

Abstract
Objective: To explore the influence of SALL4 in cardiac fibroblasts on the progression of myocardial infarction. Methods: Analysis of genes specifically expressed in myocardial infarction by bioinformatics methods; The impact of SALL4 on myocardial infarction was assessed using mouse ultrasound experiments and Masson staining; The effect of SALL4 on the expression levels of collagen-I and collagen-III in myocardial tissue was examined by immunohistochemical staining; The migration ability of cardiac fibroblasts was evaluated using a Transwell assay; The proliferative ability of cardiac fibroblasts was tested using a CCK-8 assay; The relative fluorescence intensity of α-SMA and CTGF in cardiac fibroblasts were checked through immunofluorescence staining experiment; The expression of SALL4, DOT1L, H3K79me2, P53, SHP2, YAP, nucleus-YAP, collagen-I, α-SMA, CTGF, and PAI-1 in myocardial tissues or cardiac fibroblasts was detected using western blot analysis. Results: SALL4-specific high expression in myocardial infarction; SALL4 intensified the alterations in the heart structure of mice with myocardial infarction and worsened the fibrosis of myocardial infarction; SALL4 also promoted the expression of SALL4, DOT1L, H3K79me2, P53, SHP2, YAP, nucleus-YAP, collagen-I, collagen-III, α-SMA, CTGF, and PAI-1 in myocardial infarction tissues and cardiac fibroblasts; Subsequently, SALL4 could enhance the immunofluorescence intensity of α-SMA and CTGF; Moreover, SALL4 could promote the proliferation and migration of cardiac fibroblasts. Conclusion: In cardiac fibroblasts, SALL4 mediates the DOT1L/H3K79me2 signaling pathway to inhibit SHP2, which then promotes the YAP/TAZ signaling pathway, thereby facilitating the progression of myocardial infarction.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-81815-y.
Keywords: Myocardial infarction, Cardiac fibroblasts, SALL4, SHP2, DOT1L/H3K79me2
Subject terms: Cell biology, Medical research
Introduction
Myocardial infarction (MI), as the most common fatal cardiovascular disease, has a steadily increasing incidence and mortality rate. According to the China Cardiovascular Report, about 3.5 million people die from cardiovascular diseases each year, with approximately 2.5 million cases of MI. Myocardial fibrosis, an important pathological process in ventricular remodeling after MI, is characterized by the excessive accumulation of collagen fibers and the deposition of the extracellular matrix (ECM)1,2. Initially, myocardial fibrosis serves as a mechanism of repair to protect the heart from rupture. However, during the sub-acute and chronic phases, excessive fibrotic response leads to abnormal heart function and ventricular wall hardening, eventually increasing the likelihood of heart failure3,4. Therefore, identifying key targets for myocardial fibrosis and halting the fibrotic process is crucial for delaying the progression of ischemic heart disease and improving patient prognosis.
It is confirmed that cardiac fibroblasts (CFs) play a key role in the process of myocardial fibrosis. Under physiological conditions, cardiac fibroblasts are in a quiescent state, playing an important role in maintaining the normal structure, electrical conduction, mechanical contraction, and relaxation functions of the heart. However, when stimulated by MI, cardiac fibroblasts proliferate, migrate, and differentiate into cardiac myofibroblasts, leading to excessive deposition of the extracellular matrix and interstitial fibrosis of myocardial cells, thereby decreasing the compliance of the ventricular wall, reducing or losing elasticity, and resulting in compensatory loss of cardiac systolic function, ultimately leading to heart failure or even severe outcomes like cardiac rupture. Therefore, by starting from the functional characteristics of cardiac fibroblasts and their role in the MI repair process, exploring new targets that can inhibit the excessive proliferation and transformation of cardiac fibroblasts to reduce excessive myocardial fibrosis after MI is essential5–7.
SALL4 is a gene that plays a significant role during human development. It belongs to a family of genes known as SAL-like (SALL) which are transcription factors involved in regulating developmental processes. These genes are characterized by having multiple zinc finger structural domains, a DNA-binding mode used by transcription factors to regulate the expression of other genes. In genetics and clinical medicine, mutations in SALL4 are associated with the occurrence of various congenital diseases, especially Duane-radial ray syndrome (DRRS) and Okihiro syndrome. These diseases typically present a complex array of symptoms including limb abnormalities, limited eye movement, reduced hearing, and kidney issues, with symptoms varying significantly among patients. In the research field, SALL4 has garnered considerable attention, particularly in stem cell research and oncology8,9. As a pluripotency factor, SALL4 is crucial for maintaining the differentiation potential of embryonic stem cells, which has potential applications in regenerative medicine and developing new therapeutic strategies for diseases.
DOT1L, short for "Disruptor of telomeric silencing 1-like," is a protein that is a histone methyltransferase, specifically catalyzing di-methylation (me2) and tri-methylation (me3) on the 79th lysine residue of histone H3 (H3K79). This methylation is crucial for regulating gene expression, cell cycle control, and DNA damage repair among other biological processes. DOT1L is closely associated with specific types of cancer, particularly in some cases of acute myeloid leukemia (AML), where abnormal activation of DOT1L due to chromosomal translocations is related to the disease’s progression. In these cases, DOT1L promotes cancer cell proliferation and survival by aberrantly methylating H3K79, leading to the activation of incorrect gene expression programs. Furthermore, DOT1L and its methylation activity on H3K79 play roles in various biological processes such as cell memory and developmental processes, indicating its complex and multifaceted role in cellular biology10,11. Research has also found that SALL4 can promote the DOT1L/H3K79me2 signaling pathway. Hence, in this study, we explored the effect of SALL4 in cardiac fibroblasts on the progression of myocardial infarction.
