

Abstract
Purpose
Among patients with autoimmune myositis, associated interstitial lung disease (MA-ILD) is a known contributor of excess morbidity and mortality. Recent data on survival in idiopathic inflammatory myopathies originate primarily in Asia and Europe and vary widely. We sought to examine mortality in a large U.S. myositis cohort focusing in particular on the impact of associated ILD.
Methods
A cross-sectional analysis of participants from the Johns Hopkins Myositis Center with autoimmune myositis (polymyositis [PM], dermatomyositis [DM], or clinically amyopathic dermatomyositis [CADM]) was conducted. The primary outcome assessed was all-cause mortality. Cumulative mortality rates were estimated using the Kaplan–Meier test; the Cox proportional hazards model was used to compare group differences in survival.
Results
Eight hundred and thirty-one participants were included with a median follow-up time of 4.5 years. Four hundred thirty-eight (53 %) had PM, 362 (43 %) had DM, and 31 (4 %) had CADM. Ninety-four (11 %) participants had clinically evident ILD. Overall, 51 participants died (6 %). In those without ILD, the survival rates at 1, 5, and 10 years were 99, 95, and 90 %, respectively. In those with ILD, the survival rates at 1, 5, and 10 years were 97, 91, and 81 %, respectively. The risk of death was statistically significantly higher among participants with ILD compared to those without ILD (HR 2.13. 95 % CI 1.06–4.25; p = 0.03).
Conclusions
We analyzed one of the largest known cohorts of patients with autoimmune myositis and found significantly higher mortality rates among those with clinically evident ILD compared to those without clinically evident ILD. Our results suggest that ILD remains an important and significant source of mortality in patients with inflammatory myopathies and as such should be screened for and treated aggressively.
Keywords: Survival, Interstitial lung disease, Idiopathic inflammatory myopathy, Dermatomyositis, Polymyositis, Clinically amyopathic dermatomyositis
Introduction
Polymyositis (PM) and dermatomyositis (DM) are autoinflammatory muscle diseases characterized by progressive symmetric proximal muscle weakness and, in DM, rash. Clinically, amyopathic DM (CADM) is a subset of DM which demonstrates characteristic cutaneous findings in the absence of proximal muscle weakness and serum muscle enzyme abnormalities [1]. A significant proportion of patients with inflammatory myositis have associated interstitial lung disease (MA-ILD) and parenchymal lung damage with varying degrees of inflammation and/or fibrosis [2–4]. Depending on the method of ascertainment, the reported prevalence of MA-ILD ranges between 5 and 65 % [5–7]. In a study of incident myositis cases where all participants underwent high-resolution computed tomography examinations of the chest, 65 % had some evidence of concomitant ILD [3].
Reported survival in idiopathic inflammatory myopathies varies widely depending on the population studied with overall mortality rates as high as 26 % reported [8, 9]. MA-ILD is known to significantly increase morbidity and mortality [6–11]. Modern studies evaluating the impact of MA-ILD on mortality are small and/or derived from Asian and European cohorts [11–14]. We sought to examine mortality in a large U.S. myositis cohort focusing in particular on the impact of associated ILD.
Methods
Study Population
We conducted a retrospective cross-sectional analysis of consecutive patients with autoimmune myositis evaluated at the Johns Hopkins Myositis Center between January 2006 and December 2014. Autoimmune myositis cases included those with polymyositis (PM), dermatomyositis (DM), or clinically amyopathic dermatomyositis (CADM). All cases were diagnosed based on the Bohan and Peter criteria [15, 16] or the modified Sontheimer definition [1]. Participants with inclusion body myositis were excluded. All participants provided written informed consent; the present study was approved by the Johns Hopkins University IRB (NA_00007454).
ILD Characterization
Pulmonary function tests (PFTs) including spirometry, lung volumes measured by helium dilution, and diffusing capacity by single breath carbon monoxide based on American Thoracic Society (ATS) criteria were reviewed [17]. Myositis-associated interstitial lung disease (MA-ILD) was ascertained by screening all individuals by ATS criteria for restriction or diffusing capacity deficits [2, 18]. Only those with abnormal PFTs based on those criteria and evidence of diffuse parenchymal lung disease on high-resolution computed tomography (HRCT) were designated as having clinically evident MA-ILD. HRCTs were interpreted by dedicated lung radiologists and reviewed by a pulmonologist (CJ or SD).
