

Abstract
Engaging in regular exercise and physical activity contributes to delaying the onset of cardiovascular diseases (CVDs). However, the physiological mechanisms underlying the benefits of regular exercise or physical activity in CVDs remain unclear. The disruption of mitochondrial homeostasis is implicated in the pathological process of CVDs. Exercise training effectively delays the onset and progression of CVDs by significantly ameliorating the disruption of mitochondrial homeostasis. This includes improving mitochondrial biogenesis, increasing mitochondrial fusion, decreasing mitochondrial fission, promoting mitophagy, and mitigating mitochondrial morphology and function. This review provides a comprehensive overview of the benefits of physical exercise in the context of CVDs, establishing a connection between the disruption of mitochondrial homeostasis and the onset of these conditions. Through a detailed examination of the underlying molecular mechanisms within mitochondria, the study illuminates how exercise can provide innovative perspectives for future therapies for CVDs.
Keywords: cardiovascular diseases, exercise, exerkines, mitochondrial homeostasis
Subject Categories: Exercise, Cardiovascular Disease, Mechanisms, Oxidant Stress
Nonstandard Abbreviations and Acronyms
- AMPK
adenosine5′‐monophosphate‐activated protein kinase
- BNIP3
BCL2 adenovirus E1B 19kDa interacting protein 3
- DCM
dilated cardiomyopathy
- DRP1
dynamin‐related protein 1
- FGF21
fibroblast growth factor 21
- FSTL1
follistatin like 1
- FUNDC1
FUN14 domain containing 1
- IRI
ischemia–reperfusion injury
- MCU
mitochondrial calcium uniporter
- MFN
mitochondrial fusion protein
- mPTP
mitochondrial permeability transition pore
- PGC‐1α
peroxisome proliferator‐activated receptor‐gamma coactivator 1alpha
- ROS
reactive oxygen species
- WHO
World Health Organization
Cardiovascular diseases (CVDs) are a batch of conditions affecting the heart or blood vessels, ranking as the primary causes of global mortality according to the World Health Organization. 1 Currently, despite extensive research on diet, drugs, surgery, and environmental factors for CVDs, effective interventions remain scarce, resulting in a low cure rate and high medical costs, which imposes a significant economic burden on most patients affected by these conditions. Regular exercise has long been acknowledged as a crucial intervention to avert and defer the advancement of these diseases. 2 Consistent engagement in aerobic activities with adequate intensity and duration can markedly enhance cardiorespiratory fitness, contributing to decelerating the progression and reducing mortality associated with CVDs. 3 The World Health Organization 2020 guidelines advocate moderate to vigorous intensity physical activity for individuals across all age groups. 4
Mitochondrial dysfunction is acknowledged as a prevalent metabolic disorder linked to the pathogenesis of CVDs. Consistent exercise or physical activity is beneficial for enhancing metabolic health in individuals with CVDs. 5 However, the physiological mechanisms underlying the benefits of physical exercise on mitochondrial metabolism and function in CVDs remain poorly understood. Hence, it is essential to precisely comprehend and elucidate the role of exercise in ameliorating mitochondrial dysfunction and its physiological mechanisms to generate novel insights for the treatment of CVDs. This review provides a comprehensive analysis of how regular exercise can effectively address CVDs and explores the potential physiological mechanisms.
MITOCHONDRIAL HOMEOSTASIS IN CARDIOVASCULAR DISEASES
Mitochondria, often referred to as the powerhouses of the cell, play a crucial role in both catabolism and anabolism. They actively participate in regulating intracellular calcium (Ca2+) homeostasis, instigating inflammatory responses, and ultimately, overseeing a multitude of pathways that govern regulated cell death. When cellular homeostasis is disrupted, mitochondria can regulate metabolism and maintain equilibrium through processes such as synthesis, fusion, fission, and mitophagy. Cardiomyocytes heavily depend on the oxidative phosphorylation of mitochondria to generate ATP. Mitochondria provide cardiomyocytes with a sustained energy supply and adequate calcium buffering, crucial for their dependency during repetitive, calcium‐mediated contractile activity. Hence, a reduction in the bioenergy efficiency of the mitochondrial network can directly impair myocardial contraction. When the mitochondrial network is damaged, the healthy calcium balance and inflammation equilibrium are disrupted, leading to alterations in typical heart performance, including impairment cardiac systolic and diastolic function, arrhythmias, arterial stenosis, and so forth.
MITOCHONDRIAL DYNAMICS IN CARDIOVASCULAR DISEASES
Mitochondria are vigorously active cellular structures that experience a perpetual cycle of fusion and fission, altering the shape, size, and position of mitochondria. Because of the opposing functions of various proteins that facilitate fission, such as DRP1 (dynamin‐related protein 1), mitochondrial FIS1 (fission 1) protein, MFF (mitochondrial fission factor), and DNM1L (dynamin 1‐like) protein, as well as fusion‐promoting proteins like MFN1/2 (mitochondrial fusion protein 1/2) and OPA1 (optic atrophy protein 1), the mitochondrial network undergoes constant remodeling. 6 Division at the periphery allows damaged material to be shed into smaller mitochondria destined for mitophagy, mediated by FIS1, and division in the central zone leads to the proliferation of mitochondria, mediated by MFF. Both types are mediated by DRP1. By overseeing mitochondrial morphology and reshaping the mitochondrial network, the quality and quantity of mitochondria are modified, allowing them to quickly adapt to the energy demands of cells. Disruption of mitochondrial dynamics initiates chronic inflammation and mitochondrial dysfunction via interorganelle communication and mislocalization of mitochondrial DNA.
In patients with diabetes, alterations in mitochondrial morphology, heightened fragmentation, and elevated expression of DRP1 in endothelial cells have been observed. 7 In addition, it has been discovered that phosphorylation of DRP1 can enhance phenylephrine‐induced cardiac hypertrophy, and the inhibition of DRP1 by mitochondrial division inhibitor‐1 can mitigate cardiac hypertrophy. 8 The Notch1‐DRP1/MFN1 axis diminishes myocardial infarction (MI) size by reducing mitochondrial fission and inhibiting ventricular remodeling. 9 The simultaneous deficiency of MFN1/2 in cardiomyocytes results in a severe impairment in mitochondrial fusion, leading to cardiac dysfunction linked to the swift progression of dilated cardiomyopathy (DCM). 10 Downregulation of OPA1 promotes cardiac hypertrophy after transverse aortic contraction. 11 Simultaneously, the activation of overlapping activity with m‐AAA protease can excessively hydrolyze OPA1, promoting mitochondrial division and fragmentation, thereby exacerbating DCM and heart failure (HF). 12 From the abnormal expression of mitochondrial dynamic proteins, it can be inferred that aberrant mitochondrial dynamics exacerbate CVDs by impairing myocardium and leading to abnormal cardiac function, playing a significant role in the onset and progression of CVDs.
MITOPHAGY IN CARDIOVASCULAR DISEASES
To uphold the stability of the mitochondrial network and intracellular environment, cells selectively envelop and degrade damaged and dysfunctional mitochondria, a process known as mitophagy. Seventeen autophagy‐related proteins are involved in the mitophagy process, including ULK1 (unc‐51‐like autophagy‐activating kinase 1), along with FIP200, VPS34 (vacuolar protein sorting 34), BNIP3 (BCL2 adenovirus E1B 19kDa interacting protein 3), BECN1 (Beclin 1), and AMBRA1 (autophagy and beclin 1 regulator 1).
DRP1 can not only promote mitochondrial fission but also induce mitophagy. Disruption of DRP1 led to mitochondrial prolongation, inhibition of mitophagy, accumulation of damaged mitochondria, and increased apoptosis in cardiomyocytes at baseline, thereby contributing to cardiac dysfunction and increasing susceptibility to ischemia/reperfusion. 13 Mice exposed to cardiomyocyte‐specific deletion of ATG5 (autophagy protein 5) under time‐controlled conditions exhibited disorganized sarcomere structure, collapsed mitochondria, impaired mitochondrial respiratory function and cardiac dysfunction. 14 TBK1 (TANK‐binding kinase 1) is a crucial kinase that regulates mitophagy. Deletion of TBK1 specifically in cardiomyocytes exacerbated chronic doxorubicin cardiotoxicity by inhibiting mitophagy. 15 TRP53 (transformation‐related protein 53), commonly known for p53, binds to Parkin, impeding its translocation to damaged mitochondria and the subsequent clearance via mitophagy. Thereby, inhibiting cytosolic p53 to activate mitophagy may enhance cardiac functional capacity with advancing age. 16 It has been reported that BNIP3L‐mediated mitophagy facilitated the cardiac progenitor cells to undergo proper mitochondrial network reorganization during differentiation, ultimately repairing and regenerating the injured heart. 17 Additionally, the ablation of BECN1 alleviated abnormal cardiac remodeling in a murine model of pressure overload, emphasizing the crucial role of mitophagy in the progression of the disease. 18 Enhancing mitochondrial autophagy to clear accumulated harmful substances in pathological heart can promote cardiac renewal and metabolism, effectively improving heart function and alleviating CVDs.
MITOCHONDRIAL ENERGY METABOLISM IN CARDIOVASCULAR DISEASES
Vibrant cardiomyocytes fulfill their heightened energy demands by metabolizing fatty acids, branched‐chain amino acids, and a limited amount of glucose to power the tricarboxylic acid cycle, generating ATP through the electron transport chain. Sustaining mitochondrial fatty acid oxidation can inhibit the rise of oxidative stress, diminish cardiac hypertrophy and fibrosis, and alleviate angiotensin II‐induced diastolic dysfunction. 19 Moreover, the lymphatic system plays a crucial role in CVDs by transporting fluid from the endocardium to the epicardium during myocardial contraction. Impaired myocardial contraction and lymphatic obstruction can lead to myocardial edema, inflammation, and fibrosis. 20 Normal fatty acid oxidation maintains the expression circuit of the prospero homeobox 1 (PROX1) gene in mitochondria, thus safeguarding the response of lymphatic endothelial cells to lymphangiogenic mediators and lymphangiogenesis, 21 maintaining normal heart function. The metabolic deletion of branched‐chain amino acids contributes to HF associated with oxidative stress and metabolic disorders induced by mechanical overload. 22 Alterations in the metabolic composition proportion of mitochondrial metabolism (fatty acid oxidation, branched‐chain amino acids metabolism and glucose metabolism) can lead to related diseases. For example, hypoxia‐inducing factor 1α triggers a gene expression program involving PPARγ (peroxisome proliferator‐activated receptor γ), amplifying the proportion of glucose uptake, subsequently resulting in cardiac systolic dysfunction. 23 Myocardial succinyl‐CoA (coenzyme A) levels were significantly reduced in mice with MI, impairing mitochondrial oxidative phosphorylation capacity. Subsequently, the accumulation of succinate in ischemic myocardium induces vigorous oxidative bursts, exacerbating ischemia–reperfusion injury (IRI), 24 whereas the restoration of succinyl‐CoA and oxidative phosphorylation capacity prevented HF progression in MI mice. 25 Mitochondria serve as metabolic hubs, and the proper functioning of mitochondrial metabolism is crucial to maintain normal cellular activities essential for life, effectively preventing the onset of some CVDs.
