptosis is most likely integral to lupus immune and parenchymal cell dysfunction, inflammation, and tissue damage. A continuum of publications indicates that ferroptosis is not only a novel therapeutic target in SLE/LN but understanding why cells progress to ferroptosis may shed light on fundamentals of cellular biology. For example, infections are common in SLE [173], and neutrophils release large amounts of ROS and undergo netosis during infections [174]. Since the hepcidin-ferroportin axis actively sequesters iron in neutrophils [175], whether its chronic activation impacts neutrophil ferroptosis to provide additional autoantigens remains to be investigated. Additionally, the understanding of the crosstalk between metabolism and ferroptosis is limited. The metabolic needs of proliferative and autoreactive B cells that drive a preference for fatty acid uptake and active breakdown of lipid droplets to generate ATP also generate intermediate metabolites essential for ferroptosis. How cells utilize iron for enhanced metabolic need, while putting a brake on ferroptosis may help identify novel cellular mechanisms in the pathogenesis of SLE. Thus, new generation ferroptosis inhibitors like FerroLOXINs that block enzyme-protein complexes [176], unlike archetype ferroptosis inhibitors like Liproxstatin-1 (that suppress nonenzymatic lipid peroxidation-induced ferroptosis by trapping the lipid derived free radicals) [100] hold promise as adjunct therapy to reduce dose and dependency of toxic immunosuppressants.
Funding
Supported by grants from Vifor Pharma (P0213104) and NIH (RO1DK136011) to Yogesh Scindia and Laurence Morel.
Footnotes
CRediT authorship contribution statement
Laurence Morel: Writing – review & editing. Yogesh Scindia: Conceptualization, Writing – original draft, Writing – review & editing.
Declaration of competing interest
The authors have declared that no conflict of interest exists.
Data availability
No data was used for the research described in the article.
References
- [1].Brunner HI, Gladman DD, Ibanez D, Urowitz MD, Silverman ED, Difference in disease features between childhood-onset and adult-onset systemic lupus erythematosus, Arthritis Rheum. 58 (2008) 556–562. [DOI] [PubMed] [Google Scholar]
- [2].Liu Z, Davidson A, Taming lupus-a new understanding of pathogenesis is leading to clinical advances, Nat. Med 18 (2012) 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Pieterse E, van der Vlag J, Breaking immunological tolerance in systemic lupus erythematosus, Front. Immunol 5 (2014) 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tsokos GC, Systemic lupus erythematosus, N. Engl. J. Med 365 (2011) 2110–2121. [DOI] [PubMed] [Google Scholar]
- [5].Rullo OJ, Tsao BP, Recent insights into the genetic basis of systemic lupus erythematosus, Ann. Rheum. Dis 72 (Suppl. 2) (2013) ii56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Almaani S, Meara A, Rovin BH, Update on lupus nephritis, Clin. J. Am. Soc. Nephrol.: CJASN 12 (2017) 825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Madaio MP, The role of autoantibodies in the pathogenesis of lupus nephritis, Semin. Nephrol 19 (1999) 48–56. [PubMed] [Google Scholar]
- [8].Londono Jimenez A, Mowrey WB, Putterman C, Buyon J, Goilav B, Broder A, Brief report: tubulointerstitial damage in lupus nephritis: a comparison of the factors associated with tubulointerstitial inflammation and renal scarring, arthritis, Rheumatol 70 (2018) 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rijnink EC, Teng YKO, Wilhelmus S, Almekinders M, Wolterbeek R, Cransberg K, Bruijn JA, Bajema IM, Clinical and histopathologic characteristics associated with renal outcomes in lupus nephritis, Clin. J. Am. Soc. Nephrol.: CJASN 12 (2017) 734–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hong S, Healy H, Kassianos AJ, The emerging role of renal tubular epithelial cells in the immunological pathophysiology of lupus nephritis, Front. Immunol 11 (2020) 578952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wilson PC, Kashgarian M, Moeckel G, Interstitial inflammation and interstitial fibrosis and tubular atrophy predict renal survival in lupus nephritis, Clin. Kidney J 11 (2018) 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Smith CD, Cyr M, The history of lupus erythematosus. From Hippocrates to Osler, Rheum. Dis. Clin. N. Am 14 (1988) 1–14. [PubMed] [Google Scholar]
- [13].Robinson GA, Wilkinson MGL, Wincup C, The role of Immunometabolism in the pathogenesis of systemic lupus erythematosus, Front. Immunol 12 (2021) 806560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang A, Zhao J, Qin Y, Zhang Y, Xing Y, Wang Y, Yu Z, Yan J, Han M, Yuan J, Hui Y, Guo S, Ning X, Sun S, Alterations of the gut microbiota in the lupus nephritis: a systematic review, Ren. Fail 45 (2023) 2285877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Piga M, Arnaud L, The Main challenges in systemic lupus erythematosus: where do we stand? J. Clin. Med 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Davidson A, Aranow C, Mackay M, Lupus nephritis: challenges and progress, Curr. Opin. Rheumatol 31 (2019) 682–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wlazlo E, Mehrad B, Morel L, Scindia Y, Iron metabolism: An under investigated driver of