Other Measures
Other data extracted included demographic information (age, gender, and race by self-report) and myositis-specific or myositis-associated autoantibody profiles when available.
Mortality Assessment
All-cause mortality was confirmed through the Social Security Death Index. Follow-up time was established as the date of diagnosis with autoimmune myositis (which may have predated enrollment in the Johns Hopkins Myositis Database) to the date of death or last follow-up visit.
Analysis
Chi-square test or Fisher’s exact test was used for group comparisons of binary data. Cumulative mortality rates were estimated using the Kaplan–Meier test. Univariate and multivariate Cox regression analyses were used to compare survival of participants with and without ILD and to assess for clinical associations with mortality. The full multivariate model included age at diagnosis, ILD, sex, race/ethnicity, and inflammatory myopathy subtype. All calculations were performed using intercooled Stata 12 (StataCorp, College Station, TX).
Results
Eight hundred and thirty-one participants with a median follow-up time of 4.5 years were included in the study. Women outnumbered men and most participants were Caucasian. Four hundred thirty-eight (53 %) had PM, 362 (43 %) had DM, and 31 (4 %) had CADM. Ninety-four (11 %) participants had confirmed ILD. Those with ILD were more likely to be African American than those without ILD (35 vs. 17 %, respectively; p < 0.01). African Americans made up 19 % of the entire cohort but represented 35 % of those with clinically evident ILD. A summary of participant characteristics is included in Table 1.
Table 1.
Participant characteristicsa
| Interstitial lung disease | p value | ||
|---|---|---|---|
| Present n = 94 | Absent n = 737 | ||
| Age, mean ± SD | 49 ± 11 | 50 ± 16 | 0.63 |
| Women | 60 (64 %) | 511 (69 %) | 0.29 |
| Race | <0.01 | ||
| Caucasian | 55 (59 %) | 558 (76 %) | |
| African American | 33 (35 %) | 128 (17 %) | |
| Asian | 5 (5 %) | 17 (2 %) | |
| Other | 1 (1 %) | 20 (3 %) | |
| Unknown | – | 14 (2 %) | |
| Diagnosis | |||
| Polymyositis | 44 (47 %) | 394 (54 %) | 0.12 |
| Dermatomyositis (DM) | 49 (52 %) | 313 (42 %) | |
| Clinically amyopathic DM | 1 (1 %) | 30 (4 %) | |
| Died | 12 (13 %) | 39 (5 %) | 0.01 |
Data are n (%) unless otherwise specified
Mean baseline PFT values in MA-ILD participants revealed mild restrictive and moderate carbon monoxide diffusion capacity deficits (Table 2). The mean total lung capacity percent predicted was 67 % and the mean diffusing capacity percent predicted was 60 %. Myositis-specific or myositis-associated autoantibody data were available for a third of all participants (Supplemental Table 1) and 63 % of participants with MA-ILD (Table 2). Most MA-ILD participants with a detectable autoantibody had myositis-specific antibodies, the vast majority of which were antisynthetase autoantibodies (Table 2).
Table 2.
Baseline characteristics of participants with interstitial lung diseasea
| Baseline PFTs (N = 94) | |
| FVC % predicted, mean (SD) | 65 (20) |
| TLC % predicted, mean (SD) | 67 (18) |
| DLCO % predicted, mean (SD) | 60 (20) |
| Autoantibody (N = 80) | |
| Myositis specific | |
| Antisynthetases | |
| Anti-Jo-1 | 29 (36) |
| Anti-PL-7 | 2 (3) |
| Anti-PL-12 | 4 (5) |
| Anti-EJ | 5 (6) |
| Anti-MDA-5 | 5 (6) |
| Anti-SRP | 1 (1) |
| Anti-HMGCR | 1 (1) |
| Anti-TIF1-γ | 1 (1) |
| Myositis associated | |
| Anti-PM/Scl | 6 (8) |
| Anti-Ku | 2 (3) |
| Anti-Ro | 2 (3) |
| Anti-U1RNP | 1 (1) |
| None detected | 21 (26) |
Number (percent) unless otherwise specified
Overall, 51 participants died (6 %) during the follow-up time period. Median overall survival rates at 1, 5, and 10 years were 97, 95, and 90 % and 97, 91, and 81 % in the myositis alone and MA-ILD groups, respectively (Table 3). In the full multivariate model, the risk of death was statistically significantly higher among participants with ILD compared to those without ILD (HR 2.13. 95 % CI 1.06–4.25; p = 0.03; Fig. 1). Mortality was not influenced by sex, race/ethnicity, or type of autoimmune myositis.
