

Abstract
Purpose
Heterozygous pathogenic variants in SPAST are known to cause Hereditary Spastic Paraplegia 4 (SPG4), the most common form of HSP, characterized by progressive bilateral lower limbs spasticity with frequent sphincter disorders. However, there are very few descriptions in the literature of patients carrying biallelic variants in SPAST.
Methods
Targeted Sanger sequencing, panel sequencing and exome sequencing were used to identify the genetic causes in 9 patients from 6 unrelated families with symptoms of HSP or infantile neurodegenerative disorder.
Results
We describe 5 patients with pure HSP with a variable age of onset, mostly in infancy, and 4 patients with profound intellectual disability and progressively worsening tetrapyramidal syndrome. The patients' parents, heterozygous carriers of pathogenic SPAST variants, included both asymptomatic carriers and patients with classic forms of SPG4.
Conclusion
Biallelic variants of SPAST may explain cases of hereditary spastic paraplegia with autosomal recessive inheritance. Furthermore, some biallelic variants may also cause psychomotor regression with an infantile neurodegenerative disorder, associated with a tetrapyramidal syndrome, a new phenotype associated with the SPAST gene.
Keywords: homozygous, neurodegenerative disorder, SPAST, spastic paraplegia, SPG4
INTRODUCTION
Hereditary spastic paraplegia (HSP) is a heterogeneous group of genetic disorders characterized by progressive spasticity of lower limbs. The most common type of HSP, sporadic or with autosomal dominant transmission is spastic paraplegia 4 (SPG4, OMIM# 182601) [1], caused by heterozygous pathogenic variants in SPAST [2]. Most frequent symptoms include progressive bilateral spasticity of the lower limbs, sphincter disturbance and impaired vibration sense at the ankles defining pure HSP [3, 4]. Complex forms of HSP with associated seizures, cerebellar atrophy, intellectual disability, peripheral neuropathy and other neurological symptoms have also been described in SPG4 [5, 6]. The age at onset vary both between and within families, with two peaks between birth and the first decade and between the third and fifth decades of life [7, 8]. Intellectual disability can sometimes be associated with SPG4, more frequently in patients with an earlier age at onset [7, 9]. SPAST gene codes for spastin, a microtubule‐associated protein (MAP), which is a microtubule‐severing enzyme [10]. Some genotype–phenotype correlations have been proposed in SPG4, with earlier onset for missense variants located in the AAA domain [7].
Although the phenotype of autosomal dominant SPG4 has been widely reported, there are only a few descriptions of carriers of biallelic variants. The hypomorphic variant S44L [11] has been described as a severity factor when found in trans of a pathogenic variant in SPAST, with an earlier onset of symptoms [11]. To our knowledge, there are only three descriptions of homozygous pathogenic variants in the literature, associated with a typical pure phenotype of SPG4 in one case [12] and with a more severe phenotype, including epilepsy and progressive psychomotor deterioration, in two families [8, 13].
We report here a series of nine patients carrying biallelic variants in SPAST, with the aim of better describing the phenotypic spectrum associated with this rare condition. Indeed, the phenotypes of these patients vary dramatically in severity, ranging from classical pure HSP to infantile neurodegenerative disorder with spastic quadriplegia, an under‐recognized phenotype associated with the SPAST gene.
MATERIALS AND METHODS
Patient's collection, variants detection and analyses are detailed in Data S1. Informed consent was obtained from all individual participants included in the study. This study has been approved by the appropriate ethics committee (CHU de Bordeaux, CPP Sud Ouest et Outre Mer III) and has, therefore, been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki.
CLINICAL AND GENETIC FINDINGS
Family A—Patient 1,2
Patients 1 and 2 were born from first‐degree cousin consanguineous parents (Figure 1a) in France, from gypsy community. Patient 1, the elder brother, presented with psychomotor delay and spastic paraplegia. At 3 years, he presented generalized tonic–clonic seizures, with electroencephalographic abnormalities (generalized diffuse spike–wave paroxysms) and was treated with sodium valproate and clobazam. He experienced psychomotor regression since the age of 18 months, with communication regression, losing the ability to sit, never acquiring the ability to walk and evolving into spastic tetraparesis, leaving him bedridden with swallowing difficulties and profound intellectual disability. He died of aspiration pneumonia at the age of 21. Cerebral MRI performed at age 7 revealed diffuse cerebral atrophy and T2 white matter hyperintensities at the level of the lenticulostriate nuclei, controlled at 17 years showing diffuse cerebral atrophy and lenticulostriate nuclei lesions stability.
