

Summary
The accumulation of damaged mitochondria in the heart is associated with heart failure. Mitophagy is an autophagic degradation system that specifically targets damaged mitochondria. We have reported previously that Bcl2-like protein 13 (Bcl2-L-13) mediates mitophagy and mitochondrial fission in mammalian cells. However, the in vivo function of Bcl2-L-13 remains unclear. Here, we demonstrate that Bcl2-L-13-deficient mice and knockin mice, in which the phosphorylation site (Ser272) on Bcl2-L-13 was changed to Ala, showed left ventricular dysfunction in response to pressure overload. Attenuation of mitochondrial fission and mitophagy led to impairment of ATP production in these mouse hearts. In addition, we identified AMPKα2 as the kinase responsible for the phosphorylation of Bcl2-L-13 at Ser272. These results indicate that Bcl2-L-13 and its phosphorylation play an important role in maintaining cardiac function. Furthermore, the amplitude of stress-stimulated mitophagic activity could be modulated by AMPKα2.
Keywords: Bcl2-L-13, mitochondria, mitophagy, heart failure
Graphical abstract
Highlights
-
•
Bcl2-L-13 plays a protective role in the pressure-overloaded heart
-
•
Bcl2-L-13 is involved in mitophagy and mitochondrial fission in vivo
-
•
AMPKα2 is the responsible kinase to phosphorylate Bcl2-L-13 at Ser272
Murakawa et al. demonstrate that pressure overload to the heart induces Bcl2-L-13-mediated mitochondrial fission and mitophagy. Furthermore, its activation by phosphorylation is essential for maintaining cardiac function. The kinase library screening revealed that AMPKα2 is the responsible kinase. These findings elucidate the cardioprotective role of Bcl2-L-13.
Introduction
Heart failure is a major cause of morbidity and mortality despite advances in its management.1 Mitochondria are abundant in energy-demanding cardiac tissue, and defects in mitochondrial structure and function are associated with heart failure.2 Under pathological conditions, clearance of dysfunctional mitochondria is critical for maintaining the homeostasis of cardiomyocytes.3 Autophagy is a highly conserved process of protein and organelle degradation in response to nutrient shortages and cytotoxic insults.4 While autophagy nonselectively sequesters its cargos, selective degradation of damaged mitochondria relies on an autophagy-related transport system: mitophagy. In yeast, the Atg32 protein plays an essential role in mitophagy.5,6 In mammalian cells, several types of mitophagy receptors or receptor-related factors, including NIP3-like protein X (NIX), BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), FUN14 domain containing 1 (FUNDC1), and phosphatase and tensin homolog-induced putative kinase protein 1 (PINK1)/Parkin have been reported.7 We have previously identified Bcl2-L-13 as a functional mammalian homolog of Atg32.8 Bcl2-L-13 induces both mitochondrial fission and mitophagy through its binding with microtubule-associated protein 1A- or 1B-light chain 3B (LC3B) in HEK293 cells. Phosphorylation and dephosphorylation serve as regulatory mechanisms to modulate the activity of mitophagy-related molecules.9,10,11,12 We have also previously reported that phosphorylation of Ser272 on Bcl2-L-13 results in the enhancement of mitophagic activity.8 However, the kinase responsible for this phosphorylation has not been identified. Furthermore, the in vivo function of Bcl2-L-13 in the heart is poorly understood.
The heart is an organ that demonstrates high mRNA expression levels of Bcl2-L-13.13 Therefore, this study aimed to determine the in vivo functional role of Bcl2-L-13 in cardiac function. For this, we generated Bcl2-L-13-deficient mice. The mice showed attenuation of pressure overload-induced mitochondrial fission and mitophagy to protect the heart against pressure overload. In addition, analyses on knockin mice, in which the phosphorylation site Ser272 was changed to Ala (Bcl2l13S272A/S272A), indicated that the activation of Bcl2-L-13 by its phosphorylation at Ser272 is essential for maintaining mitochondrial dynamics and cardiac function under pressure overload. Furthermore, screening of the kinase library revealed that 5′-AMP-activated protein kinase catalytic subunit alpha-2 (AMPKα2) is the kinase responsible for the phosphorylation of Ser272 in Bcl2-L-13.
Results
Ablation of Bcl2l13 led to reduced cardiac function after pressure overload
To investigate the in vivo role of Bcl2-L-13 in the heart, we generated Bcl2-L-13-deficient (Bcl2l13−/−) mice. First, we designed a gene targeting strategy to conditionally inactivate the Bcl2l13 gene by inserting loxP sites in introns 2 and 4 (Figure S1A). Homologous recombinants were identified using Southern blot analysis (Figure S1B). We then crossed homozygous floxed Bcl2l13 mice (Bcl2l13flox/flox) with transgenic mice expressing Cre recombinase under control of the β-actin promoter (βActin-Cre mice) to obtain conventional Bcl2l13−/− mice because global knockin mice were used in the following experiment. Mating between Bcl2l13+/− mice produced offspring with the expected Mendelian ratio (male Bcl2l13+/+:male Bcl2l13+/−:male Bcl2l13−/−:female Bcl2l13+/+:female Bcl2l13+/−:female Bcl2l13−/− = 23:46:12:25:41:20). Bcl2l13−/− mice were viable, fertile, and normal in appearance. The efficiency of Bcl2-L-13 ablation in the heart was determined by immunoblot analysis. Bcl2-L-13 was totally abrogated in Bcl2l13−/− hearts (Figure S1C). There were no significant differences in physiological or echocardiographic parameters at 10 weeks of age between Bcl2l13−/− and Bcl2l13+/+ mice (Table S1).
To examine the in vivo role of Bcl2-L-13 in the heart during cardiac stress, Bcl2l13−/− and Bcl2l13+/+ mice were subjected to pressure overload via transverse aortic constriction (TAC) surgery. We used a 25G needle to exact mild pressure overload, which does not induce cardiac dysfunction and heart failure in Bcl2l13+/+ mice but does induce cardiac hypertrophy. Four weeks after surgery, echocardiographic analysis revealed an increase in end-systolic left ventricular (LV) internal dimension (LVIDs) and a decrease in LV fractional shortening (FS), which is an index for systolic function, in the TAC-operated Bcl2l13−/− group, while the LV dimension and function of the TAC-operated Bcl2l13+/+ group remained the same (Figures 1A and 1B). The thickness of the end-diastolic interventricular septum wall (IVSd) and LV posterior wall (LVPWd) was elevated in TAC-operated Bcl2l13+/+ and Bcl2l13−/− mice compared to the corresponding sham-operated mice. The IVSd and LVPWd in TAC-operated Bcl2l13−/− mice were significantly shorter than in TAC-operated Bcl2l13+/+ mice. The LV weight-to-tibia length ratio, an index for cardiac hypertrophy, was increased in TAC-operated groups compared to the corresponding sham groups; however, the extent of this increase was less in the TAC-operated Bcl2l13−/− group. There was no significant increase in lung-to-body weight ratio, an index for lung congestion, in either Bcl2l13−/− or Bcl2l13+/+ mice (Figure 1C). The mRNA expression level of Nppa, which reflects cardiac hypertrophy, was increased in the TAC-operated Bcl2l13+/+ and Bcl2l13−/− groups; however, levels were higher in the TAC-operated Bcl2l13+/+ group. The mRNA expression level of Nppb, which primarily reflects LV dysfunction, was increased significantly in the TAC-operated Bcl2l13−/− group compared to all other groups (Figure 1D). Histological analysis revealed an increased fibrosis fraction in both TAC-operated groups; however, cellular infiltration was not observed in any group (Figures S1D and S1E). The cardiomyocyte cross-sectional area was larger in the TAC-operated Bcl2l13+/+ group compared to the Bcl2l13−/− group (Figure 1E). These data suggest that ablation of Bcl2l13 results in cardiac dysfunction with decreased hypertrophic responses against pressure overload.
Figure 1.

Pressure overload-induced cardiac dysfunction in Bcl2l13−/− mice
Bcl2l13+/+ (WT) and Bcl2l13−/− (KO) mice were subjected to pressure overload by means of transverse aortic constriction (TAC). The mice were analyzed 4 weeks after TAC.
(A) Representative images of M-mode echocardiographic tracings from sham- or TAC-operated WT or KO mice. Scale bars: 0.1 s and 2 mm.
(B) Echocardiographic parameters. n = 8 (WT sham), 8 (KO sham), 9 (WT TAC), or 9 (KO TAC) per group. LVIDd, end-diastolic left ventricular internal dimension; LVIDs, end-systolic left ventricular internal dimension; FS, fractional shortening; IVSd, end-diastolic interventricular septum wall thickness; LVPWd, end-diastolic left ventricular posterior wall thickness.
(C) Physiological parameters. n = 8 (WT sham), 8 (KO sham), 9 (WT TAC), or 9 (KO TAC) per group. LV/TL, left ventricular weight/tibia length; lungW/TL, lung weight/tibia length.
(D) mRNA expression of Nppa and Nppb (n = 6). Gapdh mRNA was used as the loading control. The average value in the WT sham group was set to 1.
(E) Wheat germ agglutinin-stained heart sections. Scale bar: 50 μm. Cardiomyocyte cross-sectional areas were measured by tracing the outline of 100 myocytes in the non-fibrotic area on each section (n = 3). Results are shown as mean with 95% confidence intervals (CIs). Statistical analysis included one-way ANOVA followed by Tukey-Kramer’s post hoc test. All pairwise comparisons were performed. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figure S1.
Mitochondrial fission and mitophagy were attenuated in Bcl2l13−/− hearts after the TAC operation
To explore the molecular mechanisms underlying the abnormal cardiac phenotypes observed in Bcl2l13−/− mice, we analyzed the mice at the earlier time course after TAC. Cardiac function decreased in TAC-operated Bcl2l13−/− mice over time (Figure S2A). We chose to perform the analyses 5 days after TAC to minimize the contribution of operation-related events secondary to the initial and essential events that induced the cardiac phenotypes in Bcl2l13−/− mice. Five days after the operation, the TAC-operated Bcl2l13−/− hearts demonstrated LV systolic dysfunction, while the LV function of the TAC-operated Bcl2l13+/+ group was maintained (Figure S2B). The IVSd, LVPWD, and LV weight-to-tibia length ratios were increased significantly to a similar extent in both TAC groups, and there was no significant increase in lung-to-body weight ratio (Figure S2C). In line with the echocardiographic and physiological analyses, the cardiomyocyte cross-sectional area was increased to a similar degree in both TAC groups (Figure S2D). In addition, we analyzed the contribution of cell death to the cardiac phenotypes. Apoptosis was detected by terminal transferase dUTP nick end labeling (TUNEL) staining, and necrosis was detected by the translocation of HMGB1 from the nucleus. Both apoptosis and necrosis were increased significantly by TAC operation, but there was no significant difference between TAC-operated Bcl2l13+/+ and TAC-operated Bcl2l13−/− mice, suggesting that cell death is not involved in the cardiac dysfunction observed in Bcl2l13−/− mice (Figures S2E and S2F).
We have reported previously that Bcl2-L-13 induces both mitochondrial fission and mitophagy in mammalian cells.8 In the current study, we examined whether Bcl2-L-13 performs these functions in in vivo hearts. First, we performed ultrastructural analysis. Using the Freehand selection tool, inter-myofibrillar mitochondria were traced. The mitochondrial long diameter, which is the primary axis of the best-fitting ellipse to the traced pixels, and the mitochondrial area were measured. The population of mitochondria with shorter long diameters or smaller areas was increased significantly after TAC in Bcl2l13+/+ mice, whereas it was significantly decreased in Bcl2l13−/− mice (Figures 2A–2C and S3A). In addition, we evaluated the protein levels of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) to examine the contribution of mitochondrial biogenesis to mitochondrial morphological changes (Figure S3B). The protein levels of PGC1α were similar in all groups. These data suggest that pressure overload-induced mitochondrial fission was attenuated in Bcl2l13−/− hearts. We subsequently analyzed protein levels to examine the involvement of the molecules related to mitochondrial fusion and fission in this phenotype (Figures S3C and S3D). Dynamin-related protein 1 (Drp1) induces mitochondrial fission by interacting with fission-related protein 1.14 Phosphorylation of Drp1 at Ser616 promotes mitochondrial fission, while phosphorylation of Drp1 at Ser637 inhibits mitochondrial fission.15,16 In contrast, mitofusin 1 (Mfn1) and Mfn2 and optic atrophy 1 increase the formation of elongated mitochondrion networks. The protein level of phosphorylated Drp1 (Ser616) was upregulated significantly in TAC-operated Bcl2l13−/− mice compared to the sham-operated control; however, this can be considered a compensatory change. The expression of other proteins did not show significant differences among groups.