Table 3.
Autoimmune myositis estimated median overall survival rates, % (SE)
| 1 year | 3 years | 5 years | 10 years | |
|---|---|---|---|---|
| Interstitial lung disease | ||||
| Absent | 99 (0.4) | 97 (0.7) | 95 (0.9) | 90 (2.2) |
| Present | 97 (1.8) | 91 (3.0) | 91 (3.0) | 81 (6.3) |
Fig. 1.
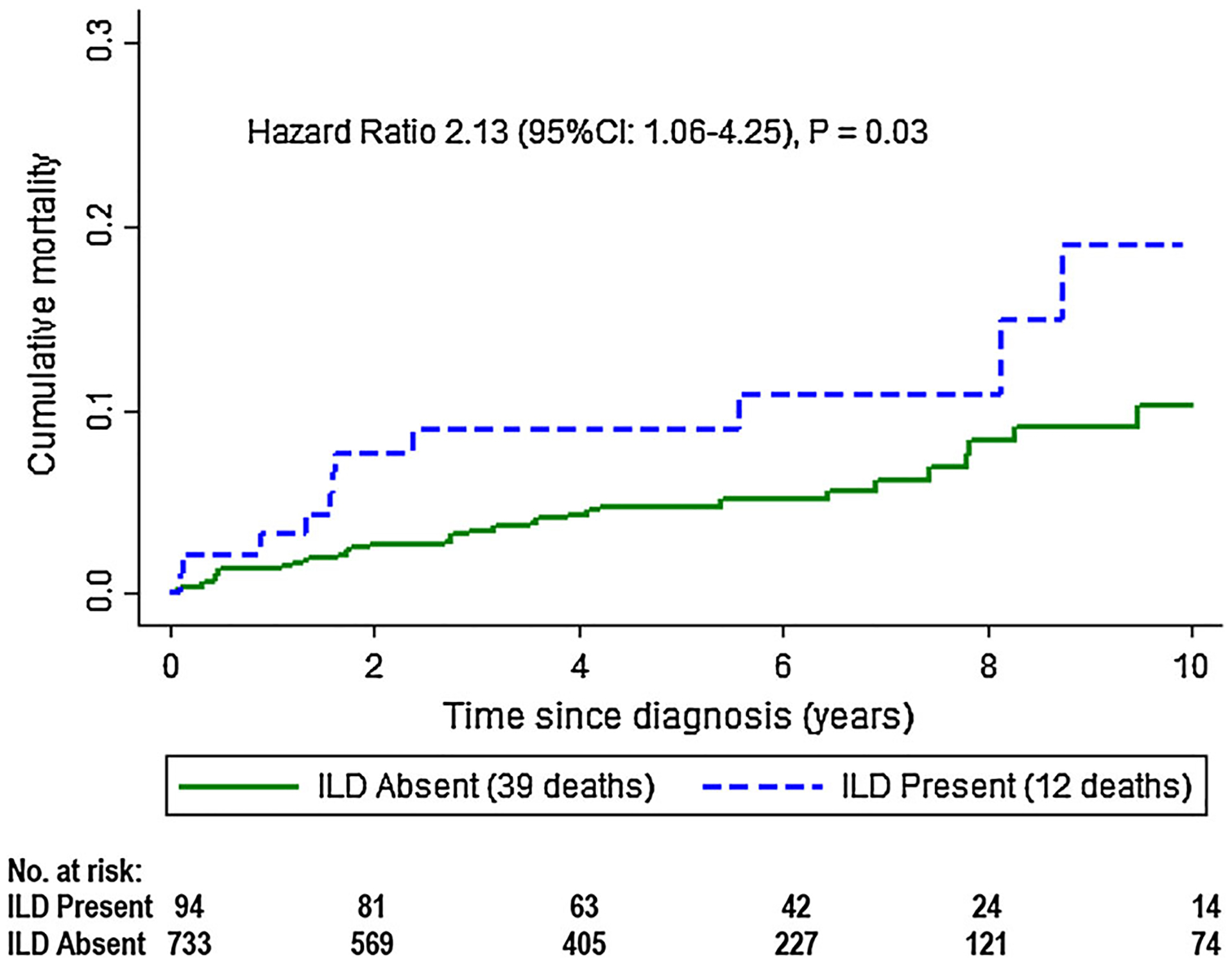
Estimated cumulative survival in participants with autoimmune myositis
Discussion
We analyzed one of the largest known cohorts of patients with autoimmune myositis from a single academic center in the US and found significantly higher mortality rates among those with clinically evident ILD compared to those without clinically evident ILD. The distribution of myopathy subtype, age, and gender were similar to other cohorts described in the literature [3, 14]. Only 11 % of our cohort had ILD which is lower than other historical series [11]. This may reflect the method of ILD characterization in our study, including only clinically evident MA-ILD. Similar to other studies, we did not see any significant differences in mortality between the groups with PM or DM [8]. Unlike prior studies we did see a significant difference in mortality in patients with and without ILD [19]. Our overall survival rates are better than historical reports [11, 20–22] but consistent with more recent studies [9].