FIGURE 1.
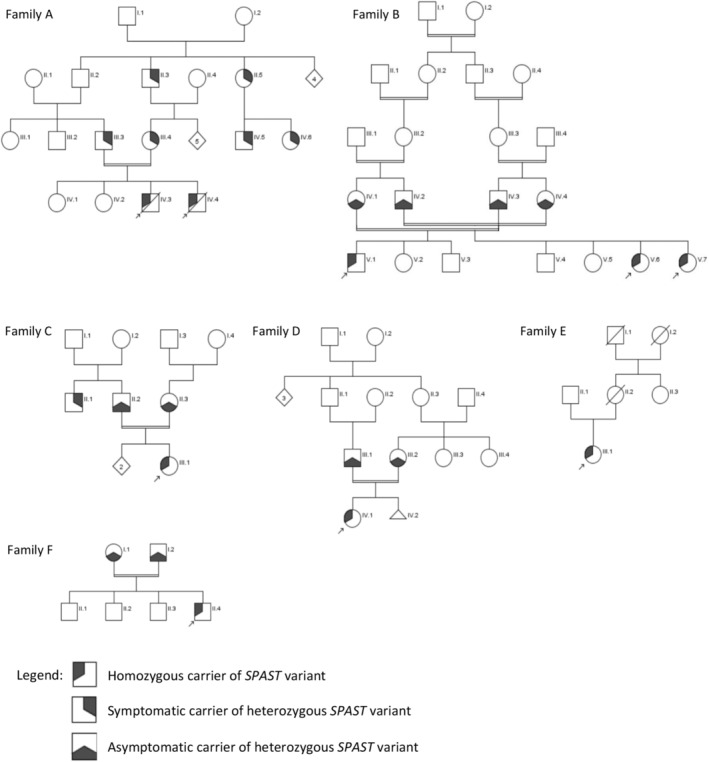
Pedigree of families A, B, C, D, E, and F.
His brother, patient 2, was monitored during pregnancy with in‐utero cerebral MRI because of his brother's pathology, which showed normal results. His neurological examination was normal at the age of 3 and 6 months, with only an extensor plantar response noticed at 6 months, which can be physiological at this age. He had a psychomotor delay, acquiring sitting at 11 months and progressed to spastic tetraplegia with psychomotor regression from the age of 18 months. At the age of 2 years, a cerebral MRI showed T2 white matter hyperintensities posterior to the ventricular junctions and at the level of the centrum semiovale, without subsequent imaging control. At the age of 14, he developed paroxysmal abnormal movements of the right lower limb, suggestive of myoclonia, although these episodes were not confirmed by an EEG recording. Interictal EEG showed slow background activity without paroxysms, but fast rhythms due to treatment by diazepam. He died at the age of 15 from pneumonia, likely related to swallowing difficulties.
Ten years later, their parents presented signs of pure spastic paraplegia around 45 years for the father, and 53 years for the mother, leading to the identification of the same heterozygous SPAST variant c.447 T > A (p.Tyr149*) in both parents. This variant had previously been identified at the heterozygous state in a cousin who also displayed progressive spastic paraplegia. Subsequently, Sanger sequencing was performed on stored DNA from the two deceased brothers revealing homozygous status for the familial variant. Whole exome sequencing (WES) was also performed and revealed no other variants to explain their phenotype.
This variant was previously described as pathogenic in ClinVar (VCV000989091.1). This nonsense variant, located in the MIT domain and absent from the control population, is pathogenic according to the ACMG classification (PVS1, PM2_supp, PM3_supp, PP1) (Table 1) [14].
TABLE 1.