Figure 2.

Mitochondrial morphology and mitophagy in TAC-operated Bcl2l13−/− mice
(A) Electron micrographs of mouse hearts 5 days after TAC. Scale bar: 1 μm.
(B and C) Violin plots visualizing the distribution of long diameters and areas of mitochondria measured in (A). Using the Freehand selection tool, inter-myofibrillar mitochondria were traced. The long diameter is the primary axis of the best-fitting ellipse to the traced pixels. The area enclosed by the dotted line in the violin plots is enlarged and shown on the right, with two groups overlaid. The histogram of the distribution of long diameters is shown in Figure S3A.
(D) Immunostaining of LC3B and ATP synthase in the heart 5 days after TAC. Scale bar: 20 μm. Images in the box at higher magnification are shown on the right. The white arrow indicates the colocalization of LC3B and ATP synthase double-positive dot. The number of LC3B and ATP synthase double-positive dots per 1 mm2 is shown in the bar graph (n = 3). Results are shown as mean with 95% CI. Statistical analysis by Kruskal-Wallis test in (B) and (C) and one-way ANOVA followed by Tukey-Kramer’s post hoc test in (D). All pairwise comparisons were performed. ∗∗∗p < 0.001. See also Figures S2 and S3.
Mitophagy was evaluated using immunohistochemical analysis, whereby mitophagy was identified by colocalizing the autophagosome marker LC3B and mitochondrion marker ATP synthase. Five days after TAC, mitophagy was upregulated significantly in Bcl2l13+/+ hearts, while there was no significant difference between sham-operated and TAC-operated Bcl2l13−/− hearts (knockout [KO] sham vs. KO TAC: p = 0.8446; relative values for each group to WT sham group: KO sham group 0.476, wild-type [WT] TAC group 2.31, KO TAC group 0.636) (Figure 2D).
Parkin has been reported to be upregulated due to cardiac pressure overload.17,18 Therefore, we examined the involvement of Parkin in cardiac dysfunction in TAC-operated Bcl2l13−/− hearts. In agreement with previous findings, the protein level of Parkin was upregulated in both TAC groups; however, there was no significant difference between TAC-operated Bcl2l13+/+ and Bcl2l13−/− mice (Figure S3E). Furthermore, there were no significant differences in PINK1 expression between the groups. To examine the contribution of Parkin-dependent mitophagy to the cardiomyopathy seen in TAC-operated Bcl2l13−/− mice, we generated double KO mice of Bcl2-L-13 and Parkin. If Parkin has a pivotal role in maintaining cardiac function under pressure overload independent of Bcl2-L-13, then Bcl2l13−/−Prk2−/− mice would show worse cardiac function than Bcl2l13−/−Prk2+/+ mice. TAC-operated Bcl2l13−/−Prk2−/− mice showed a similar level of cardiac dysfunction as Bcl2l13−/−Prk2+/+ mice, suggesting that Parkin does not play a pivotal role in cardiac reaction against pressure overload in Bcl2l13−/− mice (Figures S3F and S3G).
Mitochondrial reactive oxygen species production was increased and ATP production was reduced in TAC-operated Bcl2l13−/− hearts
Downregulation of mitophagy might result in attenuation of the removal of dysfunctional mitochondria, leading to increased reactive oxygen species (ROS) production and reduced ATP production in cardiomyocytes. We isolated cardiomyocytes 5 days after TAC and evaluated ROS using the mitochondrial superoxide indicator MitoSOX (Figure 3A). We identified mitochondria strongly stained with MitoSOX only in cardiomyocytes isolated from TAC-operated Bcl2l13−/− mice. Furthermore, we analyzed mitochondrial DNA (mtDNA) damage by evaluating the level of replicated 8.2 kb mtDNA using a mouse real-time PCR mitochondrial DNA damage analysis kit. The damage to the mtDNA results in the inhibition of PCR of 8.2 kb mtDNA. In agreement with the results from the MitoSOX analysis, the amount of 8.2 kb-PCR product was decreased significantly in TAC-operated Bcl2l13−/− hearts, indicating increased mtDNA damage (Figure S3H). Next, we evaluated the mitochondrial respiration of isolated cardiomyocytes 5 days after TAC using a Seahorse XFe24 extracellular flux analyzer (Figure 3B). The mitochondrial stress test demonstrated that the baseline oxygen consumption rate (OCR) of Bcl2l13+/+ cardiomyocytes was upregulated significantly in response to pressure overload, while it was unchanged in Bcl2l13−/− mice. Furthermore, maximum OCR and ATP concentration were decreased significantly in TAC-operated Bcl2l13−/− hearts (Figures 3B and 3C). These data suggest that damaged mitochondria escaped from mitophagic degradation produced more ROS and less ATP and were accumulated in TAC-operated Bcl2l13−/− hearts.
Figure 3.

Mitochondrial dysfunction in TAC-operated Bcl2l13−/− mice
(A) Mitochondrial reactive oxygen species (ROS) production in cardiomyocytes. Cardiomyocytes were isolated 5 days after the TAC operation and stained with MitoSOX and MitoTracker Green for confocal microscopy (n = 3). MitoSOX-positive mitochondria in the boxed area are shown at higher magnification in the inset. Scale bar: 10 μm. The number of MitoSOX-positive mitochondria per cell is shown in the bar graph. At least 30 cells were observed in each experiment.
(B) The oxygen consumption rate (OCR) of isolated cardiomyocytes 5 days after TAC was assessed using the Seahorse XF24 extracellular flux analyzer. n = 3 (WT sham), 3 (KO sham), 3 (WT TAC), or 5 (KO TAC) per group. Reagents were injected sequentially during the assay to yield final concentrations of 1 μM oligomycin, 2 μM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP), 2 μM rotenone, and 4 μM antimycin A. Basal respiration and maximal respiration are shown in the bar graphs.
(C) Tissue ATP levels were measured using the left ventricle from mice 5 days after TAC. n = 7 (WT sham), 7 (KO sham), 6 (WT TAC), or 6 (KO TAC) per group. Results are shown as mean with 95% CI. Statistical analysis by one-way ANOVA followed by Tukey-Kramer’s post hoc test. All pairwise comparisons were performed. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
A phosphorylation-silencing mutation of Bcl2-L-13 led to reduced cardiac function in response to pressure overload
We have shown previously that the phosphorylation of Bcl2-L-13 at Ser272 is important for the mitophagic activity of Bcl2-L-13.8 To evaluate the level of phosphorylation, we generated an antibody against phospho-Bcl2-L-13 (Ser272). The antibody recognized a band in the lysate isolated from HEK293 cells expressing WT Bcl2-L-13, whereas it recognized a weak signal from the lysates of cells expressing Bcl2-L-13 (S272A) (Figure S4A). In Bcl2l13+/+ mice, the phosphorylation level of Bcl2-L-13 at Ser272 increased 4 weeks after the TAC operation, suggesting that the phosphorylation of Bcl2-L-13 was upregulated in response to pressure overload (Figure 4A). To investigate the in vivo role of the phosphorylation of Bcl2-L-13 at Ser272 in the heart, we generated knockin mice (Bcl2l13S272A/S272A), in which the phosphorylation site (Ser272) on Bcl2-L-13 was changed to Ala (Figure S4B). Bcl2l13WT/S272A mice were crossed to obtain Bcl2l13S272A/S272A mice. Mice were born at the expected Mendelian ratios (male Bcl2l13w/w:male Bcl2l13WT/S272A:male Bcl2l13S272A/S272A:female Bcl2l13w/w:female Bcl2l13WT/S272A:female Bcl2l13S272A/S272A = 14:36:9:11:27:12). Bcl2l13S272A/S272A mice showed similar physiological parameters and cardiac function compared to WT mice (Table S2). A western blot analysis indicated that the protein level of phospho-Bcl2-L-13 (Ser272) was reduced significantly in Bcl2l13S272A/S272A (Figure S4C).
Figure 4.

Pressure overload-induced cardiac dysfunction in Bcl2-L-13 (S272A) knockin mice
(A) Western blot analysis of phospho- Bcl2-L-13 (Ser272) in the Bcl2l13+/+ (WT) and Bcl2l13−/− (KO) mouse hearts 4 weeks after TAC (n = 3). The bar graph shows densitometric analysis.
(B) Representative images of M-mode echocardiographic tracings from sham- or TAC-operated wild-type (WT) or Bcl2-L-13 (S272A) knockin (KI) mice 4 weeks after surgery. Scale bars, 0.1 s and 2 mm.
(C) Echocardiographic parameters (n = 10).
(D) Physiological parameters (n = 10).
(E) mRNA expression of Nppa and Nppb (n = 8). Gapdh mRNA was used as the loading control. The average value in the WT sham group was set to 1.
(F) Wheat germ agglutinin-stained heart sections. Scale bar: 50 μm. Cardiomyocyte cross-sectional areas were measured by tracing the outline of 100 myocytes in a non-fibrotic area in each section (n = 4). Results are shown as mean with 95% CI. Statistical analysis by one-way ANOVA followed by Tukey-Kramer’s post hoc test. All pairwise comparisons were performed. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figure S4.
We subsequently subjected those mice to pressure overload using a 25G needle. The Bcl2l13S272A/S272A mice exhibited LV chamber dilation and cardiac dysfunction 4 weeks after surgery, while the LV function of the TAC-operated Bcl2l13w/w group was maintained (Figures 4B and 4C). Although TAC increased the LV weight-to-tibia length ratio in both groups, this ratio was significantly higher in Bcl2l13S272A/S272A mice (Figure 4D). There was no significant increase in lung-to-body weight ratio in either TAC group. The mRNA levels of Nppa and Nppb were significantly higher in TAC-operated Bcl2L13S272A/S272A hearts compared to all other groups (Figure 4E). From histological analysis, cellular infiltration was not observed in any group (Figure S4D). The fibrosis fraction (Figure S4E) and cardiomyocyte cross-sectional area (Figure 4F) were increased in both TAC-operated groups. TAC-operated Bcl2l13S272A/S272A hearts demonstrated larger fibrosis fractions and cross-sectional areas than TAC-operated WT mice. These data suggest that the phosphorylation of Bcl2-L-13 at Ser272 plays an important role in maintaining cardiac function under pressure overload.
We examined mitochondrial morphology and mitophagic activity 5 days after TAC. As the Bcl2-L-13 (S272A) mutant maintained its ability to induce mitochondrial fission in the in vitro study,8 we expected TAC-operated Bcl2l13S272A/S272A mice to display a mitochondrial morphology similar to TAC-operated Bcl2l13w/w. However, contrary to our expectations, the population of mitochondria with longer lengths and smaller mitochondrial areas was increased in TAC-operated Bcl2l13S272A/S272A mice (Figures 5A–5C and S5A). Fusion/fission-related molecules showed no differences in protein or phosphorylation levels between TAC-operated groups (Figures S5B and S5C). Immunohistochemical analysis revealed that TAC-operated Bcl2l13S272A/S272A mice had significantly lower levels of mitophagy than TAC-operated Bcl2l13w/w mice (Figure 5D). Furthermore, the cytosolic ATP concentration in TAC-operated Bcl2l13S272A/S272A hearts was lower than in Bcl2l13w/w hearts (Figure 5E). While the protein level of Parkin was upregulated in both TAC groups, there was no significant difference in the level between TAC-operated WT and knockin mice (Figure S5D). In addition, there were no significant differences in PINK1 expression between the groups. Therefore, Bcl2l13 ablation and the S272A mutation had detrimental effects on the cardiac phenotype and comparable impacts on the mitochondria dynamics in response to pressure overload.
