

Abstract
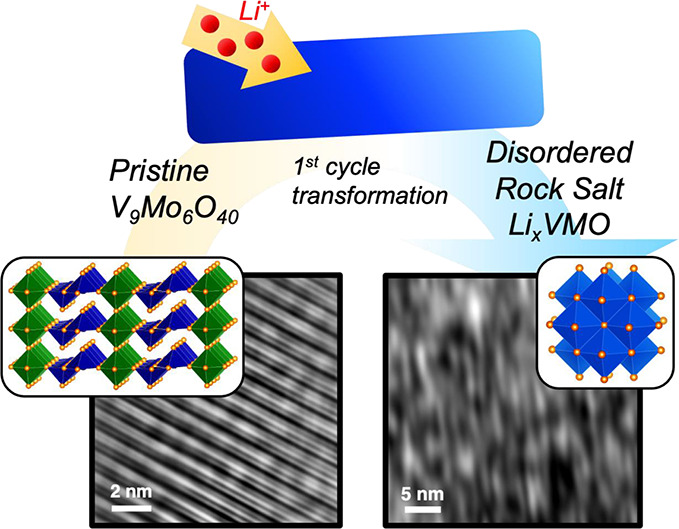
Electrochemically-formed disordered rock salt compounds are an emerging class of Li-ion electrode materials for fast-charging energy storage. However, the specific factors that govern the formation process and the resulting charge storage performance are not well understood. Here, we characterize the transformation mechanism and charge storage properties of an electrochemically-formed disordered rock salt from V9Mo6O40 (VMO). The crystal structure of VMO has similar motifs to that of α-V2O5, a well-studied analogue, but VMO has less mechanical flexibility due to additional corner-sharing octahedra in its structure. As a result, VMO undergoes a single-step transformation pathway, which we characterize through operando X-ray diffraction, and forms an unusual highly distorted lamellar microstructure, as we show with high-resolution transmission electron microscopy. The resulting LixVMO material shows fast charging and other electrochemical characteristics and performance typical of many nanomaterials, even though the material is composed of relatively large particles.
Introduction
Electrochemical energy storage devices with both high energy density and high power density are essential for a number of important technologies, including electric vehicles and grid-scale storage.1,2 While next-generation energy storage chemistries show great promise, traditional insertion-based Li-ion batteries currently show the best balance of capacity, rate capability, and stability with extended cycling.3 Despite these advantages, Li-ion batteries still have significant rate limitations, especially with the high mass loadings used in commercial cells.4,5 During charging, stored charge in the device is balanced by solid-state redox reactions between a host material and a Li+ electrolyte, and achieving full capacity requires transport of both Li+ and electrons throughout the full volume of all grains of host material in the electrode. Typically, this is difficult to achieve at high cycling rates since the diffusion of Li+ in the solid state is several orders of magnitude slower than that in the liquid electrolyte, even in the best-performing host materials.6 As such, a fundamental understanding of how the crystal structure and particle morphology influence both ionic and electronic transport is necessary in the search for ever better electrode materials.7,8
One class of compounds of particular interest as electrodes in Li-ion batteries is the rock-salt-derived family of materials. The base rock salt structure, which comprises a densely packed edge-sharing network of cations and anions extending in three-dimensions, serves as a basis for a variety of relevant materials, including several conversion-type anodes,9−12 the layered Ni-based and disordered rock salt cathodes,13−19 and spinel LiMn2O420 and Li4Ti5O12.21 Accordingly, extensive experimental and theoretical work has focused on understanding the details of charge storage in this family. While the dense bonding environment in the rock salt crystal structure typically leads to high activation energies for Li+ hopping and thus poor ionic diffusion, an appropriate distribution of weakly repulsive cation species (typically Li+) can lower the activation energies dramatically.15,22,23 For example, in the layered Ni-based cathodes, cations order into alternating slabs of smaller (Li+) and larger (Ni3+, Co3+, Mn3+, etc.) cations, and this well-defined ordering is essential for good ionic transport throughout the structure.22 Alternatively, in disordered rock salt structures, a large amount of weakly repulsive Li+ cations can still provide sufficient ionic diffusion, provided that they form a percolating network of hopping pathways in the crystal structure.15,23
Rock salt structures can also be formed electrochemically during initial ion insertion and often remain electrochemically active in subsequent cycles. Most famously, this process occurs for V2O5, which undergoes a series of reversible phase transitions at moderate Li+ contents (0 < x < 1.5), and then transforms irreversibly to a disordered-rock-salt-type ω-LixV2O5 phase at deep lithiation (2 < x < 3).24,25 The ω-LixV2O5 phase has been extensively studied as a potential cathode material cycled between 2.0–4.0 V vs Li/Li+ (0 < x < 2) due to its excellent rate capability when nanostructured, especially in composite structures with conductive carbons.26−29 Recently, it was observed that ω-LixV2O5 could be lithiated significantly further, and the resulting disordered rock salt Li3+xV2O5 showed exceptional performance in a voltage region ideal for a fast-charging anode material (0.01–2.0 V vs Li/Li+).30,31 Importantly, these results firmly establish the potential for the application of electrochemically-formed disordered rock salt compounds in commercial cells.
The electrochemical formation of disordered rock salt structures is not unique to V2O5. Previous work has shown that this process occurs upon Li+ insertion in Li2MoO4,32 Li2W2O7,33 and TiO2.34,35 TiO2 also undergoes a similar transformation on Na+ insertion.36,37 Most recently, it was demonstrated that amorphous films of Nb oxide crystallize into a rock salt phase when cycled with Li+; this work stands in contrast to previous cases where crystalline precursor materials were used.35 Broadly, these examples all utilize deep lithiation and produce fairly disordered materials since they are formed at room temperature. Beyond these trends, however, relatively little is known about this wide-ranging class of materials since they have only recently gained greater attention. In particular, there is a clear need for design principles regarding the role of precursor structure and composition in governing the transformation process, the structure of the resulting disordered rock salt material, and its electrochemical performance.
In this work, we thus characterize the electrochemical formation of a new disordered rock salt phase formed from the crystalline compound V9Mo6O40 (VMO). VMO was first reported in 1982 and has received relatively little attention aside from basic characterization of its charge storage performance.38−40 While a range of vanadium molybdenum oxides with lower Mo content form solid-solutions in the α-V2O5 crystal structure, which consists of layers of edge-sharing pyramids held together by van der Waals forces, VMO and the related phase V2MoO841 make the lowest Mo content structures that are not isostructural with α-V2O5. The crystal structure of VMO possesses the same edge-sharing pyramids that are seen in α-V2O5, but instead of extending infinitely in two-dimensions, they alternate with planes of corner-sharing octahedra.39 Importantly, this change imparts rigidity into the crystal structure and makes VMO a three-dimensionally connected tunnel structure instead of a layered van der Waals compound.
Here, we study the Li+ insertion properties of micron-scale particles of VMO. Using operando synchrotron X-ray diffraction (SXRD), we demonstrate that VMO transforms directly into a disordered rock salt structure upon lithiation, in contrast to the multiphase pathway seen for α-V2O5. Subsequent cycling in the resulting disordered rock salt LixVMO shows good rate capability and cycling longevity, with clear increases in the diffraction intensity upon insertion, followed by a decrease in the diffraction intensity and formation of a new disordered diffraction signal during deinsertion of Li+. High-resolution transmission electron microscopy (TEM) shows that the disordered rock salt LixVMO has an unusual microstructure consisting of a collection of highly distorted, loosely coherent atomic planes. These data suggest that the increased rigidity in the pristine VMO crystal structure relative to α-V2O5 dramatically changes the formation pathway and microstructure of the resulting disordered rock salt material and more broadly highlights the role of precursor structure and composition in this family of compounds.