Variants molecular characteristics table.
| Variant | NM | Genomic position | CADD Phred | REVEL | GnomAD frequency | Domain | Conservation | PhyloP 100 | Clinvar | Publications |
|---|---|---|---|---|---|---|---|---|---|---|
| c.153C > G, p.(Tyr51*) | NM_014946 | g.32289053C > G (Hg19) | 36.00 | NA | 0 | N‐ter | NA | NA | Pathogenic ** (VCV001369382.7) | PMID 20932283, 20562464 |
| c.166C > T, p.(Pro56Ser) | NM_014946 | g.32289066C > T (Hg19) | 26.00 | 0.65 | 0 | N‐ter | Zebrafish | 5.16 | Absent | NA |
| c.447 T > A, p.(Tyr149*) | NM_014946.3 | g.32087523 T > A (Hg19) | 35.00 | NA | 0 | MIT | NA | NA | Pathogenic * (VCV000989091.1) | NA |
| c.467 T > C, p.(Leu156Pro) | NM_014946.4 | g.32087543 T > C (Hg38) | 27.60 | 0.879 | 0 | MIT | Zebrafish | 5.34 | Absent | NA |
| c.1076 T > C, p.(Ile359Thr) | NM_014946.3 | g.32116190 T > C (Hg38) | 26.50 | 0.928 | 0 | AAA | Zebrafish | 6.05 | Absent | NA |
Note: CADD Phred and REVEL scores are metrics used to predict the deleteriousness of genetic variants by integrating various genomic annotations. A threshold for pathogenicity has been set at greater than 20 for the CADD score. Variants with REVEL scores greater than 0.5 are considered potentially pathogenic. PhyloP100 is a metric used to assess the evolutionary conservation of nucleotide positions within genomic sequences. The commonly used threshold for conservation is set at 1, with higher scores indicating greater conservation and potential functional importance of genomic regions.
Abbreviations: AAA, AAA domain; MIT, Microtubule Interacting and Trafficking domain; MTBD, Microtubules binding domain; NA, not available; N‐ter, N‐terminal domain.
Family B—Patient 3,4,5
Patients 3, 4 and 5 belong to a large family with multiple consanguinity loops (Figure 1b) from Algeria. Patients 3 and 4 are siblings. They developed spastic paraplegia at the age of 7 years for patient 3 and 6 years for patient 4, with secondary involvement of the upper limbs. They both had cerebral MRI, which showed no abnormalities. Patient 3 presented with dysphonia and patient 4 presented with dysphagia (age at onset is unknown). Their parents did not show any neurological signs or gait disturbance at 63 years for their father and 64 years for their mother.
WES identified a homozygous variant c.1076 T > C, p.(Ile359Thr) in SPAST in patients 3 and 4. This variant is rare, absent from control population, located in the AAA domain of the protein and affects an amino acid conserved up to Zebrafish. In silico predictors are in favor of pathogenicity (CADD Phred 26,50, REVEL 0,928) (Table 1). This variant is probably pathogenic according to the ACMG classification (PM1, PM2_supp, PM3_supp, PP1_strong, PP3_mod).
At the age of 1 year, patient 5 developed spasticity of the lower limbs, with pyramidal syndrome. His neurological examination was normal in the upper limbs. He was able to sit up at 9 months and walk at 15 months, and no psychomotor delay was reported until now. He did not have epilepsy. He had a normal brain MRI, but never had an electroencephalogram as he never presented clinical seizures. His parents report no symptoms of HSP, but they have not been examined by a neurologist. He had NGS panel sequencing of HSP genes, showing the same homozygous variant than his cousins, patients 3 and 4.
Family C—Patient 6
Some partial clinical and biological information of patient 6 have already been published by Varghaei et al [8]. Here, we provide further details of the clinical data. She was born to consanguineous parents (Figure 1c) from Canada. She showed no delay in motor acquisition, with sitting acquisition at 6 months and walking acquisition at 1 year, but spasticity and leg weakness were already reported during walking acquisition. Later in childhood, she developed hypophonia, dysarthria and dysphagia leading to swallowing difficulties. She had Special Education Needs school, without cognitive testing done. She had upper limb spasticity from the age of 6. At age 18, ability to walk and speak was lost, with lasting gradual worsening of motor and cognitive features. Brain MRI was performed, normal (age unknown), but no EEG. WES identified the pathogenic homozygous variant c.153C > G, p.(Tyr51*) in SPAST. This nonsense variant is reported as pathogenic in Clinvar (VCV001369382.7), absent from controls. This variant is likely pathogenic according to the ACMG classification (PVS1, PM2_supp) (Table 1).