Methods
Bioinformatics analysis
Myocardial infarction-related dataset GSE66360 was screened from the Gene Expression Omnibus (GEO) database.RNA sequencing data were normalised and transformed using log2.The processed data were corrected for batch effects using the Combat method. Differential expression analysis was performed using the limma package with the screening criteria of |log2 Fold Change|> 2 and p-value < 0.05. Multiple hypothesis tests were corrected for p-values using the Benjamini–Hochberg method. The paired-samples t-test statistical method was selected for correlation analysis of the expression of SALL4 and DOTIL among the differential genes.
Establishment of mouse model
8-week-old C57BL/6 male mice were purchased from Henan SCBS Bioscience Co., Ltd.. Mice weighed 20 ± 2 g and were studied in all experiments. All mice were housed in Plexiglas cages, maintained on a 12/12-h light/dark cycle, and received food and water ad libitum in a temperature- and humidity-controlled room. They were divided into 5 groups: Sham, Model, SALL4-NC, SALL4-KD, and SALL4-OE, with 8 mice per group. The mice were anesthetized with 5% isoflurane, placed in a supine position, and the left anterior chest area was prepared by making a longitudinal incision approximately 1.5 cm in length after disinfection. A vertical mattress suture was prepared at the incision site for later closure. Subsequently, the chest wall muscles were bluntly separated layer by layer, and the chest cavity was rapidly entered through the 4th intercostal space. Hemostatic forceps were used to open the intercostal space, and the heart was gently squeezed with the left hand in sync with its beating, to pop it out through the intercostal space. Next, a suture needle was used to ligate the heart both 2 mm below the left atrial appendage and 0.5 mm from the lung cone passing through the anterior descending branch of the left coronary artery. After ligation, the heart was gently put back into the chest cavity, air was squeezed out, and the reserved sutures at the incision site were tightened to close the incision. Mice modelling mortality rate of 20.8%. Finally, the mice were revived and administered glucose saline. For the Sham group, only the incision was made without ligation of the heart. SALL4-shRNA and SALL4-overexpressing lentivirus as well as negative controls (purchased from Genomeditech Co., Ltd., Shanghai, China) were injected into the myocardium of mice in the SALL4-NC, SALL4-KD, and SALL4-OE groups, respectively. Mice are placed in the IVC cage box, and the density of animals should not be too high. Before putting the animals into the euthanasia box, do not fill CO2 into the euthanasia box; after putting the animals into the cage, fasten the lid of the cage, connect the CO2 gas pipe at the entrance where the water bottle is placed, open the valve of the gas bottle, and fill CO2 into the box at the rate of replacing 10% ~ 30% of the euthanasia box’s volume per minute, so that CO2 fills the cage box. After the mouse faints and loses the ability to move, increase the gas flow, the maximum flow rate should not exceed 0.5kPa, make sure that the mouse does not move, does not breathe, and the pupil is dilated, then turn off the CO2 and observe for two minutes to make sure that the mouse is dead. After euthanasia, close the gas valve and remove the body and store it in the designated freezer. The animal experiment was approved by the Medical Ethics Committee of Hebei Provincial People’s Hospital.(2024-DW-056). All methods in this experiment were performed in accordance with the relevant guidelines and regulations and conformed to THE RULES OF 3R and ARRIVE guidelines.
Echocardiography
After successful modeling and continuous culture for 3 days, the mice in each group were anesthetized with isoflurane, and their chest hair was removed. The cardiac structure post-treatment was assessed using Vevo 2100. Subsequently, two-dimensional echocardiographic outcomes were acquired. The ejection fraction (EF) and shortening fraction (FS), left ventricular diastolic diameter (LVID; d) and left ventricular systolic diameter (LVID; s), systolic left anterior wall thickness (LVAW; s), and diastolic left anterior wall thickness (LVAW; d) were measured to represent the cardiac structure and function.
Masson’s trichrome staining
Mice were dissected 4 weeks after modelling to collect heart tissue, which was then fixed with 4% paraformaldehyde and embedded in paraffin. Sections of 4 μm thickness were cut and stained with an affinity reagent. After washing, sections were stained with hematoxylin for 3 min, differentiated with hydrochloric acid-alcohol, and stained with Masson’s acid fuchsin solution (containing ponceau and acid magenta) for 10 min. Differentiation was conducted with 1% phosphomolybdic acid solution for 4 min, followed by staining with light green solution for 5 min, and mounted with neutral resin. The sections were then observed under a microscope.
Immunohistochemical staining
Myocardial tissue paraffin sections were routinely deparaffinised to water, and tissue antigen repair was performed by autoclaving. 3% H2O2 was added and incubated at room temperature for 5 min to remove endogenous peroxidase. The sections were washed with PBS, then the secondary antibody was added and incubated for 40 min at 37 ℃. After the sections were washed with PBS, DAB chromogen was added and the colour was developed for 5 min at room temperature. The sections were stained with hematoxylin stain for 40 s and washed with water for 1 min. 1% hydrochloric acid was used for alcohol differentiation for 3 to 5 h. Sections were dehydrated with gradient alcohol, clarified with xylene, and sealed with neutral gum. lmageJ software was used for analysis.