Experimental Section
Material Preparation
Micron-scale bulk V9Mo6O40 was prepared through a modified aqueous sol–gel route based on freeze-drying. In a typical synthesis, 200 mg of NH4VO3 (Sigma-Aldrich) and 200 mg of (NH4)6Mo7O24·4H2O (Alfa Aesar) were dissolved with stirring in 5 mL of deionized water in a 20 mL scintillation vial at 85 °C. Once the precursors were fully dissolved, the precursor solution was vitrified by dropwise addition to liquid N2, then lyophilized in a custom-built vacuum chamber on a Schlenk line for 12 to 24 h at pressures <200 mTorr. The resulting dried precursor powder was calcined in a muffle furnace in air at 550 °C (30 min ramp, 1 h. hold). A variety of calcination temperatures were found to result in phase pure V9Mo6O40, and this procedure was chosen since it provides a balance between moderate particle size and good crystallinity. After calcination and prior to electrode fabrication, the as-synthesized VMO products were ground in a mortar and pestle to yield a fine powder.
Structural Characterization
Scanning electron microscopy (SEM) images and energy-dispersive spectroscopy (EDS) spectra were obtained by using a model JEOL JSM-6700F field emission electron microscope. Imaging was conducted with a 5 kV accelerating voltage and 6 mm working distance. EDS spectra were taken with a 15 kV accelerating voltage at a 15 mm working distance. TEM was performed using an FEI Technai G2 T20 high-resolution EM operating at 200 kV. Sample purity was assessed using laboratory X-ray diffraction collected with a PANalytical X’Pert Pro diffractometer operating with Cu Kα (λ = 1.5418 Å) using a 0.05° step size, an accelerating voltage of 45 kV, and a current of 40 mA. X-ray photoelectron spectroscopy (XPS) analysis was performed by using a Kratos Axis Ultra DLD with a monochromatic Al Kα radiation source. A charge neutralizer filament was used to control charging of the sample, a 20 eV pass energy was used with a 0.1 eV step size, and scans were calibrated using the C 1s peak shifted to 284.8 eV. The integrated areas of the XPS peaks and atomic ratios were found using CasaXPS software. The atomic sensitivity factors used were from the Kratos library within Casa software. All samples were loaded into the instrument without exposure to air. For ex situ characterization of materials after cycling, including SEM, TEM, and XPS, electrodes were cycled in coin cells, which were subsequently opened inside of a glovebox. The electrodes were then washed multiple times in dimethyl carbonate (DMC) and dried under vacuum before sample preparation for the respective techniques.
Electrochemical Testing
VMO powders were assembled into composite slurry electrodes for electrochemical testing. Electrodes had an overall composition of 80% active material, 5% multiwalled carbon nanotubes, 5% vapor-grown carbon fibers, and 10% poly(vinylidene fluoride) (PVDF) binder. Prior to slurry preparation, the dry slurry components and current collector were heated at 100 °C under vacuum overnight. The active material was ground with the carbons in a mortar and pestle dry, then a PVDF binder solution (2.5 wt % in N-methyl pyrrolidone) was added and mixed to produce a homogeneous paste that was cast onto a carbon-coated aluminum current collector with a doctor blade. The electrodes were dried under vacuum at 100 °C overnight. Electrodes with 0.7 mm diameter were punched out for electrochemical testing with mass loadings of active material of 1–1.5 mg/cm2. These moderate mass loadings were chosen to study the intrinsic performance of the active material as they allow for reproducible electrode performance with minimal optimization of the slurry. High mass loadings, such as those used in most practical devices, lead to rate limitations from electrode composition and distribution of active materials that require extensive optimization to overcome.
All electrochemical measurements were conducted in stainless-steel 2032 coin cells (MTI) with a stainless-steel conical spring, a 0.5 mm stainless-steel spacer for the anode, a 0.5 mm aluminum spacer for the cathode, and a glass microfiber separator (Whatman). The electrolyte was 1 M LiPF6 in 1:1 ethylene carbonate (EC): DMC with ∼50 μL of the electrolyte per cell. All cycling experiments were performed between 1.5 and 3.5 or 4.0 V vs Li/Li+ on a VMP3 potentiostat/galvanostat (Bio-Logic). For testing purposes, VMO electrodes were studied in a half cell configuration versus a Li metal anode. When calculating C-rates, the theoretical capacity used was 240 mAh/g, based on the 1 e– Li+ insertion reaction into V9Mo6O40 (1C = 240 mA/g, 5C = 1.20 A/g, etc.). Before all cycling experiments shown, including the long-term cycling and cyclic voltammetry, VMO electrodes went through galvanostatic rate testing. The first cycle, which includes conversion of pristine VMO to the disordered rock salt phase, was performed at 1C.
Operando Synchrotron X-ray Diffraction
All operando SXRD measurements were performed at beamline 11-ID-C at the Advanced Photon Source at Argonne National Laboratory at 105.7 keV in transmission geometry with an area detector. For this experiment, freestanding pellet electrodes of VMO were made with 6:1:1:2 mass ratio of VMO: vapor-grown carbon fibers: multiwalled carbon nanotubes: PTFE binder. These electrodes were loaded into AMPIX cells in an argon glovebox with Li metal counter electrodes, glass fiber separators, and electrolyte (1 M LiPF6 in 1:1 EC:DMC).42 All two-dimensional SXRD data were calibrated with a LaB6 external standard and integrated into one-dimensional diffraction patterns using GSAS-II.43 Background subtraction was performed on the diffraction data to remove scattering from the electrolyte. Crystal structures were visualized with the VESTA software package.44
Results and Discussion
Synthesis and Structure of V9Mo6O40
The chemical formula and crystal structure of V9Mo6O40 (VMO) were originally identified in the 1980s using X-ray diffraction and confirmed in a subsequent study with neutron powder diffraction.38,39 The crystal structure consists of alternating planes of edge-sharing pyramids and corner-sharing octahedra along the a-axis, and these planes extend infinitely in the b-axis direction (Figure 1A, middle).39 The corner-sharing octahedra also connect parallel to the c-axis, while the pyramids repeat without connecting in that direction. V9Mo6O40 has a composition near the middle of the oxidized V–Mo oxides, of which the binary oxides α-V2O5 and α-MoO3 are the end members (Figure 1A). Both α-V2O5 and α-MoO3 are layered van der Waals compounds, although the different size, charge, and preferred coordination number of V5+ and Mo6+ lead them to have different arrangements of atoms within that structure type. Whereas α-V2O5 layers are made of edge-sharing square pyramid dimers that corner share with alternating orientation along the layer direction,45 α-MoO3 consists of a bilayer network of edge-sharing octahedra, with bilayers separated by the van der Waals gap.46,47 The α-V2O5 phase can accommodate the addition of Mo in a solid-solution manner over an extended range; V2−xMoxO5 is stable (0 ≤ x ≤ 0.6) with only minor changes in the structure.48 For example, in V2–xMoxO5, Mo5+ can exist to better fit within the structure with a more stable coordination environment similar to that of V5+. However, by x = 1, the greater stability of Mo6+ favors the inclusion of corner-sharing octahedra, in which each transition metal (TM) cation is coordinated by six oxygen atoms, with decreased electrostatic repulsion between sites compared to the edge-sharing pyramid dimers in α-V2O5. This transition leads to the middle region of the composition range, which includes V9Mo6O40 (VMO), the focus of this study, and the isostructural V2MoO8. Importantly, the three-dimensional connectivity of the planes of corner-sharing octahedra makes VMO a tunnel structure without a van der Waals gap, although the structure still retains some layered character due to the presence of edge-sharing pyramid dimer units. Additional discussion about the chemical formula and charge neutrality in the VMO crystal structure can be found in the Supporting Information.
Figure 1.

(A) Structure space of the fully oxidized V–Mo oxides, with the crystal structures of V9Mo6O40 (middle) and the two end members α-V2O5 (left) and α-MoO3 (right) highlighted. The dashed boxes indicate edge-sharing pyramid dimers, a structural feature shared by α-V2O5 and V9Mo6O40. (B) Powder X-ray diffraction pattern and (C) scanning electron micrograph of the bulk V9Mo6O40 in this study.
