

Abstract
Background
Hypoxia‐induced pulmonary hypertension (HPH) is a severe vascular disorder that is characterized by the involvement of endothelial‐to‐mesenchymal transition (EndMT) in its pathogenesis. Our previous research has suggested that the gene versican may have a crucial role in the development of HPH. However, the exact function of versican in HPH requires further investigation.
Methods and Results
The expression of versican and markers of EndMT was assessed using Western blot, immunohistochemistry, and immunofluorescence. Vascular remodeling and right ventricular hypertrophy in patients with HPH and mice were evaluated through hematoxylin and eosin staining, Masson's staining, and hemodynamic measurements. Protein interactions were validated using co‐immunoprecipitation, and the DNA methylation level of versican was examined using methylation‐specific polymerase chain reaction. Compared with the control, EndMT was observed in patients with HPH, HPH mouse models, and hypoxia‐treated human pulmonary artery endothelial cells, accompanied by a significant increase of versican. Endothelium‐specific knockdown of versican reversed HPH progression and effectively prevented EndMT in mouse models and human pulmonary artery endothelial cells. We further confirmed that versican participated in EndMT by targeting the key transcription factor Twist1. Additionally, the upregulation of versican may be attributed to promoter hypomethylation, which was mediated by reduced DNA methyltransferases activity under hypoxic conditions.
Conclusions
This study provides the initial evidence showcasing the role of promoter hypomethylation‐mediated versican upregulation in promoting EndMT by targeting Twist1, which facilitates vascular remodeling and the progression of HPH. These findings offer a promising new target for the treatment of HPH.
Keywords: DNA methylation, endothelial‐to‐mesenchymal transition, hypoxia‐induced pulmonary hypertension, Twist1, versican
Nonstandard Abbreviations and Acronyms
- CD31
platelet endothelial cell adhesion molecule‐1
- DNMT
deoxyribonucleic acid methyltransferase
- α‐SMA
α‐smooth muscle actin
Research Perspective.
What Is New?
Versican is critical in the regulation of endothelial‐to‐mesenchymal transition in patients with pulmonary hypertension.
The transcription factor Twist1 has been identified as a key downstream target of versican and is a necessary regulator of endothelial‐to‐mesenchymal transition.
Upregulation of versican in pulmonary hypertension may be due to the reduction of DNA methyltransferases under hypoxic conditions, which mediated DNA promoter hypomethylation of versican.
What Question Should Be Addressed Next?
As a key endothelial‐to‐mesenchymal transition–related regulatory gene in pulmonary hypertension, questions remain regarding the specifics of how versican interacts with Twist1 to exert its effect.
Future studies are needed to address the specific mechanism by which DNA methyltransferases regulate versican DNA methylation levels under hypoxic conditions.
Hypoxia‐induced pulmonary hypertension (HPH) is a progressive cardiopulmonary disorder commonly associated with chronic hypoxic lung diseases, which is defined as a resting mean pulmonary arterial pressure >20 mm Hg. 1 With an estimated impact on ≈1% of the global population, pulmonary hypertension has become a significant global public health concern, particularly among individuals aged 65 years and older, where its prevalence can reach up to 10%. 2 Given the rising prevalence of chronic lung diseases such as chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis, HPH has become the second common cause of pulmonary hypertension. 3 , 4 It is characterized by hypoxia‐induced pulmonary vasoconstriction and subsequent pulmonary vascular remodeling, 5 ultimately leading to right heart failure and even death. 6 Unfortunately, the mechanisms causing increased pulmonary vascular resistance in HPH are poorly understood, and current treatment strategies are limited, leading to a generally poor prognosis for patients with HPH. 7 Thus, urgent development of novel diagnostic and therapeutic strategies is necessary.
Pulmonary endothelial cells, located on the inner wall of vessels, protect vascular cells from pathological stimuli and damage. 8 Normal endothelial cell function and the adhesion of extracellular matrix (ECM) are crucial for the structural integrity of the endothelial barrier. 9 Chronic hypoxia can disrupt endothelial homeostasis and cause excessive deposition of ECM, which drives disease progression by inducing a process called endothelial‐to‐mesenchymal transition (EndMT). 10 , 11 EndMT involves the transformation of endothelial cells into mesenchymal cells. 12 Accumulating evidence suggests that EndMT plays a crucial role in the development and progression of HPH, 13 but its underlying mechanisms are not yet fully elucidated and deserve further investigation.
Versican is a large chondroitin sulfate proteoglycan predominantly found in the ECM. 14 It plays a critical role in promoting endothelial cell proliferation, adhesion, and differentiation by participating in the regulation of the transforming growth factor β/bone morphogenetic protein signaling pathway, binding with vascular endothelial growth factor. 15 Previous studies have demonstrated that the activation of versican induced by transforming growth factor β in tumor cells can trigger epithelial‐to‐mesenchymal transition, facilitating increased cell migration and invasion. 16 Through bioinformatics analysis, our preliminary research has identified versican as a potential crucial gene involved in the development of HPH. Furthermore, we have observed a significant upregulation of versican in the endothelium of pulmonary blood vessels in patients with HPH. 17 These findings provide initial evidence supporting the hypothesis that versican may contribute to the progression of HPH through regulation of EndMT. Previous research has also suggested the involvement of versican in modulating the epithelial‐to‐mesenchymal transition process through its interaction with the transcription factor Snail. 18 Based on this knowledge, our investigation aims to identify the downstream targets of versican among these transcription factors (TFs), thereby enhancing our understanding of versican's contribution to EndMT. However, it is important to note that further research is needed to fully elucidate the specific mechanisms underlying versican involvement in EndMT and determine whether versican interacts with specific TFs in this regulatory context.