Extraction and isolation of mouse cardiomyocytes and cardiac fibroblasts
After disinfection, remove the apical part of the mouse heart, use ophthalmic scissors to cut it into small pieces of tissue about 1 mm3, then add DMEM medium containing 10% phosphate buffer solution, use a rubber-tipped buret to suck up the pieces of tissue and spread them evenly on the side wall of the 50 ml cell culture flasks, and then place the side with the pieces of tissue facing up in a carbon dioxide incubator at 37℃, 5% CO2, and then turn the flask over to make the pieces of tissue submerged in the culture medium. After 4 ~ 6h of incubation, the bottle was turned over so that the tissue blocks were immersed in the culture medium.Prepare high-density (density of 1.017 gm) and low-density (density of 0.407 g/mL) Percoll separately, and slowly spread the low-density Percol on top of the high-density Percol. High-density (density 1.017 gm) and low-density (density 0.407 g/mL) Percoll were prepared separately, and the low-density Percoll was slowly spread on top of the high-density Percoll. The combined cell resuspension was counted and added slowly on top of the Percoll, and centrifuged at 1700 g for 30 min to collect the upper fibroblasts and lower cardiomyocytes separately. The cells were washed twice with DMEM complete medium containing 10% FBS, the first time by centrifugation at 600 g for 5 min, and the second time by centrifugation at 250 g for 5 min. The cell sediment was resuspended with overnight medium and counted, and then transferred to appropriate culture dishes and incubated overnight at 37℃ with 5% CO2. On the next day, the culture medium was changed to the appropriate specialised medium for continuous incubation.
Immunofluorescence staining
Cells were spread on coverslips overnight. After fixation in 4% paraformaldehyde, the cells were permeabilized with 0.5% Triton X-100. Then, the coverslips were submerged in blocking solution for 1 h, followed by incubation with primary antibodies against collagen-I and CTGF overnight. The coverslips were rinsed twice with PBS before observation and analyzed with a fluorescence microscope.
Cell culture
Human cardiac fibroblasts were purchased from Wuhan Puno Biotech Co., Ltd., and cultured in low-glucose, low-serum DMEM, under a 1% O2 environment. Based on the SALL4 (NM-020436) genetic data from GenBank, an overexpressing lentivirus targeting SALL4 and a negative control were used. The synthetic gene was inserted into the pGMLV-SC5 lentiviral core vector, which contains the CMV promoter-driven enhanced green fluorescent protein (GFP) reporter gene. Human cardiac fibroblasts were infected with concentrated virus in serum-free medium. After 24 h, the supernatant was replaced with complete culture medium. Cells were treated with the DOT1L inhibitor pinometostat (5 nM) and the SHP2 inhibitor PHPS1 (30 μM).
Extraction of nuclear proteins
Cultured cells were washed with PBS to remove medium residues. Resuspend the cell precipitate in an appropriate amount of ice-cold Lysis Buffer I (containing protease inhibitors). Gently homogenise 20 times using a Dounce homogeniser to mechanically fragment cell membranes without disrupting nuclei. The homogenate is centrifuged at low temperature (800 g, 4°C, 5 min) and the precipitated nuclei are resuspended in Lysis Buffer II (also containing protease inhibitors). The resuspension is placed on ice for approximately 30 min and gently vortexed intermittently to ensure complete lysis of the nuclei. Samples can be sonicated or further assisted in lysis using mechanical means to ensure maximum release of nuclear proteins. The lysed sample is centrifuged at high speed (12,000 g) at low temperature for approximately 10 min to remove insoluble debris. The supernatant is the extracted nuclear proteins and can be used directly in subsequent experiments.
CCK-8
Cardiac fibroblasts in the logarithmic growth phase were digested, diluted, and counted using a cell counting plate. After cell adherence, they were cultured for 24h, 48h and 72h. The culture medium was then replaced, and 10μl of CCK-8 reagent was added to each well. The plate was incubated in the dark for 2h before the cell viability was measured at 450 nm using a microplate reader. The procedure was repeated three times, and data were recorded.
Transwell
The cell invasion ability was evaluated using the Transwell assay. 1 × 10^5 cells suspended in 200μl serum-free DMEM/F-12 were seeded into the upper chamber (8μm pore size, Millipore) coated with matrix gel, and the lower chamber received 500μl DMEM/F-12 containing 10% FBS. After incubation for 24 h, cells in the upper chamber were gently removed with a cotton swab. The migrating cells on the bottom surface were fixed with 4% paraformaldehyde and stained with crystal violet. Photographs were taken of the cells, and manual cell counting was conducted under a microscope at 200 × magnification.
Western blot
Mix 490μl RIPA lysis buffer, 5μl protease inhibitor PMSF, and 5μl phosphatase inhibitor, and place on ice until use. Total protein was extracted. The protein concentration of the samples was determined using a BCA protein assay kit. Separating gel and stacking gel were prepared. Protein samples were mixed with 5 × SDS-PAGE Loading Buffer to a final concentration of 1 × and boiled at 100℃ for 5 min, then placed on ice. 30μg of protein sample was loaded into each well, and electrophoresis was conducted. After transfer, the PVDF membrane was incubated in 5% skim milk in TBST blocking solution at room temperature on a shaker for 2h. Antibody incubation and ECL chemiluminescence: The PVDF membrane was placed in a hybridization bag, incubated with diluted primary antibody on a room temperature shaker for 1h, then stored in a 4℃ refrigerator for 24h. The PVDF membrane was removed and washed with 1 × TBST on a shaker (3 times, 10min each). The membrane was then incubated with secondary antibody at room temperature for 2h. After washing the membrane with TBST 3 times, each for 10 min, the membrane was soaked in chemiluminescence liquid and detected using the ECL method.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 9.0 software. Data collected from three independent repeats were presented as mean ± standard deviation or mean ± SEM. The t-test was used to evaluate differences between two groups. One-way ANOVA was applied to analyze differences among more than two groups. P < 0.05 indicated statistically significant differences.