Figure 5.

Mitochondrial morphology, mitophagy, and ATP concentration in TAC-operated Bcl2-L-13 (S272A) KI hearts
(A) Electron micrographs of mouse hearts 5 days after TAC. Scale bar: 1 μm.
(B and C) Violin plots visualizing the distribution of long diameters and areas of mitochondria measured in (A). Using the Freehand selection tool, inter-myofibrillar mitochondria were traced. The long diameter is the primary axis of the best-fitting ellipse to the traced pixels. The area enclosed by the dotted line in the violin plots is enlarged and shown on the right, with two groups overlaid. The histogram of the distribution of long diameters is shown in Figure S5A.
(D) Immunostaining of LC3B and ATP synthase in the heart 5 days after TAC. Scale bar: 20 μm. Images in the box at higher magnification are shown on the right. The white arrows indicates the colocalization of LC3B and ATP synthase double-positive dots. The number of LC3B and ATP synthase double-positive dots per 1 mm2 is shown in the bar graph (n = 3).
(E) Tissue ATP levels measured using the left ventricle from mice 5 days after TAC (n = 6). Results are shown as mean with 95% CI. Statistical analysis by Kruskal-Wallis test in (B) and (C) and one-way ANOVA followed by Tukey-Kramer’s post hoc test in (D) and (E). All pairwise comparisons were performed. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figure S5.
Kinase siRNA library screening revealed that AMPKα2 is the kinase responsible for Bcl2-L-13 phosphorylation at Ser272
To further explore the mechanism of Bcl2-L-13 activation, we attempted to determine the kinase responsible for the phosphorylation of Bcl2-L-13 at Ser272. To establish an assay suitable for manual screening using the small interfering RNA (siRNA) library of kinases, we tested whether phospho-Bcl2-L-13 is detectable by fluorescence immunocytochemistry using HEK293 cells that stably expressed HA-Bcl2-L-13. Carbonyl cyanide m-chlorophenylhydrazone (CCCP), a mitochondrial oxidative phosphorylation uncoupler, induces mitophagy. Phospho-Bcl2-L-13-positive dots were detected after administration of bafilomycin A1 and CCCP, and they were colocalized with signals from the anti-hemagglutinin (HA) antibody (Figure S6A). To confirm the colocalization between HA and phospho-Bcl2-L-13, we performed quantification analysis using the JACoP plugin of ImageJ to obtain the Manders’ coefficients of the images presented in Figure S6A19,20 The results showed that, in DMSO-treated cells, the Manders’ coefficient was 0.938, while that in CCCP-treated cells was 0.967. These results strongly suggest that the green signals from the HA antibody colocalize with the red signals from the phospho-Bcl2-L-13 antibody. A primary screen using the Silencer Human Kinase siRNA Library (708 genes, three siRNAs per gene) was carried out (Figures 6A and S6B). For quantification of phospho-Bcl2-L-13 (Ser272)-positive dots, local maxima were determined using the “find maxima” function of ImageJ. Candidates that were able to reduce the number of phospho-Bcl2-L-13 (Ser272)-positive dots induced by CCCP treatment were selected. The criteria for selection, which were determined in the preliminary experiments, were an over 60% reduction in phospho-Bcl2-L-13 (Ser272)-positive dots compared with positive controls in at least one of three siRNAs or an over 40% reduction in at least two siRNAs. From this, a collection of 74 genes were identified (Table S3). Furthermore, Z scoring was used to confirm our screening criteria. We obtained 64 genes with a Z score greater than 1.5 as hits. All of them were included in the identified 74 genes (Figure S6C; Table S6).
Figure 6.

AMPKα2 is the kinase responsible for Bcl2-L-13 phosphorylation at Ser272
(A) Schematic of the responsible kinase screening workflow.
(B) In vitro kinase assay in the third screening. Bacterially synthesized HA-Bcl2-L-13 was mixed with purified candidate proteins and ATP. After incubation at 37°C for 30 min, the reaction mix was subjected to western blotting using an anti-phospho-Bcl2-L-13 (Ser272) antibody.
(C) The effect of AMPKα2 knockdown in CCCP-induced Bcl2-L-13 phosphorylation. HEK293A cells stably expressing HA-Bcl2-L-13 were transfected with control siRNA or siAMPKα2 for 72 h. Then, the cells were treated with DMSO or 15 μM CCCP for the indicated times, and cell lysates were subjected to western blot analysis. Densitometric analysis of phospho-Bcl2-L-13 (Ser272) is shown in the bar graph. The value for the group with control siRNA (siCtrl) transfection and 15 min of DMSO treatment in each experiment was set to 1 (n = 3).
(D) Upregulation of AMPKα2 activity by CCCP treatment. To analyze AMPKα2-specific activity, HEK293A cells stably expressing HA-Bcl2-L-13 were transfected with siAMPKα1 for 72 h and then treated with DMSO or 15 μM CCCP. The value for the group with 15-min DMSO treatment in each experiment was set to 1 (n = 4).
(E and F) HEK293A cells were transfected with control siRNA (siCtrl) or siAMPKα2 for 72 h, followed by transfection with an empty vector or HA-Bcl2-L-13. Forty-four hours after transfection, cells were treated with 100 nM bafilomycin A1 for 4 h and immunostained with anti-LC3B and anti-ATP synthase antibodies. Images in the box at higher magnification are shown on the right. White arrows indicate the puncta recognized as colocalized by the software. The number of LC3B dots colocalized with ATP synthase dots per cell is shown in (F). At least 20 cells were counted for each group (n = 3). Scale bar: 10 μm.
(G) Upregulation of AMPKα2 phosphorylation 5 days after TAC operation. To analyze AMPKα2-specific phosphorylation, lysates from the left ventricle were subjected to immunoprecipitation with an anti-AMPKα2 antibody followed by immunoblotting with an anti-phospho-AMPKα (Thr172) antibody. Densitometric analysis of phospho-AMPKα (Thr172) is shown in the right bar graph. The value for the WT sham group in each experiment was set to 1 (n = 3).
(H) Interaction between Bcl2-L-13 and AMPKα2 5 days after TAC. Lysates from the left ventricle were subjected to immunoprecipitation with an anti-AMPKα2 antibody. Co-precipitated Bcl2-L-13 was detected by immunoblotting. Densitometric analysis of Bcl2-L-13 is shown in the graph below. The value for the WT sham group in each experiment was set to 1 (n = 3). Results are shown as mean with 95% CI. Statistical analysis by unpaired, two-tailed t tests in (C), (D), (F), and (H) and one-way ANOVA followed by Tukey-Kramer’s post hoc test in (G). All pairwise comparisons were performed. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. ns, not significant. See also Figures S6 and S7.
To narrow down the candidates, we conducted a secondary screen (Figure S6D). As the knockdown of the responsible kinase should reduce Bcl2-L-13-mediated mitophagy, we evaluated mitophagy induced by CCCP via fluorescence immunocytochemistry using anti-ATP synthase and anti-LC3B antibodies after knockdown of the candidate genes. Mitophagy was evaluated manually by counting the number of ATP synthase dots colocalized with LC3B dots. At least 20 cells were quantified. We carried forward 18 genes that demonstrated a more than 40% reduction of ATP synthase- and LC3B-colocalized dots compared with the positive control (Table S4). The criteria for selection were determined in the preliminary experiment. We also evaluated mitophagy using the JACoP plugin of ImageJ for colocalization analysis.19,20 We identified 9 genes with a Z score greater than 1.0 as hits (Figure S6E; Tables S4 and S6). As seven candidates obtained from counting with software overlapped with those from manual counting, we carried forward 20 genes in total. Since the phosphorylation target is a serine residue, we selected 14 genes that have been reported to have serine/threonine-protein kinase activity. We tested the kinase activity of these 14 candidates by in vitro kinase assay. For this assay, purified candidate kinases were mixed with bacterially synthesized HA-Bcl2-L-13 and 1 mM ATP. The reaction mix was subjected to western blot analysis. Of 14 candidates, AMPKα2 was found to significantly upregulate the phospho-Bcl2-L-13 protein level (Figure 6B). Synthesized HA-Bcl2-L-13 (S272A) was not phosphorylated by purified AMPKα2 protein (Figure S6F).
To validate the screening results, we evaluated Bcl2-L-13 phosphorylation in AMPKα2 knockdown cells. The protein level of phospho-Bcl2-L-13 increased 1 h after CCCP administration (Figure 6C). While knockdown of AMPKα2 significantly reduced the phosphorylation level of Bcl2-L-13, knockdown of the isoform of AMPKα, AMPKα1, did not (Figures 6C and S6G). To confirm the upregulation of AMPKα2 activity by CCCP, we induced knockdown of AMPKα1 and measured AMPKα activity (Figure 6D). CCCP significantly upregulated AMPKα2 activity for 1 h. We tested the effect of a known AMPK inhibitor, compound C, and an AMPK activator, quercetin, on CCCP-induced phosphorylation of Bcl2-L-13. Compound C significantly reduced the protein level of phospho-Bcl2-L-13, while quercetin significantly upregulated it (Figures S7A and S7B). To explore upstream stimuli to activate AMPKα2 other than decreased ATP level, we assessed the effect of a ROS inhibitor, N-acetyl-L-cysteine (NAC), and a calcium chelator, O,O'-Bis(2-aminophenyl)ethyleneglycol-N,N,N',N'-tetraacetic acid, tetraacetoxymethyl ester (BAPTA-AM), on CCCP-induced phosphorylation of Bcl2-L-13 (Figures S7C and S7D). NAC had no effect on the phosphorylation of Bcl2-L-13, while BAPTA-AM inhibited CCCP-induced phosphorylation. Because calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2) is an upstream kinase of AMPK, and its activity depends on intracellular calcium concentration, we conducted a knockdown experiment (Figure S7E). Knockdown of CaMKK2 significantly reduced the phosphorylation level of Bcl2-L-13, suggesting that intracellular calcium signaling through CaMKK2 is important for Bcl2-L-13 phosphorylation. Next, we evaluated Bcl2-L-13-induced mitophagy using the JACoP plugin of ImageJ for colocalization analysis.19,20 As we reported previously, overexpression of Bcl2-L-13 increased mitophagy (Figures 6E and 6F). Knockdown of AMPKα2 significantly reduced ATP synthase and LC3B double-positive dots. Finally, we evaluated the activity of AMPKα2 and AMPKα2-Bcl2-L-13 interaction in TAC-operated hearts. To quantify AMPKα2-specific phosphorylation, we immunoprecipitated AMPKα2 and immunoblotted with an anti-phospho-AMPKα Thr172 antibody (Figure 6G). The phosphorylation of AMPKα2 was increased significantly in TAC-operated Bcl2l13+/+ mice, while phosphorylation of AMPKα2 was suppressed in Bcl2l13−/− hearts 5 days after TAC. To investigate whether Bcl2-L-13 forms a complex with AMPKα2 in the heart after TAC, an immunoprecipitation experiment was conducted (Figure 6H). AMPKα2 precipitated Bcl2-L-13, and the interaction was upregulated significantly following TAC operation. We concluded that AMPKα2 is the kinase responsible for phosphorylating Ser272 on Bcl2-L-13.
Discussion
Our data indicate that Bcl2-L-13 is not required for normal embryonic development. Because the heart is one of the organs that shows the highest mRNA expression levels of Bcl2-L-13, we primarily analyzed the heart in this study. We demonstrated that Bcl2-L-13-deficient mice showed normal cardiac structure and function under basal conditions, indicating that Bcl2-L-13 is not essential for the development or postnatal growth of the heart. In stressed hearts, Bcl2-L-13 plays a protective role in the development of cardiac dysfunction induced by pressure overload.