renal pathology in lupus nephritis, Front. Med. (Lausanne) 8 (2021) 643686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wincup C, Sawford N, Rahman A, Pathological mechanisms of abnormal iron metabolism and mitochondrial dysfunction in systemic lupus erythematosus, Expert Rev. Clin. Immunol 17 (2021) 957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen Q, Wang J, Xiang M, Wang Y, Zhang Z, Liang J, Xu J, The potential role of ferroptosis in systemic lupus erythematosus, Front. Immunol 13 (2022) 855622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang W, Lin Z, Feng J, Liang Q, Zhao J, Zhang G, Chen R, Fu R, Identification of ferroptosis-related molecular markers in glomeruli and tubulointerstitium of lupus nephritis, Lupus 31 (2022) 985–997. [DOI] [PubMed] [Google Scholar]
- [21].Hershko C, Control of disease by selective iron depletion: a novel therapeutic strategy utilizing iron chelators, Baillieres Clin. Haematol 7 (1994) 965–1000. [DOI] [PubMed] [Google Scholar]
- [22].Lederman HM, Cohen A, Lee JW, Freedman MH, Gelfand EW, Deferoxamine: a reversible S-phase inhibitor of human lymphocyte proliferation, Blood 64 (1984) 748–753. [PubMed] [Google Scholar]
- [23].Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr., HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing, Science 292 (2001) 464–468. [DOI] [PubMed] [Google Scholar]
- [24].Templeton DM, Liu Y, Genetic regulation of cell function in response to iron overload or chelation, Biochim. Biophys. Acta 1619 (2003) 113–124. [DOI] [PubMed] [Google Scholar]
- [25].Rouault TA, The indispensable role of mammalian iron sulfur proteins in function and regulation of multiple diverse metabolic pathways, Biometals 32 (2019) 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Winterbourn CC, Toxicity of iron and hydrogen peroxide: the Fenton reaction, Toxicol. Lett 82-83 (1995) 969–974. [DOI] [PubMed] [Google Scholar]
- [27].Schaich KM, Borg DC, Fenton reactions in lipid phases, Lipids 23 (1988) 570–579. [DOI] [PubMed] [Google Scholar]
- [28].Galy B, Conrad M, Muckenthaler M, Mechanisms controlling cellular and systemic iron homeostasis, Nat. Rev. Mol. Cell Biol 25 (2) (2023) 133–155. [DOI] [PubMed] [Google Scholar]
- [29].Nemeth E, Ganz T, Hepcidin-Ferroportin interaction controls systemic Iron homeostasis, Int. J. Mol. Sci 22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Aschemeyer S, Qiao B, Stefanova D, Valore EV, Sek AC, Ruwe TA, Vieth KR, Jung G, Casu C, Rivella S, Jormakka M, Mackenzie B, Ganz T, Nemeth E, Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin, Blood 131 (2018) 899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J, Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization, Science 306 (2004) 2090–2093. [DOI] [PubMed] [Google Scholar]
- [32].Gao G, Li J, Zhang Y, Chang YZ, Cellular iron metabolism and regulation, Adv. Exp. Med. Biol 1173 (2019) 21–32. [DOI] [PubMed] [Google Scholar]
- [33].Hentze MW, Muckenthaler MU, Galy B, Camaschella C, Two to tango: regulation of mammalian iron metabolism, Cell 142 (2010) 24–38. [DOI] [PubMed] [Google Scholar]
- [34].Cronin SJF, Woolf CJ, Weiss G, Penninger JM, The role of Iron regulation in Immunometabolism and immune-related disease, Front. Mol. Biosci 6 (2019) 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Atkinson MA, Joo S, Sule S, Hepcidin and arterial stiffness in children with systemic lupus erythematosus and lupus nephritis: a cross-sectional study, PLoS One 14 (2019) e0214248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Marks ES, Bonnemaison ML, Brusnahan SK, Zhang W, Fan W, Garrison JC, Boesen EI, Renal iron accumulation occurs in lupus nephritis and iron chelation delays the onset of albuminuria, Sci. Rep 7 (2017) 12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR, Ferroptosis: an iron-dependent form of nonapoptotic cell death, Cell 149 (2012) 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Scindia Y, Wlazlo E, Ghias E, Cechova S, Loi V, Leeds J, Ledesma J, Helen C, Swaminathan S, Modulation of iron homeostasis with hepcidin ameliorates spontaneous murine lupus nephritis, Kidney Int. 98 (2020) 100–115. [DOI] [PubMed] [Google Scholar]
- [39].Perl A, Oxidative stress in the pathology and treatment of systemic lupus erythematosus, Nat. Rev. Rheumatol 9 (2013) 674–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wojcik P, Gegotek A, Zarkovic N, Skrzydlewska E, Oxidative stress and lipid mediators modulate immune cell functions in autoimmune diseases, Int. J. Mol. Sci 22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cheng Q, Mou L, Su W, Chen X, Zhang T, Xie Y, Xue J, Lee PY, Wu H, Du Y, Ferroptosis of CD163(+) tissue-infiltrating macrophages and CD10(+) PC(+) epithelial cells in lupus nephritis, Front. Immunol 14 (2023) 1171318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Alli AA, Desai D, Elshika A, Conrad M, Proneth B, Clapp W, Atkinson C, Segal M, Searcy LA, Denslow ND, Bolisetty S, Mehrad B, Morel L, Scindia Y, Kidney tubular epithelial cell ferroptosis links glomerular injury to tubulointerstitial pathology in lupus nephritis, Clin. Immunol 248 (2023) 109213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yang B, Hou S, Huang S, Li H, Li Y, Ferroptosis inhibitor regulates the disease progression of systematic lupus erythematosus mice model through Th1/Th2 ratio, Curr. Mol. Med 23 (2023) 799–807. [DOI] [PubMed] [Google Scholar]