We synthesized micrometer-scale powders of VMO through a modified sol–gel-type method based on freeze-drying. In this process, aqueous solutions of NH4VO3 and (NH4)6Mo7O24 precursors were vitrified in liquid N2 and dried through sublimation on a Schlenk line. The resulting dried V–Mo precursor powder was then calcined in air at 550 °C to crystallize VMO. We note that freeze-drying is a well-established route to powders with high surface area and porosity, and, here, helps improve purity by preventing the precipitation of NH4VO3, which often occurs with traditional sol–gel synthesis.49−52 Powder X-ray diffraction on as-synthesized powders shows formation of the VMO phase with greater than 99% purity, confirming the benefits of our approach (Figure 1B). Rietveld refinement of this laboratory diffraction data, as well as SXRD data of pristine VMO pellet electrodes before cycling, confirms a close match with the previously reported lattice parameters (Figures S1, S2 and Tables S1, S2). SEM characterization of the VMO shows anisotropic particles with a distribution of sizes and shapes (Figure 1C). Most commonly observed is a bar-shape morphology, where particles are about 2–10 μm long, 1–2 μm wide, and only about 100–500 nm deep, which clearly indicates a preferred axis of growth for VMO crystals. These bar-like particles often possess L-shaped bends, which presumably form where two sections of crystal extend in different directions within the same particle. Additional SEM characterization of the pristine VMO particle morphology is available in Figure S3 of the Supporting Information.
Electrochemistry of V9Mo6O40
To evaluate the electrochemical behavior of VMO when cycled with Li+, these VMO powders were assembled into composite electrodes with conductive carbon and binder and tested in a half-cell configuration in coin cells. For galvanostatic cycling experiments, we calculated a theoretical capacity of 240 mAh/g for VMO, which assumes the insertion of 1 Li+ per TM into the VMO structure. All C-rates used were based on this capacity (1C = 0.24 A/g, 5C = 1.2 A/g, etc.).
Initial charge storage in VMO occurs through a sloping profile with a few small features until about 0.5 Li+/TM is inserted, at which point, a large plateau occurs (Figure 2A). This plateau encompasses a considerable range of stoichiometry (about 0.9 Li+/TM) and corresponds to the transformation to a disordered rock salt phase, as we show later. After the plateau is complete, little additional charge storage is observed until the voltage cutoff at 1.5 V. On delithiation, and during subsequent cycles, charge storage occurs through a highly sloped profile without recurrence of the plateau seen during the first lithiation. During the first delithiation, more than 1 Li+/TM is removed (255 mAh/g), but this capacity is not fully sustained in subsequent cycles, during which a moderate capacity of about 0.75–0.85 Li+/TM, or 180–200 mAh/g, is reversibly achieved.
Figure 2.

Electrochemical performance of LixVMO. (A) Galvanostatic charge storage profiles of VMO electrodes for the first three cycles. During the first lithiation, the material undergoes an irreversible transformation to a disordered rock salt phase, as shown by the large plateau. (B) Galvanostatic cycling performance of VMO electrodes at different rates. (C) Long-term galvanostatic cycling at 10C. (D) Cyclic voltammetry of VMO electrodes at different rates. (E) Power-law (log–log) plot of the variation of the peak current of the main redox peak as a function of the scan rate. The high value of the slope, approaching 1, suggests a pseudocapacitive charge storage mechanism.
Galvanostatic rate testing was performed to evaluate the rate capability of the VMO electrodes within two different voltage windows, 1.5–4.0 V vs Li/Li+ and 1.5–3.5 V vs Li/Li+ (Figure 2B). LixVMO shows good retention of its capacity at high rates despite the relatively large size of the particles tested. For the 1.5–4.0 V window, nearly 150 mAh/g (0.63 Li+/TM), or about 75% of the slow rate capacity, is stored at 5C, while at 20C, 95 mAh/g (0.40 Li+/TM), or about 48% of slow rate capacity, is accessed. At 60C, which corresponds to less than a 1 min charge or discharge, a respectable capacity of 48 mAh/g (0.2 Li+/TM) or about 25% of the slow rate capacity is obtained. At these higher rates, the sloping GV profile is maintained (Figure S4). When the narrower window is used (1.5 vs 3.5 V vs Li/Li+), similar performance is observed at slow and moderate rates, with a greater difference at the highest rates tested. For example, at 5C and 20C, 140 mAh/g and 77 mAh/g are stored, but at 60C, the narrower voltage window leads to greater losses from polarization, and only about 14 mAh/g is retained. Overall, this charge storage performance demonstrates fast kinetics in relatively large particles of VMO, and even better performance can likely be achieved with optimization of particle shape and size, as has been shown extensively with V2O5.28
To assess the reversibility and stability of charge storage processes in LixVMO electrodes, long-term galvanostatic cycling was performed over 500 cycles at 10C after initial rate testing (Figure 2C). In the narrower voltage window up to 3.5 V vs Li/Li+, LixVMO shows good retention of its capacity, with 77% of its initial capacity maintained after 500 cycles. In many bulk battery materials, Li+ insertion is accompanied by first-order phase transitions, which decrease cycle life considerably due to repeated discontinuous changes in volume.53 LixVMO, on the other hand, shows a sloping galvanostatic charge storage profile, which suggests the absence of a first-order phase transition, and this is likely the reason for the comparatively better capacity retention. When the voltage window is expanded up to 4.0 V vs Li/Li+, the improved cycling kinetics are accompanied by a trade-off in stability, since in this window LixVMO shows a lower capacity retention of only 61% after 500 cycles. Notably, the galvanostatic profiles remain sloping and show no change in shape over the extended cycling, regardless of the voltage window (Figure S5).
Cyclic voltammetry, collected at sweep rates from 0.1 to 1.0 mV/s, was also performed on LixVMO electrodes after they had undergone galvanostatic rate testing (Figure 2D). The cyclic voltammogram for LixVMO shows a single broad redox peak centered around 2.2 V for the anodic sweep and 2.4 V for the cathodic sweep. Additionally, an even broader shoulder feature is seen for the higher voltage region (>2.5 V). The broad peak signatures of redox seen here for LixVMO are unlike those for most bulk battery materials, which typically show sharp peaks in cyclic voltammetry. This behavior can be attributed to the disordered structure of the electrochemically-formed rock salt LixVMO, which we characterize later. Notably, even though both the V and Mo are undergoing redox, there is only one broad redox peak in the CV; this is commonly observed for disordered rock salt structure materials and may arise because all TM sites, on average, have the same local bonding environment.15 To further characterize the charge storage behavior of LixVMO, we analyzed the cyclic voltammograms using the power-law dependence of each peak current (ipeak) as a function of the sweep rate (ν), which is expected to vary according to the equation ipeak = aνb where a and b are constants. The value of b, determined by fitting the slope of log(ipeak) versus log(ν), indicates whether the reaction is diffusion-limited (b = 0.5) or capacitive (b = 1.0) (Figure 2E).2,4,54 Intermediate values correspond to diffusion behavior that is between semi-infinite diffusion and purely capacitive behavior. This analysis has been used frequently in the literature to distinguish diffusion-controlled or “battery-like” and pseudocapacitive redox reactions, which appear capacitive due to fast Li+ diffusion and the absence of large first-order phase transitions.4,55−58 The values of b at the main redox peaks of LixVMO are 0.94 and 0.93 for the anodic and cathodic sweep, respectively, which suggests a highly capacitive charge storage process, despite the relatively large particle sizes studied here. This result is likely due to the absence of a first-order phase transition during Li+ insertion in LixVMO, along with fast Li+ diffusion kinetics.