In recent years, there has been increasing attention to the role of epigenetics in the regulatory mechanisms of HPH. 19 , 20 DNA methylation is a common epigenetic modification that plays a crucial role in gene transcription regulation. 21 Hypoxia has been found to affect the expression of DNA methyltransferases (DNMTs), leading to aberrant DNA methylation, which regulates the transcription and expression of genes involved in disease progression and potentially serving as a therapeutic target. 22 , 23 We hypothesize that hypoxia can also result in changes in the levels of DNMTs in pulmonary endothelium. Previous study by Luo et al identified the presence of CpG islands in versican sequence and demonstrated that DNA hypomethylation‐mediated overexpression of versican played a significant role in the pathogenesis of upper urinary tract urothelial carcinoma. 24 Therefore, we speculated that hypoxia may influence the methylation levels of versican by modulating the expression of DNMTs, ultimately regulating the EndMT process in HPH.
In this study, we demonstrated the upregulation of versican in patients with HPH, mice, and cell models for the first time. Knockdown of versican effectively reversed EndMT both in vitro and vivo, highlighting the significant role of versican in HPH through its regulation of EndMT. Additionally, unlike the EndMT process in other diseases, we identified transcription factor Twist1 as a key downstream target of versican in HPH. Furthermore, we found that the elevated expression of versican in HPH may be due to hypoxia‐induced reduction of DNMTs, leading to DNA promoter hypomethylation. These unprecedented findings provide valuable insights into the involvement of versican in the pathogenesis of HPH and present it as an intriguing therapeutic target.
METHODS
Data Availability Statement
All data and supporting materials have been provided with the published article.
Ethics Statement
This study was conducted with the approval of the Medical Ethics Committee of the Affiliated Wuxi People's Hospital of Nanjing Medical University (Approval No. KS000024) and was carried out in accordance with the principles outlined in the Declaration of Helsinki. Informed consent was obtained from all patients participating in this study. Animal experiments were conducted following the guidelines provided in the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. The study protocols were approved by the Animal Ethics Committee of Nanjing Medical University (Approval No. KY21033).
Sample Collection
We selected a total of 6 patients for our study, consisting of 3 patients with HPH and 3 patients without HPH. Lung tissue samples of 2 groups were obtained from lobectomies for benign lung nodules at the Affiliated Wuxi People's Hospital of Nanjing Medical University. Chronic obstructive pulmonary disease was selected as a representative disease of HPH, which was diagnosed by postbronchodilator forced expiratory volume in 1 second/forced vital capacity ratio lower than 70% and without other lung diseases, such as bronchiectasis, diffuse interstitial lung disease, cystic fibrosis, or lung cancer. The assessment of HPH was conducted through right heart catheterization during surgery, with mean pulmonary arterial pressure higher than 20 mm Hg. 2 Additionally, other types of pulmonary hypertension and systemic diseases were excluded in the evaluation process. The screening criteria for the normal group are patients similar to the HPH group in sex and age, without smoking history, and exclusion of chronic lung disease. Table 1 presents the demographic and clinicopathological characteristics of all the subjects included in the study.
Table 1.
Demographic and Clinicopathological Characteristics of the Participants
| Group | HPH (n=3) | Normal (n=3) | P value |
|---|---|---|---|
| Age | 77.00±2.65 | 73.67±7.57 | 0.51 |
| FEV1/pred, % | 51.70±2.87 | 91.83±1.79 | <0.001 |
| FEV1/FVC, % | 48.90±2.62 | 84.97±2.12 | <0.001 |
| mPAP, mm Hg | 59.67±13.87 | 17.33±2.08 | <0.001 |
Mean±SD. P<0.05 is considered to be significant. FEV1 indicates forced expiratory volume in 1 s; FEV1/pred, forced expiratory volume in 1 s as percentage of predicted value; FVC, forced vital capacity; HPH, hypoxia‐induced pulmonary hypertension; and mPAP, mean pulmonary artery pressure.
Establishment of HPH Mouse Model and Hemodynamic Evaluation
The male C57BL/6 mice aged 6 to 8 weeks were obtained from Changzhou Cavens Experimental Animal Co., Ltd. All mice had free access to water and a standard laboratory diet, and they were kept in a 12‐hour light/dark cycle at room temperature. The mice were randomly assigned to 4 groups (n=8 per group), including the normoxia group, hypoxia group, hypoxia+sh‐NC group, and hypoxia+sh‐versican group. At the beginning of the experiment, on day 0, the hypoxia+sh‐NC and hypoxia+sh‐versican groups were intratracheally administered adeno‐associated virus serotype 5 containing the expression of NC and versican inhibitors, respectively. To minimize interference with versican expression in epithelial and other cells within lung tissue, we used adeno‐associated virus‐sh‐versican labeled with the vascular endothelial‐specific Tie promoter. An aliquot of the vector at 1011 genome equivalents was prepared in 20 to 30 μL of phosphate buffered saline for intratracheal administration, and the control group received an equal volume of phosphate buffered saline. After 3 weeks, the effectiveness of versican interference was confirmed, and the normoxia and hypoxia treatment began. The mice in the hypoxia groups were housed in a hypobaric chamber containing 10% O2 for exposure to hypoxia, while the mice in the normoxia group were continuously housed under normal conditions (21% O2).
Four weeks later, relevant indicators were measured in different groups of mice. The mice were weighed and anesthetized by intraperitoneal injection of 50 mg/kg 3% pentobarbital sodium. Right ventricular systolic pressure was measured using a pressure transducer attached to a catheter inserted into the right ventricle and collected using the PowerLab data acquisition system and LabChart 8.0 software. The Fulton index, calculated by determining the ratio of right ventricle weight to left ventricle weight and septum weight [RV/(LV+S)], was also determined as an indicator of pulmonary hypertension. Additionally, lung and heart tissues from the mice were collected for further experiments.