- [44].Chen Q, Xiang M, Gao Z, Lvu F, Sun Z, Wang Y, Shi X, Xu J, Wang J, Liang J, The role of B-cell ferroptosis in the pathogenesis of systemic lupus erythematosus, Clin. Immunol 256 (2023) 109778. [DOI] [PubMed] [Google Scholar]
- [45].Li P, Jiang M, Li K, Li H, Zhou Y, Xiao X, Xu Y, Krishfield S, Lipsky PE, Tsokos GC, Zhang X, Glutathione peroxidase 4-regulated neutrophil ferroptosis induces systemic autoimmunity, Nat. Immunol 22 (2021) 1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Distefano AM, Martin MV, Cordoba JP, Bellido AM, D’Ippolito S, Colman SL, Soto D, Roldan JA, Bartoli CG, Zabaleta EJ, Fiol DF, Stockwell BR, Dixon SJ, Pagnussat GC, Heat stress induces ferroptosis-like cell death in plants, J. Cell Biol 216 (2017) 463–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bogacz M, Krauth-Siegel RL, Tryparedoxin peroxidase-deficiency commits trypanosomes to ferroptosis-type cell death, Elife 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wong-Ekkabut J, Xu Z, Triampo W, Tang IM, Tieleman DP, Monticelli L, Effect of lipid peroxidation on the properties of lipid bilayers: a molecular dynamics study, Biophys. J 93 (2007) 4225–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gaschler MM, Stockwell BR, Lipid peroxidation in cell death, Biochem. Biophys. Res. Commun 482 (2017) 419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Conrad M, Pratt DA, The chemical basis of ferroptosis, Nat. Chem. Biol 15 1137–1147. [DOI] [PubMed] [Google Scholar]
- [51].Kagan VE, Tyurina YY, Sun WY, Vlasova II, Dar H, Tyurin VA, Amoscato AA, Mallampalli R, van der Wel PCA, He RR, Shvedova AA, Gabrilovich DI, Bayir H, Redox phospholipidomics of enzymatically generated oxygenated phospholipids as specific signals of programmed cell death, Free Radic. Biol. Med, 147 (2020) 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bayir H, Anthonymuthu TS, Tyurina YY, Patel SJ, Amoscato AA, Lamade AM, Yang Q, Vladimirov GK, Philpott CC, Kagan VE, Achieving life through death: redox biology of lipid peroxidation in Ferroptosis, cell, Chem. Biol 27 (2020) 387–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yin H, Xu L, Porter NA, Free radical lipid peroxidation: mechanisms and analysis, Chem. Rev 111 (2011) 5944–5972. [DOI] [PubMed] [Google Scholar]
- [54].Kuhn H, Banthiya S, van Leyen K, Mammalian lipoxygenases and their biological relevance, Biochim. Biophys. Acta 2015 (1851) 308–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, Kapralov AA, Amoscato AA, Jiang J, Anthonymuthu T, Mohammadyani D, Yang Q, Proneth B, Klein-Seetharaman J, Watkins S, Bahar I, Greenberger J, Mallampalli RK, Stockwell BR, Tyurina YY, Conrad M, Bayir H, Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis, Nat. Chem. Biol 13 (2017) 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].von Krusenstiern AN, Robson RN, Qian N, Qiu B, Hu F, Reznik E, Smith N, Zandkarimi F, Estes VM, Dupont M, Hirschhorn T, Shchepinov MS, Min W, Woerpel KA, Stockwell BR, Identification of essential sites of lipid peroxidation in ferroptosis, Nat. Chem. Biol 19 (2023) 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Mortensen MS, Ruiz J, Watts JL, Polyunsaturated fatty acids drive lipid peroxidation during Ferroptosis, Cells 12 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Dalleau S, Baradat M, Gueraud F, Huc L, Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance, Cell Death Differ. 20 (2013) 1615–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Niedernhofer LJ, Daniels JS, Rouzer CA, Greene RE, Marnett LJ, Malondialdehyde, a product of lipid peroxidation, is mutagenic in human cells, J. Biol. Chem 278 (2003) 31426–31433. [DOI] [PubMed] [Google Scholar]
- [60].Jiang X, Stockwell BR, Conrad M, Ferroptosis: mechanisms, biology and role in disease, Nat. Rev. Mol. Cell Biol 22 (2021) 266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Stockwell BR, Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications, Cell 185 (2022) 2401–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, Sandoval-Gomez G, Clish CB, Doench JG, Schreiber SL, Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis, Nat. Chem. Biol 16 (2022) 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Tu H, Tang LJ, Luo XJ, Ai KL, Peng J, Insights into the novel function of system xc- in regulated cell death, Eur. Rev. Med. Pharmacol. Sci 25 (2021) 1650–1662. [DOI] [PubMed] [Google Scholar]
- [64].Li FJ, Long HZ, Zhou ZW, Luo HY, Xu SG, Gao LC, System X(c) (−)/GSH/GPX4 axis: An important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy, Front. Pharmacol 13 (2022) 910292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Seibt TM, Proneth B, Conrad M, Role of GPX4 in ferroptosis and its pharmacological implication, Free Radic. Biol. Med 133 (2019) 144–152. [DOI] [PubMed] [Google Scholar]
- [66].Ayala A, Munoz MF, Arguelles S, Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal, Oxidative Med. Cell. Longev 2014 (2014) 360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR, Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis, Proc. Natl. Acad. Sci. USA 113 (2016). E4966–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Yuan H, Li X, Zhang X, Kang R, Tang D, Identification of ACSL4 as a biomarker and contributor of ferroptosis, Biochem. Biophys. Res. Commun 478 (2016) 1338–1343. [DOI] [PubMed] [Google Scholar]