Results
Myocardial infarction leads to specific upregulation of SALL4 and DOT1L in myocardial tissue
Bioinformatics analysis showed that the GSE66360 dataset contained a total of 49 myocardial infarction-related samples and 50 healthy samples. 339 significantly up-regulated genes and 103 down-regulated genes were screened by limma package analysis. Among them, SALL4 and DOTIL were significantly expressed up-regulated genes, and SALL4 was found to promote DOT1L expression by correlation expression scatter plot. We then extracted cardiomyocytes and cardiac fibroblasts from normal and myocardial infarcted mice. Changes in SALL4 in them were detected using western blot. The results showed that the relative protein expression of SALL4 in cardiomyocytes and cardiac fibroblasts of myocardial infarction mice was significantly elevated relative to normal mice (Fig. 1A,B).
Fig. 1.

Impact of SALL4 on Cardiac Function in Mice with Myocardial Infarction. A: Differential gene volcano plots and horizontal scatter plots of SALL4 and DOT1L expression; B: Western blot detection of SALL4 expression in core myofibroblasts of cardiomyocytes in normal and myocardial infarcted mice; GAPDH as control protein; C: Ultrasound images of mice and statistics of EF, FS, LVAW;d, LVAW;s, LVID;d, and LVID;s. Data are expressed as mean + standard deviation. N = 8; **P < 0.01; *P < 0.05; nsP > 0.05.
SALL4 can exacerbate myocardial infarction-induced structural and functional heart damage in mice
Cardiac function of the various mouse groups was detected using ultrasound equipment. Results showed a significant decrease in the expression of EF, FS, LVAW; d, and LVAW; s, and an increase in LVID; d and LVID; s in the Model group, SALL4-NC group, SALL4-KD (knockdown) group, and SALL4-OE (overexpression) group compared to the sham group. There were no significant differences in EF, FS, LVAW; d, LVAW; s, LVID; d and LVID; s between the Model and SALL4-NC groups. In contrast, SALL4-KD mice showed increased EF, FS, LVAW; d, and LVAW; s expression and decreased LVID; d and LVID; s expression compared to the SALL4-NC group. SALL4-OE mice exhibited significant exacerbation with lower EF, FS, LVAW; d, and LVAW; s expression, and higher LVID; d and LVID; s expression (Fig. 1C). This suggests that SALL4 can worsen heart structure and function damage in mice with myocardial infarction.
SALL4 can aggravate myocardial tissue fibrosis in myocardial infarction in mice
The results of Masson’s trichrome staining demonstrated that myocardial cells were swollen, with nuclear necrosis, severe inflammatory response, and cell lysis in the Model, SALL4-NC, SALL4-KD and SALL4-OE groups, compared to the sham group. Infarct size, cardiomyocytes, and morphology were indistinguishable between the Model and SALL4-NC groups. The infarct area was notably reduced, and myocardial cells and morphology appeared relatively normal in the SALL4-KD group; However, cell swelling, nuclear necrosis, inflammation, and cell lysis were more pronounced in the SALL4-OE group.
The results of immunohistochemical staining showed that the relative protein expression levels of collagen-I and collagen-III in the model group, SALL4-NC group, SALL4-KD group, and SALL4-OE group were significantly increased compared with the sham group. There was no significant difference in the relative protein expression levels of collagen-I and collagen-III between the model group and SALL4-NC group. Compared with the SALL4-NC group, the relative protein expression levels of collagen-I and collagen-III in the SALL4-KD group decreased, while those in the SALL4-OE group significantly increased (Fig. 2).
Fig. 2.

Impact of SALL4 on Fibrosis in Mice with Myocardial Infarction. A: Masson staining results; B: Immunohistochemical staining results and statistical data on relative protein expression of collagen-I and collagen-III. Data are expressed as mean + standard deviation. N = 8; **P < 0.01; *P < 0.05; nsP > 0.05.
SALL4 can mediate the DOT1L/H3K79me2 signaling pathway to inhibit SHP2, thus promoting the expression of YAP/TAZ signaling pathway-related proteins in myocardial infarction tissue
Western blot results showed that the relative protein expression levels of SALL4, DOT1L, H3K79me2, P53, YAP, collagen-I, α-SMA, CTGF, and PAI-1 were significantly elevated, while SHP2 expression was significantly decreased in the Model, SALL4-KD, and SALL4-OE groups compared to the sham group. Relative to the Model group, the SALL4-KD group showed a significant decrease in these protein levels, while SHP2 levels were significantly increased. In contrast, in the SALL4-OE group, all the aforementioned protein levels were markedly higher, and SHP2 levels were significantly lower (Fig. 3), indicating that SALL4 can mediate the DOT1L/H3K79me2 pathway to suppress SHP2, which in turn boosts the expression of proteins related to the YAP/TAZ signaling pathway.
Fig. 3.