MITOCHONDRIAL OXIDATIVE STRESS IN CARDIOVASCULAR DISEASES
Appropriate amounts of reactive oxygen species (ROS) can promote immunity, repair, survival, and growth. In cardiac mitochondria, the main ROS generated is the superoxide radical anion (), which can undergo dismutation to form hydrogen peroxide. Hydroxyl radicals (OH•) are also produced through the decomposition of hydroperoxides or by the reaction of excited atomic oxygen with water. The mitochondrial respiratory chain serves as a significant endogenous source of , a toxic by‐product of oxidative phosphorylation. Electrons derived from nicotinamide adenine dinucleotide and reduced flavine adenine dinucleotide pass through the electron transport chain to ultimately reduce oxygen, forming H2O. However, significant amounts of , are produced when oxygen undergoes incomplete reduction, primarily due to electron leakage at complex I and III. Oxidative stress occurs when there is an excess production of ROS, resulting in the buildup of inflammation, mitochondrial damage and cell death. Monoamine oxidase stands as a notable generator of ROS within mitochondria and has been recognized as a key factor influencing the redox balance in human atrial myocardium, linked to postoperative atrial fibrillation. 26 MI induces oxidative stress and leads to ventricular hypertrophy and fibrosis. Additionally, oxidative stress exacerbates atherosclerosis, a chronic inflammatory disease. 27 During oxidative stress, reactive oxygen species target endothelial cell lipids, initiating a nonenzymatic cascade that culminates in the formation of bioactive molecule known as 8‐iso‐prostaglandin F2alpha. The concentration of 8‐iso‐prostaglandin F2alpha, a particular quantitative indicator of oxidative stress, increases with the severity of HF and is associated with ventricular dilation. 28 To mitigate the detrimental effects of ROS, organisms have developed a sophisticated 3‐tiered antioxidant defense system. 29 The first‐line defense mechanism is the most effective, involving antioxidant enzymes such as superoxide dismutase, catalase, and glutathione peroxidase. 29 These antioxidant enzymes are all detectable within mitochondria. The third line of antioxidant defense relies on multiple enzyme systems that repair or eliminate oxidized proteins and other biomolecules. 29 Oxidative stress heightens the likelihood of protein misfolding and toxic aggregation, contributing to cardiac abnormalities such as atherosclerosis, IRI, cardiomyopathy, and HF, which can be alleviated by the protein quality control system, including the ubiquitin‐proteasome system and autophagy‐lysosomal pathways initiated at mitochondria. 30 Activation of nuclear factor erythroid 2‐related factor 2/heme oxygenase 1 signaling, regarded as the main regulatory pathway of intracellular defense against oxidative stress, protected vascular endothelial cells from oxidative stress in the treatment of atherosclerosis. 31 Furthermore, SIRT3 (sirtuin 3), located in mitochondria, mediates anti‐inflammatory, antiautophagic, and antioxidant functions of PCSK9 (proprotein convertase subtilisin‐kexin type 9) inhibitors in endothelial cells. 32 Mitochondria produce cellular energy and are vulnerable to oxidative stress, which damages their structure and function, impairing cardiac function. Activating mitochondrial antioxidant enzymes and autophagy‐lysosomal pathways helps remove damaged components, quickly alleviating oxidative stress damage.
Ca2+ HOMEOSTASIS IN CARDIOVASCULAR DISEASES
Ca2+ is involved in mitochondrial ATP synthesis and the initiation of mPTP (mitochondrial permeability transition pore). Under physiological conditions, an increased uptake of mitochondrial Ca2+ enhances ATP production. However, under pathological conditions, increased mitochondrial Ca2+ uptake induces mitochondrial Ca2+ overload, triggering mPTP opening. The mPTP enables the unrestricted passage of small molecules and ions (<1.5 kDa) through the inner mitochondrial membrane. This leads to dissipation of the membrane potential, resulting in an imbalance in ATP production, swelling of mitochondria, and rupture of the OMM (outer mitochondrial membrane). These events contribute to regulated cell death, involving either apoptosis or necrosis. 33 Therefore, to prevent mitochondrial Ca2+ overload and subsequent activation of regulated cell death, precise control over Ca2+ uptake is essential. Mitochondria takes up Ca2+ through the mitochondrial calcium uniporter complex, which consists of the MCU (mitochondrial calcium uniporter), MICU1 (mitochondrial calcium uptake 1), MICU2 and EMRE (essential MCU regulator). 34 The MCU protein forms tetramers to create the channel's transmembrane Ca2+ pore, which is regulated by 2 distinct mechanisms. Initially, the MICU1 protein dimerizes with MICU2 in the intermembrane space and binds via electrostatic interactions to the cytoplasmic entrance of the MCU pore, thereby closing the uniporter. Subsequently, when intermembrane space Ca2+ levels rise to low micromolar concentrations, MICUs undergo conformational changes that unblock the MCU pore, allowing Ca2+ to permeate. The second regulatory mechanism of MCU is Ca2+‐independent and involves the metazoan‐specific EMRE subunit, a small single‐pass membrane protein in the inner mitochondrial membrane. It has been established that EMRE binding is necessary for metazoan MCU to exhibit transport activity. The primary regulation of Ca2+ export from the mitochondrial matrix is contingent upon the Na+/Ca2+ antiporter SLC8B1 (solute carrier family 8 member B1), also recognized as NCLX. 33
MCUB, a paralog of the pore‐forming subunit MCU, integrated into MCU, restricts Ca2+ overload during cardiac injury and reduces infarct size after ischemia–reperfusion. 35 Blocking CaMKII (calmodulin‐dependent protein kinase II)‐induced mPTP opening mitigates mitochondrial damage and cardiac hypertrophy. 36 Additionally, the leaky RyR2 (ryanodine receptor 2), a channel on the cardiac sarcoplasmic reticulum responsible for Ca2+ release, contribute to mitochondrial Ca2+ overload, dysmorphology, and malfunction, ultimately leading to life‐threatening arrhythmias. 37 Loss of Slc8b1 in adult mouse cardiomyocytes causes mitochondrial Ca2+ overload, leading to sudden death with extensive mitochondrial permeability transition‐driven myocardial necrosis. In contrast, overexpression of Slc8b1 mediates a potent cardioprotective effect in a murine model of cardiac IRI. 38 The protein matrix can bind neutral, uncharged sites of Ca2+ to interact with organic anions such as sulfated mucopolysaccharides, thereby delaying atherosclerosis. 39 However, the accumulation of Ca2+ and lipids in the vascular endothelium promotes the formation of calcified plaque, ultimately leading to atherosclerosis. The exchange and balance of mitochondrial calcium ions regulate myocardial contraction, contribute to normal cardiac rhythm, influence myocardial cell membrane potential, and are crucial for maintaining normal cardiac physiological activities.
ENDURANCE EXERCISE REGULATES MITOCHONDRIAL QUALITY CONTROL
Mitochondrial quality control is a comprehensive network for monitoring mitochondrial quality and an endogenous cellular protective program that is essential for maintaining mitochondrial homeostasis and function. Mitochondrial quality control regulates and sustains mitochondrial homeostasis by coordinating various processes, including mitochondrial biogenesis, fission, fusion, autophagy, and protein homeostasis. Exercise enhances mitochondrial function through regulating mitochondrial quality control.
ENDURANCE EXERCISE PROMOTES MITOCHONDRIAL BIOGENESIS
During exercise, the recruitment of myofibril motor units generates an action potential that induces the release of Ca2+ by sarcoplasmic reticulum and the subsequent increase in cytosolic Ca2+ promotes ATP synthesis and ROS in mitochondria, thereby promoting the translocation of PGC‐1α (peroxisome proliferator‐activated receptor‐gamma coactivator 1alpha), 40 , 41 considered as a vital regulator of mitochondrial biogenesis, and its family members, PGC‐1β and PGC‐related co‐activator, upregulate gene transcription by docking with transcription factors and additional proteins on DNA promoters to regulate NuGEMPs (nuclear genes encoding mitochondrial proteins). Nuclear‐derived proteins newly expressed are imported into mitochondria via transferases present located in the OMM and inner mitochondrial membrane to promote the activation of mitochondrial unfolded protein response and mitochondrial biogenesis for protection and repair of damaged mitochondria. 42
Participation in an 8‐week swimming regimen improved skeletal muscle mitochondrial biogenesis in mice by boosting the expression of PGC‐1α and key mitochondrial biogenesis markers, such as SDH (succinate dehydrogenase) and COX IV (cytochrome c oxidase‐IV). 43 TFAM (mitochondrial transcription factor A), encoded by nucleus extracellular, was also found to form complexes with SIRT1, p53, and Polγ to stabilize and maintain the integrity of mitochondria. Gestational exercise stimulated protein expression of PGC‐1α and TFAM, promoting liver mitochondrial biogenesis in female offspring. 44 Additionally, a single 3‐hour wheel‐running exercise was adequate to prompt structural alterations in the mitochondrial network of the skeletal muscle in 8‐week‐old female mice. 45 A 12‐week program of combined aerobic and resistance training increased specific lipid intermediates in skeletal muscle, such as cardiolipin, phosphatidylcholine, and phosphatidylethanolamine, along with mitochondrial complex I‐V, thereby enhancing mitochondrial respiratory capacity and promoting mitochondrial biogenesis. 46 (Figure 1).
Figure 1. Endurance exercise regulates mitochondrial quality control.
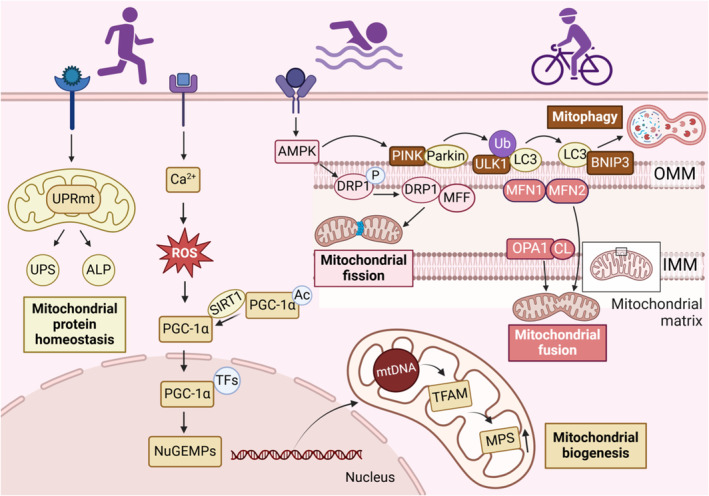
Exercise maintains mitochondrial function by promoting mitochondrial biogenesis, dynamic balance, autophagy, and protein homeostasis. Endurance exercise stimulates the PGC‐1α pathway to drive mitochondrial biogenesis. Physical exercise also activates AMPK, which promotes PINK‐dependent mitophagy and facilitates mitochondrial fission through DRP1 and MFF. The fusion of the OMM is mediated by MFN1/2, and OPA1 facilitates the fusion of the IMM, together driving mitochondrial fusion. In addition, exercise training activates the mitochondrial unfolded protein response, which regulates the ubiquitin‐proteasome system and the autophagy‐lysosomal pathway to degrade damaged or misfolded proteins and maintain mitochondrial protein homeostasis. Ac indicates acetylation; ALP, autophagy‐lysosomal pathway; AMPK, adenosine5′‐monophosphate‐activated protein kinase; BNIP3, BCL2 adenovirus E1B 19kDa interacting protein 3; DRP1, dynamin‐related protein 1; IMM, inner mitochondrial membrane; LC3, microtubule‐associated protein 1 light chain 3; MFF, mitochondrial fission factor; MFN1/2, mitochondrial fusion protein 1/2; NuGEMP, nuclear genes encoding mitochondrial protein; OMM, outer mitochondrial membrane; OPA1, optic atrophy protein 1; P, phosphorylation; PGC‐1α, peroxisome proliferator‐activated receptor‐gamma coactivator 1alpha; PINK, PTEN‐induced putative kinase; ROS, reactive oxygen species; SIRT1, sirtuin 1; TFs, transcription factors; TFAM, mitochondrial transcription factor A; Ub, ubiquitin; ULK1, unc‐51‐like autophagy‐activating kinase 1; UPRmt, mitochondrial unfolded protein response; and UPS, ubiquitin‐proteasome system.