- [69].Chaitidis P, Adel S, Anton M, Heydeck D, Kuhn H, Horn T, Lipoxygenase pathways in Homo neanderthalensis: functional comparison with Homo sapiens isoforms, J. Lipid Res 54 (2013) 1397–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Haschka D, Hoffmann A, Weiss G, Iron in immune cell function and host defense, Semin. Cell Dev. Biol 115 (2021) 27–36. [DOI] [PubMed] [Google Scholar]
- [71].Wang Y, Guo F, Guo Y, Lu Y, Ji W, Lin L, Chen W, Xu T, Kong D, Shen Q, Zhu Y, Liu P, Su J, Wang L, Li Y, Gao P, Shan J, Liu S, Untargeted lipidomics reveals specific lipid abnormalities in systemic lupus erythematosus, Clin. Exp. Rheumatol 40 (2022) 1011–1018. [DOI] [PubMed] [Google Scholar]
- [72].Ferreira HB, Pereira AM, Melo T, Paiva A, Domingues MR, Lipidomics in autoimmune diseases with main focus on systemic lupus erythematosus, J. Pharm. Biomed. Anal 174 (2019) 386–395. [DOI] [PubMed] [Google Scholar]
- [73].Lightfoot YL, Blanco LP, Kaplan MJ, Metabolic abnormalities and oxidative stress in lupus, Curr. Opin. Rheumatol 29 (2017) 442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kienhofer D, Boeltz S, Hoffmann MH, Reactive oxygen homeostasis - the balance for preventing autoimmunity, Lupus 25 (2016) 943–954. [DOI] [PubMed] [Google Scholar]
- [75].Gong M, Choi SC, Park YP, Zou X, Elshikha AS, Gerriets VA, Rathmell JC, Mohamazadeh M, Morel L, Transcriptional and metabolic programs promote the expansion of follicular helper T cells in lupus-prone mice, iScience 26 (2023) 106774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Abboud G, Choi SC, Zhang X, Park YP, Kanda N, Zeumer-Spataro L, Terrell M, Teng X, Nundel K, Shlomchik MJ, Morel L, Glucose requirement of antigen-specific autoreactive B cells and CD4+ T cells, J. Immunol 210 (2023) 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Cornaby C, Elshikha AS, Teng X, Choi SC, Scindia Y, Davidson A, Morel L, Efficacy of the combination of metformin and CTLA4Ig in the (NZB x NZW)F1 mouse model of lupus nephritis, Immunohorizons 4 (2020) 319–331. [DOI] [PubMed] [Google Scholar]
- [78].Morel L, Immunometabolism in systemic lupus erythematosus, Nat. Rev. Rheumatol 13 (2017) 280–290. [DOI] [PubMed] [Google Scholar]
- [79].Zhao M, Li MY, Gao XF, Jia SJ, Gao KQ, Zhou Y, Zhang HH, Huang Y, Wang J, Wu HJ, Lu QJ, Downregulation of BDH2 modulates iron homeostasis and promotes DNA demethylation in CD4(+) T cells of systemic lupus erythematosus, Clin. Immunol 187 (2018) 113–121. [DOI] [PubMed] [Google Scholar]
- [80].Gao X, Song Y, Wu J, Lu S, Min X, Liu L, Hu L, Zheng M, Du P, Yu Y, Long H, Wu H, Jia S, Yu D, Lu Q, Zhao M, Iron-dependent epigenetic modulation promotes pathogenic T cell differentiation in lupus, J. Clin. Invest 132 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Scindia Y, Mehrad B, Morel L, Labile iron accumulation augments T follicular helper cell differentiation, J. Clin. Invest 132 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Lai ZW, Hanczko R, Bonilla E, Caza TN, Clair B, Bartos A, Miklossy G, Jimah J, Doherty E, Tily H, Francis L, Garcia R, Dawood M, Yu J, Ramos I, Coman I, Faraone SV, Phillips PE, Perl A, N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: a randomized, double-blind, placebo-controlled trial, Arthritis Rheum. 64 (2012) 2937–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Fortner KA, Blanco LP, Buskiewicz I, Huang N, Gibson PC, Cook DL, Pedersen HL, Yuen PST, Murphy MP, Perl A, Kaplan MJ, Budd RC, Targeting mitochondrial oxidative stress with MitoQ reduces NET formation and kidney disease in lupus-prone MRL-lpr mice, Lupus Sci. Med 7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Nairz M, Ferring-Appel D, Casarrubea D, Sonnweber T, Viatte L, Schroll A, Haschka D, Fang FC, Hentze MW, Weiss G, Galy B, Iron regulatory proteins mediate host resistance to Salmonella infection, Cell Host Microbe 18 (2015) 254–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Xiao X, Yeoh BS, Vijay-Kumar M, Lipocalin 2: An emerging player in Iron homeostasis and inflammation, Annu. Rev. Nutr 37 (2017) 103–130. [DOI] [PubMed] [Google Scholar]
- [86].Arnhold J, Furtmuller PG, Obinger C, Redox properties of myeloperoxidase, Redox Rep. 8 (2003) 179–186. [DOI] [PubMed] [Google Scholar]
- [87].Nakashige TG, Zhang B, Krebs C, Nolan EM, Human calprotectin is an iron-sequestering host-defense protein, Nat. Chem. Biol 11 (2015) 765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Legrand D, Lactoferrin, a key molecule in immune and inflammatory processes, Biochem. Cell Biol 90 (2012) 252–268. [DOI] [PubMed] [Google Scholar]
- [89].Ma S, Jiang W, Zhang X, Liu W, Insights into the pathogenic role of neutrophils in systemic lupus erythematosus, Curr. Opin. Rheumatol 35 (2023) 82–88. [DOI] [PubMed] [Google Scholar]