Her parents, carriers of the pathogenic variant in a heterozygous state, had normal neurological examination in adulthood (precise age at examination unknown).
Family D—Patient 7
Patient 7 comes from India and presented with global developmental delay, epilepsy onset at 4 months of age with general sharp wave discharges on EEG at 7 months, severe spasticity since 8 months of age and microcephaly (49 cm, −2.9SD, manual measurement). At 10 years of age, she presented with severe intellectual disability, developmental delay and epilepsy; she was unable to stand and could only speak a bisyllables. Trio WES showed a homozygous missense variant c.467 T > C p.(Leu156Pro) in SPAST, inherited from each parent (Figure 1d). This missense variant is absent in the control population, affects an amino acid conserved up to Zebrafish, is located in the MIT domain and is predicted to be pathogenic by several in silico predictors (CADD Phred 27.6; REVEL 0.879) (Table 1). According to the ACMG classification, this variant is likely pathogenic (PM1, PM2_supp, PM3_supp, PP3_mod).
Family E—Patient 8
Patient 8 was born in Algeria and moved to France at 8. She acquired walking at a normal age but presented with spastic diplegia in infancy (the exact age of onset is unknown). She lost her ability to walk at the age of 17 and is now wheelchair‐bound (Figure 1f). She had a normal education. Clinical examination at the age of 38 years shows increased and brisk tendon reflexes, as well as Babinski sign. Possible cognitive deterioration was also suspected, without available neuropsychological testing. Panel sequencing of 57 genes of HSP revealed the homozygous probably pathogenic variant c.1076 T > C, p.(Ile359Thr), already identified in patients 3, 4 and 5 (no familial link was found between them).
Family F—Patient 9
Patient 9, born from consanguineous parents who are first cousins in France (Figure 1f), showed abnormal walking at 16 months, characterized by tiptoeing and inwardly turned feet, accompanied by frequent falls. He had normal language development with first words at 10 months and complete sentences at 3 years. At 4 years, etiological investigations including cerebral MRI, somatosensory evoked potentials and biomarker assays (including immunoglobulin levels, alpha‐fetoprotein, tocopherol and very long‐chain fatty acids) showed normal results. At 5 years, neurological examination revealed predominant spasticity in the adductor and triceps sural muscles, knee flexion contractures and exaggerated osteotendinous reflexes in both upper and lower limbs. However, no upper limb spasticity was observed at age 11, with lower limb symptoms remaining relatively stable.
Genetic analysis by panel sequencing of 54 genes associated with HSP at age 5 revealed a homozygous SPAST variant c.166C > T, p.(Pro56Ser), inherited from both parents. This variant, located in the N‐terminal domain, is absent in control populations and predicted to be pathogenic by multiple in silico predictors (CADD Phred 23.9, SIFT Deleterious, Polyphen 2 probably damaging) (Table 1). The variant's significance remains uncertain according to the ACMG classification (PM2_supp, PM3_supp, PP3_supp) but affects a conserved amino acid up to Zebrafish.
DISCUSSION
We present the largest series of nine patients carriers of biallelic variants in SPAST, including three patients with severe intellectual disability, associated with epilepsy, a new phenotype associated with SPAST. All nine patients had progressive lower limb spasticity and the majority (6/9) had upper limb spasticity (Table 2). Four of the nine patients had severe intellectual disability, with some cognitive regression. They all had an early age of onset, ranging from 6 months to 7 years.
TABLE 2.