Our patients with MA-ILD had mild to moderate disease at the time of autoimmune myositis diagnosis. Based on the distribution of African Americans in our cohort, African Americans were disproportionally represented in the group with clinically evident ILD. Additionally, previous data suggest that African Americans have more severe MA-ILD than Caucasians [23]. We did not, however, detect a significant difference in the mortality rate in African Americans. This most likely reflects our sample size and low number of deaths overall and warrants further study. Most of our MA-ILD patients with a tested or detectable autoantibody had antisynthetase autoantibodies. However, similar to other series, a large proportion of our MA-ILD patients had autoantibodies other than anti-Jo-1 [11]. Interestingly, only a small proportion of our patients with CADM, a subset of DM without apparent muscle involvement often associated with rapidly progressive ILD, had MA-ILD [13]. Mortality rates are expected to be higher among this group and could provide a potential explanation for our higher than previously reported survival rates [13, 24]. Additionally, the size of our study, duration and decade(s) of follow-up, and modern treatment protocol could account for our overall and MA-ILD mortality rates being lower than previously reported [11, 20–22, 25].
This study has several strengths including its size, a large proportion of often understudied African Americans, and robust pulmonary involvement characterization. Several weaknesses, however, merit discussion. The exact cause of death is unknown. It is reasonable to speculate that many of the patients with MA-ILD died due to progressive lung disease but the data were not available. Similarly, the presence or absence of malignancy was not completely captured. Cancer-associated myositis is a significant driver of mortality, and the inclusion or exclusion of this group accounts for a large proportion of the variability seen in autoimmune myositis survival studies [11, 22]. The time between autoimmune myositis and MA-ILD diagnoses, an important potential influencer of mortality, was not fully accessible for the analysis. Likewise, the percentage of incident versus prevalent cases of autoimmune myositis and MA-ILD at the time of referral to our center was not abstracted. Longitudinal PFT and cigarette smoke exposure data were unattainable for a significant proportion of the cohort. Finally, a large number of participants had no available autoantibody data; therefore, an accurate analysis of the effect of autoantibody status on mortality could not be performed.
In conclusion, our results suggest that ILD remains an important and significant source of mortality in patients with inflammatory myopathy, and should be screened for and treated aggressively.
Supplementary Material
Acknowledgments
The authors would like to thank William Kelly for his contribution to improvements in the Myositis Center Clinical Database.
Funding
This study was financially supported by The Huayi and Siuling Zhang Discovery Fund.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00408-016-9896-x) contains supplementary material, which is available to authorized users.
Conflict of interest The authors have no known conflicts of interest to disclose.