In our previous study, we hypothesized that a mammalian mitophagy receptor would share certain molecular features with Atg32, which is essential for mitophagy in yeast, and identified Bcl2-L-13 as a functional homolog of Atg32.8 Bcl2-L-13 mediates mitochondrial fission and mitophagy in HEK293 cells. The interaction between Bcl2-L-13 and LC3B is required for Bcl2-L-13-mediated mitophagy, suggesting that Bcl2-L-13 is involved in the canonical mitophagy pathway. From in vivo analysis, our data showed that ablation of Bcl2l13 decreased mitochondrial fission and mitophagy induced by TAC operation in the heart. Parkin is a well-characterized molecule related to the regulation of mitophagy and has been reported to have a protective or detrimental role under pressure overload in the heart.17,18 We examined its involvement in the cardiac dysfunction observed in TAC-operated Bcl2l13−/− mice. However, ablation of Parkin did not demonstrate additional effects on cardiac function after TAC operation, suggesting that Parkin is dispensable for maintaining cardiac function in TAC-operated Bcl2l13−/− hearts. Then, we analyzed mitochondrial respiration and ATP levels. In agreement with previous reports, oligomycin addition did not decrease the OCR in adult mouse cardiomyocytes in this study.21,22,23 This may be explained by decreased ATP demand of noncontracting adult cardiomyocytes and a relatively high rate of proton leak. Bcl2l13−/− hearts could not upregulate basal mitochondrial respiration in response to pressure overload and produced significantly less ATP than Bcl2l13+/+ hearts. These results suggest that attenuation of mitochondrial fission and mitophagy by Bcl2l13 ablation leads to the accumulation of damaged mitochondria, which produce less ATP, ultimately resulting in cardiac dysfunction.
Receptor-mediated mitophagy depends on the phosphorylation of mitochondrial outer membrane-anchored receptors, such as NIX, BNIP3, and FUNDC1. NIX and BNIP3 have been reported to be phosphorylated at Ser34 or Ser17 and Ser24 flanking the LC3-interacting region (LIR), respectively,11,24 while FUNDC1 is phosphorylated at Tyr18, Ser13, and Ser17.10,11,12 We have reported previously that the phosphorylation of Ser272 on Bcl2-L-13, flanking the LIR, is essential for the mitophagic activity of Bcl2-L-13 in HEK293 cells. In this study, our data showed that the protein level of phospho-Bcl2-L-13 (Ser272) was upregulated in response to pressure overload. To investigate the importance of Bcl2-L-13 phosphorylation in pressure-overloaded hearts, we generated Bcl2-L-13 phosphorylation site-deficient knockin mice. The knockin mice demonstrated cardiac dysfunction 4 weeks post-TAC, suggesting that the activation of Bcl2-L-13 by phosphorylation is vital for maintaining cardiac function.
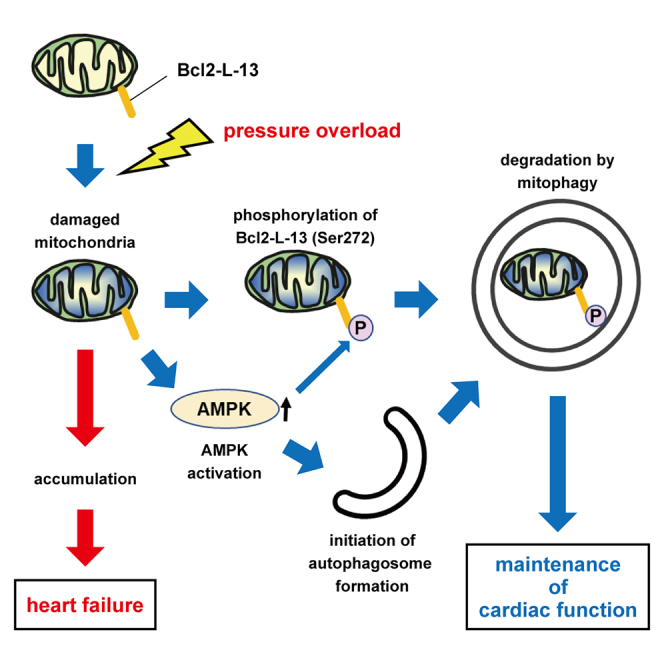
While the kinases responsible for BNIP3 and NIX activation remain unknown, FUNDC1 has been reported to be phosphorylated by Src kinase, casein kinase 2, and ULK1. In this study, we attempted to identify a kinase responsible for Ser272 phosphorylation on Bcl2-L-13. A series of screenings using siRNA or in vitro kinase assays identified AMPKα2 as the responsible kinase. The in vitro kinase assay and co-immunoprecipitation studies from endogenous proteins demonstrated that AMPKα2 binds to Bcl2-L-13 in the stressed heart and directly phosphorylates Bcl2-L-13. Previous studies have reported that AMPK regulates mitophagy through the phosphorylation of ULK1.25 Furthermore, we previously identified a mechanism of Bcl2-L-13-mediated mitophagy induction whereby Bcl2-L-13 recruits the ULK1 complex to recruit mitophagy machinery.26 Myristoylation of the β subunit of AMPK has been shown to localize AMPK to mitochondria during mitophagy.27 Thus, AMPK activates ULK1 to initiate autophagosome formation and migrates to mitochondria to directly phosphorylate Bcl2-L-13 for mitophagy induction. Therefore, we propose a model where pressure overload damages mitochondria, leading to a decrease in ATP production, which activates AMPK. AMPK subsequently phosphorylates Bcl2-L-13 to activate mitophagy for the degradation of damaged mitochondria, which may produce detrimental ROS (Figure 7). Moreover, our in vivo analysis revealed that the phosphorylation of AMPKα2 was upregulated significantly by pressure overload. Unexpectedly, the phosphorylation was attenuated in Bcl2l13−/− hearts. This suggests that Bcl2-L-13 itself might be involved in AMPKα2 activation in the stressed heart.
Figure 7.

A model of the role of AMPK in Bcl2-L-13-mediated mitophagy under pressure overload
Pressure overload damages mitochondria, leading to decreased ATP production and subsequent activation of AMPK. Activated AMPK initiates autophagosome formation and directly phosphorylates Bcl2-L-13 for mitophagy induction.
AMPK is a heterotrimeric complex comprising a catalytic α subunit and two regulatory subunits, β and γ. The two α subunits, α1, and α2, are encoded by the discrete genes PRKAA1 and PRKAA2. Although the α1 and α2 isoforms are 90% identical within the kinase domains, they have been reported to have distinct functions. While α1 KO mice show severe anemia due to less deformability of erythrocytes and higher sensitivity to erythrophagocytosis,28 α2 KO mice are insulin resistant and glucose intolerant.29 A protective role of AMPKα2 in the heart under pressure overload has also been reported.30,31 PRKAA2−/− mice demonstrated significantly exacerbated TAC-induced ventricular hypertrophy and decreased LV ejection fraction. These results are consistent with the phenotype observed in TAC-operated knockin mice in this study.
Interestingly, TAC-operated Bcl2l13−/− mice demonstrated milder hypertrophy than Bcl2l13+/+ mice, while Bcl2-L-13 (S272A) knockin mice showed more severe hypertrophy. Previous studies have reported that the promotion of mitochondrial fission resulted in cardiomyocyte hypertrophy.32,33 Our previously reported in vitro study showed that Bcl2-L-13 knockdown induced mitochondrial elongation, while Bcl2-L-13 overexpression induced mitochondrial fission and mitophagy.8 Although a recent study has shown that Bcl2-L-13 promotes mitophagy through Drp1-mediated mitochondrial fission in glioblastoma,34 Bcl2-L-13 induced mitochondrial fission even in Drp1 knockdown HEK293 cells in our previous report.8 The discrepancy might be due to differences in cell types. In addition, Bcl2-L-13 (S272A) overexpression had no effect on mitochondrial dynamics but inhibited mitophagy, suggesting that Bcl2-L-13 is involved in mitochondrial fission and mitophagy and that its phosphorylation regulates mitophagy but not fission. However, in the current study, TAC-operated knockin mice demonstrated elongated mitochondria, contrary to our expectations. In addition, sham-operated Bcl2l13−/− mice had an increased number of mitochondria with shorter long diameters compared to Bcl2l13+/+ mice. We could not explain these changes based on the results of the protein analyses of fusion/fission-related molecules. Therefore, further studies regarding the mechanisms of Bcl2-L-13 mediated mitochondrial fission, including its relationship to Ser272 phosphorylation, are needed to explain the discrepancies in the mitochondrial dynamics between in vitro and in vivo models and how mitochondrial dynamics are related to pressure overload-induced cardiac hypertrophy.
In summary, Bcl2-L-13 deficiency induced the development of cardiac dysfunction, presumably due to the impairment of mitochondrial fission and mitophagy, and subsequent accumulation of damaged mitochondria. Phosphorylation of Bcl2-L-13 at Ser272 by AMPKα2 plays an important role in this process. Our findings provide the first evidence that Bcl2-L-13 has protective effects on the heart, and activation of Bcl2-L-13 through the phosphorylation at Ser272 could be a potential therapeutic target for heart failure patients. Since the involvement of calcium signaling in AMPKα2-dependent Bcl2-L-13-phosphorylation is suggested in this study, and AMPK is implicated in a wide range of metabolic pathways, it is important to clarify the entire upstream intracellular signaling cascades of AMPKα2 for the specific targeted therapy. In addition, the combination of subunit isoforms in the AMPK complex should also be taken into consideration.
Limitations of the study
We used global KO mice of Bcl2-L-13 in this study to compare the phenotype with global knockin mice used in the following experiment. However, the use of global KO mice is a limitation of the study in that we cannot rule out the effects of Bcl2-L-13 ablation in non-cardiomyocyte cells or in other organs on observed cardiac phenotypes. Cardiomyocyte-specific KO mice would help us to obtain a better understanding of the exact roles of Bcl2-L-13 in cardiomyocytes.
Resource availability
Lead contact
Requests for further information, resources, and reagents should be directed to and will be fulfilled by the lead contact, Kinya Otsu (otsu.kinya@ncvc.go.jp).
Materials availability
The plasmids and mice generated in this study are available upon request.
Data and code availability
-
•
All data supporting the findings of this study are available within the article and its supplemental information. Source data, including images, have been deposited in Figshare: https://doi.org/10.6084/m9.figshare.25284883 and DRYAD: https://doi.org/10.5061/dryad.9cnp5hqsx.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
The work was supported by the European Research Council (692659), the British Heart Foundation (CH/11/3/29051 and RG/16/15/32294), the Fondation Leducq (RA15CVD04), JSPS KAKENHI grant 18H02807 (to K.O.), JSPS KAKENHI grants 22K16102 and 24K11215 (to T.M.), and JSPS KAKENHI grant 21K21350 (to S.O.).
Author contributions
T.M. was responsible for research design, execution, data analysis, and manuscript preparation. J.I., M.-C.R., M.T., J.M.-A., C.N., R.S., H.N., and K.M. were responsible for performing experiments and manuscript preparation. M.Z. was responsible for data analysis and manuscript preparation. S.O. was responsible for conducting and supervising experiments, data analysis, and manuscript preparation. K.N. was responsible for developing the mouse model and the supervision of experiments, data analysis, and manuscript preparation. R.F., A.M.S., O.Y., and Y.S. were responsible for data interpretation and manuscript preparation. K.O. was responsible for the supervision and design of the research, data interpretation, and manuscript preparation.