Summary table of clinical data for the nine patients.
| Family A | Family B | Family C | Family D | Family E | Family F | TOTAL | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | Patient 9 | 9 | ||
| Genetic | SPAST variant | c.447 T > A,p.(Tyr149*) Homozygous | c.1076 T > C, p.(Ile359Thr) Homozygous | c.153C > G, p.(Tyr51*) Homozygous | c.467 T > C, p.(Leu156Pro) Homozygous | c.1076 T > C, p.(Ile359Thr) Homozygous | c.166C > T, p.(Pro56Ser) Homozygous | ||||
| Neurodeveloppement | Neonatal hypotonia | NA | + | − | − | + | − | − | NA | − | 2/7 |
| Sitting acquisition | + | + (11m) | + | + | + (9m) | + (6m) | 2y with support | + | + (5 m) | 9/9 | |
| Walking acquisition | − | − | + | + | + (15m) | + (1y) | − | + | + (16m) | 6/9 | |
| Developpemental regression | + | + | − | − | − | − | − | +/− (possible cognitive decline) | − | 2/9 | |
| Intellectual disabily | Severe | Severe | − | − | − | Severe | Severe | − | − | 4/9 | |
| Neurology | Age of onset | < 3y | 6 m | 7y | 6y | 1y | 1y | 8 m | Unknown, loss of gait at 17y | < 16 m | |
| Lower limbs spasticity (AO) | + | + (11 m) | + | + | + | + (12 m) | + (8 m) | + | 16 m | 9/9 | |
| Upper limbs spasticity | + (3y) | + | + | + | − | + (6y) | − | + | 6/9 | ||
| Cerebral MRI abnomalies | + | + | − | − | − | − | − | NA | NA | 2/7 | |
| Epilepsy | + | + | − | − | − | − | + (onset at 4 m) | − | − | 3/9 | |
| Microcephay | + (‐3DS) | + | NA | NA | NA | NA | + | NA | − | 3/4 | |
| Father | Genetic | c.447 T > A heterozygous | c.1076 T > C heterozygous | NA | c.153C > G heterozygous | c.467 T > Cheterozygous | NA | c.166C > T heterozygous | |||
| Symptomatic (age of onset) | Pure HSP at 50 y | Asymptomatic at 63 y | Normal examination in adulthood | Not symptomatic | 1/3 | ||||||
| Mother | Genetic | c.447 T > A heterozygous | c.1076 T > C heterozygous | NA | c.153C > G heterozygous | c.467 T > C heterozygous | NA | c.166C > T heterozygous | |||
| Symptomatic (age of onset) | Pure HSP at 55y | Asymptomatic at 64 y | Normal examination in adulthood | Not symptomatic | 1/3 | ||||||
Abbreviations: HSP, Hereditary Spastic Paraplegia; m, months; NA, not available; y, years.
These different phenotypes are consistent with the descriptions available in the literature. Cruz‐Camino et al. described two siblings of consanguineous parents, with epilepsy, degenerative psychomotor regression, limb spasticity with extensor plantar responses and axial hypotonia [13]. They also had corneal opacity and dysostotic bones. WES was performed in only one child, showing a homozygous nonsense variant in SPAST (c.1634C > G, p.(Ser545*)). Their parents, heterozygous carriers of the variants, showed no signs of HSP, but older family members described signs of HSP, with autosomal dominant transmission in the family. WES showed no other variant explaining dysostotic bones or corneal opacity but show a variant of unknown significance in GABRA1. De Bot et al. described two brothers carrying a homozygous missense variant in the AAA domain (c.1600C > G, p.Leu534Val). They both presented with pure spastic paraparesis, both with an onset at the age of 39 years. Their parents remain asymptomatic until an advanced age [12].
The SPAST gene encodes Spastin, a microtubule‐associated protein (MAP), which is a microtubule‐severing enzyme. Spastin, like other members of the AAA family, has ATPase activity through its AAA domain. The microtubule (MT) network is an essential structure for the proper functioning of neurons, particularly at the axon level, where it helps to guide exchanges between the cell body and axonal terminals [15]. By cutting MT, Spastin contributes to the formation of smaller MT fragments, allowing dynamics at the end of the MT network for synaptogenesis [16, 17]. It also helps to improve MT stability by incorporating fragments of GTP tubulin, thereby limiting depolymerisation of the network. Its localization to endoplasmic reticulum (ER) and lipid droplets (LD) has been associated to LD metabolism, membrane remodelling and ER shaping as well [18, 19].