Electrochemical Formation of Disordered Rock Salt ω-LixVMO
To characterize the transformation of VMO during its first lithiation, we performed operando SXRD at beamline 11-ID-C at the Advanced Photon Source. In this experiment, the active material is assembled with conductive carbon and binder into freestanding pellets, which are cycled in AMPIX cells at a C/5 rate.42 The operando SXRD data from Q = 1 to 5 Å–1 are shown in Figure 3A along with the galvanostatic cycling profile collected during the experiment, while selected diffraction patterns are highlighted in Figure 3B. The pristine VMO initially shows shifts of the diffraction peaks to lower Q during the first sloping region of the cycling profile, which suggest some minor expansion of the lattice upon lithiation. As the plateau begins, shifting stops, and instead a steady decrease in the intensity of the initial VMO phase is observed as its transforms into a different structure. Two broad peaks emerge at 3.1 and 4.3 Å–1 by the middle of the plateau and continue to increase in intensity until the end of the cycle, by which point, the initial VMO phase has completely disappeared. These new peaks are characteristic of a rock salt phase, which we denote here as ω-LixVMO, after the convention used for V2O5. As discussed previously, rock salt phases consist of densely packed cations and anions that form a network of edge-sharing octahedra in three dimensions. In ω-LixVMO, the O2– ions make up the anion sublattice, while the cation sites likely have a disordered mixture of V, Mo, and Li ions. For an ideal rock salt structure, there are equal numbers of cations and anions, but during electrochemical cycling of ω-LixVMO, the amount of Li and thus the total number of cations are dependent on the state-of-charge. In the fully lithiated state, ω-LixVMO appears to have a nearly equal number of cations and anions: VMO electrodes showed charge storage corresponding to the insertion of 1.5 Li+ during the first cycle transformation, which would give a nominal formula of Li22V9Mo6O40 (37 cations and 40 anions) for the fully lithiated ω-LixVMO. A schematic of the transformation from pristine VMO to the ω-LixVMO phase is shown in Figure 3C. After the transformation, the initial pristine VMO structure is not recovered upon delithiation, confirming that the process is irreversible. During delithiation, Li+ is removed from ω-LixVMO, which leads to fewer cations within the structure and likely disrupts the stability of the rock salt phase. This effect is reflected in the operando SXRD data, which show a considerable decrease in the intensity of the rock salt phase peaks during delithiation. We will examine this process more closely in a subsequent section. As we discussed previously, α-V2O5, which shares its edge-sharing pyramid structural motif with VMO, also transforms into a disordered rock salt phase upon deep lithiation. However, prior to becoming the rock salt phase, α-V2O5 goes through three additional discrete structural phase transformations involving puckering of the layers to better coordinate the inserted Li+ (Figure S6).25,30 VMO, on the other hand, shows minimal change in structure before it directly transitions to the rock salt phase. We hypothesize that this change in the pathway originates from the additional rigidity imparted by the planes of corner-sharing octahedra in the VMO crystal structure. Whereas the edge-sharing pyramid dimers can distort relatively freely in V2O5, their connection to the octahedra in VMO pins their movement and prevents significant distortion until the full-scale transformation of the crystal structure to the rock salt phase.
Figure 3.

Transformation of VMO to a disordered rock salt phase during its first cycle. (A) Operando SXRD data on VMO plotted alongside the galvanostatic profile during the first cycle. The pristine VMO transforms during the first lithiation into a disordered rock salt phase. Asterisks indicate peaks from cell components. (B) Selected patterns from the data in (A) showing the pristine VMO and the disordered rock salt phase after the first lithiation and delithiation. (C) Schematic of the transformation.
We further characterized the disordered rock salt LixVMO using electron microscopy to analyze how the dramatic change in the atomic-scale crystal structure affects the morphology and microstructure of the material. Particles with the bar-like morphology seen for pristine VMO (Figures 4A and 4B) were readily observed throughout a cycled LixVMO electrode that had transformed to the rock salt phase (Figures 4D, 4E, and S7). Although there was some evidence of cracks forming, these data indicate that the particle morphology is largely preserved through the structural transition during cycling. Additionally, EDS on the cycled electrode confirmed the presence of V and Mo (Figure S8). To characterize the microstructure of disordered rock salt LixVMO, we performed high-resolution transmission electron microscopy (HRTEM) on particles from a VMO electrode cycled through the first delithiation. Unlike the rigid, crystalline planes seen for pristine VMO (Figure 4C), the cycled LixVMO showed a collection of short, distorted planes of atoms that loosely fit together (Figure 4F). This highly distorted “lattice” frequently appeared as nanoscale domains (Figure S9), although it was also observed to extend throughout entire particles (Figure 4E). We confirmed its rock salt atomic-scale crystal structure with selected area electron diffraction (Figure 4F, inset, Figure S10). No evidence of the pristine VMO phase was observed, which confirms the complete conversion of the material, as shown by the operando SXRD data.
Figure 4.

Characterization of the disordered rock salt LixVMO. Electron microscopy of the (A–C) pristine VMO and (D,E) delithiated disordered rock salt LixVMO. Panels (A,D) show SEM images, while panels (B,E) and (C,F) show low- and high-resolution TEM images, respectively. The particle morphology is retained after transformation, but the cycled LixVMO shows an unusual microstructure consisting of highly distorted planes of atoms. The inset of (C) shows the fast Fourier transform of the pristine VMO, while the inset of (F) shows the selected area electron diffraction pattern confirming the rock salt crystal structure for the cycled material.
To our knowledge, the unusual distorted microstructure of rock salt VMO is unique among the family of electrochemically formed rock salt materials and may result from its more direct formation pathway. We hypothesize, for example, that the structural rigidity in the pristine VMO crystal structure likely leads to its direct transformation to the rock salt structure while simultaneously preventing full randomization of the cations in the newly formed rock salt phase.
Since Li has much lower electron scattering contrast than V or Mo, the microstructure shown here may reflect segregated domains of Li-rich and transition-metal-rich regions, despite an overall rock salt structure, leading to the lamellar morphology of the final lattice. It may also reflect strain-induced distortion of the rock salt structure to form very small crystalline domains, or some combination of the two. While the disordered nature of the rock salt VMO prevented us from obtaining sufficient resolution to see individual planes of atoms in the material, based on the size of individual lamellae (<1 nm), it is clear that the length scale for domain formation is very small along at least one axis. Because of this nonuniform atom distribution, the resulting material effectively has nanoscale crystalline domains within a dense micron-scale particle. The small domain size is likely responsible for the sloping galvanostatic profile and highly capacitive electrochemical kinetics of the ω-LixVMO that are shown in Figure 2. More importantly, previous work has highlighted the need of a percolating network of Li within disordered rock salt structures for long-range ion diffusion, and any formation of Li-rich domains may enhance diffusion in ω-LixVMO and contribute to its high rate capability.15 Notably, we found that ω-LixV2O5 cycled in the same voltage window showed slower charge storage kinetics (Figure S6), and several previous reports indicate much worse cycling stability of ω-LixV2O5 compared to that of ω-LixVMO.27,59 As such, the microstructure shown here may enhance both the rate capability and the cycling stability of the ω-LixVMO material.
Charge Storage Mechanism in Disordered Rock Salt LixVMO
To investigate the mechanism for charge storage in the disordered rock salt LixVMO, we performed operando SXRD during the second lithiation–delithiation cycle after the material had already transformed into the disordered rock salt phase. The operando SXRD data are shown in Figure 5A for the (200) and (220) regions, the two most intense reflections of the phase, along with the galvanostatic cycling profile. As the lithiation proceeds, no shifting of either peak is observed, but a clear increase in the intensity of the phase is observed. During delithiation, the intensity decreases back to a similar level to that at the beginning of the cycle. Selected diffraction patterns at the beginning, middle, and end of the lithiation-delithiation cycle are highlighted in Figure 5B. The increase in intensity of the rock salt LixVMO as it is lithiated is clearly shown. The individual line plots in the delithiated state also help explain the decreasing intensity seen during delithiation: in both delithiated patterns, a very broad scattering feature from Q = 1.5 to 2.5 Å–1 is observed in the delithiated state, which then disappears during lithiation. This result indicates that removal of Li+ from the rock salt VMO produces an even more disordered phase, which makes sense based on the need for near-equal numbers of cations and anions in a rock salt crystal structure. As LixVMO is delithiated, about half of the originally inserted lithium is removed from the structure, so there would then be considerably more anions than cations. Accordingly, upon delithiation, LixVMO forms a distorted structure to better accommodate this imbalance. The transition between these two phases and the instability of the delithiated phase likely lead to degradation of the material and capacity loss during extended cycling; because more delithiation occurs when cycling VMO with the higher 4.0 V voltage cutoff, cycling under these conditions increases the amount of degradation per cycle.