Declaration of interests
The authors declare no competing interests.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal Anti-HA tag (clone C29F4) | Cell Signaling Technology | Cat# 3724; RRID: AB_1549585 |
| Rabbit polyclonal anti-LC3B | Cell Signaling Technology | Cat# 2775; RRID: AB_915950 |
| Mouse monoclonal anti-Myc Tag (clone 9B11) | Cell Signaling Technology | Cat# 2776; RRID: AB_390779 |
| Rabbit polyclonal anti-Phospho-DRP1 (Ser637) | Cell Signaling Technology | Cat#4867; RRID: AB_10622027 |
| Rabbit polyclonal anti-Phospho-DRP1 (Ser616) | Cell Signaling Technology | Cat# 3455; RRID: AB_2085352 |
| Rabbit monoclonal anti-Mitofusin2 (clone D2D10) | Cell Signaling Technology | Cat# 9482; RRID: AB_2716838 |
| Rabbit monoclonal anti-Mitofusin1 (clone D6E2S) | Cell Signaling Technology | Cat# 14739; RRID: AB_2744531 |
| Rabbit monoclonal anti-OPA1 (clone D6U6N) | Cell Signaling Technology | Cat# 80471; RRID: AB_2734117 |
| Mouse monoclonal anti-GAPDH (clone D4C6R) | Cell Signaling Technology | Cat# 97166; RRID: AB_2756824 |
| Mouse monoclonal anti-α-Tubulin (clone DM1A) | Cell Signaling Technology | Cat# 3873; RRID: AB_1904178 |
| Rabbit polyclonal anti-Parkin | Cell Signaling Technology | Cat# 2132; RRID: AB_10693040 |
| Rabbit monoclonal anti-PINK1 (clone D8G3) | Cell Signaling Technology | Cat# 6946; RRID: AB_11179069 |
| Rabbit polyclonal anti-AMPKα1 | Cell Signaling Technology | Cat# 2795; RRID: AB_560856 |
| Rabbit polyclonal anti-AMPKα2 | Cell Signaling Technology | Cat# 2757; RRID: AB_560858 |
| Rabbit polyclonal anati-AMPKα2 | Proteintech | Cat# 18167-1-AP; RRID: AB_10695046 |
| Rabbit polyclonal anti-BCL2L13 | Proteintech | Cat# 16612-1-AP; RRID: AB_1850928 |
| Mouse monoclonal anti-PGC1-α (1C1B2) | Proteintech | Cat# 66369-1-Ig; RRID: AB_2828002 |
| Mouse monoclonal anti-FLAG tag (clone M2) | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| Mouse monoclonal anti-α-sarcomeric actin | Sigma-Aldrich | Cat# A2172; RRID: AB_476695 |
| Rabbit polyclonal anti-TTC11 | abcam | Cat# ab96764; RRID: AB_10679033 |
| Rabbit polyclonal anti-HMGB1 | abcam | Cat# ab18256; RRID:AB_444360 |
| Mouse monoclonal anti-ATP synthase | Life Technologies | Cat# A-21351; RRID: AB_221512 |
| Mouse monoclonal anti-Drp1 (clone 22) | BD Transduction Laboratories | Cat# 611738; RRID: AB_399214 |
| Sheep Anti-Mouse IgG HRP Conjugated | Amersham | Cat#NA931; RRID: AB_772210 |
| Donkey Anti-Rabbit IgG HRP Conjugated | Amersham | Cat# NA934; RRID: AB_772206 |
| IRDye 800CW Donkey anti-Goat IgG | LI-COR Biosciences | Cat# 926–32214; RRID: AB_621846 |
| IRDye 680LT Goat anti-Mouse IgG | LI-COR Biosciences | Cat# 926–68020; RRID: AB_10706161 |
| IRDye 680RD Donkey anti-Rabbit IgG | LI-COR Biosciences | Cat# 926–68073; RRID: AB_10954442 |
| IRDye 800CW Donkey anti-Mouse IgG | LI-COR Biosciences | Cat# 926–32212; RRID: AB_621847 |
| IRDye 800CW Goat anti-Rabbit IgG | LI-COR Biosciences | Cat# 926–32211; RRID: AB_621843 |
| Goat anti-Mouse IgM (Heavy chain) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 568 | Thermo Fisher Scientific | Cat# A-21043; RRID:AB_2535712 |
| Goat anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 488 | Thermo Fisher Scientific | Cat# A-11034; RRID:AB_2576217 |
| Donkey anti-Goat IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 568 | Thermo Fisher Scientific | Cat# A-11057; RRID:AB_2534104 |
| Chicken Anti-Goat IgG (H + L) Antibody, Alexa Fluor 647 Conjugated | Thermo Fisher Scientific | Cat# A21469; RRID:AB_10374877 |
| Donkey anti-Mouse IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 568 | Thermo Fisher Scientific | Cat# A10037; RRID:AB_11180865 |
| Bacterial and virus strains | ||
| Genlantis TurboCells BL21Competent Cells | Gelantis | AMS.C302020 |
| Chemicals, peptides, and recombinant proteins | ||
| RNAi MAX | Invitrogen | Cat# 13778030 |
| ScreenFect A | Wako | Cat# 299-73203 |
| Penicillin-Streptomycin-Glutamine | Gibco | Cat# 10378016 |
| geneticin | Gibco | Cat# 10131-027; CAS: 108321-42-2 |
| Bafilomycin A1 | LC Laboratories | Cat# B-1080; CAS: 88899-55-2 |
| ECL Prime Western Blotting Detection Reagent | GE Healthcare Life Science | Cat# RPN2232 |
| Lumigen ECL Ultra | Lumigen | Cat# TMA-100 |
| CCCP | Sigma-Aldrich | Cat# C2759; CAS: 555-60-2 |
| ProLong Gold Antifade Mountant with DAPI | Thermo Fisher Scientific | Cat# P36935 |
| MitoSox | Life Technologies | Cat# M36008 |
| fluorescein isothiocyanate-conjugated lectin | Sigma | L4895 |
| protease inhibitor cocktail | Cell Signaling Technology | #5871 |
| phosphatase inhibitor cocktail | Cell Signaling Technology | #5870 |
| Dynabeads Protein A for Immunoprecipitation | Thermo Fisher Scientific | Cat# 1001D |
| GST Spin Purification Kit | Thermo Scientific Pierce | Cat# 11804025 |
| PreScission Protease | GE Healthcare Lifescience | – |
| AMPK A2/B1/G1 Recombinant Human Protein | Thermo Fisher Scientific | Cat# PV4674 |
| CAMK2B (CaMKII Beta) Recombinant Human Protein | Thermo Fisher Scientific | Cat# PV4205 |
| ERK4 Recombinant Protein | SignalChem | Cat# M30-34G-20 |
| FASTK Recombinant Protein | SignalChem | Cat# F01-10G-05 |
| Haspin Recombinant Protein | SignalChem | Cat# G10-11G-05 |
| IRAK2 Recombinant Protein | SignalChem | Cat# I10-10BG-05 |
| NLK Recombinant Human Protein | Thermo Fisher Scientific | Cat# PV4309 |
| CDC42 BPA (MRCKA) Recombinant Human Protein | Thermo Fisher Scientific | Cat# PV4398 |
| Recombinant human PKM2 protein | abcam | Cat# ab89364 |
| MAP3K7/MAP3K7IP1 (TAK1-TAB1) Recombinant Human Protein | Thermo Fisher Scientific | Cat# PV4394 |
| ACVR2A Human Protein | Thermo Fisher Scientific | Cat# PV6124 |
| AAK1 Recombinant Human Protein | Thermo Fisher Scientific | Cat# A30967 |
| RIPK3 Recombinant Protein | SignalChem | Cat# R09-10G-05 |
| PKAc beta, active | Eurofins Pharma Discovery | Cat# 15-007 |
| BAPTA-AM | TCI chemicals | Cat# T2845 |
| Dorsomorphin | FUJIFILM Wako | Cat# 044-33751 |
| Quercetin | Sigma-Aldrich | Cat# Q4951 |
| N-acetylcysteine | Sigma-Aldrich | Cat# A9165: CAS: 616-91-1 |
| Critical commercial assays | ||
| ATP Assay Kit | abcam | Cat# ab83355 |
| In situ Apoptosis Detection Kit | Takara Bio | Cat# MK500 |
| AMPK Kinase Assay Kit | Cyclex | Cat# CY-1182 |
| DNeasy Blood & Tissue Kit | QIAGEN | Cat# 69504 |
| mouse Real-time PCR Mitochondrial DNA Damage Analysis Kit | Detroit R&D | Cat# DD2M |
| Deposited data | ||
| Raw and analyzed data | This paper; Figshare | Tables S5, S6 and S7; https://doi.org/10.6084/m9.fig.share.25284883 |
| Images from primary and second screening | This paper; Dryad | https://doi.org/10.5061/dryad.9cnp5hqsx |
| Experimental models: Cell lines | ||
| Human: HEK293A Cell line | Invitrogen | Cat# R70507; RRID: CVCL_6910 |
| Experimental models: Organisms/strains | ||
| Bcl2-L-13 knockout mice | This paper | N/A |
| Bcl2-L-13 S272A knockin mice | This paper | N/A |
| Oligonucleotides | ||
| Silencer™ Human Kinase siRNA Library | ThermoFisher Scientific | Cat# A30079 |
| siRNA: AMPKα1 | ThermoFisher Scientific | Cat# AM51551, siRNA ID: 768 |
| siRNA: AMPKα2 | ThermoFisher Scientific | Cat# AM51331, siRNA ID: 771 |
| Negative Control No. 1 siRNA | ThermoFisher Scientific | Cat# 4390843 |
| Recombinant DNA | ||
| pDONR223-PRKAA2 | Johannessen et al.35 | Addgene; Cat# 23671 |
| HA-Bcl2-L-13-pcDNA3.1 | Murakawa et al.8 | N/A |
| myc-AMPKα2-pcDNA3.1 | This paper | N/A |
| pGEX-Bcl2-L-13 | This paper | N/A |
| Software and algorithms | ||
| ImageJ (Version 1.52p) | National Institue of Health, USA | https://imagej.nih.gov/ij/index.html |
| Image Studio | LI-COR Biosciences | N/A |
| ImageQuantTL v7.0 | GE Healthcare Life Sciences | – |
| Graphpad Prism 9 | GraphPad Software | N/A |
Experimental model and study participant details
Cell culture
HEK293A cells (RRID: CVCL_6910) were obtained from Invitrogen and were grown in Dulbecco’s modified Eagle’s medium (D5671, Sigma) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin-glutamine (10378016, Gibco) at 37°C under 5% CO2. The sex of the cells was female.
Animal study
All animal studies were approved by the King’s College London Ethical Review Process Committee and UK Home Office (project license PPL70/7260) and the Animal Research Committee of Osaka University. All experiments were carried out in accordance with the U.K. Animals (Scientific Procedures) Act 1986, and the associated guidelines, Directive 2010/63/EU for animal experiments and in accordance with the Guidelines for Animal Experiments of Osaka University and the Japanese Animal Protection and Management Law. The authors complied with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines.
Generation of conventional Bcl2l13 knockout mice
The mouse C57BL/6J BAC genomic library (BACPAC Resources Center) was used to generate a homology sequence for the Bcl2l13 gene targeting vector. The homology sequence was generated by high-fidelity polymerase chain reaction (PCR) and cloned into the targeting vector containing the DT-loxP-PGK-Neo-loxP cassette. The targeting vector was electroporated into ES cells (F1; SVJ129 and C57BL/6J). The transfected ES clones were subsequently selected for neomycin resistance. Southern blotting and karyotyping analyses were performed to obtain ES clones exhibiting the desired homologous recombination and normal karyotype. These targeted ES clones were injected into blastocyst mouse embryos to generate chimeric mice. The chimeric mice were crossed with C57BL/6J mice to validate germ-line transmission. The offspring with floxed Bcl2l13 mice were crossed with transgenic mice expressing Cre recombinase under the control of the β-actin promoter (βActin-Cre mice) in the C57BL/6J background to generate conventional Bcl2l13 gene deletion. Mice were backcrossed onto a C57BL/6J background for six generations. Genotyping was determined using the following primer sets: 5′- TGT GGA TAC CAT TCT CTT CCT GT-3’ and 5′-AAG TTG GCT TTT GAG ACG TAC C-3′ to amplify the deleted allele, and 5′- GGT TTT TAT TGA CCT GGT GAG C -3' and 5′- AAG TTG GCT TTT GAG ACG TAC C -3' to amplify the wild-type allele.