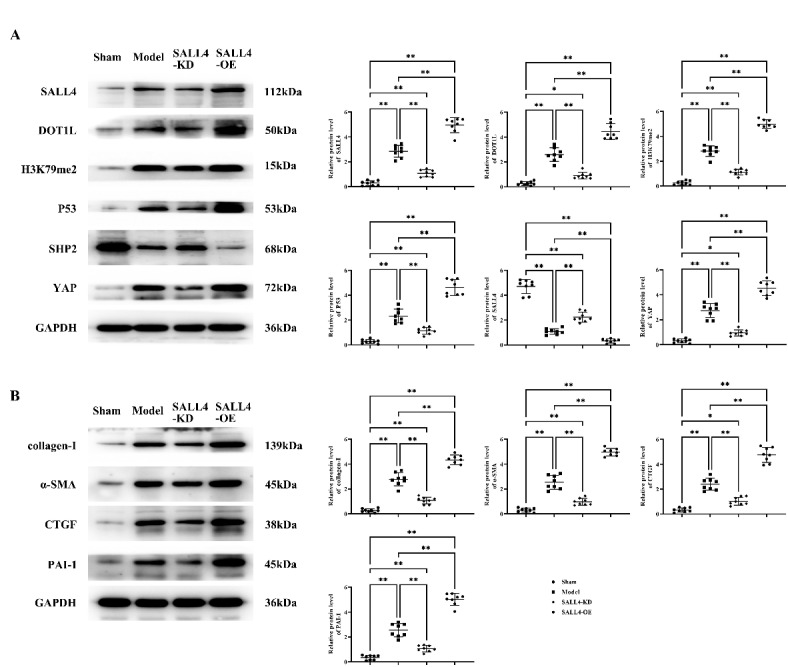
Impact of SALL4 on the DOT1L/H3K79me2 Signaling Pathway in Cardiac Tissue. A: Protein bands and statistical data for the relative protein expression of SALL4, DOT1L, H3K79me2, P53, SHP2, and YAP; B: Protein bands and statistical data for the relative protein expression of collagen-I, α-SMA, CTGF, and PAI-1. GAPDH as control protein. Data are expressed as mean + standard deviation. N = 8; **P < 0.01; *P < 0.05; nsP > 0.05.
SALL4 can mediate the DOT1L/H3K79me2 signaling pathway to inhibit SHP2, thus promoting the expression of YAP/TAZ signaling pathway-related proteins in cardiac fibroblasts
Western blot results indicated that, compared to the NC (negative control) group, the SALL4-OE group displayed a significant increase in the relative protein expression of SALL4, DOT1L, P53, nucleus-YAP, collagen-I, α-SMA, and CTGF; The relative protein levels of p-SHP2 and t-SHP2 were substantially decreased. However, after the addition of the DOT1L inhibitor pinometostat, except for SALL4, there was a significant reduction in the expression levels of DOT1L, P53, nucleus-YAP, collagen-I, α-SMA, and CTGF in both NC and SALL4-OE groups with no significant differences, while p-SHP2 and t-SHP2 levels significantly increased with no significant differences. After further addition of PHPS1, the relative protein expression of DOT1L, P53, and t-SHP2 in the NC and SALL4-OE groups reverted to trends observed prior to pinometostat addition; The level of p-SHP2 significantly decreased with no significant differences, and the protein levels of nucleus-YAP, collagen-I, α-SMA, and CTGF significantly increased with no significant differences.
The results of immunofluorescence staining showed that the relative fluorescence intensities of CTGF and α-SMA in the SALL4-OE group were significantly increased compared with those in the NC group; However, the relative fluorescence intensities of CTGF and α-SMA in both the NC and SALL4-OE groups were significantly decreased after the addition of pimonostat, with no significant difference. After further addition of PHPS1, the relative fluorescence intensities of CTGF and α-SMA were significantly increased in both NC and SALL4-OE groups with no significant difference (Fig. 4).
Fig. 4.

Impact of SALL4 on the DOT1L/H3K79me2 Signaling Pathway in Myocardial Fibroblasts. A: Protein bands and statistical data for the relative protein expression of SALL4, DOT1L, P53, p-SHP2, t-SHP2, and YAP; B: Protein bands and statistical data for the relative protein expression of collagen-I, α-SMA, and CTGF; GAPDH as control protein; C: Immunofluorescence staining results and statistical data on relative fluorescence intensity of CTGF and α-SMA. Data are expressed as mean + standard deviation. N = 3; **P < 0.01; *P < 0.05; nsP > 0.05.
SALL4 can promote the proliferation and invasion capability of cardiac fibroblasts
The results of the CCK-8 assay indicated that the OD value was significantly increased in the SALL4-OE group compared to the NC group. However, after the addition of pinometostat, the OD values significantly decreased in both NC and SALL4-OE groups with no significant differences. After further addition of PHPS1, OD values significantly increased in both groups with no significant differences.
The results of the Transwell assay demonstrated that, compared to the NC group, the number of invading cells was significantly increased in the SALL4-OE group. However, after the addition of pinometostat, the number of invading cells significantly decreased in both groups with no significant differences. After further addition of PHPS1, the number of invading cells significantly increased in both groups with no significant differences (Fig. 5).
Fig. 5.

Impact of SALL4 on the Proliferation and Invasion Capabilities of Myocardial Fibroblasts. A: Results for the CCK-8 assay; B: Results for the Transwell assay and statistical data for the number of invading cells. Data are expressed as mean + standard deviation. N = 3; **P < 0.01; ; nsP > 0.05.
The above experimental results illustrate that SALL4 mediates the DOT1L/H3K79me2 signaling pathway to inhibit SHP2, thereby promoting the YAP/TAZ signaling pathway, which in turn promotes the progression ofAppended myocardial infarction (Fig. 6).
Fig. 6.

SALL4 mediates the inhibition of SHP2 through the DOT1L/H3K79me2 signaling pathway in myocardial fibroblasts, thereby promoting the YAP/TAZ signaling pathway and contributing to the progression of myocardial infarction.
Discussion
Among the various factors involved in ventricular remodeling following myocardial infarction (MI), myocardial fibrosis plays a dominant role. Research has identified three main stages of post-MI myocardial fibrosis: initially, the inflammatory phase occurs, followed gradually by the proliferative phase and finally developing into the remodeling phase, with each stage presenting different pathological states. In the initial stage, the activation of the fibrosis pathway plays a cardioprotective role, preventing cardiac rupture caused by left ventricular wall thinning due to the infarction. However, during the proliferative or remodeling phase, excessive fibrosis can lead to reduced contractile and diastolic capabilities of the heart, causing deterioration in heart function and ultimately leading to heart failure or even cardiac sudden death. Investigating targeted treatment for cardiac fibrosis after MI is an important therapeutic strategy to improve the prognosis of patients with MI12–14. Therefore, discovering and exploring new targets for the regulation of post-MI myocardial fibrosis is key to this research.