Establishment of Hypoxic HPAEC Model and Cellular Transfection
Human pulmonary artery endothelial cells (HPAECs) obtained from iCell (Shanghai, China) were cultured in endothelial cell medium (iCell, #002) supplemented with 5% fetal bovine serum, 1% endothelial cell growth supplement, and 1% penicillin and streptomycin (iCell, Shanghai, China). HPAECs at passages 3 to 8 were used for independent replication experiments. To establish an in vitro model of HPH, HPAECs in the hypoxia group were incubated in a hypoxic chamber maintained at 1% O2, 4% CO2, and 95% N2. The normoxia group was cultured at 37 °C in a humidified atmosphere of 5% CO2.
Small interfering RNAs (siRNAs) targeting versican and Twist1 were purchased from RiboBio (Guangzhou, China). A nontargeted control siRNA (siRNA‐NC) was used as a negative control. The sequences of the constructs used in this study are listed in Table 2. HPAECs were divided into 5 groups: normoxia group, hypoxia group, hypoxia+si‐NC group, hypoxia+si‐versican group, and hypoxia+si‐Twist1 group. Transfection of siRNAs was performed using Lipofectamine 3000 (Invitrogen, Carlsbad, CA) following the manufacturer's protocols when HPAECs reached 30% to 50% confluency. The transfection efficiency was assessed by quantitative polymerase chain reaction (qPCR). After 24 hours of transfection, HPAECs were cultured at 37 °C for an additional 48 hours either in a normoxic environment (21% O2 and 5% CO2 incubator) or under hypoxic conditions (1% O2 and 5% CO2 incubator). After 48 hours of culture, cell migration, invasion, and the expression levels of EndMT‐related proteins were evaluated.
Table 2.
Sequences of siRNAs and PCR Primers
| Name | Sequence |
|---|---|
| si‐VCAN‐1 | CCAGTAAATTCACCTTCGA |
| si‐VCAN‐2 | GTAGGCTGTTATGGAGATA |
| si‐VCAN‐3 | GATGAAACCTCGTTATGAA |
| si‐Twist1‐1 | TGGCAAGCTGCAGCTATGT |
| si‐Twist1‐2 | GCAAGATTCAGACCCTCAA |
| si‐Twist1‐3 | TGAGCAACAGCGAGGAAGA |
| β‐Actin‐Forward | 5'‐ATCATGTTTGAGACCTTCAACA‐3' |
| β‐Actin‐Reverse | 5'‐CATCTCTTGCTCGAAGTCCA‐3' |
| VCAN‐Forward | 5'‐CCATCTCACAAGCATCCTGTCTCAC‐3' |
| VCAN‐Reverse | 5'‐CTGCCATCAGTCCAACGGAAGTC‐3' |
| Twist1‐Forward | 5'‐GCCAGGTACATCGACTTCCTCT‐3' |
| Twist1‐Reverse | 5'‐TCCATCCTCCAGACCGAGAAGG‐3' |
PCR indicates polymerase chain reaction; siRNA, small interfering RNA; and VCAN, versican.
Hematoxylin and Eosin, Masson's Trichrome, and Immunohistochemical Staining
The lung tissues were fixed in 4% paraformaldehyde (biosharp, BL539A) and then embedded in paraffin. The paraffin‐embedded tissues were cut into 4‐μm‐thick sections. Afterwards, the sections were dewaxed and rehydrated. Hematoxylin and eosin staining kit (Solarbio, G1120) and Masson's trichrome staining kit (Nanjing Jiancheng, D026) were used according to the provided instructions to assess the thickness of the pulmonary artery media and the extent of perivascular collagen deposition fibrosis. Immunohistochemical staining was performed to examine the protein levels of versican and Twist1. The antibodies used were anti‐versican (Abcam, ab270445, 1:200) and anti‐Twist1 antibodies (Proteintech, 25 465‐1‐AP, 1:200). The stained tissues were observed using a light microscope (Olympus Corporation, Tokyo, Japan).
Immunofluorescence and Immunocytochemistry
The lung tissue sections or cells cultured in confocal dishes (whb‐bio, WHB‐35‐14‐1) were washed and fixed in 4% paraformaldehyde. They were then incubated with 0.2% Triton X‐100 (Beyotime, P0096) to permeabilize the membranes. After serum blocking, the samples were incubated with primary antibodies overnight. The antibodies used were platelet endothelial cell adhesion molecule‐1 (CD31) (proteintech, 28 083‐1‐AP, 1:200) and α‐smooth muscle actin (α‐SMA) (proteintech, 67 735‐1‐Ig, 1:200), or versican (abcam, ab270445, 1:200) and von Willebrand Factor (proteintech, 66 682‐1‐Ig, 1:200). The next day, appropriate fluorescent secondary antibodies (Alexa Fluor 647 Goat Anti‐Rabbit IgG H&L, abcam, ab150079, 1:1000; Alexa Fluor 488 Goat Anti‐Mouse IgG H&L, abcam, ab150113, 1:1000) and Dapi (Beyotime, P0131) were applied. Confocal laser microscopy (Leica, TCS SP8 DLS, Germany) was used for fluorescence detection.
Western Blot Analysis and Co‐IP
Total protein from lung tissues and cultured HPAECs was extracted and quantified using the bicinchoninic acid protein assay kit (BCA, Beyotime, P0012). Equal amounts of protein (20 μg) were loaded onto 10% SDS‐PAGE gels and transferred to polyvinylidene fluoride membranes (Millipore, IPVH00010). The membranes were blocked with 5% nonfat milk at room temperature and then incubated with primary antibodies overnight at 4 °C. Primary antibodies used included anti‐versican (abcam, ab270445, 1:200), anti‐CD31 (proteintech, 28 083‐1‐AP, 1:1000), anti‐vascular endothelial‐cadherin (proteintech, 66 804‐1‐Ig, 1:1000), anti‐Vimentin (CST, #5741, 1:1000), anti‐α‐SMA (abcam, ab32575, 1:3000), anti‐Twist1 (Proteintech, 25 465‐1‐AP, 1:500), anti‐DNMT3a (proteintech, 20 954‐1‐AP, 1:5000) and anti‐DNMT3b (proteintech, 26 971‐1‐AP, 1:1000). The next day, the membranes were incubated with corresponding secondary antibodies [Goat anti‐rabbit IgG(H+L), Beyotime, A0208, 1:3000; Goat anti‐mouse IgG(H+L), Beyotime, A0216, 1:3000] at room temperature for 1 hour. Chemiluminescence signals were detected using an ECL kit (Vazyme, E411‐04/05), and an imager machine (G:Box Chem XRQ, Syngene, UK) was used for imaging. ImageJ software was utilized for quantification.