ENDURANCE EXERCISE ENHANCES MITOCHONDRIAL DYNAMICS BALANCE
Exercise maintains an optimal balance between mitochondrial fusion and fission by promoting the correct expression of mitochondrial fusion proteins and fission proteins, ensuring mitochondrial and cellular homeostasis. Inhibition of MFN‐mediated mitochondrial fusion leads to mitochondrial fragmentation and metabolic reprogramming. In reality, fragmented mitochondria are more inclined to induce oxidative stress, reduce ATP synthesis, and trigger apoptosis through mitochondrial permeability transition. 47 Regular exercise activates AMPK (adenosine5′‐monophosphate [AMP]‐activated protein kinase), optimizing mitochondrial fission and fusion. In worms' body‐wall muscle, a single 4‐hour swimming session induced a cycle of mitochondrial fragmentation followed by fusion after a recovery period, and long‐term swimming sessions delayed mitochondrial fragmentation and the decline in physical fitness associated with age. 48
The changes in mitochondrial dynamics depend on training frequency and duration. Engaging in 12‐week swimming training (4 hour/day, 5 day/week), enhanced mitochondrial morphology and dynamics, increasing the expression of OPA1, DRP1, and MFN2 proteins in zebrafish liver. 49 Additionally, 12 weeks of moderate‐intensity treadmill exercise also ameliorated mitochondrial dynamic damage in skeletal muscle of obese mice. 50 Similarly, an 8‐week moderate‐intensity treadmill training regimen diminished excessive mitochondrial fission and inflammation in the soleus muscle of diabetic rats. 51 After undergoing 12 weeks of supervised aerobic exercise training (5 day/week, 85% heart rateMAX), skeletal muscle of sedentary adults exhibited a significant alteration in the expression of mitochondrial fusion and fission proteins, leading to the development of a more integrated, tubular mitochondrial network. 52 Sixteen weeks of supervised training in sedentary people accelerated fusion to increase mitochondrial content to meet the metabolic demands of exercise. Meanwhile, lifelong exercise primarily stimulated skeletal muscle mitophagy, aligning with heightened fusion and reduced fission, indicating an increased mitochondrial turnover. 53 In the physiological state, mitochondrial fusion and division check each other, maintaining a dynamic balance within the mitochondria. When this balance is disturbed, mitochondrial function becomes impaired, eventually leading to a variety of diseases. (Figure 1).
ENDURANCE EXERCISE ACTIVATES MITOPHAGY
Under stress, such as oxidative stress, mtDNA mutations gradually accumulate, leading to a decrease in mitochondrial membrane potential and depolarized damage, eventually culminating in cell death. To maintain mitochondrial and cellular homeostasis and protect cells from damage caused by dysfunctional mitochondria, cells selectively envelop damaged or dysfunctional mitochondria within the cell and degrade them—a process known as mitophagy. Organelles that cannot meet the metabolic needs of the proteins will undergo division through the interaction of the proteins FIS1 and MFF with DRP1, which are eventually cleared by mitophagy.
Following acute treadmill running, mitophagy occurs at 6 hours post exercise in mice skeletal muscle by activating AMPK and ULK1 signaling pathways. 54 The same activation effect was also detected in the quadriceps muscles of those who exercised after 2 hours. 55 Parkin is an E3 ubiquitin ligase implicated in mitophagy. Acute endurance running‐induced skeletal muscle mitophagy is dependent on Parkin and attenuated with age in aged mice. 56 Additionally, mitochondrial membrane‐tethered BNIP3 plays a role in initiating mitophagy, potentially by interacting with LC3 (light chain 3). 57 Elevated levels of BNIP3 were observed in rodent skeletal muscle following acute eccentric exercise. 58 This increase is consistent with findings in human skeletal muscle after 8 weeks of continuous moderate cycling. 59 (Figure 1).
ENDURANCE EXERCISE PROMOTES MITOCHONDRIAL PROTEIN HOMEOSTASIS
Exercise can induce the expression of nuclear coding genes and mitochondrial coding genes, thereby promoting mitochondrial protein synthesis. For instance, an acute session of high‐intensity aerobic exercise led to a prolonged increase in mitochondrial proteins. 60 Furthermore, exercise activates mitochondrial unfolded protein response to facilitate the degradation of damaged or misfolded proteins by regulating the ubiquitin‐proteasome system and autophagy‐lysosomal pathways. 61 Additionally, exercise can trigger the expression of mitochondrial protein kinase and promote posttranslational modification of mitochondrial proteins. 62 Endurance training increases the expression of SIRT3 to reduce excessive mitochondrial protein acetylation, thereby maintaining mitochondrial protein homeostasis. 63 In conclusion, regular exercise maintains mitochondrial protein homeostasis by promoting mitochondrial protein synthesis, degradation and posttranslational modification. (Figure 1).
Mitokines are signaling molecules, facilitating communication between mitochondria experiencing local stress and those in distant cells and tissues, mainly involving FGF21 (fibroblast growth factor 21), GDF15 (growth differentiation factor 15) and some mitochondria‐derived peptides. 64 Exercise can also preserve mitochondrial protein homeostasis by regulating the expression levels of these mitokines. The 8‐week endurance exercise training regimen boosted FGF21 expression, exercise capacity, and altered the distribution of skeletal muscle fiber sizes. 65 GDF15, also considered as a myokine and cardiokine, is highly responsive to cellular stress, with elevated circulating GDF15 released from skeletal muscle in humans during intensive submaximal exercise. 66 Mitochondria‐derived peptides are microproteins encoded by the small open reading frames of mtDNA. Eight mitochondria‐derived peptides including humanin, mitochondrial open reading frames of the 12 S‐c, and small humanin‐like peptides 1–6 have been discovered and there are more yet to be discovered. After acute high‐intensity exercise, the increased abundance of humanin in muscle and plasma contributes to inhibiting apoptosis and calcium overload, enhancing mitochondrial biogenesis, ultimately leading to reduced ROS production and alleviation of oxidative stress. 67 , 68 Additionally, exercising on a stationary bicycle triggered the endogenous expression of mitochondrial open reading frames of the 12 S‐c in both skeletal muscle and circulation, improving muscle metabolism and delaying aging. 69
EXERCISE ENHANCES MITOCHONDRIAL HOMEOSTASIS IN CARDIOVASCULAR DISEASES
Atherosclerosis
Atherosclerosis is an inflammatory process that results in the accumulation of fatty or necrotic residues in the vessel wall. This leads to fibroplasia and calcinosis, causing thickening and hardening of the arterial wall and subsequently narrowing of the vessel lumen. The rupture of an atherosclerotic plaque and the subsequent formation of a thrombus in the bloodstream can lead to ischemic events, such as MI or stroke. Based on the characteristics of atherosclerosis, exercise can effectively stimulate the antioxidant pathway in blood vessels, reduce vascular damage and inflammation, and promote angiogenesis, ultimately mitigating atherosclerosis.
After exhaustive exercise, TLR9 (toll‐like receptor 9), interacting with beclin1, activates mitochondrial UCP2 (uncoupling protein 2)‐AMPK‐Akt (protein kinase 3)‐FoxO1 (forkhead box O1) axis, 70 triggering activation of antioxidant pathways in mice endothelial cells, ultimately alleviating atherosclerosis. 71 Endurance swim training staves off endothelial aging in coronary arteries by boosting the regulator FUNDC1 (FUN14 domain containing 1)‐induced mitophagy in a PPARγ‐dependent manner. As a result, it shields aged mice from IRI. 72 Moderate‐intensity treadmill exercise enhanced cardiac hydrogen sulfide biosynthesis and attenuated pyroptosis, promoting angiogenesis via STAT3 (signal transducer and activator of transcription 3) and Ca2+/CaMKII signaling pathways 73 , 74 and decreasing endothelial–mesenchymal transition. 75 PGC‐1α was significantly activated by 1 bout of swimming (3 hour/session, 2 times, separated by 45 minutes). 41 Given its robust angiogenesis potential in skeletal muscle, PGC‐1α likely facilitates exercise‐induced angiogenesis, thereby aligning mitochondrial biogenesis, consuming oxygen and nutrient, with angiogenesis, delivering oxygen and nutrient. Additionally, consistent physical exercise regulates the formation of atherosclerotic plaques through the modulation of various myokines, including IL‐6 (interleukin‐6), ADPN (adiponectin), and FGF21. 76 High‐intensity interval training and moderate‐intensity continuous training alike stimulate irisin secretion in skeletal muscles to boost circulating irisin level. By activating the PGC‐1a‐FNDC5 (fibronectin type III domain‐containing protein 5)‐irisin axis, this process stimulates the AMPK‐PI3K (phosphoinositide 3‐kinase)‐Akt‐eNOS (endothelial nitric oxide synthase) pathway, thereby mitigating endothelial damage and reducing pathological inflammation in atherosclerotic blood vessel of diabetic mice. 77 (Table) (Figure 2).
Table 1.