The pathogenic mechanisms of SPAST variants are not yet fully understood, the relative predominance of loss‐of‐function variants in SPG4 [7, 20] suggests that haploinsufficiency is a mode of pathogenicity in SPG4. However, missense variants are also common and cluster in the AAA domain, suggesting a dominant‐negative mechanism. Indeed, functional assays have shown constitutive binding to MT and defective ATP‐ase activity, leading to deficiency in MT severing and a MT disorganization with some missense variants in the AAA domain of Spastin in vitro [10]. These differents pathogenic mechanisms could lead to genotype–phenotype correlations in patients carrying biallelic SPAST variants. Indeed, in our study, three patients diagnosed with severe intellectual disability with DEE or infantile neurodegenerative disorder showed homozygous variants, either loss‐of‐function or missense, located in the microtubule interacting and trafficking (MIT) domain (Figure 2). On the contrary, the two patients published by Cruz‐Camino with DEE had variants not located to the same domain (AAA domain). Therefore, further investigations should be carried out to verify the underlying mechanism, such as functional studies to explore the consequences on the MT network, depending on the type of variant.
FIGURE 2.
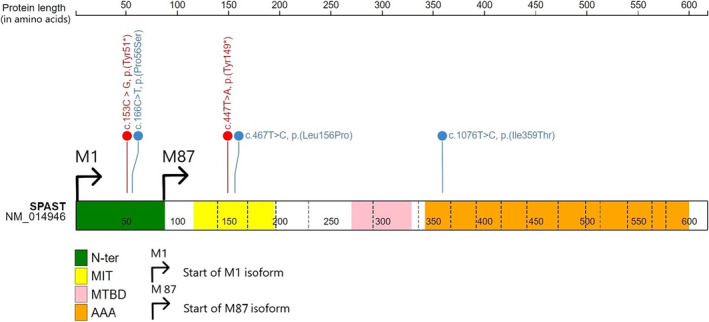
Diagrammatic representation of the SPAST gene and the localization of variants relative to the protein's functional domains of interest. The different functional domains are represented with colours: In green the N‐ter domain (N‐ter), in yellow the Microtubule Interacting and Trafficking domain (MIT), in pink the Microtubules binding domain (MTBD) and orange the AAA domain (AAA). The five different SNVs are represented according to their position in SPAST, with missense variants in blue and nonsense variants in red. The N‐ter domain, in green, is only present in the M1‐isoform of the protein.
Interestingly, our patient carrying the c.153C > G, p.(Tyr51*) homozygous variant is the first carrier with an expected complete loss of function of M1‐spastin, the brain‐enriched isoform of spastin. This variant likely results in a complete loss of M1‐spastin's ability to interact with the ER through its N‐terminal domain [21]. The M87‐spastin isoform could be unaffected in this patient, suggesting that alterations in ER and LD functions, without changes to microtubule‐severing activities, are sufficient to cause an SPG4 phenotype. However, quantitative RT‐PCR targeting each isoform in patient cells should be performed to confirm this hypothesis. It has been shown that genetic variations of SPAST have more deleterious effects on the M1 isoform than on the M87 isoform [22], and indeed we observe a severe complex HSP phenotype in our patient, including intellectual disability, neurodegeneration, and early onset at 12 months.
Most of the parents of our patients, heterozygous carriers of SPAST variants, showed no signs of HSP. However, few parents benefit from neurological examination, and with the possible late onset of SPG4, they may become symptomatic with HSP later in life. In family A, both parents and other family members, heterozygous carriers of the truncating variant, suffered from late‐onset pure form of SPG4. In this family, the homozygous brothers showed a severe phenotype with early infantile neurodegenerative condition. Interestingly, Cruz‐Camino et al. also described a family with symptomatic heterozygous carriers of a truncating variant, and severe DEE in homozygous carriers. For these two truncating variants, the severity of the phenotype was correlated with the copy number (zygosity) of the variant. Conversely, hypomorphic variants, which may not have clinical implications when heterozygous, could potentially lead to HSP when present in a biallelic state.
To further these investigations and more accurately describe the cellular effects of biallelic SPAST variants, functional assays should be performed to reveal varying effects of SPAST variants on the MT network and ER/LD functions depending on their pathogenicity when present at the heterozygous vs. homozygous state.