For Co‐IP experiments, HPAECs were immunoprecipitated with IP buffer containing IP antibody‐coupled protein A/G magnetic beads (MedChemExpress, HY‐K0202), and the protein–protein complexes were subsequently analyzed by Western blot. Rabbit IgG (Beyotime, A7016) was used as a negative control.
RNA Isolation and Quantitation
Total RNA was extracted from each sample using TRIzol reagent (Vazyme, R411‐01/02), and the concentration and purity of the RNA were determined using NanoDrop spectrophotometry (Thermo Fisher Scientific, USA). The primers used in the study were synthesized by Sangon Biotech (Shanghai, China) (Table 2). Reverse transcription reaction was performed using the HiScript II Q RT SuperMix for qPCR (+gDNA wiper) (Vazyme, R223‐01). Quantitative PCR was carried out using the ChamQ SYBR qPCR Master Mix (Vazyme, Q311‐02/03) following the manufacturer's instructions. The qPCR experiment was conducted using the ABI7500 fluorescence quantitative PCR instrument (Applied Biosystems, Foster City, CA). The expression levels of the target genes were calculated using the relative quantification method (2−ΔΔCt).
Cell Migration and Invasion Assay
Migration and invasion assays were performed using 8 μm Transwell chambers (Corning, Cat. No. 3422, USA). 2×104 cells/well suspended in serum‐free ECM were added to the upper chamber, while 10% fetal bovine serum ECM was added to the lower chamber. For invasion assays, a layer of matrigel (Corning, Cat. No.356234, USA) was precoated on the upper chambers at a ratio of matrigel to culture medium of 1:8 and allowed to solidify. After 48 hours of migration and invasion, the chambers were fixed with 4% paraformaldehyde and stained with crystal violet (Beyotime, C0121). Images were captured using a standard microscope (Olympus Corporation, Tokyo, Japan) to observe and analyze the migration and invasion capability between different treatment groups using ImageJ software.
Methylation‐Specific PCR
Genomic DNA was extracted from HPAECs treated at different time points using the TIANamp Genomic DNA kit (Tiangen, China) according to the manufacturer's instructions. The purity and concentration of DNA were measured using a NanoDrop spectrophotometer (Thermo Fisher Scientific, USA). Next, 500 ng of DNA was subjected to bisulfite modification using the DNA bisulfite conversion kit (Tiangen, China). Methylated and unmethylated primers for versican were designed using the MethPrimer tool (https://www.urogene.org/methprimer/) and their sequences are listed in Table 3. The modified DNA was then subjected to real‐time PCR using the methylation‐specific PCR kit (Tiangen, China) to specifically amplify the bisulfite‐converted DNA of the promoter region of versican. The amplified products were visualized by DNA agarose gel electrophoresis (Beyotime, D0163S). The relative methylation levels were analyzed using ImageJ software.
Table 3.
Sequences of Primers Used for Methylation‐Specific PCR
| Name | Sequence |
|---|---|
| Methylated primer‐Forward | 5'‐ATTTTTAGGAGTGGTTGAAGTTGC‐3' |
| Methylated primer‐Reverse | 5'‐TATCACTATACCGTCAAAACCGAA‐3' |
| Unmethylated primer‐Forward | 5′‐GATTTTTAGGAGTGGTTGAAGTTGT‐3' |
| Unmethylated primer‐Reverse | 5'‐ATATCACTATACCATCAAAACCAAA‐3' |
PCR indicates polymerase chain reaction.
Cell‐Counting‐Kit‐8 Assay
HPAECs were evenly plated in 96‐well plates and exposed to varying concentrations of SGI‐1027 (MedChemExpress, HY‐13962) for a duration of 48 hours. Subsequently, the medium in each well was replaced with 10% cell‐counting‐kit‐8 reagent (Beyotime, C0037), and the plates were incubated at 37 °C in a dark environment. The absorbance value at 450 nm was measured using a microplate reader (Thermo Fisher Scientific, Multiskan GO, USA) to determine cell viability of HPAECs in the different treatment groups. The optical density at 450 nm was recorded for each well.
Statistical Analysis
All data were expressed as mean±SD and analyzed using GraphPad Prism version 8.0. An unpaired 2‐tailed Student t test was performed for comparisons between 2 groups, while 1‐way ANOVA with the Tukey test was performed for multiple comparisons. Statistical significance was considered at P<0.05.
RESULTS
EndMT, Vascular Remodeling, and Elevated Versican Expression in Patients With HPH
As shown in Figures 1A and 1B, the lung tissues of patients with HPH exhibited significant thickening of the pulmonary vascular wall and increased deposition of collagen around the blood vessels, as revealed by hematoxylin and eosin and Masson's trichrome staining. Since EndMT plays a crucial role in the vascular remodeling in HPH, we examined the expression of EndMT markers in the lung tissue of both groups. Immunofluorescence staining was performed using CD31 and α‐SMA, labeled with red and green fluorescence, respectively. The merged yellow fluorescence indicated colocalization of endothelial and mesenchymal markers in the pulmonary vascular endothelium of patients with HPH (Figure 1C). Consistent with our previous bioinformatics analysis, immunohistochemistry results (Figures 1D and 1E) revealed a significantly elevated expression of versican in the endothelium of pulmonary blood vessels in patients with HPH, accompanied by a reduced expression of the CD31 marker. Furthermore, Western blot analysis showed significantly higher expression levels of versican and mesenchymal markers, such as vimentin and α‐SMA, and reduced expression levels of endothelial markers, including CD31 and vascular endothelial cadherin, in the HPH group compared with the normal group (Figures 1F and 1G). These findings indicate the objective presence of EndMT in HPH. These results suggest that vascular remodeling and EndMT occur in the pulmonary arteries of patients with HPH compared with the normal group, and versican may be involved in the initiation of EndMT in HPH.