Exercise Regulates Cytokines in Cardiovascular Diseases
| Cytokines | Subjects | Exercise intervention program | Main mechanism | Main biological action | Cardiovascular diseases | Reference | |||
|---|---|---|---|---|---|---|---|---|---|
| Model | Sample size | Type | Intensity | Duration | |||||
| UCP2 | Mice | ND | Treadmill running | Exhausted | ND | UCP2/AMPK/Akt/FoxO1 | Autophagy↑ and oxidation resistance↑ | Atherosclerosis | 71 |
| FUNDC1 | Mice | ND | Endurance swim training | 70% maximal power | 5 d/wk; 90 min/d; 4 wk | PPARγ/FUNDC1/LC3 | Mitophagy↑ and IRI↓ | Atherosclerosis | 72 |
| PGC‐1α | Rats | ND | Swimming training | 3 h/session, 2 times, separated by 45 min | 1 d | P38γMAPK/ATF2/PGC‐1α/VEGF | Mitochondrial biogenesis↑ and angiogenesis↑ | Atherosclerosis | 41 |
| Irisin | Mice | 45 | Treadmill running | HIIT and MICT | 3 times/wk; 23 min/set; 6 wk | PGC‐1a/FNDC5/irisin | Mitochondrial oxidative damage↓ and blood lipid ↓ | Atherosclerosis | 77 |
| FGF21 | Mice | 40 | Treadmill running and ladder‐climbing | 76% of VO2max and 75% of the maximum load | 5 d/wk; 4 wk and 3 times/set, 9 sets/d, 4 wk | FGF21/TGF‐β1/Smad2/3/MMP2/9 | Mitochondrial biogenesis↑, oxidative stress↓, cell apoptosis↓ and cardiac fibrosis↓ | Ischemic heart disease | 83 |
| Musclin | Mice | 40 | Treadmill running | 15 m/min at 5° inclination, 45 min | 5 d/wk, 3 wk | cGMP/PKGI/CREB/PGC‐1α | Mitochondrial biogenesis↑ | Ischemic heart disease | 82 |
| SIRT1 | Human; rats | 70 | Treadmill running | 10m/min, 5 min (40%–50% VO2max); 13 m/min, 5 min; 16 m/min, 50 min(60%–70% VO2max) | 5d/wk, 3wk | SIRT1/PGC‐1α/PI3K/Akt | Mitochondrial integrity and biogenesis↑ | Ischemic heart disease | 78 |
| HSP70 | Healthy mAu: ale | 11 | Cycling | 30 min at 55% VO2max, 20 min at 70% VO2max and until exhaustion (10 min) at 80% of VO2max | 60 min, 1 session | HSP70/TLR4/ERK/p38MAPK/HSP27 | Mitochondrial protein folding↑, cell proliferation and angiogenesis↑ | Ischemic heart disease | 92, 93 |
| FGF21 | Mice | 20 | Treadmill running | 9 m/min | 5 d/wk, 1 h/d, 6 wk | AMPK/FOXO3/SIRT3 | Toxic lipid‐induced mitochondrial dysfunction↓ and oxidative stress↓ | Diabetic cardiomyopathy | 99 |
| Mhrt779 | mice | 72 | Swimming training | 15 min/session, 2 times/d, +15min/2 d → 90 min/session, 90 min/session, 2 times/d, 7 d | 21 d | Mhrt779/Brg1/Hdac2/Akt/p‐GSK3β | Mitochondrial protein posttranslation modification↑ and antihypertrophic effects↑ | Hypertrophic cardiomyopathy | 97 |
| c‐Ddx | Mice | 72 | Swimming training | 15 min/session, 2 times/d, +15 min/2 d → 90 min/session, 90 min/session, 2 times/day, 7 d | 21 d | c‐Ddx/AMPK/eEF2 | Mitochondrial protein degradation↑ and antihypertrophic memory pretreatment↑ | Hypertrophic cardiomyopathy | 98 |
| IL‐6 | healthy male | 6 | One‐legged knee extensor exercise | 40% W max, ke | 5 h | IL‐6/SERCA2a | Cardiotoxicity of doxorubicin and mitochondrial oxidative stress↓ | Dilated cardiomyopathy | 108 |
“↑” indicates improve; “↓”, decrease; +15 min/2 d → 90 min/session, increased by 15 min every 2 d until 90 min/session; 38MAPK: p38 mitogen‐activated protein kinase; Akt, protein kinase B; AMPK, adenosine5′‐monophosphate‐activated protein kinase; ATF2, activating transcription factor 2; Brg1, brahma‐related gene 1; c‐Ddx, circ‐Ddx60; cGMP, cyclic guanosine monophosphate; CREB, cyclic adenosine monophosphate response element binding; d, day(s); eEF2, eukaryotic translation elongation factor 2; ERK, extracellular signal‐regulated protein kinases; FGF21, fibroblast growth factor 21; FNDC5, fibronectin type III domain‐containing protein 5; FoxO1, forkhead box O1; FUNDC1, FUN14 domain containing 1; Hdac2, histone deacetylase 2; HIIT, high‐intensity interval training; HSP, heat shock protein; IL‐6, interleukin‐6; IRI, ischemia–reperfusion injury; ke, knee extensor peak power output; LC3, light chain 3; Mhrt779, long noncoding myosin heavy chain‐associated RNA transcript; MICT, moderate‐intensity continuous training; MMP, matrix metalloproteinase; ND, not determined; PGC‐1α, peroxisome proliferator‐activated receptor‐gamma coactivator 1alpha; p‐GSK3β, phosphorylated glycogen synthase kinase 3β; PI3K, phosphatidylinositol‐3‐hydroxykinase; PKGI, type I cGMP‐dependent protein kinases; PPARγ; peroxisome proliferator‐activated receptor γ; SERCA2a, sarcoplasmic reticulum calcium ATPase 2a; SIRT, sirtuin; Smad2/3, phospho Thr8; TGF‐β, transforming growth factor‐beta; TLR4: toll‐like receptor 4; VEGF, vascular endothelial growth factor; and W max: maximal watts.
Figure 2. Exercise improves mitochondrial homeostasis in atherosclerosis.
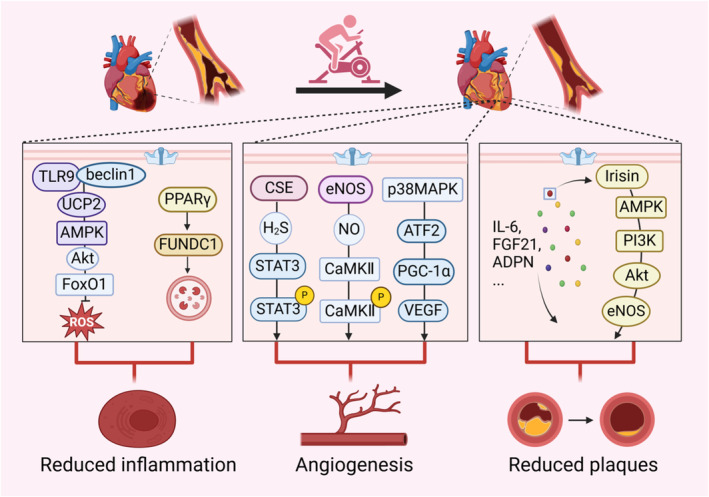
Atherosclerosis exhibits vascular inflammation, vascular damage and accumulation of plaques. Regular exercise can reduce atherosclerotic plaques and inflammatory responses, promoting the formation of endothelial vessels through the improvement of mitochondria‐related signaling pathways. Exhaustive exercise activates antioxidant pathways within the mitochondria of skeletal muscle in mice, while endurance swim training induces FUNDC1‐mediated mitophagy to reduce inflammatory responses in aged mice. Moderate‐intensity treadmill exercise and swimming sessions stimulate STAT3/CaMKII signaling pathway within the mitochondria of mice with diabetic cardiomyopathy and PGC‐1α signaling pathway within the skeletal muscle of rats, promoting angiogenesis. Moreover, physical exercise regulates the formation of atherosclerotic plaques by modulating various myokines, such as IL‐6, FGF21, and irisin. In this figure, exerkine irisin is taken as an example to illustrate its function of reducing endothelial damage and plaques in diabetic mice. ADPN indicates adiponectin; Akt, phosphorylated serine/threonine kinase; AMPK, adenosine5′‐monophosphate‐activated protein kinase; ATF2, activating transcription factor 2; CaMKII, calmodulin‐dependent protein kinase II; CSE, cystathionine‐γ‐lyase; eNOS, endothelial nitric oxide synthase; FGF21, fibroblast growth factor 21; FoxO1, forkhead box O1; FUNDC1, FUN14 domain containing 1; IL‐6, interleukin‐6; NO, nitric oxide; p38MAPK, p38 mitogen‐activated protein kinase; PGC‐1α, peroxisome proliferator‐activated receptor‐gamma coactivator 1alpha; PI3K, phosphatidylinositol‐3‐hydroxykinase; PPARγ; peroxisome proliferator‐activated receptor γ; ROS, reactive oxygen species; STAT3, signal transducer and activator of transcription 3; TLR9, toll‐like receptor 9; UCP2, uncoupling protein 2; and VEGF, vascular endothelial growth factor.
Ischemic Heart Disease
Ischemic heart disease is a heart disease caused by myocardial ischemia, which is the leading cause of death from CVDs, according to the World Heart Report 2023. The primary cause of ischemic heart disease is atherosclerosis. The accumulation of atherosclerotic plaque gradually narrows coronary arteries, restricting blood flow and leading to MI. After ischemia, cell apoptosis occurs, leading to cardiac fibrosis and exacerbating inflammatory responses during reperfusion. Exercise training can mitigate apoptosis, inflammation, and myocardial fibrosis following ischemia–reperfusion, while also promoting vascular regeneration through improved mitochondrial redox balance.
Four weeks of treadmill training enhanced mitochondrial integrity and biogenesis, concurrently reducing myocardial damage by activating the SIRT1/PGC‐1α/PI3K/Akt pathway in post‐MI myocardium. 78 In MI mice, there is an enhanced conversion of LC3‐I to LC3‐II accompanied by increased levels of cargo protein p62. 79 Remarkably, after 8 weeks of 15‐minute swimming training, there was a reduction in LC3‐II and p62, coupled with elevated expression of PINK (PTEN‐induced putative kinase)/Parkin to modulate mitophagy. 79 Furthermore, there was a significantly increased SIRT3 level in myocardial mitochondria after moderate training, which mitigated ROS production under hypoxic conditions, consequently decreasing apoptosis and fibrosis in MI mice. 79 , 80 The results showed that the implementation of exercise programs can alleviate cardiac fibrosis, pathological remodeling and dysfunction caused by ischemia though modulating exerkines, including musclin, FGF21, FSTL1 (follistatin like 1) and myonectin. 81 For instance, musclin, an exerkine and an important mediator for mitochondrial adaptations to endurance training, augmented treadmill training (15 m/minute, 45 minutes, 5 day/week, 3 weeks)‐induced cardiac mitochondrial biogenesis and cardiac protection by enhancing cGMP/PKG/CREB/PGC1α‐dependent signaling. 82 Both aerobic and resistance exercise training can enhance FGF21 protein expression, deactivate the TGF‐1β (transforming growth factor beta)‐Smad2/3‐MMP2/9 (matrix metalloproteinase 2/9) signaling pathway, alleviate cardiac fibrosis, oxidative stress, and cell apoptosis, thereby ultimately enhancing cardiac function in mice with MI. 83 Four weeks of dynamic resistance training enhanced the expression of FSTL1 derived from skeletal muscle, potentially compensating for the deficiency of cardiac FSTL1, promoting myocardial angiogenesis, and inhibiting pathological remodeling via the DIP2A‐Smad2/3 signaling pathway in MI rats. 84 Endurance training reduced infarct size following ischemia/reperfusion in wild‐type mice but not in myonectin‐knockout mice. Conversely, transgenic overexpression of myonectin alone effectively mitigated myocardial damage after ischemia/reperfusion. 85
Reperfusion after MI can aggravate the structural damage and dysfunction of tissues and organs, which is called IRI. 86 IRI is usually caused by free radical damage, calcium overload and white blood cell activation. 86 Too much calcium entering the mitochondria results in the dysfunction of cytochrome oxidase system, an increase in ROS and damage to mitochondrial function. 87 In particular, elevated levels of Ca2+ can stimulate the multiprotein inflammasome, thereby facilitating the maturation of the proinflammatory cytokine IL‐1β and triggering an inflammatory response. 88 During endurance exercise, the ratio of ADP/ATP increases, which helps to promote the oxidative phosphorylation of mitochondria and reduce oxidative stress. 89 Moderate physical exercise can improve the endogenous antioxidant defense system, stimulating the expression of superoxide dismutase 1, superoxide dismutase 2, glutathione peroxidase, and glutathione reductase, while lowering the levels of several inflammatory markers, including IL‐6, homocysteine and TNF‐α (tumor necrosis factor‐alpha), reducing the inflammatory response. 90 Exercise triggers the release of molecules, packaged mainly in extracellular vesicles called exosomes, into the bloodstream, emphasizing the crucial role of intertissue signaling proteins as mediators in adapting to exercise. 91 After a single bout of endurance exercise (30′ treadmill, 70% heart rate), exosomes specific marker protein—HSP70 (heat shock protein 70) can rapidly activate TLR4/ERK (extracellular regulated protein kinase)/p38 MAPK pathway to promote HSP27 phosphorylation, facilitating antioxidative effects through the increased expression of mitochondrial antioxidant enzymes, ultimately inhibiting vascular endothelial cells apoptosis and exerting cardioprotective effect. 92 , 93 In addition to the aforementioned, high‐intensity treadmill exercise promotes vascular regeneration through the NAD(+)/SIRT1 pathway in skeletal muscle. 94 (Table 1) (Figure 3).
Figure 3. Exercise improves IHD by facilitating intercellular communication.