To generate double knockout mice of Bcl2l13 and Park2, Bcl2l13−/− mice were crossed with Park2−/− mice in the C57BL/6J background. Park2−/− mice were purchased from The Jackson Laboratory.
Generation of Bcl2-L-13 (S272A) knock-in mice
Bcl2-L-13 (S272A) knock-in phospho-deficient mice were generated by genOway. The S272A mutation was introduced by replacing the TCC (serine) codon with the GCA (alanine) codon, located in exon 8 of the Bcl2l13 gene. The targeting vector containing a Neo expression cassette for positive selection and a DTA expression cassette for negative selection was electroporated in ES cells. Selected ES cell clones were then assessed by PCR and Southern blot analysis to validate the presence of the correct recombination event. These ES clones were injected into blastocysts to create chimeric mice. Chimeric mice were subsequently mated with C57BL/6 Cre deleter mice to excise the Neo selection cassette and generate heterozygous mice carrying the Neo-excised knock-in point mutant allele. The obtained progeny were genotyped by PCR, and the recombinase-mediated excision event was further validated by Southern blot analysis. Genotyping was done using the forward primer 5′- GAA AAT CAT TCT TAG CTA ACC TCA GTT TCA GGA G -3' and the reverse primer 5′- TGA GGC TGG ACA GAA TAT TGA GAC TGT TG -3'. For all experiments, wild-type and homozygotes were generated exclusively by breeding heterozygotes. To avoid potential sex-specific differences, all experiments used male mice. Mice were randomly assigned to control or TAC groups, and studies were performed unblinded regarding mouse genotypes.
Method details
TAC and echocardiography
The 8 to 12-week-old mice were subjected to TAC using a 25-gauge needle.36 Sham-operated mice underwent the same operation without aortic constriction. A 6–0 silk suture was placed around the transverse aorta (between the innominate and left common carotid artery) and tied loosely in a single knot. A pre-sterilized, blunt-end 25-gauge needle was placed within the knot alongside the transverse aorta. The knot was tightened fully with an additional knot, and the blunt-end needle was removed.
Echocardiography was conducted using the Vevo 2100 system (Visual Sonics) on conscious mice.37 Non-invasive measurements of the tail blood pressure were also performed on conscious mice using a BP Monitor for rats and mice (Muromachi Kikai).
Antibodies
The following primary antibodies were purchased: anti-HA tag (#3724), anti-myc tag (#2276), anti-LC3B (#2775), anti-pDrp1 Ser637 (#4867), anti-pDrp1 Ser616 (#3455), anti-Mfn2 (#9482), anti-Mfn1 (#14739), anti-OPA1 (#80471), anti-GAPDH (#97166), α-tubulin (#3873), anti-Parkin (#2132), anti-PINK1 (#6946), anti-AMPKα1 (#2795), and anti-AMPKα2 (#2757) from Cell Signaling Technology, anti-Bcl2-L-13 (16612-1-AP), anti-PGC1α (66369-1-AP) and anti-AMPKα2 (18167-1-AP) from Proteintech, anti-FLAG (F1804), and anti-α-sarcomeric actin (A2172) from Sigma-Aldrich, anti-Fis1 (ab96764), and anti-HMGB1 (ab18256) from Abcam, anti-ATP synthase (A-21351, Life Technologies), and anti-Drp1 (611738, BD Transduction Laboratories). Anti-phospho-Bcl2-L-13 (Ser272) was generated by the Medical Research Council. A peptide corresponding to amino acid residues 267–277 of mouse Bcl2-L-13, in which phosphorylation site (serine) is substituted to aspartic acid (SLGPEDWQQIA), was injected into a sheep. The antisera were affinity-purified on phosphopeptide–agarose, followed by chromatography on the corresponding dephospho peptide–agarose column to remove antibodies that recognize dephosphorylated Bcl2-L-13.
Secondary antibodies used were the following: Amersham ECL Mouse IgG (NA931) and Amersham ECL Rabbit IgG (NA934) from GE Healthcare, and IRDye 800CW Donkey anti-Goat IgG (H + L) (926–32214), IRDye 680LT Goat anti-Mouse IgG (H + L) (926–68020), IRDye 680RD Donkey anti-Rabbit IgG (H + L) (926–68073), IRDye 800CW Donkey anti-Mouse IgG (H + L) (926–32212) and IRDye 800CW Goat anti-Rabbit IgG (H + L) (926–32211) from LI-COR Biosciences, and Goat anti-mouse IgM (Heavy chain) Alexa Fluor 568 (A21043), Goat anti-rabbit IgG (H + L) Alexa Fluor 488 (A11034), Donkey anti-goat IgG (H + L) Alexa Fluor 568 (A11057), Donkey anti-Mouse IgG (H + L) Alexa Fluor 568 (A10037), and Chicken anti-goat IgG (H + L) Alexa Fluor 647 (A21469) from Invitrogen.
Transfection
Transient transfections were performed using ScreenFect A (299–73203, Wako). After 48 h, cells were subjected to analysis unless otherwise indicated. For siRNA-mediated knockdown, cells were transfected with 20 nM AMPKα1 siRNA (AM51551, siRNA ID: 768) and 20 nM AMPKα2 siRNA (AM51331, siRNA ID: 771) from Thermo Fisher using 3.75 μL/mL of RNAiMAX (13778150, Invitrogen). The non-targeting siRNA control (4390843) was obtained from Thermo Fisher. After 72 h of transfection, cells were subjected to analysis unless otherwise indicated.
Construction of plasmids
To generate HA-Bcl2-L-13-pcDNA3.1, N-terminal hemagglutinin (HA) -tagged mouse Bcl2-L-13 was cloned into pcDNA3.1.8 The pDONR223-PRKAA2 was a gift from William Hahn & David Root (Addgene plasmid # 23671).35 To generate myc-AMPKα2-pcDNA3.1, human AMPKα2 was amplified from pDONR223-PRKAA2 with forward (5′- AGC AGA ATT CGA TGG CTG AGA AGC AGA AGC AC -3') and reverse (5′- AGC AGG ATC CTC AAC GGG CTA AAG TAG TAA T -3') primers. The amplified fragment was directly inserted into pCR-Blunt II-TOPO and then subcloned into pcDNA3.1. To obtain pGEX-Bcl2-L-13, Bcl2-L-13 (1–407) including HA tag was amplified from HA-Bcl2-L-13-pcDNA3.1 with forward (5′- AGC AGG ATC CGC CGC CAT GGA GTA CCC ATA CGA CGT A -3') and reverse (5′- AGC ACT CGA GTC AGG CCT TGC CCT CGG CGG GCA GGC CAC T -3') primers. The amplified fragment was directly inserted into pCR-Blunt II-TOPO and subcloned into the BamHI/XhoI site of pGEX-6P-2.
Construction of HA-Bcl2-L-13 stable cell line
The HA-Bcl2-L-13-pcDNA3.1 plasmid was transfected into HEK293A cells using ScreenFect A. After 48 h, the cells were passaged, and 1 mg/mL G418 for selection was added 24 h later. After 14 days, the single colonies were transferred to 24-well plates and expanded.
Southern blot analysis
Genomic DNAs (gDNAs) were purified from embryonic stem cells using the Qiagen DNeasy kit (Qiagen) according to the manufacturer’s instructions. Approximately 10 μg of gDNA was digested with restriction enzymes and ran in a 1% agarose gel for 20 h at 30 V. Genomic DNA fragments were transferred from the gel via capillary action to a nylon membrane (0.45 μm pore) using alkaline transfer. The membrane was then exposed to a source of UV light for 3 min for cross-linking. Probes for hybridization were labeled with 32P-dCTP using the Amersham Megaprime DNA labeling system (GE Healthcare). Hybridization was performed at 68°C overnight using the QuikHyb Rapid Hybridization solution (Agilent).
Histological analysis
Left ventricle samples were embedded in an OCT compound (Thermo Fisher Scientific Inc) and then immediately frozen in liquid nitrogen. The samples were cut into 5 μm-thick sections. The sections were fixed with acetone for hematoxylin–eosin and Masson’s trichrome staining and with 4% paraformaldehyde for wheat germ agglutinin staining. For wheat germ agglutinin staining, heart samples were stained with fluorescein isothiocyanate-conjugated lectin (L4895, Sigma) to measure the cross-sectional area of cardiomyocytes.
Images were captured by a fluorescence microscope (BZ-X700, Keyence). The fibrosis fraction and cardiomyocyte cross-sectional area were examined using ImageJ (National Institutes of Health; Version 1.52p).
Quantitative RT–PCR
Total RNA was isolated from the left ventricles using the TRIzol reagent (Thermo Fisher Scientific). The mRNA expression levels were determined by quantitative reverse transcription PCR using a SuperScript IV reverse transcriptase (Thermo Fisher Scientific) for reverse transcription and a PowerUp SYBR Green PCR Master Mix (Thermo Fisher Scientific) for the quantitative reverse transcription PCR reaction with the following PCR primers: forward 5′- TCG TCT TGG CCT TTT GGC T -3′ and reverse 5′- TCC AGG TGG TCT AGC AGG TTC T -3′ for Nppa, forward 5’ -AAG TCC TAG CCA GTC TCC AGA -3′ and reverse 5′- GAG CTG TCT CTG GGC CAT TTC -3′ for Nppb, and forward 5′- ATG ACA ACT TTG TCA AGC TCA TTT -3′ and reverse 5′- GGT CCA CCA CCC TGT TGC T -3′ for Gapdh. All data were normalized to the Gapdh mRNA content and expressed as a fold increase over the control group.
SDS–PAGE and western blotting
Cells were washed in ice-cold PBS and lysed in lysis buffer (50 mM Tris-HCl, 137 mM NaCl, 1 mM EDTA, 10% glycerol, 1% Triton X-100, a protease inhibitor cocktail (#5871, Cell Signaling), and a phosphatase inhibitor cocktail (#5870, Cell Signaling), pH 8.0) on ice.
For mouse tissue, samples were homogenized and lysed in lysis buffer (25 mM Tris-HCl, 150 mM NaCl, 1% deoxycholic acid sodium salt, 0.1% SDS, 1.0% NP-40 substitute, pH 7.6) on ice. Proteins were subjected to SDS–PAGE and transferred to a nitrocellulose membrane. Membranes were incubated with primary antibodies overnight at 4°C, followed by incubations with secondary antibodies at room temperature (RT) for 1 h. The blot was developed using an infrared imaging system (ODYSSEY CLx, LI-COR Biosciences) or ImageQuant LAS4000mini (GE Healthcare Life Sciences). Image Studio software (LI-COR Biosciences) or ImageQuantTL v7.0 (GE Healthcare Life Sciences) was used for quantitative analysis to evaluate protein expression levels.
Immunoprecipitation
To investigate if Bcl2-L-13 forms a complex with AMPKα2, an immunoprecipitation experiment was conducted. Lysates from LV were precleared with 30 μL of magnetic beads-coupled protein A (1001D, Invitrogen) and 1 μg of rabbit immunoglobulin G. Precleared lysates were subjected to immunoprecipitation using 1 μg of the anti-AMPKα2 antibody or rabbit immunoglobulin G and 30 μL of magnetic beads-coupled protein A at 4°C for 4 h. The precipitated complexes were washed three times with lysis buffer for immunoblotting by an anti-Bcl2-L-13 antibody.