Figure 1. Vascular remodeling, EndMT and increased VCAN in pulmonary vessels of HPH patients.
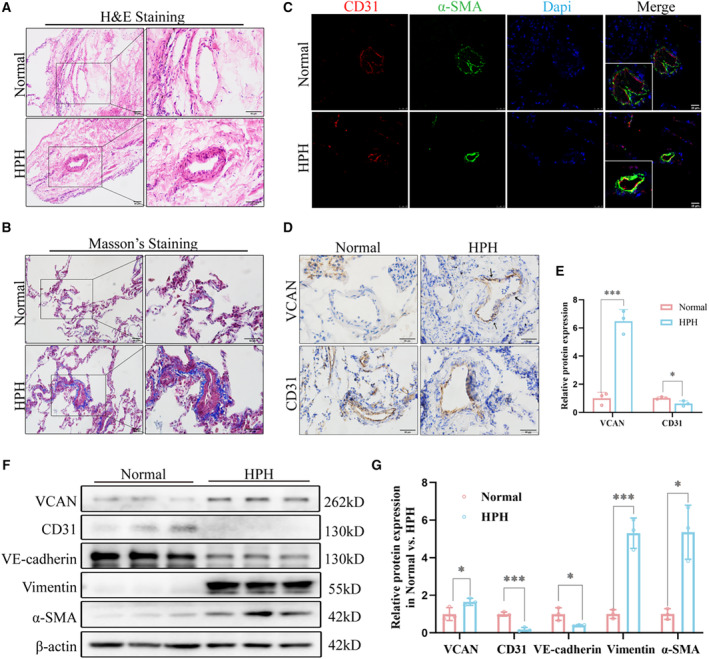
A and B, H&E staining and Masson's trichrome staining of representative lung sections from normal and HPH patients. Collagen deposition was indicated by a blue color (n=3). ×200, scale bar: 50 μm; ×400, Scale bar: 50 μm. (C) Representative immunofluorescence staining showed that CD31 (red) was colocalized with α‐SMA (green) in patients with HPH compared with the normal (n=3). ×400, scale bar: 25 μm. (D and E) Representative immunohistochemistry staining and quantitative analysis of VCAN and CD31 in normal vs HPH. Positive staining was indicated by a brown color and indicated with black arrow (n=3). ×400, scale bar: 50 μm. *P<0.05. (F and G) The protein levels and densitometric quantification of VCAN, CD31, VE‐cadherin, vimentin, and α‐SMA in lung tissues of 2 groups were determined by Western blot analysis. β‐Actin was housekeeping gene (n=3). *P<0.05, ***P<0.001. An unpaired 2‐tailed Student t test was performed for comparisons between 2 groups. CD31, Platelet endothelial cell adhesion molecule‐1; H&E, hematoxylin–eosin; HPH, hypoxia‐induced pulmonary hypertension; α‐SMA, α‐smooth muscle Actin; VCAN, versican; and VE‐cadherin, vascular endothelial cadherin.
EndMT, Vascular Remodeling, and Elevated Versican Expression in HPH Mouse Models
Subsequently, we generated HPH mouse models by subjecting them to chronic hypoxia and investigated the underlying mechanism of hypoxia‐induced EndMT in vivo. Consistent with successful model establishment, we observed a significant increase in right ventricular systolic pressure (Figures 2A and 2B) and higher RV/(LV+S) ratio (Figure 2C) in hypoxia‐exposed mice. Histological analysis using hematoxylin and eosin and Masson's trichrome staining revealed muscularization of pulmonary arteries and perivascular collagen deposition in HPH mice (Figures 2D and 2E). Immunofluorescence staining of CD31 and α‐SMA confirmed the presence of EndMT in the pulmonary endothelium of HPH mice (Figure 2F), which was further supported by the results of Western blot analysis (Figures 2I and 2J). Furthermore, our observations indicated a marked upregulation of versican expression in the pulmonary endothelium of mice with HPH when compared with the control group, as evidenced in Figures 2G and 2H. Collectively, these findings demonstrate the successful establishment of the HPH mouse model and reveal that hypoxia leads to pulmonary vascular remodeling, EndMT, and elevated versican expression in HPH mice.
Figure 2. Vascular remodeling, EndMT, and increased VCAN in pulmonary vessels of successfully constructed HPH mouse models.