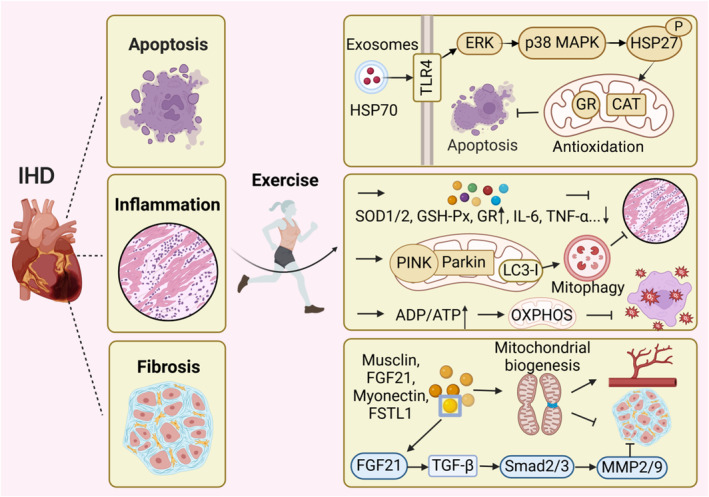
Following ischemia, cell apoptosis ensues, cardiac fibrosis develops, and the inflammatory response is exacerbated during reperfusion. Exercise training can reduce apoptosis, inflammation and myocardial fibrosis following IR and promote vascular regeneration by improving mitochondrial redox balance. During exercise, exerkines are transported via exosomes to specific sites where they exert antiapoptotic functions through the increased expression of mitochondrial antioxidant enzymes, such as glutathione reductase, catalase. After exercise, the increase of antioxidant enzymes and the decrease inflammatory factors can reduce inflammation. Remarkably, swimming training make an elevated expression of PINK/Parkin and LC3‐I, which can modulate mitophagy to exert a similar anti‐inflammatory effect. Endurance exercise also enhances the ADP/ATP ratio, thereby promoting mitochondrial OXPHOS and reducing mitochondrial oxidative stress. The release of exerkines such as musclin, FGF21, FSTL1, and myonectin promote mitochondrial dynamics, biogenesis, and autophagy, which collectively contribute to angiogenesis and inhibit myocardial fibrosis. In this figure, exerkine FGF21 is taken as an example to illustrate its antifibrotic effect. Notably, exerkines play a crucial role in augmenting intercellular communication and ameliorating cardiovascular diseases. CAT indicates catalase; ERK, extracellular signal‐regulated protein kinase; FGF21, fibroblast growth factor 21; FSTL1, follistatin like 1; GR, glutathione reductase; GSH‐Px, glutathione peroxidase; HSP70, heat shock protein 70; IHD, ischemic heart disease; IL‐6, interleukin‐6; IR, ischemia–reperfusion; LC3, light chain 3; MMP2/9, matrix metalloproteinase‐2/9; p38 MAPK, p38 mitogen‐activated protein kinase; OXPHOS, oxidative phosphorylation; P, phosphorylation; PINK, PTEN‐induced putative kinase; Smad2/3, phospho Thr8; SOD, superoxide dismutase; TGF‐β, transforming growth factor‐beta; TLR4, toll‐like receptor 4; and TNF‐α, tumor necrosis factor‐alpha.
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy is a type of cardiomyopathy characterized by left ventricle thickening, asymmetric ventricular septum hypertrophy, left ventricle cavity shrinkage, and diastolic dysfunction, with left ventricle ejection fraction retained or increased, which can lead to HF. Cardiac muscle hypercontractility and diastolic dysfunction are key pathophysiological abnormalities in hypertrophic cardiomyopathy.
For the majority of patients with hypertrophic cardiomyopathy, engaging in moderate‐ and high‐intensity exercise training is advantageous for enhancing cardiorespiratory fitness, functional capacity, and overall quality of life. 95 Exercise improves endothelium‐dependent vascular relaxation by increasing the release of nitric oxide and reducing oxidative stress. Additional mechanisms involve a better balance between prostacyclin and thromboxane levels, decreased circulating levels of endothelin‐1 and an elevation in local ADPN secretion. 96 Furthermore, swimming training significantly hinders cardiac pathological remodeling and dysfunction by activating the Mhrt779/Brg1/Hdac2/Akt/p‐GSK3β signal pathway, contributing to the post‐translational modification of mitochondrial proteins, reducing the opening of mPTP and exhibiting antihypertrophic effects. 97 Furthermore, circ‐Ddx60, a newly identified circular RNA with preferential expression in myocardial tissue, reduces mitochondrial protein synthesis to decrease the consumption of ATP and contributes to antihypertrophic memory pretreatment by inhibiting the elevation of phosphorylation of eEF2 (eukaryotic elongation factor 2) and its upstream AMPK. This discovery provides a potential avenue for identifying novel therapeutic targets for pathological myocardial hypertrophy. 98 Diabetic cardiomyopathy, a type of hypertrophic cardiomyopathy, is a cardiac condition induced by hyperglycaemia and insulin resistance. Research shows that treadmill exercise can enhance the cardioprotective effect of FGF21, which prevents toxic lipid‐induced mitochondrial dysfunction and oxidative stress by activating AMPK/FOXO3 (forkhead box O1)/SIRT3 signaling axis in in mice with diabetic cardiomyopathy 99 (Table 1, Figure 4). It can be seen from this discussion that exerkines are released into the bloodstream through exercise, influencing cardiac vessels and cardiomyocytes. They enhance mitochondrial function, promote vascular relaxation, and contribute to antimyocardial hypertrophy effects.
Figure 4. Exercise improves mitochondrial function in HCM.
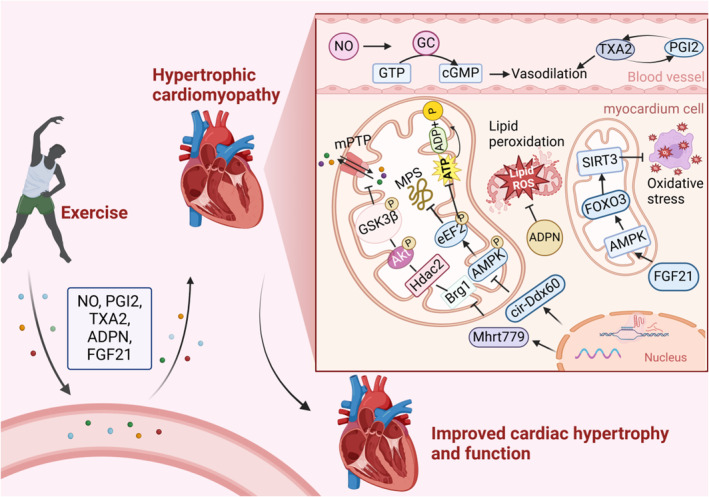
Exerkines are released into the bloodstream through exercise, influencing cardiac vessels and cardiomyocytes. They enhance mitochondrial function, promote vascular relaxation, and contribute to antimyocardial hypertrophy effects. Increased NO activates GC to catalyze the synthesis of GTP into cGMP, promoting vascular relaxation, and the balance between TXA2 and prostacyclin also contributes to this effect.
Dilated Cardiomyopathy
DCM is typically defined by the enlargement of the left ventricle or both ventricles, or systolic dysfunction, without abnormal loading conditions or significant coronary artery disease that could cause ventricular remodeling. The causes include genetic factors (primary dilated cardiomyopathy) and acquired factors (secondary dilated cardiomyopathy). Therefore, diagnostic tests and treatment strategies should always account for both genetic predisposition and acquired risk factors. 100
Endurance exercise for 3 months improved ventricular ejection fraction, mitigated fibrosis, and decreased cardiomyocyte apoptosis in aged mice with DCM due to gene mutation. 101 However, not all exercise is beneficial for improving DCM. For mice lacking nuclear DNA ANT1 (adenine nucleotide translocase 1), endurance exercise actually exacerbated the DCM. 102 Meanwhile, Danon disease, resulting from radical mutations in the LAMP2 gene, presents cardiac hypertrophy followed by left ventricular dilatation and systolic dysfunction in terms of cardiac phenotype. Compared with sedentary lifestyle, exercise training stimulated autophagy and aggravated the DCM phenotype in Danon mice. 103 In terms of DCM caused by acquired risk factors, its occurrence is linked to Ca2+ imbalance, mitochondrial dysfunction, inflammation, and metabolic abnormalities. Treadmill exercise for 6 weeks promotes Ca2+ homeostasis and arrhythmia remodeling in arrhythmogenic right ventricular cardiomyopathy, a type of DCM. 104 , 105 Exercise can promote mitochondrial biogenesis and normal metabolism, reducing inflammation and metabolic disorders caused by DCM 106 , 107 (Figure 5). Doxorubicin accumulated in the mitochondria, induces oxidative stress and inhibits the SERCA2a (sarcoplasmic reticulum calcium ATPase 2a), which, together with myocardial fibrosis and vascular dysfunction, induces DCM. One‐legged dynamic knee extensor exercise for 5 hours can reduce the cardiotoxicity of doxorubicin and improve DCM by releasing exerkines, such as irisin and IL‐6. 108 Inflammation and immune system disorders are still important pathological mechanisms causing DCM. 109 Exhaustive exercise has been shown to trigger PINK1‐dependent mitophagy, reduce the release of damage‐associated molecular patterns, and consequently, mitigate inflammatory responses. 110 (Table 1) Exercise can effectively reduce the vicious cycle between mitochondrial dysfunction, Ca2+ homeostasis disruption, and inflammatory imbalance to mitigate DCM through enhancing mitochondrial biogenesis, promoting mitophagy, and improving mitochondrial metabolism and Ca2+ balance.
Figure 5. Exercise mitigates DCM.
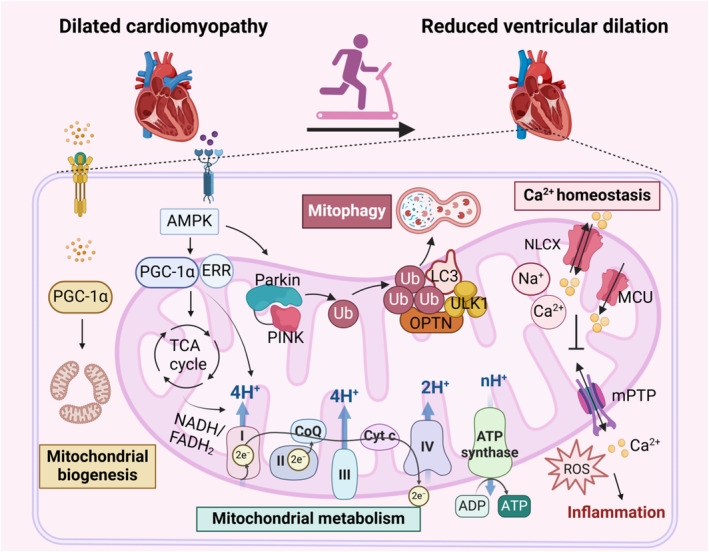
In DCM, disruptions in Ca2+ regulation, mitochondrial dysfunction, and inflammation coexist, mutually reinforcing and exacerbating the condition. Exercise promotes mitochondrial biogenesis, enhances mitochondrial metabolism, supports proper mitochondrial oxidative phosphorylation, and ensures energy supply and mitochondrial function. Additionally, exercise stimulates PINK‐dependent mitophagy, reducing inflammation. It also facilitates the normal transport of mitochondrial Ca2+, maintains Ca2+ homeostasis, and helps prevent oxidative stress and inflammation caused by mPTP opening. Through effective exercise intervention, the progression of DCM is significantly inhibited, leading to reduced ventricular dilation and improved cardiac function. AMPK indicates adenosine5′‐monophosphate‐activated protein kinase; CoQ, coenzyme Q; Cyt C, cytochrome C; DCM, dilated cardiomyopathy; ERK, extracellular regulated protein kinase; ERR, estrogen‐related receptor; FADH, flavine adenine dinucleotide; LC3, light chain 3; MCU, mitochondrial calcium uniporter; mPTP, mitochondrial permeability transition pore; NADH, nicotinamide adenine dinucleotide; NLCX, mitochondrial Na+/Ca2+ exchanger; OPTN, optineurin; PGC‐1α, peroxisome proliferator‐activated receptor‐gamma coactivator 1alpha; PINK, PTEN‐induced putative kinase; ROS, reactive oxygen species; TCA cycle, tricarboxylic acid cycle; Ub, ubiquitin; and ULK1, unc‐51‐like autophagy‐activating kinase 1.