CONCLUSION
In this study, we show that biallelic variants in SPAST lead to SPG4 classical presentation. Alternatively, some SPAST biallelic variants can lead to infantile onset neurodegenerative disorder, a new severe phenotype associated with this gene, which is a crucial information for genetic counselling. Finally, a complete loss of function of the M1‐spastin is associated with a complex HSP phenotype.
AUTHOR CONTRIBUTIONS
Manon Degoutin: Writing – original draft; conceptualization; methodology; data curation; software. Chloé Angelini: Writing – original draft; writing – review and editing; conceptualization. Claire Bar: Data curation; writing – review and editing. Wahiba Amer El Khedoud: Investigation. Christine Barnerias: Investigation. Razika Boulariah‐Hadjou: Writing – review and editing; investigation. Mehrdad A. Estiar: Writing – review and editing; investigation. Claire Ewenczyk: Writing – review and editing; investigation. Ziv Gan‐Or: Writing – review and editing; investigation. Didier Lacombe: Investigation. Claire Lefeuvre: Investigation. Purvi Majethia: Writing – review and editing; investigation. Mouna Messaoud‐Khelifi: Investigation. Dhanya Lakshmi Narayanan: Investigation. Guy A. Rouleau: Writing – review and editing; investigation. Oksana Suchowersky: Investigation; writing – review and editing. Anju Shukla: Investigation. Marine Guillaud‐Bataille: Conceptualization; software; investigation. Giovanni Stevanin: Conceptualization; methodology; writing – review and editing; writing – original draft. Cyril Goizet: Writing – original draft; writing – review and editing; conceptualization; methodology.
CONFLICT OF INTEREST STATEMENT
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Supporting information
Data S1: Supporting Information.
ACKNOWLEDGMENTS
We are indebted to the patients and their families for participation in this study. This research did not receive any specific grant. Part of this study has been funded by the Association Strumpell‐Lorrain‐HSP‐France, CIHR Emerging Team Grant, in collaboration with the Canadian Organization for Rare Disorders (CORD) [grant number RN127580–260005] and National Institutes of Health, United States, and “Genetic Diagnosis of Neurodevelopmental Disorders in India” (1R01HD093570‐01A1). Figure 1 has been created thanks to CeGaT Pedigree Chart Designer.
Degoutin M, Angelini C, Bar C, et al. From spastic paraplegia to infantile neurodegenerative disorder: Expanding the phenotypic spectrum associated with biallelic SPAST variants. Eur J Neurol. 2025;32:e70025. doi: 10.1111/ene.70025
DATA AVAILABILITY STATEMENT
Data are available on request from the authors.
REFERENCES
- 1. Méreaux JL, Banneau G, Papin M, et al. Clinical and genetic spectra of 1550 index patients with hereditary spastic paraplegia. Brain. 2022;145(3):1029‐1037. doi: 10.1093/brain/awab386 [DOI] [PubMed] [Google Scholar]
- 2. Hazan J, Fontaine B, Bruyn RPM, et al. Linkage of a new locus for autosomal dominant familial spastic paraplegia to chromosome 2p. Hum Mol Genet. 1994;3(9):1569‐1573. doi: 10.1093/hmg/3.9.1569 [DOI] [PubMed] [Google Scholar]
- 3. Dürr A, Davoine CS, Paternotte C, et al. Phenotype of autosomal dominant spastic paraplegia linked to chromosome 2. Brain. 1996;119(5):1487‐1496. doi: 10.1093/brain/119.5.1487 [DOI] [PubMed] [Google Scholar]
- 4. Lo Giudice T, Lombardi F, Santorelli FM, Kawarai T, Orlacchio A. Hereditary spastic paraplegia: clinical‐genetic characteristics and evolving molecular mechanisms. Exp Neurol. 2014;261:518‐539. doi: 10.1016/j.expneurol.2014.06.011 [DOI] [PubMed] [Google Scholar]
- 5. Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;321(8334):1151‐1155. doi: 10.1016/S0140-6736(83)92879-9 [DOI] [PubMed] [Google Scholar]