Cardiac myofibroblasts play a crucial role in the healing process following myocardial infarction (MI), but their activation can also exacerbate cardiac damage and affect long-term recovery. After MI, the activation of cardiac myofibroblasts is part of the cardiac healing process. They form scar tissue by secreting collagen and other matrix proteins, helping to stabilize the cardiac structure. However, excessive activity of cardiac myofibroblasts can lead to an overabundance of scar tissue, known as excessive fibrosis15. This condition can reduce the elasticity of the heart, limiting its filling capacity and ultimately leading to decreased cardiac function, such as diastolic failure and heart failure. The activation of cardiac myofibroblasts is also related to the inflammatory response. Although short-term inflammation helps to clear necrotic cardiomyocytes and tissue, persistent inflammation can damage healthy cardiomyocytes, exacerbating cardiac injury4,16,17.
Isl1 (Islet-1) is a signature transcription factor that is highly expressed in cardiovascular progenitor cells. Islet-1 + cardiovascular progenitor cells have the potential to differentiate into cardiovascular cell types such as cardiomyocytes, vascular smooth muscle cells, and endothelial cells. Studies have found that the expression of SALL4 in Isl1 + cardiovascular progenitor cells not only reveals the potential and developmental characteristics of these progenitor cells, but may also provide new directions for cell therapy in the treatment of cardiovascular diseases. By regulating the expression of SALL4, the proliferation and differentiation of these progenitor cells may be enhanced, promoting the regeneration and repair of heart tissue. This indicates that SALL4 plays an important role in cardiovascular diseases. And SALL4 has mutations in tetralogy of Fallot (QAV), a congenital heart disease18,19. SALL4 (Sal-like 4) is a transcription factor belonging to the SALL family, mainly expressed during embryonic development and involved in cell fate determination and tissue formation. Although primarily associated with development, recent studies have begun to reveal its potential role in adult diseases, including heart disease. Furthermore, studies have found that in cardiac tissue, SALL1 and SALL4 may promote the proliferation of cardiomyocytes by activating genes related to cell proliferation. SALL1 and SALL4 act upstream of cyclin-dependent kinase (CDK) and cyclin genes, as well as key transcription factor genes such as cyclin (Myocd and serum response factor (SRF) key transcription factor genes are used for the development of dense cardiomyocytes. SALL4 and Myocd form a transcription complex with SRF and directly bind to the upstream regulatory regions of CDK and cyclin genes. This suggests that SALL1 and SALL4 are essential for cardiomyocyte proliferation by regulating CDK and cyclin genes that interact with Myocd and SRF. Furthermore, SALL4 and Myocd play an important role in direct cardiac reprogramming after injury to cardiac fibroblasts. Cardiac reprogramming is a technique that converts cardiac fibroblasts into cardiomyocyte-like cells to promote cardiomyocyte regeneration and replace damaged cardiomyocytes20. SALL4 can promote fibroblast proliferation. Myocd promotes the transformation of fibroblasts into cardiomyocyte-like cells through synergistic action with other transcription factors, such as Gata4 and Tbx58,21. In this study, we found that SALL4 is specifically highly expressed in myocardial infarction and that SALL4 specifically promote DOT1L. SALL4 can exacerbate damage to the cardiac structure and function in mice with myocardial infarction, as well as myocardial tissue fibrosis. SALL4 can promote the relative protein expression of SALL4, DOT1L, H3K79me2, P53, YAP, collagen-I, α-SMA, CTGF, and PAI-1 in the myocardial tissue while reducing the relative protein expression of SHP2.
Studies have discovered SALL4 dynamically recruits the histone demethylase LSD1 and the H3K79 methyltransferase DOT1L. The binding of LSD1 to the promoter of MLL-AF9 target genes reduces the ratio of H3K4me2 to H3K79me3, thereby promoting the development of the MLL-AF9 oncogene program. In addition, studies have found that in a mouse model of ischemic heart failure (HF) and in cell systems, histone 3 lysine 27 is dimethylated and lysine 36 monomethylated (H3_K27me2K36me1)22,23. Similarly, DOT1l can interact with AF9, and the degree to which DOT1L is recruited to MLL-AF9 determines the level of H3K79 methylation and transformation potential. It is possible that SALL4 and MLL-AF9 coordinate the recruitment of these epigenetic modifiers, which together regulate the local chromatin structure and control the expression of target genes, thus regulating subsequent cellular survival. DOT1L (Disruptor of Telomeric Silencing 1-like) is a histone H3K79 methyltransferase whose main function is to regulate gene expression by adding methyl groups to the 79th lysine of histone H3. This modification affects chromatin structure and gene expression, thereby impacting various cellular processes, including the cell cycle, DNA damage repair, and cell differentiation. p53 is a tumor suppressor protein playing a crucial role in the cellular response to DNA damage, cell cycle regulation, and apoptosis. p53 functions by activating or suppressing the expression of downstream target genes, which are involved in processes such as cell cycle arrest, DNA damage repair, apoptosis, and aging. H3K79me2 might enhance the transcription of DNA damage response genes, thereby promoting the activation and stabilization of p5324,25. SHP2 is a non-receptor tyrosine phosphatase that participates in the regulation of cell proliferation, differentiation, and survival via the RAS/MAPK signaling pathway. SHP2 plays a role in various cell signaling processes, including those related to cancer, metabolic disease, and cardiovascular disease. Research indicates p53 can inhibit the expression of SHP226,27.