Figure 5.

Structural change during cycling of disordered rock salt LixVMO. (A) Operando SXRD data collected on the rock salt phase LixVMO plotted alongside the galvanostatic profile for the second cycle. For clarity, data are shown only for the two most intense lattice planes of the phase. (B) Selected patterns from the data in (A). When delithiated, the diffraction peaks from the rock salt phase decrease in intensity, and a broad feature appears between Q = 1.8 and 2.5 Å–1. Asterisks indicate peaks from components of the cell.
While the low intensity of the diffraction from the disordered delithiated phase precludes identification of the phase by diffraction alone, similar behavior has been shown for the rock salt LixV2O5. A pair distribution function analysis by Christensen et al. identified the delithiated phase as a highly disordered analogue of β-LixV2O5, which consists of 2 × 2 chains of edge-sharing distorted octahedra that are connected by corner sharing with edge-sharing pyramid dimers.60 In LixV2O5, the distorted phase exists over a narrow compositional range (0.22 < x < 0.37) and is favored electrostatically at low Li+ content because the rock salt structure is too densely packed for the highly charged TM cations in the delithiated state (V5+ in the case of V2O5). Importantly, the β-LixV2O5 showed a short crystalline coherence length of only 10–15 Å, which is consistent with the very broad nature of the peak in diffraction seen here and the feature size in our HRTEM data. While we cannot verify the identity of the phase for LixVMO, several parallels in behavior are shown for LixV2O5, including the reversible change in intensity of the rock salt phase during cycling, which suggest that the atomic-scale processes for Li+ insertion and deinsertion are similar for these two disordered rock salt phases.
To characterize the changes in the oxidation states of V and Mo during cycling, we carried out ex situ XPS (Figures 6 and S11). For these experiments, VMO electrodes were cycled to the desired point, removed from the cell, washed, dried, and then transferred to the instrument for data collection. The removal and cleaning process was performed in an Ar glovebox, and the transfer to the instrument was conducted without exposure to air to prevent oxidation or other degradation of the cycled electrode. For the pristine VMO, only V5+ and Mo6+ are evident. At the end of the first lithiation, when the disordered rock salt VMO has formed and is fully lithiated, both V and Mo exist in partially reduced states, with large amounts of V4+ and Mo5+ and some doubly reduced V3+ and Mo4+.61,62 At the first delithiation, when the rock salt phase is delithiated, the doubly reduced species disappear, but significant amounts of V4+ and Mo5+ remain alongside fully oxidized V5+ and Mo6+. The presence of reduced species after delithiation suggests that some Li+ remains in the lattice as a structural component of the rock salt phase, as has been seen in other electrochemically-formed rock salt materials.30,35 When the rock salt phase is lithiated, the doubly reduced V3+ and Mo4+ return to a similar, though slightly lesser, extent, confirming the reversibility of cycling both elements in the rock salt structure. The results of the XPS data are quantified in Table 1. While the XPS technique is fully quantitative, only the near-surface region of the sample is probed, so the oxidation states could be more oxidized than those present in the bulk of the material.
Figure 6.
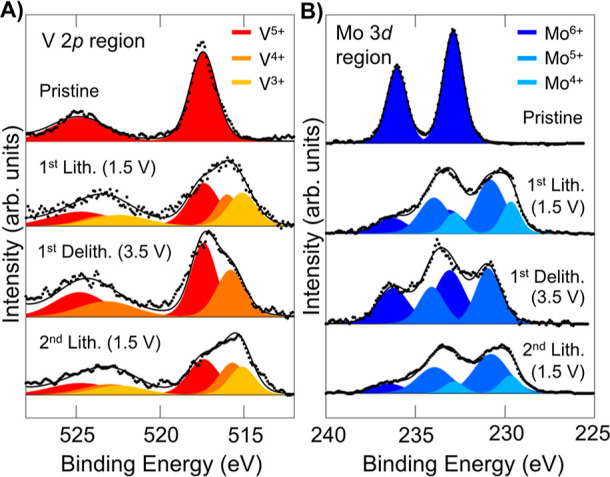
Redox activity of VMO. Ex situ XPS on VMO electrodes cycled to different points highlighting the (A) V 2p region and (B) Mo 3d region. Both elements begin in their fully oxidized state and become reduced during lithiation. The delithiated disordered rock salt shows some remnant reduced species, suggesting that some Li+ remains in the material after the first lithiation.
Table 1. Quantitative Summary of the XPS Data.
| V 2p
region |
Mo
3d region |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| % V5+ | % V4+ | % V3+ | average V+x | expected V+x (electrochem) | % Mo6+ | % Mo5+ | % Mo 44+ | average Mo+x | expected Mo+x (electrochem) | ||
| pristine | 100 | 0 | 0 | 5 | 5 | 100 | 0 | 0 | 6 | 6 | |
| 1st lith. (1.5 V) | 45 | 24 | 31 | 4.1 | 3.7 | 25 | 55 | 21 | 5 | 4.7 | |
| 1st delith. (3.5 V) | 59 | 46 | 0 | 4.6 | 4.7 | 54 | 46 | 0 | 5.5 | 5.7 | |
| 2nd lith. (1.5 V) | 41 | 32 | 27 | 4.1 | 3.8 | 20 | 63 | 16 | 5 | 4.8 | |
Finally, we evaluated the low voltage cycling behavior of the VMO (Figure S12). In recent work, the structural analogue V2O5 was demonstrated to undergo additional lithiation when cycled in a low voltage region (0.05–2.0 V vs Li/Li+) after its transformation to the disordered rock salt structure.30 While VMO does show additional capacity in this region, it appears to be accompanied by a conversion reaction, whereby VMO is reduced to V and/or Mo metal and Li2O.63 This result highlights the challenge of creating disordered rock salt phase materials that can be cycled at very low potentials, as insertion reactions in this regime are always in competition with conversion reactions.64
Conclusions
Using a combination of operando X-ray diffraction, electron microscopy, and XPS, we have characterized the electrochemical formation and reversible cycling of a disordered rock salt phase from a V9Mo6O40 precursor. Unlike the well-known example of ω-LixV2O5, VMO transforms directly into its rock salt structure during the first cycle without forming any intermediate phases. Additionally, we showed an unusual microstructure in rock salt LixVMO that consists of highly distorted nanoscale lamellae arranged in a loosely coherent lattice; this unique structure likely facilitates the improved kinetics of micrometer-scale VMO and appears to enhance cycling stability of ω-LixVMO relative to that of ω-LixV2O5 in a similar voltage range. We hypothesize that these features are due to the increased structural rigidity in the VMO lattice relative to that of α-V2O5. Overall, this work provides insight into the role of precursor structure in electrochemically formed disordered rock salt phases.