Sample preparation for electron microscopy
Before perfusion fixation, 5 U heparin and 50 mg pentobarbital/kg body weight were intraperitoneally administered to mice. Perfusion was performed using an infusion syringe pump (Harvard Apparatus). To avoid blood clots forming during fixation, the blood was washed from the circulatory system via perfusion with a buffer containing 0.1% NaNO2 and 2.5% polyvinylpyrrolidone (PVP) in 100 mM PIPES(pH 7.2). Mouse hearts were fixed by perfusion fixation using 2% glutaraldehyde (EM grade, Sigma-Aldrich) and 2% formaldehyde (EMS) in 100 mM PIPES(pH 7.2) containing 0.1% NaNO2 and 2.5% PVP. The hearts were excised and kept in a fixative solution on ice for 1 h before dissection. The left ventricular free walls were dissected from the fixed hearts and cut into 1–2 mm3 blocks. Post-fixation was carried out for 1 h at 4°C in 2% OsO4 in 100 mM sodium cacodylate (pH 7.2). Blocks were thoroughly washed with double-distilled water and then dehydrated using ethanol series (10%, 50%, 70%, and 100% ethanol) and propylene oxide. The blocks were embedded in Agar100 epoxy resin (Agar Scientific) following overnight infiltration. Tissue blocks were sectioned using a Leica Ultracut (EM UC 7) ultramicrotome. Sections were collected on Maxtaform type H6 copper finder grids (Agar Scientific) coated in-house with Pioloform and carbon films or on commercially available Formvar/Carbon slot grids (Agar Scientific). Sections were stained with UranyLess and Reynolds Lead Citrate (TAAB Laboratories Equipment Ltd.).
Electron microscopy imaging and analysis
For each experimental group, three to four animals were included in the analysis and random sampling of the left ventricle was ensured by sectioning three blocks from each mouse. Tissue sections were imaged using a 120 kV JEOL JEM-transmission electron microscope (TEM) equipped with the JEOL Ruby CCD camera. Ultrastructural assessment of the cardiac tissue was carried out on 70-nm thick sections. For assessing mitochondrial morphological parameters, areas consisting of longitudinal muscle fibers were imaged at a nominal magnification of 5000× (283.5 pixels/μm). Only inter-myofibrillar mitochondria were included in the frequency distribution analysis. Micrographs were analyzed using the open-source image processing software FIJI.38,39 Using the Freehand selection tool, inter-myofibrillar mitochondria were traced. The long diameter is the primary axis of the best-fitting ellipse to the traced pixels. In an animal, 504 to 728 mitochondria were measured and three to four animals were evaluated in each group. Frequency distribution was calculated in Microsoft Excel. GraphPad Prism was used to generate violin plots and to assess the difference between distributions of mitochondria morphological parameters. Statistical significance was evaluated using the Kruskal-Wallis statistical test. Numerical data of the measurement of mitochondria are provided in Table S5.
siRNA library screening
For the primary screen, the Silencer Human Kinase siRNA Library (three siRNAs per gene) targeting 708 genes (ThermoFisher Scientific, A30079) was used. 96-well tissue culture plates were prearrayed with 3.6 pmol of siRNA and 0.24 μL of Lipofectamine RNAiMAX (Invitrogen) per well. Reverse transfection of 3,000 HEK293A cells stably expressing HA-Bcl2-L-13 was performed with a 30 nM final concentration of siRNAs. A non-targeting siRNA control was used as the negative control.
Seventy-two hours post-transfection, 15 μM CCCP was added to induce mitophagy together with 100 nM bafilomycin A1 for 4 h. Cells were fixed with 4% paraformaldehyde in PBS and permeabilized with 0.1% Triton X-100 in PBS. Cells were then incubated with the anti-phospho-Bcl2-L-13 (Ser272) antibody overnight at 4°C, followed by incubation with anti-goat Alexa 568 for 1 h at RT. After washing, images were obtained by automated scanning using a fluorescence microscope (BZ-X700, Keyence; ×20 objective lens, ×2 digital zoom, nine view fields per well). For quantification of phospho-Bcl2-L-13 (Ser272) puncta, local maxima were determined using the “find maxima” function of the ImageJ software package. We chose target genes that demonstrated an over 60% reduction compared with positive control well in at least one out of three siRNAs or an over 40% reduction in at least two siRNAs for the secondary screen. Furthermore, Z scoring was used to confirm our screening criteria. We selected genes with a Z score greater than 1.5 as hits. We carried forward genes that fulfilled each of those criteria. The Z′ factor for the primary screening was calculated using the DMSO-treated samples (positive control for inhibition) and control siRNA-treated samples (negative control for inhibition). The Z′ factor for the primary screen was calculated to be 0.91.
For the secondary screen, HEK293A cells stably expressing HA-Bcl2-L-13 were transfected with candidate siRNA using RNAiMAX. A non-targeting siRNA control was used as the positive control. Seventy-two hours after the transfection, cells were treated with 15 μM CCCP and 100 nM bafilomycin A1 for 4 h. Cells were fixed and permeabilized with methanol for 10 min at −20°C. Cells were then incubated with rabbit anti-LC3B antibody and mouse anti-ATP synthase antibody overnight at 4°C, followed by incubation with secondary antibodies for 1 h at RT. After washing, cells were mounted with ProLong Gold Antifade Mountant and analyzed using a Nikon Ti-Eclipse inverted microscope (Nikon). Mitophagy was manually evaluated by counting the number of ATP synthase dots colocalized with LC3B dots. At least 20 cells were quantified. Target genes that demonstrated an over 40% reduction compared with the positive control were selected for the in vitro kinase assay. We also evaluated mitophagy using the JACoP plugin of ImageJ for colocalization analysis.19,20 We chose genes with a Z score greater than 1.0 as hits. The Z′ factor for the secondary screening was calculated using the DMSO-treated samples (positive coFfntrol for inhibition) and control siRNA-treated samples (negative control for inhibition). The Z′ factor for the secondary screen was calculated to be 0.76. We carried forward all genes that were selected under either of those two evaluation methods. Numerical data of the primary and secondary screens are provided in Table S6.
In vitro kinase assay
The GST-fusion proteins of HA-Bcl2-L-13 or HA-Bcl2-L-13 (S272A) were induced in the BL21 Escherichia coli strain (DE3; AMS.C302020, Genlantis) transfected with pGEX-Bcl2-L-13 or pGEX-Bcl2-L-13 (S272A) by adding 1 mM isopropyl-β-D-thiogalactoside (10724815001, Roche) for 3.5 h. Bacteria were lysed with PBS containing 1 mg/mL lysozyme (10536394, Fisher Bioreagents), 30 U/ml Benzonase (E1014-5KU, Sigma), 1 mM dithiothreitol, 1 mM EDTA, 1% Triton X-100 and protease inhibitors. GST-Bcl2-L-13 or GST-Bcl2-L-13 (S272A) were purified using the GST Spin Purification Kit (11804025, Thermo Scientific Pierce). The GST tag was cleaved using the PreScission Protease (10196324, GE Healthcare Lifescience) on a column.
For the in vitro kinase assay, 0.5 μg purified kinases were mixed with 0.5 μg bacteria-synthesized proteins from HA-Bcl2-L-13 or HA-Bcl2-L-13 (S272A), 1 mM ATP, and the kinase reaction buffer (50 mM Tris-HCl, 10 mM MgCl2, 2 mM dithiothreitol, pH7.4).
After incubation at 30°C for 30 min, the reaction mix underwent Western blotting. Purified kinases were purchased from SignalChem or Thermo Fisher Scientific.
ATP assay
Approximately 10 mg of the left ventricle was homogenized in ice-cold 2 N perchloric acid (PCA). Then, excess PCA was precipitated via the addition of ice-cold 2 M KOH, and the pH was adjusted to 6.5–8.0. The ATP concentrations of the deproteinized samples were measured using the ATP Assay Kit from Abcam (ab83355) according to the manufacturer’s instructions. Fluorometric assay (Ex/Em = 535/587 nm) was performed using the TECAN INFINITE 200 plate reader. Results were normalized to the weight of each sample.
AMPK activity assay
HEK293A cells stably expressing HA-Bcl2-L-13 were transfected with siAMPKα1 for 72 h and then treated with DMSO or 15 μM CCCP. Cells were lysed in lysis buffer (50 mM Tris-HCl, 137 mM NaCl, 1 mM EDTA, 10% glycerol, 1% Triton X-100, protease inhibitor cocktail (#5871, Cell Signaling), phosphatase inhibitor cocktail (#5870, Cell Signaling), pH 8.0). Five mg of lysate was resuspended in 90 μL AMPK kinase assay buffer (Cyclex, CY-1182). After a 30-min incubation at 30°C with the substrate peptide, IRS-1 S789, the activity of AMPK was analyzed according to the manufacturer’s instructions.
Isolation of mouse adult cardiomyocytes
The hearts were quickly excised from anesthetized mice and cannulated via the aorta. Then hearts were perfused for 1 min at 37°C with a perfusion buffer containing 120 mM NaCl, 5.4 mM KCl, 1.6 mM MgCl2, 1.2 mM NaH2PO4, 5.6 mM glucose, 20 mM NaHCO3, and 5 mM taurine (Sigma-Aldrich), followed by perfusion with collagenase buffer containing 1.2 mg/mL collagenase type 2 (Worthington Biochemical Corporation), and 0.016 mg/mL protease type XIV (P-5147, Sigma-Aldrich). Isolated cardiomyocytes were plated onto laminin (23017-015, Invitrogen)-coated plates and cultured in Minimum Essential Medium Eagle (MEM; M5650, Sigma-Aldrich) supplemented with 2.5% FBS and L-Glutamine–Penicillin–Streptomycin solution (G6784, Sigma-Aldrich).
MitoSOX analysis
Isolated mouse adult cardiomyocytes were plated onto laminin-coated glass-based dishes. Cells were stained with 5 μM MitoSOX (M36008, Life Technologies) and 150 nM MitoTracker Green for 30 min before confocal microscopic analysis was carried out using a Nikon Ti-Eclipse inverted microscope (Nikon) equipped with a Yokogawa CSU-X1-M2 spinning disk unit (Yokogawa) and an Andor Neo sCMOS camera (Andor Technology).
Assessment of mitochondrial function
The oxygen consumption of adult mouse cardiomyocyte cultures was quantified using an extracellular flux analyzer (Seahorse, Agilent Technologies). Cardiomyocytes were isolated five days after the TAC operation and cultured on laminin-coated Seahorse XFe24 culture plates (100,000 cells/well). Basal OCR and maximal OCR were analyzed with the sequential administration of 1 μM oligomycin, 2 μM Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) and 2 μM rotenone and 4 μM antimycin A.
Mitochondrial DNA damage assay
Total DNA was extracted from TAC-operated mouse LV using DNeasy Blood & Tissue Kit (69504, QIAGEN). Mitochondrial DNA damage was assessed using a mouse Real-time PCR Mitochondrial DNA Damage Analysis Kit (DD2M, Detroit R&D). The kit measures levels of mitochondrial DNA damage by replicating 8.2 kb mtDNA. If there is damage to the DNA, PCR cannot go through. Thus, higher long QPCR product formation represents less DNA damage. Samples (5 ng/uL DNA) were subjected to PCR reaction, after which PCR reaction product was subjected to real-time PCR. The quantification of the products was calculated from the standard curve using an 8.2 kb mtDNA standard supplied with the kit.
Assessment of mitophagy in in vitro
HEK293A cells were transfected with siRNA using RNAiMAX. Seventy-two hours after the initial treatment, cells were split and transfected with the expression plasmids using ScreenFect A according to the manufacturer’s instructions. Forty-four hours after the second treatment, 100 nM bafilomycin A1 was added. Four hours later, cells were fixed and permeabilized with methanol for 10 min at −20°C. Cells were incubated with primary antibodies overnight at 4°C followed by incubation with secondary antibodies for 1 h at RT. After washing, cells were mounted with ProLong Gold Antifade Mountant (P36935, Invitrogen) and analyzed using a Nikon Ti-Eclipse inverted microscope (Nikon) equipped with a Yokogawa CSU-X1-M2 spinning disk unit (Yokogawa) and an Andor Neo sCMOS camera (Andor Technology).