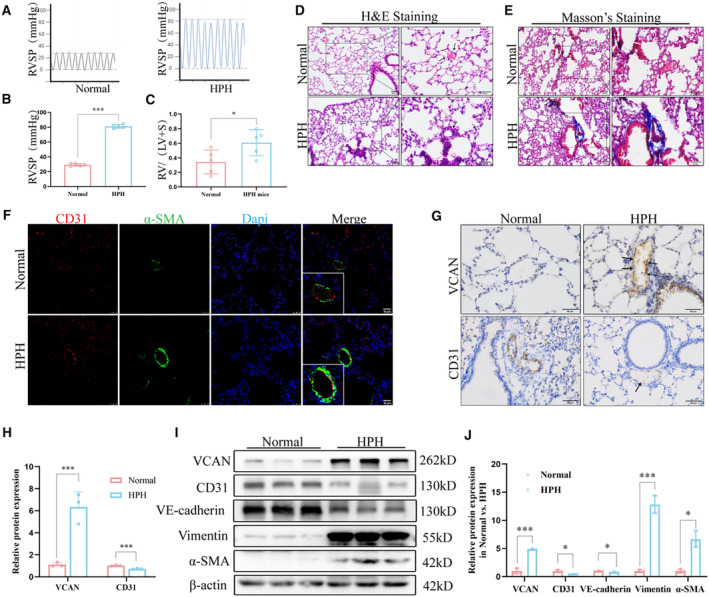
A and B, RVSP was measured using pressure sensors in the normoxia and hypoxia groups (n=5). ***P<0.001. (C) RV/(LV+S) of heart in mice of normoxia and hypoxia groups (n=5). *P<0.05. (D and E) H&E staining and Masson's trichrome staining of representative lung sections from normoxia and hypoxia groups. Arrows show normal or thickened blood vessels. Blue indicates collagen deposition (n=3). ×200, scale bar: 50 μm; ×400, Scale bar: 50 μm. (F) Co‐expression of CD31 (red) and α‐SMA(green) in the pulmonary arteries of HPH mice (n=3). ×400, scale bar: 25 μm. (G and H) Representative images of immunohistochemical staining showed that VCAN was upregulated in the pulmonary endothelium of HPH mice compared with the normal group. Positive staining was indicated by a brown color and pointed out with black arrow (n=3). ×400, scale bar: 50 μm. ***P<0.001. I and J, The protein levels and densitometric quantification of VCAN, CD31,VE‐cadherin, vimentin, and α‐SMA were determined by Western blot analysis after the hypoxia treatment (n=3). *P<0.05, ***P<0.001. An unpaired 2‐tailed Student t test was performed for comparisons between 2 groups. CD31 indicates platelet endothelial cell adhesion molecule‐1; EndMT, endothelial‐to‐mesenchymal transition; H&E, hematoxylin–eosin; HPH, hypoxia‐induced hypertension; α‐SMA, α‐smooth muscle Actin; RVSP, right ventricular systolic pressure; VCAN, versican; and VE‐cadherin, vascular endothelial cadherin.
Knockdown of Versican Attenuates EndMT and Vascular Remodeling in HPH Mice
To validate the significant role of versican in hypoxia‐induced EndMT, we conducted knockdown experiments targeting versican in vivo. Following the methods described previously, we administered intratracheal adeno‐associated virus for 3 weeks. Immunofluorescence analysis of lung sections demonstrated the presence of the virus in pulmonary vessels, confirming successful delivery to the targeted site (Figure S1A). Western blot analysis further confirmed efficient knockdown of versican in vivo (Figure S1B). The workflow diagram of different intervention groups of mice is shown in Figure 3A. Remarkably, we observed a significant reduction in elevated right ventricular systolic pressure (Figures 3B and 3C) and RV/(LV+S) ratio (Figure 3D) in HPH mice after downregulation of versican. These findings validated the protective effect of versican knockdown on HPH. Consistent with these hemodynamic changes, vascular remodeling exhibited a noticeable decline following versican knockdown, resulting in reduced pulmonary fibrosis compared with the hypoxic group (Figure 3E). Furthermore, as illustrated in Figure 3F, knocking down versican reduced the colocalization of CD31 and α‐SMA in the pulmonary vascular endothelium of HPH mice, and alleviated HPH‐induced EndMT (Figures 3G and 3H). These results suggest that the upregulation of versican in pulmonary vascular endothelium may be involved in the development of hypoxia‐induced EndMT in HPH. The inhibition of versican through knockdown may have therapeutic implications for mitigating EndMT‐associated pathologies in pulmonary hypertension.
Figure 3. Knockdown of VCAN inhibited EndMT induced by hypoxia in vivo.
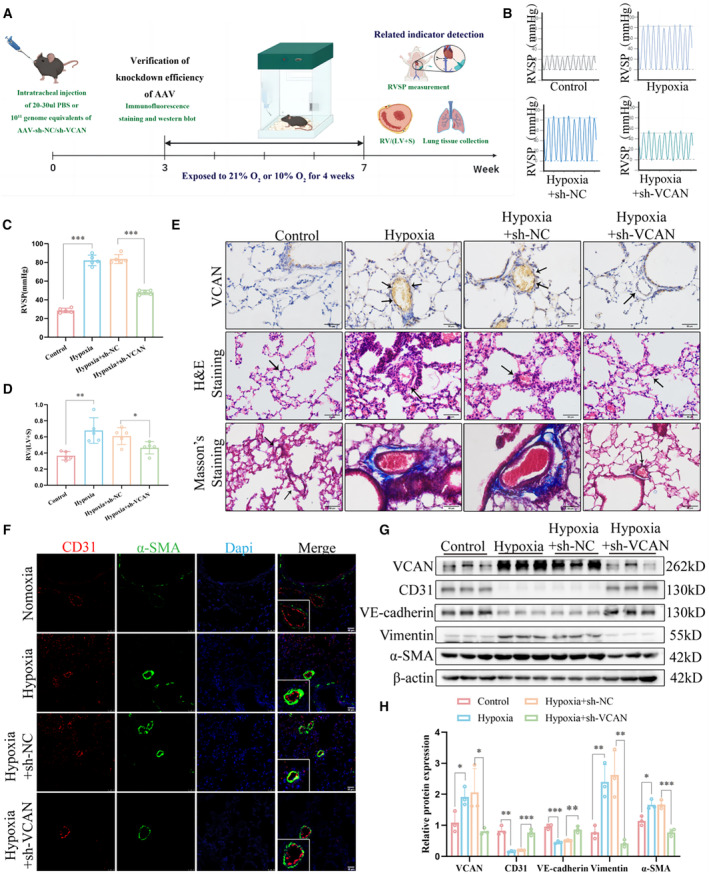
A, The workflow diagram of different intervention groups of mice. (B and C) RVSP was measured using pressure sensors in 4 groups (n=5). ***P<0.001. (D) RV/(LV+S) of heart in mice of 4 groups. *P<0.05, **P<0.05. (E) Immunohistochemical, H&E, and Masson's trichrome staining of representative lung sections from 4 groups (n=5). ×200, scale bar: 50 μm; ×400, scale bar: 50 μm. (F) Co‐expression of CD31 (red) and α‐SMA (green) in the pulmonary vascular endothelium of HPH mice, which was alleviated by VCAN knockdown (n=3). ×400, scale bar: 25 μm. (G and H) The protein levels and densitometric quantification of VCAN, CD31, VE‐cadherin, vimentin, and α‐SMA were determined by Western blot analysis (n=3). *P<0.05, **P<0.01, ***P<0.001. ANOVA with the Tukey test was performed for comparison between 4 groups. AAV indicates adeno‐associated virus; CD31, platelet endothelial cell adhesion molecule‐1; EndMT, endothelial‐to‐mesenchymal transition; H&E, hematoxylin–eosin; HPH, hypoxia‐induced hypertension; PBS, phosphate‐buffered saline; RVSP, right ventricular systolic pressure; VCAN, versican; VE‐cadherin, vascular endothelial cadherin; and α‐SMA, α‐smooth muscle Actin.