Acknowledgments
This work was supported by the US Department of Energy, Office of Basic Energy Sciences. Primary support came from Award DE-SC0014213. D.N.A. and J.N.W. acknowledge support from DE-SC0019381 for operando diffraction work at SSRL. D.D.R. and D.J.P. acknowledge support from a National Science Foundation Graduate Research Fellowship under award DGE-2034835. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. The authors thank Kamila Wiaderek and Andrey for user support. Preliminary operando SXRD studies were conducted at beamline 11-3 of the Stanford Synchrotron Radiation Lightsource (SLAC National Accelerator Laboratory). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-76SF00515. For TEM studies, the authors acknowledge the use of instruments at the Electron Imaging Center for NanoMachines supported by NIH (1S10RR23057, 1S10OD018111, and 1U24GM116792), NSF (DBI-1338135), and CNSI at UCLA.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemmater.4c01732.
Additional characterization of pristine and disordered rock salt LixVMO; Rietveld refinement and SEM images of the pristine material; galvanostatic profiles from rate and long-term cycling; SEM, TEM, electron diffraction, EDS, and XPS of the disordered rock salt VMO; comparison of VMO and V2O5; and low voltage cycling behavior of VMO (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Gür T. M. Review of electrical energy storage technologies, materials and systems: challenges and prospects for large-scale grid storage. Energy Environ. Sci. 2018, 11, 2696–2767. 10.1039/C8EE01419A. [DOI] [Google Scholar]
- Simon P.; Gogotsi Y.; Dunn B. Where do batteries end and supercapacitors begin?. Science 2014, 343, 1210–1211. 10.1126/science.1249625. [DOI] [PubMed] [Google Scholar]
- Frith J. T.; Lacey M. J.; Ulissi U. A non-academic perspective on the future of lithium-based batteries. Nat. Commun. 2023, 14, 420. 10.1038/s41467-023-35933-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C.; Ashby D. S.; Butts D. M.; DeBlock R. H.; Wei Q.; Lau J.; Dunn B. Achieving high energy density and high power density with pseudocapacitive materials. Nat. Rev. Mater. 2020, 5, 5–19. 10.1038/s41578-019-0142-z. [DOI] [Google Scholar]
- Li M.; Lu J.; Chen Z.; Amine K. 30 years of lithium-ion batteries. Adv. Mater. 2018, 30, 1800561. 10.1002/adma.201800561. [DOI] [PubMed] [Google Scholar]
- Griffith K. J.; Wiaderek K. M.; Cibin G.; Marbella L. E.; Grey C. P. Niobium tungsten oxides for high-rate lithium-ion energy storage. Nature 2018, 559, 556–563. 10.1038/s41586-018-0347-0. [DOI] [PubMed] [Google Scholar]
- Griffith K. J.; Seymour I. D.; Hope M. A.; Butala M. M.; Lamontagne L. K.; Preefer M. B.; Koçer C. P.; Henkelman G.; Morris A. J.; Cliffe M. J.; Dutton S. E.; Grey C. P. Ionic and Electronic Conduction in TiNb2O7. J. Am. Chem. Soc. 2019, 141, 16706–16725. 10.1021/jacs.9b06669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff K. E.; Robertson D. D.; Preefer M. B.; Teicher S. M. L.; Bienz J.; Kautzsch L.; Mates T. E.; Cooley J. A.; Tolbert S. H.; Seshadri R. High-Capacity Li+ Storage through Multielectron Redox in the Fast-Charging Wadsley-Roth Phase (W0.2V0.8)3O7. Chem. Mater. 2020, 32, 9415–9424. 10.1021/acs.chemmater.0c03496. [DOI] [Google Scholar]
- Poizot P.; Laruelle S.; Grugeon S.; Dupont L.; Tarascon J.-M. Nano-Sized Transition-Metal Oxides as Negative-Electrode Materials for Lithium-Ion Batteries. Nature 2000, 407, 496–499. 10.1038/35035045. [DOI] [PubMed] [Google Scholar]
- Liu H.; Wang G.; Liu J.; Qiao S.; Ahn H. Highly Ordered Mesoporous NiO Anode Material for Lithium Ion Batteries with an Excellent Electrochemical Performance. J. Mater. Chem. 2011, 21, 3046. 10.1039/c0jm03132a. [DOI] [Google Scholar]
- Sun Y.; Hu X.; Luo W.; Huang Y. Ultrathin CoO/Graphene Hybrid Nanosheets: A Highly Stable Anode Material for Lithium-Ion Batteries. J. Phys. Chem. C 2012, 116, 20794–20799. 10.1021/jp3070147. [DOI] [Google Scholar]
- Sarkar A.; Velasco L.; Wang D.; Wang Q.; Talasila G.; de Biasi L.; Kübel C.; Brezesinski T.; Bhattacharya S. S.; Hahn H.; Breitung B. High Entropy Oxides for Reversible Energy Storage. Nat. Commun. 2018, 9, 3400. 10.1038/s41467-018-05774-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabuuchi N.; Takeuchi M.; Nakayama M.; Shiiba H.; Ogawa M.; Nakayama K.; Ohta T.; Endo D.; Ozaki T.; Inamasu T.; Sato K.; Komaba S. High-Capacity Electrode Materials for Rechargeable Lithium Batteries: Li3NbO4-Based System with Cation-Disordered Rocksalt Structure. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 7650–7655. 10.1073/pnas.1504901112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Kitchaev D. A.; Kwon D. H.; Lee C. W.; Papp J. K.; Liu Y. S.; Lun Z.; Clément R. J.; Shi T.; McCloskey B. D.; Guo J.; Balasubramanian M.; Ceder G. Reversible Mn2+/Mn4+ Double Redox in Lithium-Excess Cathode Materials. Nature 2018, 556, 185–190. 10.1038/s41586-018-0015-4. [DOI] [PubMed] [Google Scholar]
- Clément R. J.; Lun Z.; Ceder G. Cation-Disordered Rocksalt Transition Metal Oxides and Oxyfluorides for High Energy Lithium-Ion Cathodes. Energy Environ. Sci. 2020, 13, 345–373. 10.1039/C9EE02803J. [DOI] [Google Scholar]
- Fan Y.; Zhang W.; Zhao Y.; Guo Z.; Cai Q. Fundamental Understanding and Practical Challenges of Lithium-Rich Oxide Cathode Materials: Layered and Disordered-Rocksalt Structure. Energy Storage Mater. 2021, 40, 51–71. 10.1016/j.ensm.2021.05.005. [DOI] [Google Scholar]
- Wu V. C.; Zhong P.; Ong J.; Yoshida E.; Kwon A.; Ceder G.; Clément R. J. The Limited Incorporation and Role of Fluorine in Mn-Rich Disordered Rocksalt Cathodes. ACS Energy Lett. 2024, 9, 3027–3035. 10.1021/acsenergylett.4c01075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunariwal N.; Dogan F.; Roy I.; Kumar K.; Nordlund D.; Ma Q.; Cabana J. Structural and Chemical Evolution of Highly Fluorinated Li-Rich Disordered Rocksalt Oxyfluorides as a Function of Temperature. Adv. Funct. Mater. 2024, 34, 2312010. 10.1002/adfm.202312010. [DOI] [Google Scholar]