CONCLUSIONS
Exercise can enhance cardiovascular health through promoting the release of hormones, improving body metabolism, reducing blood lipids, and other aspects. What is noteworthy is that exerkines released after exercise, an aspect of improved physical fitness caused by exercise, may play a role in reducing diseases and improving cardiovascular function by addressing mitochondrial dysfunction. Here, we offer a exhaustive review of the possible impacts of physical activity and regular exercise in CVDs, establishing a connection between mitochondrial dysfunction and the initiation and progression of CVDs. Exercise regulates mitochondrial biogenesis, dynamic balance, autophagy, and protein homeostasis to achieve quality control and enhance mitochondrial homeostasis, thereby improving cardiovascular health. By delving into the mitochondria‐related molecular mechanisms underlying the effects of exercise on CVDs, this review provides new insights for future treatments of CVDs.
Sources of Funding
This work supported in part by a grant from the National Natural Science Foundation of China (31971097, 32271226), the National Key R&D Program of China (2020YFA0803800), research project of Shanghai University of Sport (2023STD023).
Disclosures
None.
Acknowledgments
Author contributions: Huijie Zhang: Writing‐original draft; writing‐review and editing; visualization; conceptualization. Yuxuan Zhang: Investigation. Jiaqiao Zhang: Investigation. Dandan Jia: Writing‐review and editing; supervision; conceptualization. All named authors have seen and approved the final version of the article. All authors contributed to the design and the writing of the article.
This article was sent to Neel S. Singhal, MD, PhD, Associate Editor, for review by expert referees, editorial decision, and final disposition.
For Sources of Funding and Disclosures, see page 14.
References
- 1. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, et al. Heart disease and stroke Statistics‐2017 update: a report from the American Heart Association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chow LS, Gerszten RE, Taylor JM, Pedersen BK, van Praag H, Trappe S, Febbraio MA, Galis ZS, Gao Y, Haus JM, et al. Exerkines in health, resilience and disease. Nat Rev Endocrinol. 2022;18:273–289. doi: 10.1038/s41574-022-00641-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kokkinos P, Faselis C, Samuel IBH, Lavie CJ, Zhang J, Vargas JD, Pittaras A, Doumas M, Karasik P, Moore H, et al. Changes in cardiorespiratory fitness and survival in patients with or without cardiovascular disease. J Am Coll Cardiol. 2023;81:1137–1147. doi: 10.1016/j.jacc.2023.01.027 [DOI] [PubMed] [Google Scholar]
- 4. Bull FC, Al‐Ansari SS, Biddle S, Borodulin K, Buman MP, Cardon G, Carty C, Chaput JP, Chastin S, Chou R, et al. World Health Organization 2020 guidelines on physical activity and sedentary behaviour. Br J Sports Med. 2020;54:1451–1462. doi: 10.1136/bjsports-2020-102955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jia D, Tian Z, Wang R. Exercise mitigates age‐related metabolic diseases by improving mitochondrial dysfunction. Ageing Res Rev. 2023;91:102087. doi: 10.1016/j.arr.2023.102087 [DOI] [PubMed] [Google Scholar]
- 6. Chen W, Zhao H, Li Y. Mitochondrial dynamics in health and disease: mechanisms and potential targets. Signal Transduct Target Ther. 2023;8:333. doi: 10.1038/s41392-023-01547-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Makino A, Scott BT, Dillmann WH. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia. 2010;53:1783–1794. doi: 10.1007/s00125-010-1770-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chang YW, Chang YT, Wang Q, Lin JJ, Chen YJ, Chen CC. Quantitative phosphoproteomic study of pressure‐overloaded mouse heart reveals dynamin‐related protein 1 as a modulator of cardiac hypertrophy. Mol Cell Proteomics. 2013;12:3094–3107. doi: 10.1074/mcp.M113.027649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dai SH, Wu QC, Zhu RR, Wan XM, Zhou XL. Notch1 protects against myocardial ischaemia‐reperfusion injury via regulating mitochondrial fusion and function. J Cell Mol Med. 2020;24:3183–3191. doi: 10.1111/jcmm.14992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen Y, Liu Y, Dorn GW II. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. 2011;109:1327–1331. doi: 10.1161/CIRCRESAHA.111.258723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Piquereau J, Caffin F, Novotova M, Prola A, Garnier A, Mateo P, Fortin D, le Huynh H, Nicolas V, Alavi MV, et al. Down‐regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc Res. 2012;94:408–417. doi: 10.1093/cvr/cvs117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wai T, Garcia‐Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, Ruperez FJ, Barbas C, Ibanez B, Langer T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science. 2015;350:aad0116. doi: 10.1126/science.aad0116 [DOI] [PubMed] [Google Scholar]
- 13. Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015;116:264–278. doi: 10.1161/CIRCRESAHA.116.303356 [DOI] [PubMed] [Google Scholar]
- 14. Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, Oka T, Tamai T, Oyabu J, Murakawa T, et al. Inhibition of autophagy in the heart induces age‐related cardiomyopathy. Autophagy. 2010;6:600–606. doi: 10.4161/auto.6.5.11947 [DOI] [PubMed] [Google Scholar]
- 15. Yu W, Deng D, Li Y, Ding K, Qian Q, Shi H, Luo Q, Cai J, Liu J. Cardiomyocyte‐specific Tbk1 deletion aggravated chronic doxorubicin cardiotoxicity via inhibition of mitophagy. Free Radic Biol Med. 2024;222:244–258. doi: 10.1016/j.freeradbiomed.2024.06.009 [DOI] [PubMed] [Google Scholar]
- 16. Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai‐Kanai E, Ueyama T, Ikeda K, Ogata T, Matoba S. Cytosolic p53 inhibits parkin‐mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 2013;4:2308. doi: 10.1038/ncomms3308 [DOI] [PubMed] [Google Scholar]
- 17. Lampert MA, Orogo AM, Najor RH, Hammerling BC, Leon LJ, Wang BJ, Kim T, Sussman MA, Gustafsson ÅB. BNIP3L/NIX and FUNDC1‐mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy. 2019;15:1182–1198. doi: 10.1080/15548627.2019.1580095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–1793. doi: 10.1172/JCI27523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Choi YS, de Mattos AB, Shao D, Li T, Nabben M, Kim M, Wang W, Tian R, Kolwicz SC Jr. Preservation of myocardial fatty acid oxidation prevents diastolic dysfunction in mice subjected to angiotensin II infusion. J Mol Cell Cardiol. 2016;100:64–71. doi: 10.1016/j.yjmcc.2016.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Laine GA, Allen SJ. Left ventricular myocardial edema. Lymph flow, interstitial fibrosis, and cardiac function. Circ Res. 1991;68:1713–1721. doi: 10.1161/01.res.68.6.1713 [DOI] [PubMed] [Google Scholar]
- 21. Meçe O, Houbaert D, Sassano ML, Durré T, Maes H, Schaaf M, More S, Ganne M, García‐Caballero M, Borri M, et al. Lipid droplet degradation by autophagy connects mitochondria metabolism to Prox1‐driven expression of lymphatic genes and lymphangiogenesis. Nat Commun. 2022;13:2760. doi: 10.1038/s41467-022-30490-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, Jeyaraj D, Youn JY, Ren S, Liu Y, et al. Catabolic defect of branched‐chain amino acids promotes heart failure. Circulation. 2016;133:2038–2049. doi: 10.1161/CIRCULATIONAHA.115.020226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krishnan J, Suter M, Windak R, Krebs T, Felley A, Montessuit C, Tokarska‐Schlattner M, Aasum E, Bogdanova A, Perriard E, et al. Activation of a HIF1alpha‐PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009;9:512–524. doi: 10.1016/j.cmet.2009.05.005 [DOI] [PubMed] [Google Scholar]
- 24. Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takada S, Maekawa S, Furihata T, Kakutani N, Setoyama D, Ueda K, Nambu H, Hagiwara H, Handa H, Fumoto Y, et al. Succinyl‐CoA‐based energy metabolism dysfunction in chronic heart failure. Proc Natl Acad Sci USA. 2022;119:e2203628119. doi: 10.1073/pnas.2203628119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Anderson EJ, Efird JT, Davies SW, O'Neal WT, Darden TM, Thayne KA, Katunga LA, Kindell LC, Ferguson TB, Anderson CA, et al. Monoamine oxidase is a major determinant of redox balance in human atrial myocardium and is associated with postoperative atrial fibrillation. J Am Heart Assoc. 2014;3:e000713. doi: 10.1161/JAHA.113.000713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kattoor AJ, Pothineni NVK, Palagiri D, Mehta JL. Oxidative stress in atherosclerosis. Curr Atheroscler Rep. 2017;19:42. doi: 10.1007/s11883-017-0678-6 [DOI] [PubMed] [Google Scholar]
- 28. Mallat Z, Philip I, Lebret M, Chatel D, Maclouf J, Tedgui A. Elevated levels of 8‐iso‐prostaglandin F2alpha in pericardial fluid of patients with heart failure: a potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation. 1998;97:1536–1539. doi: 10.1161/01.cir.97.16.1536 [DOI] [PubMed] [Google Scholar]
- 29. Jomova K, Alomar SY, Alwasel SH, Nepovimova E, Kuca K, Valko M. Several lines of antioxidant defense against oxidative stress: antioxidant enzymes, nanomaterials with multiple enzyme‐mimicking activities, and low‐molecular‐weight antioxidants. Arch Toxicol. 2024;98:1323–1367. doi: 10.1007/s00204-024-03696-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martin TG, Kirk JA. Under construction: the dynamic assembly, maintenance, and degradation of the cardiac sarcomere. J Mol Cell Cardiol. 2020;148:89–102. doi: 10.1016/j.yjmcc.2020.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang Q, Liu J, Duan H, Li R, Peng W, Wu C. Activation of Nrf2/HO‐1 signaling: an important molecular mechanism of herbal medicine in the treatment of atherosclerosis via the protection of vascular endothelial cells from oxidative stress. J Adv Res. 2021;34:43–63. doi: 10.1016/j.jare.2021.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. D'Onofrio N, Prattichizzo F, Marfella R, Sardu C, Martino E, Scisciola L, Marfella L, Grotta R, Frigé C, Paolisso G, et al. SIRT3 mediates the effects of PCSK9 inhibitors on inflammation, autophagy, and oxidative stress in endothelial cells. Theranostics. 2023;13:531–542. doi: 10.7150/thno.80289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Finkel T, Menazza S, Holmstrom KM, Parks RJ, Liu J, Sun J, Liu J, Pan X, Murphy E. The ins and outs of mitochondrial calcium. Circ Res. 2015;116:1810–1819. doi: 10.1161/CIRCRESAHA.116.305484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Van Keuren AM, Tsai CW, Balderas E, Rodriguez MX, Chaudhuri D, Tsai MF. Mechanisms of EMRE‐dependent MCU opening in the mitochondrial calcium uniporter complex. Cell Rep. 2020;33:108486. doi: 10.1016/j.celrep.2020.108486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lambert JP, Luongo TS, Tomar D, Jadiya P, Gao E, Zhang X, Lucchese AM, Kolmetzky DW, Shah NS, Elrod JW. MCUB regulates the molecular composition of the mitochondrial calcium Uniporter Channel to limit mitochondrial calcium overload during stress. Circulation. 2019;140:1720–1733. doi: 10.1161/CIRCULATIONAHA.118.037968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu S, Wang P, Zhang H, Gong G, Gutierrez Cortes N, Zhu W, Yoon Y, Tian R, Wang W. CaMKII induces permeability transition through Drp1 phosphorylation during chronic beta‐AR stimulation. Nat Commun. 2016;7:13189. doi: 10.1038/ncomms13189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Venetucci L, Denegri M, Napolitano C, Priori SG. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat Rev Cardiol. 2012;9:561–575. doi: 10.1038/nrcardio.2012.93 [DOI] [PubMed] [Google Scholar]