References
- 1.Sontheimer RD (2002) Would a new name hasten the acceptance of amyopathic dermatomyositis (dermatomyositis sine myositis) as a distinctive subset within the idiopathic inflammatory dermatomyopathies spectrum of clinical illness? J Am Acad Dermatol 46(4):626–636 [DOI] [PubMed] [Google Scholar]
- 2.American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus (2002) Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med 165(2):277–304 [DOI] [PubMed] [Google Scholar]
- 3.Fathi M et al. (2004) Interstitial lung disease, a common manifestation of newly diagnosed polymyositis and dermatomyositis. Ann Rheum Dis 63(3):297–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fathi M, Lundberg IE, Tornling G (2007) Pulmonary complications of polymyositis and dermatomyositis. Semin Respir Crit Care Med 28(4):451–458 [DOI] [PubMed] [Google Scholar]
- 5.Labirua A, Lundberg IE (2010) Interstitial lung disease and idiopathic inflammatory myopathies: progress and pitfalls. Curr Opin Rheumatol 22(6):633–638 [DOI] [PubMed] [Google Scholar]
- 6.Mimori T, Nakashima R, and Hosono Y, Interstitial Lung Disease in Myositis: Clinical Subsets, Biomarkers, and Treatment. Curr Rheumatol Rep, 2012 [DOI] [PubMed] [Google Scholar]
- 7.Solomon J, Swigris JJ, Brown KK (2011) Myositis-related interstitial lung disease and antisynthetase syndrome. J Bras Pneumol 37(1):100–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Torres C et al. (2006) Survival, mortality and causes of death in inflammatory myopathies. Autoimmunity 39(3):205–215 [DOI] [PubMed] [Google Scholar]
- 9.Danko K et al. (2004) Long-term survival of patients with idiopathic inflammatory myopathies according to clinical features: a longitudinal study of 162 cases. Medicine (Baltimore) 83(1):35–42 [DOI] [PubMed] [Google Scholar]
- 10.Kalluri M, Oddis CV (2010) Pulmonary manifestations of the idiopathic inflammatory myopathies. Clin Chest Med 31(3):501–512 [DOI] [PubMed] [Google Scholar]
- 11.Marie I et al. (2002) Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum 47(6):614–622 [DOI] [PubMed] [Google Scholar]
- 12.Takada K, Nagasaka K, Miyasaka N (2005) Polymyositis/dermatomyositis and interstitial lung disease: a new therapeutic approach with T-cell-specific immunosuppressants. Autoimmunity 38(5):383–392 [DOI] [PubMed] [Google Scholar]
- 13.Moghadam-Kia S, et al. (2015) Anti-MDA5 is associated with rapidly progressive lung disease and poor survival in U.S. patients with amyopathic and myopathic dermatomyositis. Arthritis Care Res 68(5):689–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woo JH et al. (2013) Mortality factors in idiopathic inflammatory myopathy: focusing on malignancy and interstitial lung disease. Mod Rheumatol 23(3):503–508 [DOI] [PubMed] [Google Scholar]
- 15.Bohan A, Peter JB (1975) Polymyositis and dermatomyositis (second of two parts). N Engl J Med 292(8):403–407 [DOI] [PubMed] [Google Scholar]
- 16.Bohan A, Peter JB (1975) Polymyositis and dermatomyositis (first of two parts). N Engl J Med 292(7):344–347 [DOI] [PubMed] [Google Scholar]
- 17.Macintyre N et al. (2005) Standardisation of the single-breath determination of carbon monoxide uptake in the lung. Eur Respir J 26(4):720–735 [DOI] [PubMed] [Google Scholar]
- 18.Miller MR et al. (2005) General considerations for lung function testing. Eur Respir J 26(1):153–161 [DOI] [PubMed] [Google Scholar]
- 19.Grau JM et al. (1996) Interstitial lung disease related to dermatomyositis. Comparative study with patients without lung involvement. J Rheumatol 23(11):1921–1926 [PubMed] [Google Scholar]
- 20.Douglas WW et al. (2001) Polymyositis-dermatomyositis-associated interstitial lung disease. Am J Respir Crit Care Med 164(7):1182–1185 [DOI] [PubMed] [Google Scholar]
- 21.Kang EH et al. (2005) Interstitial lung disease in patients with polymyositis, dermatomyositis and amyopathic dermatomyositis. Rheumatology (Oxford) 44(10):1282–1286 [DOI] [PubMed] [Google Scholar]
- 22.Marie I et al. (2001) Polymyositis and dermatomyositis: short term and longterm outcome, and predictive factors of prognosis. J Rheumatol 28(10):2230–2237 [PubMed] [Google Scholar]
- 23.Johnson C et al. (2014) Clinical and pathologic differences in interstitial lung disease based on antisynthetase antibody type. Respir Med 108(10):1542–1548 [DOI] [PubMed] [Google Scholar]
- 24.Fujisawa T et al. (2014) Prognostic factors for myositis-associated interstitial lung disease. PLoS one 9(6):e98824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Danieli MG et al. (2014) Impact of treatment on survival in polymyositis and dermatomyositis. A single-centre long-term follow-up study. Autoimmun Rev 13(10):1048–1054 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.