- Patil S.; Darbar D.; Self E. C.; Malkowski T.; Wu V. C.; Giovine R.; Szymanski N. J.; McAuliffe R. D.; Jiang B.; Keum J. K.; Koirala K. P.; Ouyang B.; Page K.; Wang C.; Ceder G.; Clément R. J.; Nanda J. Alternate Synthesis Method for High-Performance Manganese Rich Cation Disordered Rocksalt Cathodes. Adv. Energy Mater. 2023, 13 (4), 2203207. 10.1002/aenm.202203207. [DOI] [Google Scholar]
- Tarascon J. M.; Wang E.; Shokoohi F. K.; McKinnon W. R.; Colson S. The spinel phase of LiMn2O4 as a cathode in secondary lithium cells. J. Electrochem. Soc. 1991, 138, 2859–2864. 10.1149/1.2085330. [DOI] [Google Scholar]
- Zhao B.; Ran R.; Liu M.; Shao Z. A Comprehensive Review of Li4Ti5O12-Based Electrodes for Lithium-Ion Batteries: The Latest Advancements and Future Perspectives. Mater. Sci. Eng., R 2015, 98, 1–71. 10.1016/j.mser.2015.10.001. [DOI] [Google Scholar]
- Kang K.; Meng Y. S.; Bréger J.; Grey C. P.; Ceder G. Electrodes with High Power and High Capacity for Rechargeable Lithium Batteries. Science 2006, 311, 977–980. 10.1126/science.1122152. [DOI] [PubMed] [Google Scholar]
- Lee J.; Urban A.; Li X.; Su D.; Hautier G.; Ceder G. Unlocking the Potential of Cation-Disordered Oxides for Rechargeable Lithium Batteries. Science 2014, 343, 519–522. 10.1126/science.1246432. [DOI] [PubMed] [Google Scholar]
- Delmas C.; Brèthes S.; Ménétrier M. ω-LixV2O5– a New Electrode Material for Rechargeable Lithium Batteries. J. Power Sources 1991, 34, 113–118. 10.1016/0378-7753(91)85030-Z. [DOI] [Google Scholar]
- Delmas C.; Cognac-Auradou H.; Cocciantelli J. M.; Menetrier M.; Doumerc J. P. The LixV2O5 System: An Overview of the Structure Modifications Induced by the Lithium Intercalation. Solid State Ionics 1994, 69, 257–264. 10.1016/0167-2738(94)90414-6. [DOI] [Google Scholar]
- Chen Z.; Augustyn V.; Wen J.; Zhang Y.; Shen M.; Dunn B.; Lu Y. High-Performance Supercapacitors Based on Intertwined CNT/V2O5 Nanowire Nanocomposites. Adv. Mater. 2011, 23, 791–795. 10.1002/adma.201003658. [DOI] [PubMed] [Google Scholar]
- Sathiya M.; Prakash A. S.; Ramesha K.; Tarascon J. M.; Shukla A. K. V2O5-Anchored Carbon Nanotubes for Enhanced Electrochemical Energy Storage. J. Am. Chem. Soc. 2011, 133, 16291–16299. 10.1021/ja207285b. [DOI] [PubMed] [Google Scholar]
- Yue Y.; Liang H.; Yue Y.; Liang H. Micro- and Nano-Structured Vanadium Pentoxide (V2O5) for Electrodes of Lithium-Ion Batteries. Adv. Energy Mater. 2017, 7, 1602545. 10.1002/aenm.201602545. [DOI] [Google Scholar]
- Zhong W.; Huang J.; Liang S.; Liu J.; Li Y.; Cai G.; Jiang Y.; Liu J. New Prelithiated V2O5 Superstructure for Lithium-Ion Batteries with Long Cycle Life and High Power. ACS Energy Lett. 2020, 5, 31–38. 10.1021/acsenergylett.9b02048. [DOI] [Google Scholar]
- Liu H.; Zhu Z.; Yan Q.; Yu S.; He X.; Chen Y.; Zhang R.; Ma L.; Liu T.; Li M.; Lin R.; Chen Y.; Li Y.; Xing X.; Choi Y.; Gao L.; Cho H. S.-y.; An K.; Feng J.; Kostecki R.; Amine K.; Wu T.; Lu J.; Xin H. L.; Ong S. P.; Liu P. A Disordered Rock Salt Anode for Fast-Charging Lithium-Ion Batteries. Nature 2020, 585, 63–67. 10.1038/s41586-020-2637-6. [DOI] [PubMed] [Google Scholar]
- Guo X.; Chen C.; Ong S. P. Intercalation Chemistry of the Disordered Rocksalt Li3V2O5 Anode from Cluster Expansions and Machine Learning Interatomic Potentials. Chem. Mater. 2023, 35 (4), 1537–1546. 10.1021/acs.chemmater.2c02839. [DOI] [Google Scholar]
- Mikhailova D.; Voss A.; Oswald S.; Tsirlin A. A.; Schmidt M.; Senyshyn A.; Eckert J.; Ehrenberg H. Lithium Insertion into Li2MoO4: Reversible Formation of (Li3Mo)O4 with a Disordered Rock-Salt Structure. Chem. Mater. 2015, 27, 4485–4492. 10.1021/acs.chemmater.5b01633. [DOI] [Google Scholar]
- Pralong V.; Venkatesh G.; Malo S.; Caignaert V.; Baies R.; Raveau B. Electrochemical Synthesis of a Lithium-Rich Rock-Salt-Type Oxide Li5W2O7 with Reversible Deintercalation Properties. Inorg. Chem. 2014, 53, 522–527. 10.1021/ic402543e. [DOI] [PubMed] [Google Scholar]
- Xiong H.; Yildirim H.; Shevchenko E. V.; Prakapenka V. B.; Koo B.; Slater M. D.; Balasubramanian M.; Sankaranarayanan S. K. R. S.; Greeley J. P.; Tepavcevic S.; Dimitrijevic N. M.; Podsiadlo P.; Johnson C. S.; Rajh T. Self-Improving Anode for Lithium-Ion Batteries Based on Amorphous to Cubic Phase Transition in TiO2 Nanotubes. J. Phys. Chem. C 2012, 116, 3181–3187. 10.1021/jp210793u. [DOI] [Google Scholar]
- Barnes P.; Zuo Y.; Dixon K.; Hou D.; Lee S.; Ma Z.; Connell J. G.; Zhou H.; Deng C.; Smith K.; Gabriel E.; Liu Y.; Maryon O. O.; Davis P. H.; Zhu H.; Du Y.; Qi J.; Zhu Z.; Chen C.; Zhu Z.; Zhou Y.; Simmonds P. J.; Briggs A. E.; Schwartz D.; Ong S. P.; Xiong H. Electrochemically Induced Amorphous-to-Rock-Salt Phase Transformation in Niobium Oxide Electrode for Li-Ion Batteries. Nat. Mater. 2022, 21, 795–803. 10.1038/s41563-022-01242-0. [DOI] [PubMed] [Google Scholar]
- Li W.; Fukunishi M.; Morgan B. J.; Borkiewicz O. J.; Chapman K. W.; Pralong V.; Maignan A.; Lebedev O. I.; Ma J.; Groult H.; Komaba S.; Dambournet D. A Reversible Phase Transition for Sodium Insertion in Anatase TiO2. Chem. Mater. 2017, 29, 1836–1844. 10.1021/acs.chemmater.7b00098. [DOI] [Google Scholar]
- Wei Q.; Chang X.; Butts D.; DeBlock R.; Lan K.; Li J.; Chao D.; Peng D. L.; Dunn B. Surface-Redox Sodium-Ion Storage in Anatase Titanium Oxide. Nat. Commun. 2023, 14, 7. 10.1038/s41467-022-35617-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarman R. H.; Dickens P. G.; Jacobson A. J. Preparation and Characterization of the Mixed Oxide V9Mo6O40. Mater. Res. Bull. 1982, 17, 325–328. 10.1016/0025-5408(82)90080-0. [DOI] [Google Scholar]
- Slade R. C. T.; Ramanan A.; West B. C.; Prince E. The Structure of V9Mo6O40 Determined by Powder Neutron Diffraction. J. Solid State Chem. 1989, 82, 65–69. 10.1016/0022-4596(89)90223-5. [DOI] [Google Scholar]
- Hu M.; Liang J.; Chen X.; Wei J.; Zhou Z. Preparation and electrochemical performance of Mo6V9O40 nanorods as cathode materials for Li batteries. RSC Adv. 2015, 5, 15395–15398. 10.1039/C4RA16335D. [DOI] [Google Scholar]