- 38. Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E, van Berlo JH, et al. The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature. 2017;545:93–97. doi: 10.1038/nature22082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Urry DW. Neutral sites for calcium ion binding to elastin and collagen: a charge neutralization theory for calcification and its relationship to atherosclerosis. Proc Natl Acad Sci USA. 1971;68:810–814. doi: 10.1073/pnas.68.4.810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu H, Kanatous SB, Thurmond FA, Gallardo T, Isotani E, Bassel‐Duby R, Williams RS. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science. 2002;296:349–352. doi: 10.1126/science.1071163 [DOI] [PubMed] [Google Scholar]
- 41. Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP, Holloszy JO. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC‐1. FASEB J. 2002;16:1879–1886. doi: 10.1096/fj.02-0367com [DOI] [PubMed] [Google Scholar]
- 42. Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab. 2012;23:459–466. doi: 10.1016/j.tem.2012.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ju JS, Jeon SI, Park JY, Lee JY, Lee SC, Cho KJ, Jeong JM. Autophagy plays a role in skeletal muscle mitochondrial biogenesis in an endurance exercise‐trained condition. J Physiol Sci. 2016;66:417–430. doi: 10.1007/s12576-016-0440-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stevanović‐Silva J, Beleza J, Coxito P, Rocha H, Gaspar TB, Gärtner F, Correia R, Fernandes R, Oliveira PJ, Ascensão A, et al. Exercise performed during pregnancy positively modulates liver metabolism and promotes mitochondrial biogenesis of female offspring in a rat model of diet‐induced gestational diabetes. Biochim Biophys Acta Mol basis Dis. 1868;2022:166526. doi: 10.1016/j.bbadis.2022.166526 [DOI] [PubMed] [Google Scholar]
- 45. Picard M, Gentil BJ, McManus MJ, White K, St Louis K, Gartside SE, Wallace DC, Turnbull DM. Acute exercise remodels mitochondrial membrane interactions in mouse skeletal muscle. J Appl Physiol (1985). 2013;115:1562–1571. doi: 10.1152/japplphysiol.00819.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mendham AE, Goedecke JH, Zeng Y, Larsen S, George C, Hauksson J, Fortuin‐de Smidt MC, Chibalin AV, Olsson T, Chorell E. Exercise training improves mitochondrial respiration and is associated with an altered intramuscular phospholipid signature in women with obesity. Diabetologia. 2021;64:1642–1659. doi: 10.1007/s00125-021-05430-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Joshi AU, Minhas PS, Liddelow SA, Haileselassie B, Andreasson KI, Dorn GW II, Mochly‐Rosen D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat Neurosci. 2019;22:1635–1648. doi: 10.1038/s41593-019-0486-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Campos JC, Marchesi Bozi LH, Krum B, Grassmann Bechara LR, Ferreira ND, Arini GS, Albuquerque RP, Traa A, Ogawa T, van der Bliek AM, et al. Exercise preserves physical fitness during aging through AMPK and mitochondrial dynamics. Proc Natl Acad Sci USA. 2023;120:e2204750120. doi: 10.1073/pnas.2204750120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zou YY, Tang XB, Chen ZL, Liu B, Zheng L, Song MY, Xiao Q, Zhou ZQ, Peng XY, Tang CF. Exercise intervention improves mitochondrial quality in non‐alcoholic fatty liver disease zebrafish. Front Endocrinol (Lausanne). 2023;14:1162485. doi: 10.3389/fendo.2023.1162485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Heo JW, No MH, Cho J, Choi Y, Cho EJ, Park DH, Kim TW, Kim CJ, Seo DY, Han J, et al. Moderate aerobic exercise training ameliorates impairment of mitochondrial function and dynamics in skeletal muscle of high‐fat diet‐induced obese mice. FASEB J. 2021;35:e21340. doi: 10.1096/fj.202002394R [DOI] [PubMed] [Google Scholar]
- 51. Lin J, Zhang X, Sun Y, Xu H, Li N, Wang Y, Tian X, Zhao C, Wang B, Zhu B, et al. Exercise ameliorates muscular excessive mitochondrial fission, insulin resistance and inflammation in diabetic rats via irisin/AMPK activation. Sci Rep. 2024;14:10658. doi: 10.1038/s41598-024-61415-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Axelrod CL, Fealy CE, Mulya A, Kirwan JP. Exercise training remodels human skeletal muscle mitochondrial fission and fusion machinery towards a pro‐elongation phenotype. Acta Physiol (Oxf). 2019;225:e13216. doi: 10.1111/apha.13216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Arribat Y, Broskey NT, Greggio C, Boutant M, Conde Alonso S, Kulkarni SS, Lagarrigue S, Carnero EA, Besson C, Canto C, et al. Distinct patterns of skeletal muscle mitochondria fusion, fission and mitophagy upon duration of exercise training. Acta Physiol (Oxf). 2019;225:e13179. doi: 10.1111/apha.13179 [DOI] [PubMed] [Google Scholar]
- 54. Laker RC, Drake JC, Wilson RJ, Lira VA, Lewellen BM, Ryall KA, Fisher CC, Zhang M, Saucerman JJ, Goodyear LJ, et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise‐induced mitophagy. Nat Commun. 2017;8:548. doi: 10.1038/s41467-017-00520-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schwalm C, Jamart C, Benoit N, Naslain D, Prémont C, Prévet J, Van Thienen R, Deldicque L, Francaux M. Activation of autophagy in human skeletal muscle is dependent on exercise intensity and AMPK activation. FASEB J. 2015;29:3515–3526. doi: 10.1096/fj.14-267187 [DOI] [PubMed] [Google Scholar]
- 56. Chen CCW, Erlich AT, Crilly MJ, Hood DA. Parkin is required for exercise‐induced mitophagy in muscle: impact of aging. Am J Physiol Endocrinol Metab. 2018;315:E404–e415. doi: 10.1152/ajpendo.00391.2017 [DOI] [PubMed] [Google Scholar]
- 57. Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson ÅB. Microtubule‐associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–19104. doi: 10.1074/jbc.M111.322933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lo Verso F, Carnio S, Vainshtein A, Sandri M. Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity. Autophagy. 2014;10:1883–1894. doi: 10.4161/auto.32154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Brandt N, Gunnarsson TP, Bangsbo J, Pilegaard H. Exercise and exercise training‐induced increase in autophagy markers in human skeletal muscle. Physiol Rep. 2018;6:e13651. doi: 10.14814/phy2.13651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Di Donato DM, West DW, Churchward‐Venne TA, Breen L, Baker SK, Phillips SM. Influence of aerobic exercise intensity on myofibrillar and mitochondrial protein synthesis in young men during early and late postexercise recovery. Am J Physiol Endocrinol Metab. 2014;306:E1025–E1032. doi: 10.1152/ajpendo.00487.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li Y, Xue Y, Xu X, Wang G, Liu Y, Wu H, Li W, Wang Y, Chen Z, Zhang W, et al. A mitochondrial FUNDC1/HSC70 interaction organizes the proteostatic stress response at the risk of cell morbidity. EMBO J. 2019;38:e98786. doi: 10.15252/embj.201798786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stancakova A, Goetzman E, Lam MM, Schwer B, et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell. 2011;44:177–190. doi: 10.1016/j.molcel.2011.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hinkley JM, Morton AB, Ichinoseki‐Sekine N, Huertas AM, Smuder AJ. Exercise training prevents doxorubicin‐induced mitochondrial dysfunction of the liver. Med Sci Sports Exerc. 2019;51:1106–1115. doi: 10.1249/MSS.0000000000001887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Burtscher J, Soltany A, Visavadiya NP, Burtscher M, Millet GP, Khoramipour K, Khamoui AV. Mitochondrial stress and mitokines in aging. Aging Cell. 2023;22:e13770. doi: 10.1111/acel.13770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Luo X, Zhang H, Cao X, Yang D, Yan Y, Lu J, Wang X, Wang H. Endurance exercise‐induced Fgf21 promotes skeletal muscle fiber conversion through TGF‐β1 and p38 MAPK signaling pathway. Int J Mol Sci. 2023;24:11401. doi: 10.3390/ijms241411401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kleinert M, Clemmensen C, Sjoberg KA, Carl CS, Jeppesen JF, Wojtaszewski JFP, Kiens B, Richter EA. Exercise increases circulating GDF15 in humans. Mol Metab. 2018;9:187–191. doi: 10.1016/j.molmet.2017.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cai H, Liu Y, Men H, Zheng Y. Protective mechanism of humanin against oxidative stress in aging‐related cardiovascular diseases. Front Endocrinol (Lausanne). 2021;12:683151. doi: 10.3389/fendo.2021.683151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Woodhead JST, D'Souza RF, Hedges CP, Wan J, Berridge MV, Cameron‐Smith D, Cohen P, Hickey AJR, Mitchell CJ, Merry TL. High‐intensity interval exercise increases humanin, a mitochondrial encoded peptide, in the plasma and muscle of men. J Appl Physiol (1985). 2020;128:1346–1354. doi: 10.1152/japplphysiol.00032.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Reynolds JC, Lai RW, Woodhead JST, Joly JH, Mitchell CJ, Cameron‐Smith D, Lu R, Cohen P, Graham NA, Benayoun BA, et al. MOTS‐c is an exercise‐induced mitochondrial‐encoded regulator of age‐dependent physical decline and muscle homeostasis. Nat Commun. 2021;12:470. doi: 10.1038/s41467-020-20790-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liu Y, Nguyen PT, Wang X, Zhao Y, Meacham CE, Zou Z, Bordieanu B, Johanns M, Vertommen D, Wijshake T, et al. TLR9 and beclin 1 crosstalk regulates muscle AMPK activation in exercise. Nature. 2020;578:605–609. doi: 10.1038/s41586-020-1992-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Luo JY, Cheng CK, He L, Pu Y, Zhang Y, Lin X, Xu A, Lau CW, Tian XY, Ma RCW, et al. Endothelial UCP2 is a mechanosensitive suppressor of atherosclerosis. Circ Res. 2022;131:424–441. doi: 10.1161/circresaha.122.321187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ma L, Li K, Wei W, Zhou J, Li Z, Zhang T, Wangsun Y, Tian F, Dong Q, Zhang H, et al. Exercise protects aged mice against coronary endothelial senescence via FUNDC1‐dependent mitophagy. Redox Biol. 2023;62:102693. doi: 10.1016/j.redox.2023.102693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kar S, Shahshahan HR, Hackfort BT, Yadav SK, Yadav R, Kambis TN, Lefer DJ, Mishra PK. Exercise training promotes cardiac hydrogen sulfide biosynthesis and mitigates pyroptosis to prevent high‐fat diet‐induced diabetic cardiomyopathy. Antioxidants (Basel). 2019;8:638. doi: 10.3390/antiox8120638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Xiong Y, Chang LL, Tran B, Dai T, Zhong R, Mao YC, Zhu YZ. ZYZ‐803, a novel hydrogen sulfide‐nitric oxide conjugated donor, promotes angiogenesis via cross‐talk between STAT3 and CaMKII. Acta Pharmacol Sin. 2020;41:218–228. doi: 10.1038/s41401-019-0255-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li Z, Xia H, Sharp TE III, LaPenna KB, Katsouda A, Elrod JW, Pfeilschifter J, Beck KF, Xu S, Xian M, et al. Hydrogen sulfide modulates endothelial‐mesenchymal transition in heart failure. Circ Res. 2023;132:154–166. doi: 10.1161/CIRCRESAHA.122.321326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Aengevaeren VL, Mosterd A, Bakker EA, Braber TL, Nathoe HM, Sharma S, Thompson PD, Velthuis BK, Eijsvogels TMH. Exercise volume versus intensity and the progression of coronary atherosclerosis in middle‐aged and older athletes: findings from the MARC‐2 study. Circulation. 2023;147:993–1003. doi: 10.1161/CIRCULATIONAHA.122.061173 [DOI] [PubMed] [Google Scholar]