- [90].Gupta S, Kaplan MJ, The role of neutrophils and NETosis in autoimmune and renal diseases, Nat. Rev. Nephrol 12 (2016) 402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Gupta S, Kaplan MJ, Bite of the wolf: innate immune responses propagate autoimmunity in lupus, J. Clin. Invest 131 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Mavragani CP, Kirou KA, Seshan SV, Crow MK, Type I interferon and neutrophil transcripts in lupus nephritis renal biopsies: clinical and histopathological associations, Rheumatology (Oxford) 62 (2023) 2534–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Ren Y, Tang J, Mok MY, Chan AW, Wu A, Lau CS, Increased apoptotic neutrophils and macrophages and impaired macrophage phagocytic clearance of apoptotic neutrophils in systemic lupus erythematosus, Arthritis Rheum. 48 (2003) 2888–2897. [DOI] [PubMed] [Google Scholar]
- [94].Courtney PA, Crockard AD, Williamson K, Irvine AE, Kennedy RJ, Bell AL, Increased apoptotic peripheral blood neutrophils in systemic lupus erythematosus: relations with disease activity, antibodies to double stranded DNA, and neutropenia, Ann. Rheum. Dis 58 (1999) 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Hanata N, Ota M, Tsuchida Y, Nagafuchi Y, Okamura T, Shoda H, Fujio K, Serum extracellular traps associate with the activation of myeloid cells in SLE patients with the low level of anti-DNA antibodies, Sci. Rep 12 (2022) 18397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R, Riccieri V, Bassett R, Amuro H, Fukuhara S, Ito T, Liu YJ, Gilliet M, Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus, Sci. Transl. Med 3 (2011) 73ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, Rubin CJ, Zhao W, Olsen SH, Klinker M, Shealy D, Denny MF, Plumas J, Chaperot L, Kretzler M, Bruce AT, Kaplan MJ, Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus, J. Immunol 187 (2011) 538–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Coke LN, Wen H, Comeau M, Ghanem MH, Shih A, Metz CN, Li W, Langefeld CD, Gregersen PK, Simpfendorfer KR, Arg206Cys substitution in DNASE1L3 causes a defect in DNASE1L3 protein secretion that confers risk of systemic lupus erythematosus, Ann. Rheum. Dis 80 (2021) 782–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, Zychlinsky A, Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis, Proc. Natl. Acad. Sci. USA 107 (2010) 9813–9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, Pratt DA, On the mechanism of cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the role of lipid peroxidation in Ferroptotic cell death, ACS Cent. Sci 3 (2017) 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Liossis SN, Ding XZ, Dennis GJ, Tsokos GC, Altered pattern of TCR/CD3-mediated protein-tyrosyl phosphorylation in T cells from patients with systemic lupus erythematosus. Deficient expression of the T cell receptor zeta chain, J. Clin. Invest 101 (1998) 1448–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Enyedy EJ, Nambiar MP, Liossis SN, Dennis G, Kammer GM, Tsokos GC, Fc epsilon receptor type I gamma chain replaces the deficient T cell receptor zeta chain in T cells of patients with systemic lupus erythematosus, Arthritis Rheum. 44 (2001) 1114–1121. [DOI] [PubMed] [Google Scholar]
- [103].Krishnan S, Warke VG, Nambiar MP, Tsokos GC, Farber DL, The FcR gamma subunit and Syk kinase replace the CD3 zeta-chain and ZAP-70 kinase in the TCR signaling complex of human effector CD4 T cells, J. Immunol 170 (2003) 4189–4195. [DOI] [PubMed] [Google Scholar]
- [104].Abdirama D, Tesch S, Griessbach AS, von Spee-Mayer C, Humrich JY, Stervbo U, Babel N, Meisel C, Alexander T, Biesen R, Bacher P, Scheffold A, Eckardt KU, Hiepe F, Radbruch A, Burmester GR, Riemekasten G, Enghard P, Nuclear antigen-reactive CD4(+) T cells expand in active systemic lupus erythematosus, produce effector cytokines, and invade the kidneys, Kidney Int. 99 (2021) 238–246. [DOI] [PubMed] [Google Scholar]
- [105].Murata H, Matsumura R, Koyama A, Sugiyama T, Sueishi M, Shibuya K, Tsutsumi A, Sumida T, T cell receptor repertoire of T cells in the kidneys of patients with lupus nephritis, Arthritis Rheum. 46 (2002) 2141–2147. [DOI] [PubMed] [Google Scholar]
- [106].Oexle H, Gnaiger E, Weiss G, Iron-dependent changes in cellular energy metabolism: influence on citric acid cycle and oxidative phosphorylation, Biochim. Biophys. Acta 1413 (1999) 99–107. [DOI] [PubMed] [Google Scholar]
- [107].Cronin SJ, Penninger JM, From T-cell activation signals to signaling control of anti-cancer immunity, Immunol. Rev 220 (2007) 151–168. [DOI] [PubMed] [Google Scholar]
- [108].Kuvibidila S, Nauss KM, Baliga BS, Suskind RM, Impairment of blastogenic response of splenic lymphocytes from iron-deficient mice: in vivo repletion, Am. J. Clin. Nutr 37 (1983) 15–25. [DOI] [PubMed] [Google Scholar]
- [109].Sawitsky B, Kanter R, Sawitsky A, Lymphocyte response to phytomitogens in iron deficiency, Am J Med Sci 272 (1976) 153–160. [DOI] [PubMed] [Google Scholar]
- [110].Jennifer B, Berg V, Modak M, Puck A, Seyerl-Jiresch M, Kunig S, Zlabinger GJ, Steinberger P, Chou J, Geha RS, Ohler L, Yachie A, Choe H, Kraller M, Stockinger H, Stockl J, Transferrin receptor 1 is a cellular receptor for human heme-albumin, Commun. Biol 3 (2020) 621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Batista A, Millan J, Mittelbrunn M, Sanchez-Madrid F, Alonso MA, Recruitment of transferrin receptor to immunological synapse in response to TCR engagement, J. Immunol 172 (2004) 6709–6714. [DOI] [PubMed] [Google Scholar]