Versican Knockdown Inhibits EndMT, Migration, and Invasion of Hypoxia‐Treated HPAECs
Based on our findings, we constructed a hypoxia‐induced cell model to further validate the results at the cellular level. The workflow diagram of the in vitro experiment is shown in Figure 4A. HPAECs were transfected with si‐versican‐1, si‐versican‐2, and si‐versican‐3 to achieve versican knockdown, and si‐versican‐2 was selected as the siRNA with better efficiency based on qRT‐PCR analysis (Figure S2). We observed that after hypoxia treatment, the abilities of HPAECs to migrate through the transwell chambers or invade through matrix gel were significantly enhanced, and can be remarkably inhibited by versican knockdown (Figure 4B through 4D). Then immunostaining of the endothelial cell marker von Willebrand Factor along with versican in HPAECs confirmed that hypoxia treatment significantly increased versican levels, while si‐versican transfection effectively reversed this hypoxia‐induced upregulation of versican (Figures 4E and 4G). Additionally, we observed that hypoxia induced the co‐expression of the endothelial cell marker CD31 and the mesenchymal marker α‐SMA in HPAECs, and this effect was reversed by versican knockdown (Figures 4F and 4G). Furthermore, we confirmed the occurrence of EndMT and found that si‐versican transfection significantly attenuated the loss of endothelial markers and the upregulation of mesenchymal markers induced by hypoxia using Western blot (Figures 4H and 4I). These findings indicate that the upregulation of versican in HPAECs may contribute to the development of EndMT in HPH. Knocking down versican significantly reversed hypoxia‐induced EndMT and cellular functional changes, further confirming the crucial role of versican in the pathogenesis of HPH.
Figure 4. Knockdown of VCAN inhibited hypoxia‐induced EndMT in vitro.
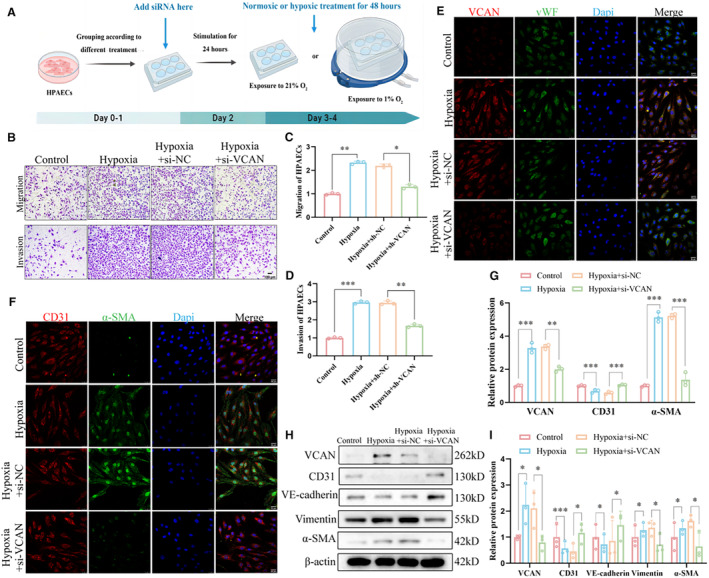
A, The workflow diagram of the in vitro experiment. (B–D) Representative images and densitometric quantification of migration and invasion assay of HPAECs (n=3). ×100, scale bar: 100 μm. ***P<0.001. (E) Co‐immunostaining of VCAN (red) with vWF(green) was performed in HPAECs (n=3). ×400, scale bar: 25 μm. (F) Co‐immunostaining of CD31 (red) with α‐SMA (green) was performed in HPAECs (n=3). ×400, scale bar: 25 μm. (G) Densitometric fluorescence intensity of VCAN, CD31, and α‐SMA in (4F and 4G) (n=3). **P<0.01, ***P<0.001. (H and I) The protein levels and densitometric quantification of VCAN, CD31,VE‐cadherin, vimentin, and α‐SMA were determined by Western blot analysis after different treatment (n=3). *P<0.05, ***P<0.001. ANOVA with the Tukey test was performed for comparison between 4 groups. CD31 indicates platelet endothelial cell adhesion molecule‐1; EndMT, endothelial‐to‐mesenchymal transition; H&E, hematoxylin–eosin; HPAECs, human pulmonary artery endothelial cells; siRNA, small interfering RNA; VCAN, versican; VE‐cadherin, vascular endothelial cadherin; vWF, von Willebrand factor; and α‐SMA, α‐smooth muscle Actin.