- 77. Lu J, Xiang G, Liu M, Mei W, Xiang L, Dong J. Irisin protects against endothelial injury and ameliorates atherosclerosis in apolipoprotein E‐null diabetic mice. Atherosclerosis. 2015;243:438–448. doi: 10.1016/j.atherosclerosis.2015.10.020 [DOI] [PubMed] [Google Scholar]
- 78. Jia D, Hou L, Lv Y, Xi L, Tian Z. Postinfarction exercise training alleviates cardiac dysfunction and adverse remodeling via mitochondrial biogenesis and SIRT1/PGC‐1α/PI3K/Akt signaling. J Cell Physiol. 2019;234:23705–23718. doi: 10.1002/jcp.28939 [DOI] [PubMed] [Google Scholar]
- 79. Zhao D, Sun Y, Tan Y, Zhang Z, Hou Z, Gao C, Feng P, Zhang X, Yi W, Gao F. Short‐duration swimming exercise after myocardial infarction attenuates cardiac dysfunction and regulates mitochondrial quality control in aged mice. Oxidative Med Cell Longev. 2018;2018:4079041. doi: 10.1155/2018/4079041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Donniacuo M, Urbanek K, Nebbioso A, Sodano L, Gallo L, Altucci L, Rinaldi B. Cardioprotective effect of a moderate and prolonged exercise training involves sirtuin pathway. Life Sci. 2019;222:140–147. doi: 10.1016/j.lfs.2019.03.001 [DOI] [PubMed] [Google Scholar]
- 81. Jin L, Diaz‐Canestro C, Wang Y, Tse MA, Xu A. Exerkines and cardiometabolic benefits of exercise: from bench to clinic. EMBO Mol Med. 2024;16:432–444. doi: 10.1038/s44321-024-00027-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Harris MP, Zeng S, Zhu Z, Lira VA, Yu L, Hodgson‐Zingman DM, Zingman LV. Myokine Musclin is critical for exercise‐induced cardiac conditioning. Int J Mol Sci. 2023;24:6525. doi: 10.3390/ijms24076525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ma Y, Kuang Y, Bo W, Liang Q, Zhu W, Cai M, Tian Z. Exercise training alleviates cardiac fibrosis through increasing fibroblast growth factor 21 and regulating TGF‐β1‐Smad2/3‐MMP2/9 signaling in mice with myocardial infarction. Int J Mol Sci. 2021;22:12341. doi: 10.3390/ijms222212341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Xi Y, Hao M, Liang Q, Li Y, Gong DW, Tian Z. Dynamic resistance exercise increases skeletal muscle‐derived FSTL1 inducing cardiac angiogenesis via DIP2A‐Smad2/3 in rats following myocardial infarction. J Sport Health Sci. 2021;10:594–603. doi: 10.1016/j.jshs.2020.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Otaka N, Shibata R, Ohashi K, Uemura Y, Kambara T, Enomoto T, Ogawa H, Ito M, Kawanishi H, Maruyama S, et al. Myonectin is an exercise‐induced myokine that protects the heart from ischemia‐reperfusion injury. Circ Res. 2018;123:1326–1338. doi: 10.1161/circresaha.118.313777 [DOI] [PubMed] [Google Scholar]
- 86. Hausenloy DJ, Yellon DM. Myocardial ischemia‐reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92–100. doi: 10.1172/JCI62874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gewirtz H, Dilsizian V. Myocardial viability: survival mechanisms and molecular imaging targets in acute and chronic ischemia. Circ Res. 2017;120:1197–1212. doi: 10.1161/CIRCRESAHA.116.307898 [DOI] [PubMed] [Google Scholar]
- 88. Hendy GN, Canaff L. Calcium‐sensing receptor, proinflammatory cytokines and calcium homeostasis. Semin Cell Dev Biol. 2016;49:37–43. doi: 10.1016/j.semcdb.2015.11.006 [DOI] [PubMed] [Google Scholar]
- 89. Mason SA, Trewin AJ, Parker L, Wadley GD. Antioxidant supplements and endurance exercise: current evidence and mechanistic insights. Redox Biol. 2020;35:101471. doi: 10.1016/j.redox.2020.101471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Koloverou E, Tambalis K, Panagiotakos DB, Georgousopoulou E, Chrysohoou C, Skoumas I, Tousoulis D, Stefanadis C, Pitsavos C; group As . Moderate physical activity reduces 10‐year diabetes incidence: the mediating role of oxidative stress biomarkers. Int J Public Health. 2018;63:297–305. doi: 10.1007/s00038-017-1052-8 [DOI] [PubMed] [Google Scholar]
- 91. Whitham M, Parker BL, Friedrichsen M, Hingst JR, Hjorth M, Hughes WE, Egan CL, Cron L, Watt KI, Kuchel RP, et al. Extracellular vesicles provide a means for tissue crosstalk during exercise. Cell Metab. 2018;27:237–251.e234. doi: 10.1016/j.cmet.2017.12.001 [DOI] [PubMed] [Google Scholar]
- 92. Lisi V, Senesi G, Bertola N, Pecoraro M, Bolis S, Gualerzi A, Picciolini S, Raimondi A, Fantini C, Moretti E, et al. Plasma‐derived extracellular vesicles released after endurance exercise exert cardioprotective activity through the activation of antioxidant pathways. Redox Biol. 2023;63:102737. doi: 10.1016/j.redox.2023.102737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Vicencio JM, Yellon DM, Sivaraman V, Das D, Boi‐Doku C, Arjun S, Zheng Y, Riquelme JA, Kearney J, Sharma V, et al. Plasma exosomes protect the myocardium from ischemia‐reperfusion injury. J Am Coll Cardiol. 2015;65:1525–1536. doi: 10.1016/j.jacc.2015.02.026 [DOI] [PubMed] [Google Scholar]
- 94. Das A, Huang GX, Bonkowski MS, Longchamp A, Li C, Schultz MB, Kim LJ, Osborne B, Joshi S, Lu Y, et al. Impairment of an endothelial NAD(+)‐H(2)S signaling network is a reversible cause of vascular aging. Cell. 2018;173:74–89 e20. doi: 10.1016/j.cell.2018.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. MacNamara JP, Dias KA, Hearon CM Jr, Ivey E, Delgado VA, Saland S, Samels M, Hieda M, Turer AT, Link MS, et al. Randomized controlled trial of moderate‐ and high‐intensity exercise training in patients with hypertrophic cardiomyopathy: effects on fitness and cardiovascular response to exercise. J Am Heart Assoc. 2023;12:e031399. doi: 10.1161/jaha.123.031399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. De Ciuceis C, Rizzoni D, Palatini P. Microcirculation and physical exercise in hypertension. Hypertension. 2023;80:730–739. doi: 10.1161/HYPERTENSIONAHA.122.19465 [DOI] [PubMed] [Google Scholar]
- 97. Lin H, Zhu Y, Zheng C, Hu D, Ma S, Chen L, Wang Q, Chen Z, Xie J, Yan Y, et al. Antihypertrophic memory after regression of exercise‐induced physiological myocardial hypertrophy is mediated by the long noncoding RNA Mhrt779. Circulation. 2021;143:2277–2292. doi: 10.1161/CIRCULATIONAHA.120.047000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zhu Y, Zheng C, Zhang R, Yan J, Li M, Ma S, Chen K, Chen L, Liu J, Xiu J, et al. Circ‐Ddx60 contributes to the antihypertrophic memory of exercise hypertrophic preconditioning. J Adv Res. 2023;46:113–121. doi: 10.1016/j.jare.2022.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Jin L, Geng L, Ying L, Shu L, Ye K, Yang R, Liu Y, Wang Y, Cai Y, Jiang X, et al. FGF21‐Sirtuin 3 Axis confers the protective effects of exercise against diabetic cardiomyopathy by governing mitochondrial integrity. Circulation. 2022;146:1537–1557. doi: 10.1161/CIRCULATIONAHA.122.059631 [DOI] [PubMed] [Google Scholar]
- 100. Heymans S, Lakdawala NK, Tschöpe C, Klingel K. Dilated cardiomyopathy: causes, mechanisms, and current and future treatment approaches. Lancet. 2023;402:998–1011. doi: 10.1016/s0140-6736(23)01241-2 [DOI] [PubMed] [Google Scholar]
- 101. Cai ZJ, Lee YK, Lau YM, Ho JC, Lai WH, Wong NL, Huang D, Hai JJ, Ng KM, Tse HF, et al. Expression of Lmna‐R225X nonsense mutation results in dilated cardiomyopathy and conduction disorders (DCM‐CD) in mice: impact of exercise training. Int J Cardiol. 2020;298:85–92. doi: 10.1016/j.ijcard.2019.09.058 [DOI] [PubMed] [Google Scholar]
- 102. Schaefer PM, Rathi K, Butic A, Tan W, Mitchell K, Wallace DC. Mitochondrial mutations alter endurance exercise response and determinants in mice. Proc Natl Acad Sci USA. 2022;119:e2200549119. doi: 10.1073/pnas.2200549119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yadin D, Petrover Z, Shainberg A, Alcalai R, Waldman M, Seidman J, Seidman CE, Abraham NG, Hochhauser E, Arad M. Autophagy guided interventions to modify the cardiac phenotype of Danon disease. Biochem Pharmacol. 2022;204:115229. doi: 10.1016/j.bcp.2022.115229 [DOI] [PubMed] [Google Scholar]
- 104. van Opbergen CJM, Bagwan N, Maurya SR, Kim JC, Smith AN, Blackwell DJ, Johnston JN, Knollmann BC, Cerrone M, Lundby A, et al. Exercise causes arrhythmogenic remodeling of intracellular calcium dynamics in Plakophilin‐2‐deficient hearts. Circulation. 2022;145:1480–1496. doi: 10.1161/CIRCULATIONAHA.121.057757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Sun B, Yao J, Ni M, Wei J, Zhong X, Guo W, Zhang L, Wang R, Belke D, Chen YX, et al. Cardiac ryanodine receptor calcium release deficiency syndrome. Sci Transl Med. 2021;13:eaba7287. doi: 10.1126/scitranslmed.aba7287 [DOI] [PubMed] [Google Scholar]
- 106. Gan Z, Fu T, Kelly DP, Vega RB. Skeletal muscle mitochondrial remodeling in exercise and diseases. Cell Res. 2018;28:969–980. doi: 10.1038/s41422-018-0078-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Barcena ML, Pozdniakova S, Haritonow N, Breiter P, Kuhl AA, Milting H, Baczko I, Ladilov Y, Regitz‐Zagrosek V. Dilated cardiomyopathy impairs mitochondrial biogenesis and promotes inflammation in an age‐ and sex‐dependent manner. Aging (Albany NY). 2020;12:24117–24133. doi: 10.18632/aging.202283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Dozic S, Howden EJ, Bell JR, Mellor KM, Delbridge LMD, Weeks KL. Cellular mechanisms mediating exercise‐induced protection against cardiotoxic anthracycline cancer therapy. Cells. 2023;12:1312. doi: 10.3390/cells12091312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Harding D, Chong MHA, Lahoti N, Bigogno CM, Prema R, Mohiddin SA, Marelli‐Berg F. Dilated cardiomyopathy and chronic cardiac inflammation: pathogenesis, diagnosis and therapy. J Intern Med. 2023;293:23–47. doi: 10.1111/joim.13556 [DOI] [PubMed] [Google Scholar]
- 110. Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP, et al. Parkin and PINK1 mitigate STING‐induced inflammation. Nature. 2018;561:258–262. doi: 10.1038/s41586-018-0448-9 [DOI] [PMC free article] [PubMed] [Google Scholar]