- [112].Zheng Y, Collins SL, Lutz MA, Allen AN, Kole TP, Zarek PE, Powell JD, A role for mammalian target of rapamycin in regulating T cell activation versus anergy, J. Immunol 178 (2007) 2163–2170. [DOI] [PubMed] [Google Scholar]
- [113].Vanoaica L, Richman L, Jaworski M, Darshan D, Luther SA, Kuhn LC, Conditional deletion of ferritin h in mice reduces B and T lymphocyte populations, PLoS One 9 (2014) e89270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M, T cell lipid peroxidation induces ferroptosis and prevents immunity to infection, J. Exp. Med 212 (2015) 555–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Yarosz EL, Ye C, Kumar A, Black C, Choi EK, Seo YA, Chang CH, Cutting edge: activation-induced Iron flux controls CD4 T cell proliferation by promoting proper IL-2R signaling and mitochondrial function, J. Immunol 204 (2020) 1708–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Grant SM, Wiesinger JA, Beard JL, Cantorna MT, Iron-deficient mice fail to develop autoimmune encephalomyelitis, J. Nutr 133 (2003) 2635–2638. [DOI] [PubMed] [Google Scholar]
- [117].Wang Z, Yin W, Zhu L, Li J, Yao Y, Chen F, Sun M, Zhang J, Shen N, Song Y, Chang X, Iron drives T helper cell pathogenicity by promoting RNA-binding protein PCBP1-mediated Proinflammatory cytokine production, Immunity 49 (2018) 80–92 e87. [DOI] [PubMed] [Google Scholar]
- [118].Devireddy LR, Hart DO, Goetz DH, Green MR, A mammalian siderophore synthesized by an enzyme with a bacterial homolog involved in enterobactin production, Cell 141 (2010) 1006–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Song W, Craft J, T follicular helper cell heterogeneity, Annu. Rev. Immunol 13 (44) (2023) 127–152. [DOI] [PubMed] [Google Scholar]
- [120].Yao Y, Chen Z, Zhang H, Chen C, Zeng M, Yunis J, Wei Y, Wan Y, Wang N, Zhou M, Qiu C, Zeng Q, Ong HS, Wang H, Makota FV, Yang Y, Yang Z, Wang N, Deng J, Shen C, Xia Y, Yuan L, Lian Z, Deng Y, Guo C, Huang A, Zhou P, Shi H, Zhang W, Yi H, Li D, Xia M, Fu J, Wu N, de Haan JB, Shen N, Zhang W, Liu Z, Yu D, Selenium-GPX4 axis protects follicular helper T cells from ferroptosis, Nat. Immunol 22 (2021) 1127–1139. [DOI] [PubMed] [Google Scholar]
- [121].Brown GJ, Canete PF, Wang H, Medhavy A, Bones J, Roco JA, He Y, Qin Y, Cappello J, Ellyard JI, Bassett K, Shen Q, Burgio G, Zhang Y, Turnbull C, Meng X, Wu P, Cho E, Miosge LA, Andrews TD, Field MA, Tvorogov D, Lopez AF, Babon JJ, Lopez CA, Gonzalez-Murillo A, Garulo DC, Pascual V, Levy T, Mallack EJ, Calame DG, Lotze T, Lupski JR, Ding H, Ullah TR, Walters GD, Koina ME, Cook MC, Shen N, de Lucas Collantes C, Corry B, Gantier MP, Athanasopoulos V, Vinuesa CG, TLR7 gain-of-function genetic variation causes human lupus, Nature 605 (2022) 349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, Marshak-Rothstein A, RNA-associated autoantigens activate B cells by combined B cell antigen receptor/toll-like receptor 7 engagement, J. Exp. Med 202 (2005) 1171–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Soni C, Wong EB, Domeier PP, Khan TN, Satoh T, Akira S, Rahman ZS, B cell-intrinsic TLR7 signaling is essential for the development of spontaneous germinal centers, J. Immunol 193 (2014) 4400–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Upasani V, Rodenhuis-Zybert I, Cantaert T, Antibody-independent functions of B cells during viral infections, PLoS Pathog. 17 (2021) e1009708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Clingan JM, Matloubian M, B cell-intrinsic TLR7 signaling is required for optimal B cell responses during chronic viral infection, J. Immunol 191 (2013) 810–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Wiggins KJ, Scharer CD, Roadmap to a plasma cell: epigenetic and transcriptional cues that guide B cell differentiation, Immunol. Rev 300 (2021) 54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Jabara HH, Boyden SE, Chou J, Ramesh N, Massaad MJ, Benson H, Bainter W, Fraulino D, Rahimov F, Sieff C, Liu ZJ, Alshemmari SH, Al-Ramadi BK, Al-Dhekri H, Arnaout R, Abu-Shukair M, Vatsayan A, Silver E, Ahuja S, Davies EG, Sola-Visner M, Ohsumi TK, Andrews NC, Notarangelo LD, Fleming MD, Al-Herz W, Kunkel LM, Geha RS, A missense mutation in TFRC, encoding transferrin receptor 1, causes combined immunodeficiency, Nat. Genet 48 (2016) 74–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Jiang Y, Li C, Wu Q, An P, Huang L, Wang J, Chen C, Chen X, Zhang F, Ma L, Liu S, He H, Xie S, Sun Y, Liu H, Zhan Y, Tao Y, Liu Z, Sun X, Hu Y, Wang Q, Ye D, Zhang J, Zou S, Wang Y, Wei G, Liu Y, Shi Y, Eugene Chin Y, Hao Y, Wang F, Zhang X, Iron-dependent histone 3 lysine 9 demethylation controls B cell proliferation and humoral immune responses, Nat. Commun 10 (2019) 2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Li J, Cowan JA, Glutathione-coordinated [2Fe-2S] cluster: a viable physiological substrate for mitochondrial ABCB7 transport, Chem. Commun. (Camb.) 51 (2015) 2253–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Pondarre C, Antiochos BB, Campagna DR, Clarke SL, Greer EL, Deck KM, McDonald A, Han AP, Medlock A, Kutok JL, Anderson SA, Eisenstein RS, Fleming MD, The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic iron-sulfur cluster biogenesis, Hum. Mol. Genet 15 (2006) 953–964. [DOI] [PubMed] [Google Scholar]