YAP (Yes-associated protein) and TAZ (transcriptional coactivator with PDZ-binding motif, also known as WWTR1) are transcription coactivators that play significant roles in cell proliferation, migration, differentiation, and survival. They are downstream effectors of the Hippo signaling pathway, primarily functioning by regulating gene expression. The Hippo signaling pathway is a key regulator of cell proliferation and apoptosis found in various organisms, crucial for maintaining organ size and tissue homeostasis. SHP2 is a non-receptor tyrosine phosphatase that regulates multiple signaling pathways through its dephosphorylation activity, affecting processes such as cell proliferation, differentiation, and migration. SHP2 plays a key role in various biological processes and diseases28–30. Studies have shown SHP2 can inhibit the activation of the YAP/TAZ signaling pathway.
YAP (Yes-associated protein) and TAZ (transcriptional coactivator with PDZ-binding motif) are downstream effectors of the Hippo signaling pathway, playing roles in controlling cell proliferation, apoptosis, migration, and organ size. They mainly function within the Hippo signaling pathway. The activity of these proteins is crucial for cell proliferation, survival, migration, and extracellular matrix production, especially in the processes of fibrosis and tumor development. Fibrosis is a pathological process involving excessive extracellular matrix (ECM) accumulation, leading to the destruction of tissue structure and function31. Collagen-I is one of the most abundant ECM proteins, essential for maintaining tissue structure and strength. The activation of YAP/TAZ can promote the expression of collagen-I, particularly evident in various fibrotic diseases (such as liver fibrosis and pulmonary fibrosis). This promotional effect may be achieved through directly enhancing the transcription of collagen genes or activating other transcription factors. α-SMA is a form of actin that is often considered a marker for cardiac fibroblasts32,33. The activation of YAP/TAZ promotes this conversion process, enhancing cell contractility, migratory capabilities, and ECM production, playing a key role in fibrotic lesions. CTGF (connective tissue growth factor) is an important ECM protein and cell proliferation regulator, involved in many biological processes, including cell proliferation, migration, adhesion, and ECM production. YAP/TAZ participate in the progression of fibrosis and tumors by promoting the expression of CTGF. PAI-1 (plasminogen activator inhibitor-1) is a major plasma protein, regulating the plasminogen system, affecting thrombus formation and ECM degradation. The activation of YAP/TAZ can increase the expression of PAI-1, thereby promoting ECM accumulation and fibrosis in some cases34. Furthermore, YAP/TAZ can mediate the production of HIF1α-induced p53, thereby inhibiting the expression of SHP2. In this study, we found that SALL4 mediates the suppression of SHP2 through the DOT1L/H3K79me2 signaling pathway in cardiac myofibroblasts, thereby promoting the expression of YAP/TAZ signaling pathway-related proteins and enhancing the proliferation and invasion of cardiac myofibroblasts.
In conclusion, SALL4 mediates the DOT1L/H3K79me2 signaling pathway in cardiac myofibroblasts, inhibiting SHP2, thereby promoting the YAP/TAZ signaling pathway, contributing to the progression of myocardial infarction, providing new targets and therapeutic methods for the treatment of myocardial infarction.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
Author contributions
YHZ conceived and designed the study. YHZ, MZ, ZPT, YQT, KNP, and HT performed the experiments and analyzed the data. YHZ wrote the manuscript. All authors reviewed and approved the final version of the manuscript.
Funding
This research was supported by Medical Science Research Project of Hebei Province (No. 20190017).
Data availability
The data that support the findings of this study are available on request from the corresponding author.
Competing interests
The authors declare no competing interests.
Ethics statement
The animal experiment was approved by the Medical Ethics Committee of Hebei Provincial People’s Hospital (2024-DW-056).
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Zu, X. et al. Risk of cardiac rupture among elderly patients with diabetes presenting with first acute myocardial infarction. Front. Endocrinol.14, 1239644 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zuo, W. et al. Macrophage-driven cardiac inflammation and healing: insights from homeostasis and myocardial infarction. Cell. Mol. Biol. Lett.28(1), 81 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zuo, P. et al. Protease-activated receptor 2 deficiency in hematopoietic lineage protects against myocardial infarction through attenuated inflammatory response and fibrosis. Biochem. Biophys. Res. Commun.526(1), 253–260 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Zhu, Y. et al. Upregulation of circular RNA CircNFIB attenuates cardiac fibrosis by sponging miR-433. Front. Genet.10, 564 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao, X. S. et al. MiR-30b-5p and miR-22-3p restrain the fibrogenesis of post-myocardial infarction in mice via targeting PTAFR. Eur. Rev. Med. Pharmacol. Sci.24(7), 3993–4004 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Zhao, J. et al. Curcumin ameliorates cardiac fibrosis by regulating macrophage-fibroblast crosstalk via IL18-P-SMAD2/3 signaling pathway inhibition. Front. Pharmacol.12, 784041 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang, Q. J. et al. Matricellular protein cilp1 promotes myocardial fibrosis in response to myocardial infarction. Circ. Res.129(11), 1021–1035 