- Eick H. A.; Kihlborg L.; Kujanpää T.; von Hofsten B.; Williams D. H.; Bunnenberg E.; Djerassi C.; Records R. The crystal structure of V2MoO8. Acta Chem. Scand. 1966, 20, 1658–1666. 10.3891/acta.chem.scand.20-1658. [DOI] [Google Scholar]
- Borkiewicz O. J.; Shyam B.; Wiaderek K. M.; Kurtz C.; Chupas P. J.; Chapman K. W. The AMPIX electrochemical cell: a versatile apparatus for in situ X-ray scattering and spectroscopic measurements. J. Appl. Crystallogr. 2012, 45, 1261–1269. 10.1107/S0021889812042720. [DOI] [Google Scholar]
- Toby B. H.; Von Dreele R. B. GSAS-II: the genesis of a modern open-source all purpose crystallography software package. J. Appl. Crystallogr. 2013, 46, 544–549. 10.1107/S0021889813003531. [DOI] [Google Scholar]
- Momma K.; Izumi F. VESTA: a three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. 10.1107/S0021889808012016. [DOI] [Google Scholar]
- Enjalbert R.; Galy J. A Refinement of the Structure of V2O5. Acta Crystallogr., Sect. C 1986, 42, 1467–1469. 10.1107/s0108270186091825. [DOI] [Google Scholar]
- Wooster N. The crystal structure of Molybdenum Trioxide, MoO3. Z. Kristallogr. Cryst. Mater. 1931, 80, 504–512. 10.1524/zkri.1931.80.1.504. [DOI] [Google Scholar]
- Kim H. S.; Cook J. B.; Lin H.; Ko J. S.; Tolbert S. H.; Ozolins V.; Dunn B. Oxygen vacancies enhance pseudocapacitive charge storage properties of MoO3-x. Nat. Mater. 2017, 16, 454–460. 10.1038/nmat4810. [DOI] [PubMed] [Google Scholar]
- Adams A. H.; Haaß F.; Buhrmester T.; Kunert J.; Ott J.; Vogel H.; Fuess H. Structure and Reaction Studies on Vanadium Molybdenum Mixed Oxides. J. Mol. Catal. A: Chem. 2004, 216, 67–74. 10.1016/j.molcata.2004.02.005. [DOI] [Google Scholar]
- Sudant G.; Baudrin E.; Dunn B.; Tarascon J.-M. Synthesis and Electrochemical Properties of Vanadium Oxide Aerogels Prepared by a Freeze-Drying Process. J. Electrochem. Soc. 2004, 151, A666. 10.1149/1.1687427. [DOI] [Google Scholar]
- Zhecheva E.; Mladenov M.; Zlatilova P.; Koleva V.; Stoyanova R. Particle size distribution and electrochemical properties of LiFePO4 prepared by a freeze-drying method. J. Phys. Chem. Solids 2010, 71, 848–853. 10.1016/j.jpcs.2010.02.012. [DOI] [Google Scholar]
- Shi S. J.; Tu J. P.; Tang Y. Y.; Yu Y. X.; Zhang Y. Q.; Wang X. L. Synthesis and electrochemical performance of Li1.131Mn0.504Ni0.243Co0.122O2 cathode materials for lithium ion batteries via freeze drying. J. Power Sources 2013, 221, 300–307. 10.1016/j.jpowsour.2012.08.031. [DOI] [Google Scholar]
- Yan Y.; Chin M. A.; Robertson D. D.; Lesel B. K.; Tolbert S. H. Tuning the Porous Structure in PMMA-Templated Mesoporous MoO2 for Pseudocapacitive Li-Ion Electrodes. J. Electrochem. Soc. 2022, 169 (4), 040545. 10.1149/1945-7111/ac63f8. [DOI] [Google Scholar]
- Robertson D. D.; Cumberbatch H.; Pe D. J.; Yao Y.; Tolbert S. H. Understanding How the Suppression of Insertion-Induced Phase Transitions Leads to Fast Charging in Nanoscale LixMoO2. ACS Nano 2024, 18, 996–1012. 10.1021/acsnano.3c10169. [DOI] [PubMed] [Google Scholar]
- Wang J.; Polleux J.; Lim J.; Dunn B. Pseudocapacitive Contributions to Electrochemical Energy Storage in TiO2 (Anatase) Nanoparticles. J. Phys. Chem. C 2007, 111 (40), 14925–14931. 10.1021/jp074464w. [DOI] [Google Scholar]
- Cook J. B.; Kim H. S.; Lin T. C.; Lai C. H.; Dunn B.; Tolbert S. H. Pseudocapacitive Charge Storage in Thick Composite MoS2 Nanocrystal-Based Electrodes. Adv. Energy Mater. 2017, 7, 1601283. 10.1002/aenm.201601283. [DOI] [Google Scholar]
- Cook J. B.; Lin T. C.; Kim H.-S.; Siordia A.; Dunn B. S.; Tolbert S. H. Suppression of Electrochemically Driven Phase Transitions in Nanostructured MoS2 Pseudocapacitors Probed Using Operando X-ray Diffraction. ACS Nano 2019, 13, 1223–1231. 10.1021/acsnano.8b06381. [DOI] [PubMed] [Google Scholar]
- Cook J. B.; Ko J. S.; Lin T. C.; Robertson D. D.; Kim H. S.; Yan Y.; Yao Y.; Dunn B. S.; Tolbert S. H. Ultrafast Sodium Intercalation Pseudocapacitance in MoS2 Facilitated by Phase Transition Suppression. ACS Appl. Energy Mater. 2023, 6, 99–108. 10.1021/acsaem.2c02368. [DOI] [Google Scholar]
- Yao Y.; Cumberbatch H.; Robertson D. D.; Chin M. A.; Lamkin R.; Tolbert S. H. On the Interplay between Size and Disorder in Suppressing Intercalation-Induced Phase Transitions in Pseudocapacitive Nanostructured MoS2. Adv. Funct. Mater. 2024, 34, 2304896. 10.1002/adfm.202304896. [DOI] [Google Scholar]
- Mai L.; Xu L.; Han C.; Xu X.; Luo Y.; Zhao S.; Zhao Y. Electrospun Ultralong Hierarchical Vanadium Oxide Nanowires with High Performance for Lithium Ion Batteries. Nano Lett. 2010, 10, 4750–4755. 10.1021/nl103343w. [DOI] [PubMed] [Google Scholar]
- Christensen C. K.; Sørensen D. R.; Hvam J.; Ravnsbæk D. B. Structural Evolution of Disordered LixV2O5 Bronzes in V2O5 Cathodes for Li-Ion Batteries. Chem. Mater. 2019, 31, 512–520. 10.1021/acs.chemmater.8b04558. [DOI] [Google Scholar]
- Silversmit G.; Depla D.; Poelman H.; Marin G. B.; De Gryse R. Determination of the V2p XPS Binding Energies for Different Vanadium Oxidation States (V5+ to V0+). J. Electron Spectrosc. Relat. Phenom. 2004, 135, 167–175. 10.1016/j.elspec.2004.03.004. [DOI] [Google Scholar]
- Scanlon D. O.; Watson G. W.; Payne D. J.; Atkinson G. R.; Egdell R. G.; Law D. S. L. Theoretical and Experimental Study of the Electronic Structures of MoO3 and MoO2. J. Phys. Chem. C 2010, 114, 4636–4645. 10.1021/jp9093172. [DOI] [Google Scholar]
- Wang W.; Qin J.; Yin Z.; Cao M. Achieving Fully Reversible Conversion in MoO3 for Lithium Ion Batteries by Rational Introduction of CoMoO4. ACS Nano 2016, 10, 10106–10116. 10.1021/acsnano.6b05150. [DOI] [PubMed] [Google Scholar]
- Liu H.; Zhu Z.; Huang J.; He X.; Chen Y.; Zhang R.; Lin R.; Li Y.; Yu S.; Xing X.; Yan Q.; Li X.; Frost M. J.; An K.; Feng J.; Kostecki R.; Xin H.; Ong S. P.; Liu P. Elucidating the Limit of Li Insertion into the Spinel Li4Ti5O12. ACS Mater. Lett. 2019, 1, 96–102. 10.1021/acsmaterialslett.9b00099. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.