- [131].Swingler S, Zhou J, Swingler C, Dauphin A, Greenough T, Jolicoeur P, Stevenson M, Evidence for a pathogenic determinant in HIV-1 Nef involved in B cell dysfunction in HIV/AIDS, Cell Host Microbe 4 (2008) 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Chen TT, Li L, Chung DH, Allen CD, Torti SV, Torti FM, Cyster JG, Chen CY, Brodsky FM, Niemi EC, Nakamura MC, Seaman WE, Daws MR, TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis, J. Exp. Med 202 (2005) 955–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Neckers LM, Yenokida G, James SP, The role of the transferrin receptor in human B lymphocyte activation, J. Immunol 133 (1984) 2437–2441. [PubMed] [Google Scholar]
- [134].Han J, Seaman WE, Di X, Wang W, Willingham M, Torti FM, Torti SV, Iron uptake mediated by binding of H-ferritin to the TIM-2 receptor in mouse cells, PLoS One 6 (2011) e23800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Kernan KF, Carcillo JA, Hyperferritinemia and inflammation, Int. Immunol 29 (2017) 401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Kell DB, Pretorius E, Serum ferritin is an important inflammatory disease marker, as it is mainly a leakage product from damaged cells, Metallomics 6 (2014) 748–773. [DOI] [PubMed] [Google Scholar]
- [137].Vanarsa K, Ye Y, Han J, Xie C, Mohan C, Wu T, Inflammation associated anemia and ferritin as disease markers in SLE, Arthritis Res. Ther 14 (2012) R182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Beyan E, Beyan C, Demirezer A, Ertugrul E, Uzuner A, The relationship between serum ferritin levels and disease activity in systemic lupus erythematosus, Scand. J. Rheumatol 32 (2003) 225–228. [DOI] [PubMed] [Google Scholar]
- [139].Indrakanti DL, Alvarado A, Zhang X, Birmingham DJ, Hinton A, Rovin BH, The interleukin-6-hepcidin-hemoglobin circuit in systemic lupus erythematosus flares, Lupus 26 (2017) 200–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Kunireddy N, Jacob R, Khan SA, Yadagiri B, Sai Baba KSS, Rajendra Vara Prasad I, Mohan IK, Hepcidin and ferritin: important mediators in inflammation associated Anemia in systemic lupus erythematosus patients, Indian, J. Clin. Biochem 33 (2018) 406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Muri J, Thut H, Bornkamm GW, Kopf M, B1 and marginal zone B cells but not follicular B2 cells require Gpx4 to prevent lipid peroxidation and Ferroptosis, Cell Rep. 29 (2019) 2731–2744 e2734. [DOI] [PubMed] [Google Scholar]
- [142].Clarke AJ, Riffelmacher T, Braas D, Cornall RJ, Simon AK, B1a B cells require autophagy for metabolic homeostasis and self-renewal, J. Exp. Med 215 (2018) 399–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Wilfling F, Haas JT, Walther TC, Farese RV Jr., Lipid droplet biogenesis, Curr. Opin. Cell Biol 29 (2014) 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Lin J, Zhang P, Liu W, Liu G, Zhang J, Yan M, Duan Y, Yang N, A positive feedback loop between ZEB2 and ACSL4 regulates lipid metabolism to promote breast cancer metastasis, Elife 12 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Hauck AK, Bernlohr DA, Oxidative stress and lipotoxicity, J. Lipid Res 57 (2016) 1976–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].van Raaij S, van Swelm R, Bouman K, Cliteur M, van den Heuvel MC, Pertijs J, Patel D, Bass P, van Goor H, Unwin R, Srai SK, Swinkels D, Tubular iron deposition and iron handling proteins in human healthy kidney and chronic kidney disease, Sci. Rep 8 (2018) 9353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Bhargava P, Schnellmann RG, Mitochondrial energetics in the kidney, Nat. Rev. Nephrol 13 (2017) 629–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Curtin JF, Donovan M, Cotter TG, Regulation and measurement of oxidative stress in apoptosis, J. Immunol. Methods 265 (2002) 49–72. [DOI] [PubMed] [Google Scholar]
- [149].Fleury C, Mignotte B, Vayssiere JL, Mitochondrial reactive oxygen species in cell death signaling, Biochimie 84 (2002) 131–141. [DOI] [PubMed] [Google Scholar]
- [150].Torti FM, Torti SV, Regulation of ferritin genes and protein, Blood 99 (2002) 3505–3516. [DOI] [PubMed] [Google Scholar]
- [151].van Swelm RPL, Wetzels JFM, Swinkels DW, The multifaceted role of iron in renal health and disease, Nat. Rev. Nephrol 16 (2020) 77–98. [DOI] [PubMed] [Google Scholar]
- [152].van Raaij SEG, Masereeuw R, Swinkels DW, van Swelm RPL, Inhibition of Nrf2 alters cell stress induced by chronic iron exposure in human proximal tubular epithelial cells, Toxicol. Lett 295 (2018) 179–186. [DOI] [PubMed] [Google Scholar]
- [153].Song J, Sheng J, Lei J, Gan W, Yang Y, Mitochondrial targeted antioxidant SKQ1 ameliorates acute kidney injury by inhibiting Ferroptosis, Oxidative Med. Cell. Longev 2022 (2022) 2223957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Khan MA, Nag P, Grivei A, Giuliani KTK, Wang X, Diwan V, Hoy W, Healy H, Gobe G, Kassianos AJ, Adenine overload induces ferroptosis in human primary proximal tubular epithelial cells, Cell Death Dis. 13 (2022) 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Kudo H, Suzuki S, Watanabe A, Kikuchi H, Sassa S, Sakamoto S, Effects of colloidal iron overload on renal and hepatic siderosis and the femur in male rats, Toxicology 246 (2008) 143–147. [DOI] [PubMed] [Google Scholar]