To visualize the mitochondria and autophagosomes, cells were immunostained with anti-ATP synthase and anti-LC3B antibodies. Mitophagy was evaluated by counting the number of ATP synthase dots colocalized with LC3B dots. The JACoP plugin of ImageJ was used for colocalization analysis. At least 20 cells were quantified for each group.19,20
Assessment of mitophagy in in vivo hearts
Left ventricle samples were embedded in the OCT compound (Thermo Fisher Scientific Inc) and immediately frozen in liquid nitrogen. The samples were sectioned into 5 μm-thickness and fixed with methanol for 10 min at −20°C. For detection of mitophagy, mitochondria, and autophagosomes were immunostained using anti-ATP synthase and anti-LC3B antibodies. Mitophagy was estimated by counting the number of ATP synthase dots colocalized with LC3B dots. More than 30 view fields per sample were obtained using the FV3000 confocal microscope (Olympus).
TUNEL staining
Frozen heart sections were stained with In situ Apoptosis Detection Kit (MK500, Takara Bio) and anti-α-sarcomeric actin antibody (A2172, Sigma-Aldrich) followed by the secondary antibody Alexa Fluor 568 Anti-Mouse IgM (A21043, Invitrogen). Samples were mounted with ProLong Gold Antifade Reagent with DAPI (P36962, Invitrogen). Five view fields per sample were obtained using a fluorescence microscope (BZ-X700, Keyence) and counted signals from TUNEL staining colocalized with nuclei.
Detection of necrosis
For immunohistochemical analysis of necrosis, frozen heart sections (5 μm-thickness) were fixed in buffered 4% paraformaldehyde. The primary and secondary antibody used was a polyclonal rabbit antibody to HMGB1 (ab18256, Abcam, 1:100 dilution) and anti-rabbit IgG Alexa Fluor 488 conjugated antibody (A11034, Invitrogen, 1:100 dilution). Heart sections were also incubated with a mouse monoclonal antibody to α-sarcomeric actin and were then mounted with ProLong Gold Antifade Reagent with DAPI. Five view fields per sample were obtained using a fluorescence microscope (BZ-X700, Keyence) and evaluated HMGB1 and nucleus to calculate the percentage of HMGB1-negative cardiomyocytes.
Quantification and statistical analysis
The number of independent biological repeats (n) is shown in the figure legends. No prior estimation of sample sizes or test for normal distribution was conducted. Results are shown as mean with 95% CI. Statistical analyses were performed using GraphPad Prism 9 (GraphPad Software). Paired data were evaluated by unpaired, two-tailed t test. A one-way analysis of variance (ANOVA) followed by Tukey–Kramer’s post hoc test was used for multiple comparisons. A value of p < 0.05 was considered statistically significant. Source data for statistical analysis are provided in Table S7.
Published: November 23, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.115001.
Supplemental information
References
- 1.McDonagh T.A., Metra M., Adamo M., Gardner R.S., Baumbach A., Böhm M., Burri H., Butler J., Čelutkienė J., Chioncel O., et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. Rev. Esp. Cardiol. 2022;75:523. doi: 10.1016/j.rec.2022.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Ramaccini D., Montoya-Uribe V., Aan F.J., Modesti L., Potes Y., Wieckowski M.R., Krga I., Glibetić M., Pinton P., Giorgi C., Matter M.L. Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front. Cell Dev. Biol. 2020;8 doi: 10.3389/fcell.2020.624216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moyzis A.G., Sadoshima J., Gustafsson Å.B. Mending a broken heart: the role of mitophagy in cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2015;308:H183–H192. doi: 10.1152/ajpheart.00708.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dikic I., Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018;19:349–364. doi: 10.1038/s41580-018-0003-4. [DOI] [PubMed] [Google Scholar]
- 5.Kanki T., Wang K., Cao Y., Baba M., Klionsky D.J. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev. Cell. 2009;17:98–109. doi: 10.1016/j.devcel.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okamoto K., Kondo-Okamoto N., Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev. Cell. 2009;17:87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 7.Onishi M., Yamano K., Sato M., Matsuda N., Okamoto K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021;40 doi: 10.15252/embj.2020104705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murakawa T., Yamaguchi O., Hashimoto A., Hikoso S., Takeda T., Oka T., Yasui H., Ueda H., Akazawa Y., Nakayama H., et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 2015;6:7527. doi: 10.1038/ncomms8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shiba-Fukushima K., Imai Y., Yoshida S., Ishihama Y., Kanao T., Sato S., Hattori N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012;2:1002. doi: 10.1038/srep01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu W., Tian W., Hu Z., Chen G., Huang L., Li W., Zhang X., Xue P., Zhou C., Liu L., et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014;15:566–575. doi: 10.1002/embr.201438501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rogov V.V., Suzuki H., Marinković M., Lang V., Kato R., Kawasaki M., Buljubašić M., Šprung M., Rogova N., Wakatsuki S., et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 2017;7:1131. doi: 10.1038/s41598-017-01258-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen G., Han Z., Feng D., Chen Y., Chen L., Wu H., Huang L., Zhou C., Cai X., Fu C., et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell. 2014;54:362–377. doi: 10.1016/j.molcel.2014.02.034. [DOI] [PubMed] [Google Scholar]
- 13.Kataoka T., Holler N., Micheau O., Martinon F., Tinel A., Hofmann K., Tschopp J. Bcl-rambo, a novel Bcl-2 homologue that induces apoptosis via its unique C-terminal extension. J. Biol. Chem. 2001;276:19548–19554. doi: 10.1074/jbc.M010520200. [DOI] [PubMed] [Google Scholar]
- 14.Yoon Y., Krueger E.W., Oswald B.J., McNiven M.A. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell Biol. 2003;23:5409–5420. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taguchi N., Ishihara N., Jofuku A., Oka T., Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 16.Cribbs J.T., Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939–944. doi: 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abudureyimu M., Yu W., Cao R.Y., Zhang Y., Liu H., Zheng H. Berberine Promotes Cardiac Function by Upregulating PINK1/Parkin-Mediated Mitophagy in Heart Failure. Front. Physiol. 2020;11 doi: 10.3389/fphys.2020.565751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao Y., Xu C., Ye J., He Q., Zhang X., Jia S., Qiao X., Zhang C., Liu R., Weng L., et al. Miro2 Regulates Inter-Mitochondrial Communication in the Heart and Protects Against TAC-Induced Cardiac Dysfunction. Circ. Res. 2019;125:728–743. doi: 10.1161/CIRCRESAHA.119.315432. [DOI] [PubMed] [Google Scholar]
- 19.Bolte S., Cordelières F.P. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc-Oxford. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 20.Chakrabarty Y., Yang Z., Chen H., Chan D.C. The HRI branch of the integrated stress response selectively triggers mitophagy. Mol. Cell. 2024;84:1090–1100.e6. doi: 10.1016/j.molcel.2024.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Readnower R.D., Brainard R.E., Hill B.G., Jones S.P. Standardized bioenergetic profiling of adult mouse cardiomyocytes. Physiol. Genom. 2012;44:1208–1213. doi: 10.1152/physiolgenomics.00129.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruas J.S., Siqueira-Santos E.S., Amigo I., Rodrigues-Silva E., Kowaltowski A.J., Castilho R.F. Underestimation of the Maximal Capacity of the Mitochondrial Electron Transport System in Oligomycin-Treated Cells. PLoS One. 2016;11 doi: 10.1371/journal.pone.0150967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scott S.R., Singh K., Yu Q., Sen C.K., Wang M. Sex as Biological Variable in Cardiac Mitochondrial Bioenergetic Responses to Acute Stress. Int. J. Mol. Sci. 2022;23 doi: 10.3390/ijms23169312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu Y., Massen S., Terenzio M., Lang V., Chen-Lindner S., Eils R., Novak I., Dikic I., Hamacher-Brady A., Brady N.R. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 2013;288:1099–1113. doi: 10.1074/jbc.M112.399345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Egan D.F., Shackelford D.B., Mihaylova M.M., Gelino S., Kohnz R.A., Mair W., Vasquez D.S., Joshi A., Gwinn D.M., Taylor R., et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murakawa T., Okamoto K., Omiya S., Taneike M., Yamaguchi O., Otsu K. A Mammalian Mitophagy Receptor, Bcl2-L-13, Recruits the ULK1 Complex to Induce Mitophagy. Cell Rep. 2019;26:338–345.e6. doi: 10.1016/j.celrep.2018.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang J., Xu Z.X., Ding Z., Lu Y., Yu Q., Werle K.D., Zhou G., Park Y.Y., Peng G., Gambello M.J., Mills G.B. Myristoylation confers noncanonical AMPK functions in autophagy selectivity and mitochondrial surveillance. Nat. Commun. 2015;6:7926. doi: 10.1038/ncomms8926. [DOI] [PubMed] [Google Scholar]
- 28.Foretz M., Guihard S., Leclerc J., Fauveau V., Couty J.P., Andris F., Gaudry M., Andreelli F., Vaulont S., Viollet B. Maintenance of red blood cell integrity by AMP-activated protein kinase alpha1 catalytic subunit. FEBS Lett. 2010;584:3667–3671. doi: 10.1016/j.febslet.2010.07.041. [DOI] [PubMed] [Google Scholar]
- 29.Viollet B., Andreelli F., Jørgensen S.B., Perrin C., Geloen A., Flamez D., Mu J., Lenzner C., Baud O., Bennoun M., et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J. Clin. Invest. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang B., Nie J., Wu L., Hu Y., Wen Z., Dong L., Zou M.H., Chen C., Wang D.W. AMPKα2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ. Res. 2018;122:712–729. doi: 10.1161/CIRCRESAHA.117.312317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang P., Hu X., Xu X., Fassett J., Zhu G., Viollet B., Xu W., Wiczer B., Bernlohr D.A., Bache R.J., Chen Y. AMP activated protein kinase-alpha2 deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction in mice. Hypertension. 2008;52:918–924. doi: 10.1161/HYPERTENSIONAHA.108.114702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song M., Mihara K., Chen Y., Scorrano L., Dorn G.W., 2nd Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metabol. 2015;21:273–286. doi: 10.1016/j.cmet.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pennanen C., Parra V., López-Crisosto C., Morales P.E., del Campo A., Gutierrez T., Rivera-Mejías P., Kuzmicic J., Chiong M., Zorzano A., et al. Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin signaling pathway. J. Cell Sci. 2014;127:2659–2671. doi: 10.1242/jcs.139394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J., Chen A., Xue Z., Liu J., He Y., Liu G., Zhao Z., Li W., Zhang Q., Chen A., et al. BCL2L13 promotes mitophagy through DNM1L-mediated mitochondrial fission in glioblastoma. Cell Death Dis. 2023;14:585. doi: 10.1038/s41419-023-06112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johannessen C.M., Boehm J.S., Kim S.Y., Thomas S.R., Wardwell L., Johnson L.A., Emery C.M., Stransky N., Cogdill A.P., Barretina J., et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu P., Zhang D., Swenson L., Chakrabarti G., Abel E.D., Litwin S.E. Minimally invasive aortic banding in mice: effects of altered cardiomyocyte insulin signaling during pressure overload. Am. J. Physiol. Heart Circ. Physiol. 2003;285:H1261–H1269. doi: 10.1152/ajpheart.00108.2003. [DOI] [PubMed] [Google Scholar]
- 37.Omiya S., Omori Y., Taneike M., Protti A., Yamaguchi O., Akira S., Shah A.M., Nishida K., Otsu K. Toll-like receptor 9 prevents cardiac rupture after myocardial infarction in mice independently of inflammation. Am. J. Physiol. Heart Circ. Physiol. 2016;311:H1485–H1497. doi: 10.1152/ajpheart.00481.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lam J., Katti P., Biete M., Mungai M., AshShareef S., Neikirk K., Garza Lopez E., Vue Z., Christensen T.A., Beasley H.K., et al. A Universal Approach to Analyzing Transmission Electron Microscopy with ImageJ. Cells. 2021;10 doi: 10.3390/cells10092177. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data supporting the findings of this study are available within the article and its supplemental information. Source data, including images, have been deposited in Figshare: https://doi.org/10.6084/m9.figshare.25284883 and DRYAD: https://doi.org/10.5061/dryad.9cnp5hqsx.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.