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao, H. et al. Sall4 and Myocd empower direct cardiac reprogramming from adult cardiac fibroblasts after injury. Front. Cell Dev. Biol.9, 608367 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou, J. et al. SALL4 correlates with proliferation, metastasis, and poor prognosis in prostate cancer by affecting MAPK pathway. Cancer Med.12(12), 13471–13485 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao, X. et al. Dot1l cooperates with Npm1 to repress endogenous retrovirus MERVL in embryonic stem cells. Nucleic Acids Res.51(17), 8970–8986 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang, Y. & Kutateladze, T. G. Methylation of histone H3K79 by Dot1L requires multiple contacts with the ubiquitinated nucleosome. Mol. Cell74(5), 862–863 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Zhuang, Y. et al. MiR-375-3p promotes cardiac fibrosis by regulating the ferroptosis mediated by GPX4. Comput. Intell. Neurosci.2022, 9629158 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Zhou, Z., Ma, D., Zhou, Y. et al. The Carthamus tinctorius L. and Lepidium apetalum Willd. drug pair inhibits EndMT through the TGFβ1/Snail signaling pathway in the treatment of myocardial fibrosis. Evid.-Based Complement. Altern. Med. eCAM2023, 6018375 (2023). [DOI] [PMC free article] [PubMed]
- 14.Zhou, Y., Richards, A. M. & Wang, P. MicroRNA-221 is cardioprotective and anti-fibrotic in a rat model of myocardial infarction. Mol. Ther. Nucleic Acids17, 185–197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang, J. et al. Salvia miltiorrhiza and Carthamus tinctorius extract prevents cardiac fibrosis and dysfunction after myocardial infarction by epigenetically inhibiting Smad3 expression. Evid. -Based Complement. Altern. Med. eCAM2019, 6479136 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zha, Y. et al. ADAMTS8 promotes cardiac fibrosis partly through activating EGFR dependent pathway. Front. Cardiovasc. Med.9, 797137 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yue, Y. et al. M2b macrophages regulate cardiac fibroblast activation and alleviate cardiac fibrosis after reperfusion injury. Circ. J.84(4), 626–635 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Monteon, A., Hughes, L., CAMBEROS, V. et al. Identification of SALL4 expressing Islet-1+ cardiovascular progenitor cell clones. Int. J. Mol. Sci. 24(2) (2023). [DOI] [PMC free article] [PubMed]
- 19.Kantaputra, P. et al. Broad spectrum of anomalies including quadricuspid aortic valve associated with a novel frameshift SALL4 variant. Clin. Genet.104(1), 133–135 (2023). [DOI] [PubMed] [Google Scholar]
- 20.Yin, C. et al. SALL4-mediated upregulation of exosomal miR-146a-5p drives T-cell exhaustion by M2 tumor-associated macrophages in HCC. Oncoimmunology8(7), 1601479 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katano, W., Mori, S., Sasaki, S. et al. Sall1 and Sall4 cooperatively interact with Myocd and SRF to promote cardiomyocyte proliferation by regulating CDK and cyclin genes. Development (Cambridge, England) 150(24) (2023). [DOI] [PubMed]
- 22.Yang, L. et al. The stem cell factor SALL4 is an essential transcriptional regulator in mixed lineage leukemia-rearranged leukemogenesis. J. Hematol. Oncol.10(1), 159 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gambardella, J. et al. Ketone bodies rescue mitochondrial dysfunction via epigenetic remodeling. JACC Basic Transl. Sci.8(9), 1123–1137 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salvati, A. et al. Combinatorial targeting of a chromatin complex comprising Dot1L, menin and the tyrosine kinase BAZ1B reveals a new therapeutic vulnerability of endocrine therapy-resistant breast cancer. Breast Cancer Res. BCR24(1), 52 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marsh, D.J., Ma, Y. & Dickson, K.A. Histone monoubiquitination in chromatin remodelling: Focus on the histone H2B interactome and cancer. Cancers12(11) (2020). [DOI] [PMC free article] [PubMed]
- 26.Siddiqui, S. S. et al. Differential dose-response effect of cyclosporine A in regulating apoptosis and autophagy markers in MCF-7 cells. Inflammopharmacology31(4), 2049–2060 (2023). [DOI] [PubMed] [Google Scholar]
- 27.Murata-Kamiya, N. & Hatakeyama, M. Helicobacter pylori-induced DNA double-stranded break in the development of gastric cancer. Cancer Sci.113(6), 1909–1918 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watkins, R.D., Buckarma, E.H., Tomlinson, J.L. et al. SHP2 inhibition enhances yes-associated protein-mediated liver regeneration in murine partial hepatectomy models. JCI Insight7(15) (2022). [DOI] [PMC free article] [PubMed]
- 29.Zmajkovicova, K. et al. GPCR-induced YAP activation sensitizes fibroblasts to profibrotic activity of TGFβ1. PloS one15(2), e0228195 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zindel, D. et al. G protein-coupled receptors can control the Hippo/YAP pathway through Gq signaling. FASEB J.35(7), e21668 (2021). [DOI] [PubMed] [Google Scholar]
- 31.Zhu, L. et al. Positive feedback loops between fibroblasts and the mechanical environment contribute to dermal fibrosis. Matrix Biol.121, 1–21 (2023). [DOI] [PubMed] [Google Scholar]
- 32.Wu, D. et al. Single-cell sequencing reveals the antifibrotic effects of YAP/TAZ in systemic sclerosis. Int. J. Biochem. Cell Biol.149, 106257 (2022). [DOI] [PubMed] [Google Scholar]
- 33.Zhu, Y. et al. The collagen matrix regulates the survival and function of pancreatic islets. Endocrine83(3), 537–547 (2024). [DOI] [PubMed] [Google Scholar]
- 34.Patel, S. et al. Rac-GTPase promotes fibrotic TGF-β1 signaling and chronic kidney disease via EGFR, p53, and Hippo/YAP/TAZ pathways. FASEB J.33(9), 9797–9810 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.