- 6. Akaba Y, Takeguchi R, Tanaka R, Takahashi S. A complex phenotype of a patient with spastic paraplegia type 4 caused by a novel pathogenic variant in the SPAST gene. Case Rep Neurol. 2021;13(3):763‐771. doi: 10.1159/000520433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Parodi L, Fenu S, Barbier M, et al. Spastic paraplegia due to SPAST mutations is modified by the underlying mutation and sex. Brain. 2018;141(12):3331‐3342. doi: 10.1093/brain/awy285 [DOI] [PubMed] [Google Scholar]
- 8. Varghaei P, Estiar MA, Ashtiani S, et al. Genetic, structural and clinical analysis of spastic paraplegia 4. Parkinsonism Relat Disord. 2022;98:62‐69. doi: 10.1016/j.parkreldis.2022.03.019 [DOI] [PubMed] [Google Scholar]
- 9. Giordani GM, Diniz F, Fussiger H, et al. Clinical and molecular characterization of a large cohort of childhood onset hereditary spastic paraplegias. Sci Rep. 2021;11(1):22248. doi: 10.1038/s41598-021-01635-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Errico A. Spastin, the protein mutated in autosomal dominant hereditary spastic paraplegia, is involved in microtubule dynamics. Hum Mol Genet. 2002;11(2):153‐163. doi: 10.1093/hmg/11.2.153 [DOI] [PubMed] [Google Scholar]
- 11. Svenson IK, Kloos MT, Gaskell PC, et al. Intragenic modifiers of hereditary spastic paraplegia due to spastin gene mutations. Neurogenetics. 2004;5(3):157‐164. doi: 10.1007/s10048-004-0186-z [DOI] [PubMed] [Google Scholar]
- 12. de Bot ST, van den Elzen RTM, Mensenkamp AR, et al. Hereditary spastic paraplegia due to SPAST mutations in 151 Dutch patients: new clinical aspects and 27 novel mutations. J Neurol Neurosurg Psychiatry. 2010;81(10):1073‐1078. doi: 10.1136/jnnp.2009.201103 [DOI] [PubMed] [Google Scholar]
- 13. Cruz‐Camino H, Vázquez‐Cantú M, Vázquez‐Cantú DL, et al. Clinical characterization of 2 siblings with a homozygous SPAST variant. Am J Case Rep. 2020;21. doi: 10.12659/AJCR.919463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med off J Am Coll Med Genet. 2015;17(5):405‐424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Millecamps S, Julien JP. Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci. 2013;14(3):161‐176. doi: 10.1038/nrn3380 [DOI] [PubMed] [Google Scholar]
- 16. Vemu A, Szczesna E, Zehr EA, et al. Severing enzymes amplify microtubule arrays through lattice GTP‐tubulin incorporation. Science. 2018;361(6404):eaau1504. doi: 10.1126/science.aau1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Costa AC, Sousa MM. The role of Spastin in axon biology. Front Cell Dev Biol. 2022;10:934522. doi: 10.3389/fcell.2022.934522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arribat Y, Grepper D, Lagarrigue S, Qi T, Cohen S, Amati F. Spastin mutations impair coordination between lipid droplet dispersion and reticulum. PLoS Genet. 2020;16(4):e1008665. doi: 10.1371/journal.pgen.1008665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Papadopoulos C, Orso G, Mancuso G, et al. Spastin binds to lipid droplets and affects lipid metabolism. PLoS Genet. 2015;11(4):e1005149. doi: 10.1371/journal.pgen.1005149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fonknechten N, Mavel D, Byrne P, et al. Spectrum of SPG4 mutations in autosomal dominant spastic paraplegia. Hum Mol Genet. 2000;9(4):637‐644. doi: 10.1093/hmg/9.4.637 [DOI] [PubMed] [Google Scholar]
- 21. Park SH, Zhu PP, Parker RL, Blackstone C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin‐1 coordinate microtubule interactions with the tubular ER network. J Clin Invest. 2010;120(4):1097‐1110. doi: 10.1172/JCI40979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Solowska JM, Rao AN, Baas PW. Truncating mutations of SPAST associated with hereditary spastic paraplegia indicate greater accumulation and toxicity of the M1 isoform of spastin. Mol Biol Cell. 2017;28(13):1728‐1737. doi: 10.1091/mbc.E17-01-0047 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Supporting Information.
Data Availability Statement
Data are available on request from the authors.