- [156].Norden AG, Lapsley M, Lee PJ, Pusey CD, Scheinman SJ, Tam FW, Thakker RV, Unwin RJ, Wrong O, Glomerular protein sieving and implications for renal failure in Fanconi syndrome, Kidney Int. 60 (2001) 1885–1892. [DOI] [PubMed] [Google Scholar]
- [157].Prinsen B, Velden M, Kaysen GA, Straver H, Rijn H, Stellaard F, Berger R, Rabelink TJ, Transferrin synthesis is increased in nephrotic patients insufficiently to replace urinary losses, J. Am. Soc. Nephrol.: JASN 12 (2001) 1017–1025. [DOI] [PubMed] [Google Scholar]
- [158].Kozyraki R, Fyfe J, Verroust PJ, Jacobsen C, Dautry-Varsat A, Gburek J, Willnow TE, Christensen EI, Moestrup SK, Megalin-dependent cubilin-mediated endocytosis is a major pathway for the apical uptake of transferrin in polarized epithelia, Proc. Natl. Acad. Sci. USA 98 (2001) 12491–12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Smith CP, Lee WK, Haley M, Poulsen SB, Thevenod F, Fenton RA, Proximal tubule transferrin uptake is modulated by cellular iron and mediated by apical membrane megalin-cubilin complex and transferrin receptor 1, J. Biol. Chem 294 (2019) 7025–7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].van Raaij SEG, Srai SKS, Swinkels DW, van Swelm RPL, Iron uptake by ZIP8 and ZIP14 in human proximal tubular epithelial cells, Biometals 32 (2019) 211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y, Regulators of Iron homeostasis: new players in metabolism, cell death, and disease, Trends Biochem. Sci 41 (2016) 274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Jenkitkasemwong S, Wang CY, Mackenzie B, Knutson MD, Physiologic implications of metal-ion transport by ZIP14 and ZIP8, Biometals 25 (2012) 643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Caruso-Neves C, Kwon SH, Guggino WB, Albumin endocytosis in proximal tubule cells is modulated by angiotensin II through an AT2 receptor-mediated protein kinase B activation, Proc. Natl. Acad. Sci. USA 102 (2005) 17513–17518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Molitoris BA, Sandoval RM, Yadav SPS, Wagner MC, Albumin uptake and processing by the proximal tubule: physiological, pathological, and therapeutic implications, Physiol. Rev 102 (2022) 1625–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Luo M, Sui Y, Tian R, Lu N, Formation of a bovine serum albumin diligand complex with rutin for the suppression of heme toxicity, Biophys. Chem 258 (2020) 106327. [DOI] [PubMed] [Google Scholar]
- [166].Pinsky M, Roy U, Moshe S, Weissman Z, Kornitzer D, Human serum albumin facilitates Heme-Iron utilization by Fungi, mBio 11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Loban A, Kime R, Powers H, Iron-binding antioxidant potential of plasma albumin, Clin. Sci. (Lond.) 93 (1997) 445–451. [DOI] [PubMed] [Google Scholar]
- [168].Soofi A, Li V, Beamish JA, Abdrabh S, Hamad M, Das NK, Shah YM, Dressler GR, Renal specific loss of Ferroportin disrupts iron homeostasis and attenuates recovery from acute kidney injury, Am. J. Physiol. Ren. Physiol 326 (2) (2023) F178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Cheng HT, Yen CJ, Chang CC, Huang KT, Chen KH, Zhang RY, Lee PY, Miaw SC, Huang JW, Chiang CK, Wu KD, Hung KY, Ferritin heavy chain mediates the protective effect of heme oxygenase-1 against oxidative stress, Biochim. Biophys. Acta 2015 (1850) 2506–2517. [DOI] [PubMed] [Google Scholar]
- [170].Hu W, Chen X, Identification of hub ferroptosis-related genes and immune infiltration in lupus nephritis using bioinformatics, Sci. Rep 12 (2022) 18826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Shoeb M, Ansari NH, Srivastava SK, Ramana KV, 4-Hydroxynonenal in the pathogenesis and progression of human diseases, Curr. Med. Chem 21 (2014) 230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, Prokisch H, Trumbach D, Mao G, Qu F, Bayir H, Fullekrug J, Scheel CH, Wurst W, Schick JA, Kagan VE, Angeli JP, Conrad M, ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition, Nat. Chem. Biol 13 (2017) 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Singh BK, Singh S, Systemic lupus erythematosus and infections, Reumatismo 72 (2020) 154–169. [DOI] [PubMed] [Google Scholar]
- [174].Trevelin SC, Shah AM, Lombardi G, Beyond bacterial killing: NADPH oxidase 2 is an immunomodulator, Immunol. Lett 221 (2020) 39–48. [DOI] [PubMed] [Google Scholar]
- [175].Renassia C, Louis S, Cuvellier S, Boussetta N, Deschemin JC, Borderie D, Bailly K, Poupon J, Dang PM, El-Benna J, Manceau S, Lefrere F, Vaulont S, Peyssonnaux C, Neutrophils from hereditary hemochromatosis patients are protected from iron excess and are primed, Blood Adv. 4 (2020) 3853–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Dar HH, Mikulska-Ruminska K, Tyurina YY, Luci DK, Yasgar A, Samovich SN, Kapralov AA, Souryavong AB, Tyurin VA, Amoscato AA, Epperly MW, Shurin GV, Standley M, Holman TR, St Croix CM, Watkins SC, VanDemark AP, Rana S, Zakharov AV, Simeonov A, Marugan J, Mallampalli RK, Wenzel SE, Greenberger JS, Rai G, Bayir H, Bahar I, Kagan VE, Discovering selective antiferroptotic inhibitors of the 15LOX/PEBP1 complex noninterfering with biosynthesis of lipid mediators, in: Proceedings of the National Academy of Sciences of the United States of America vol. 120, 2023. e2218896120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.