Versican Promoted EndMT Via Twist1
In order to investigate the regulatory mechanism of versican in EndMT, we conducted a search for potential downstream targets. Our focus was on TFs known to play crucial roles in EndMT regulation. We assessed the expression levels of 5 main TFs in HPAECs with versican knockdown and observed that only Twist1 expression was affected by changes in versican levels (Figure S3A). Further analysis revealed an increase in Twist1 expression in the pulmonary vascular endothelium of patients with HPH (Figure 5A through 5D). In vivo knockdown of versican resulted in decreased Twist1 expression in the pulmonary vessels of HPH mice (Figure 5E through 5H). Consistently, transfection of si‐versican in HPAECs led to down‐regulation of Twist1 expression, indicating that Twist1 may function as a downstream target of versican (Figures 5I and 5J). Moreover, we examined versican expression after silencing Twist1 through siRNA transfection in HPAECs and found that versican expression did not decrease (Figure S3B). Furthermore, we validated the interaction between versican and Twist1 proteins through Co‐IP (Figure 5K), and enlarged images of the immunofluorescence staining of HPAECs revealed a significant increase of versican expression in the nucleus under hypoxic conditions (Figure 5L). Collectively, these findings support the notion that Twist1 is a downstream target of versican, contributing to EndMT induction and vascular remodeling in HPH.
Figure 5. VCAN promoted EndMT via targeting transcription factor Twist1.
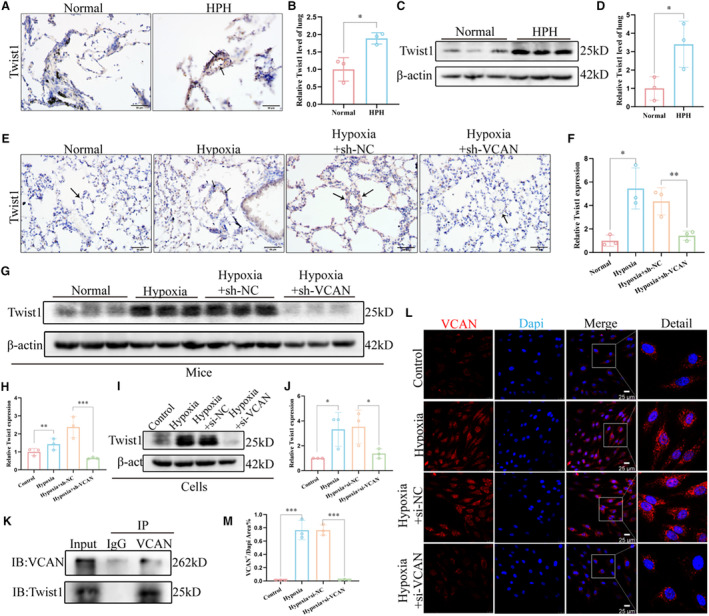
A and B, Representative images of immunohistochemical staining showed that Twist1 was upregulated in the pulmonary endothelium of patients with HPH compared with the normal group (n=3). Positive staining was indicated by a brown color and pointed out with black arrows. ×400, scale bar: 50 μm. *P<0.05. (C and D) The protein level and densitometric quantification of Twist1 in lung tissues of 2 groups were determined by Western blot analysis (n=3). *P<0.05. (E and F) Representative images of immunohistochemical staining showed that knocking down VCAN in vivo reduces upregulated Twist1 in the pulmonary vascular endothelium of HPH mice. Positive staining is indicated by a brown color and pointed out with black arrows (n=3). ×400, scale bar: 50 μm. *P<0.05, **P<0.01. (G–J) The protein levels and densitometric quantification of Twsit1 in vivo and in vitro knockdown VCAN assays were determined by Western blot analysis (n=3). *P<0.05, **P<0.01, ***P<0.001. K, The CO‐IP experiment showed that VCAN and twist1 can bind to each other in HPAECs. The target protein VCAN was immunoprecipitated with either anti‐Twist1 antibody or IgG. L, Enlarged images of the immunofluorescence staining of HPAECs nucleus. × 400, scale bar: 25 μm. An unpaired 2‐tailed Student t test was performed for comparisons between the normal group and patients with HPH, while ANOVA with the Tukey test was performed for comparison between 4 groups of in vivo and in vitro experiments. CO‐IP indicates Co‐Immunoprecipitation; EndMT, endothelial‐to‐mesenchymal transition; HPAECs, human pulmonary artery endothelial cells; HPH, hypoxia‐induced pulmonary hypertension; and VCAN, versican.
Downregulation of DNA Methyltransferase Expression and Promotion of Versican Through Promoter Hypomethylation
In order to further clarify the upstream mechanism of versican upregulation, we detected the mRNA expression of versican through qPCR and found that hypoxia can induce significant upregulation of versican mRNA (Figure 6A). We predicted the promoter region of versican and found that the CpG islands were mainly located in the 1000 bp to 1500 bp area (Figure 6B). Methylation‐specific PCR assay demonstrated that hypoxia treatment decreased the DNA methylation of versican in HPAECs and increased the DNA unmethylation of promoter region of versican (Figures 6C and 6D). Subsequently, we focused on detecting the expression of DNA methyltransferase in pulmonary endothelium under hypoxic conditions and found that compared with the control group, de novo methyltransferase DNMT3a and DNMT3b were markedly decreased in hypoxia‐treated HPAECs (Figures 6E and 6F). Further, the expression of DNMT3a and DNMT3b were also significantly decreased in HPH mice, especially in the vascular endothelium (Figure 6G through 6J). We inferred that the overexpression of versican may be mediated by the decrease in DNA methyltransferase. Then we treated HPAECs with different concentrations of SGI‐1027, an inhibitor of DNA methyltransferases, and found that SGI‐1027 reduced expression of DNMT3a and DNMT3b while it significantly increased the expression of versican (Figures 6K and 6L). Meanwhile, the methylation‐specific PCR results also showed a decrease in versican methylation after 5 μmol/L SGI‐1027 treatment (Figures 6M and 6N), which indicated that the expression of versican was regulated by the promoter hypomethylation. These findings indicate that the upregulation of versican under hypoxic conditions is predominantly governed by DNA methylation.
Figure 6. Downregulation of DNA methyltransferase expression and promotion of VCAN through promoter hypomethylation.
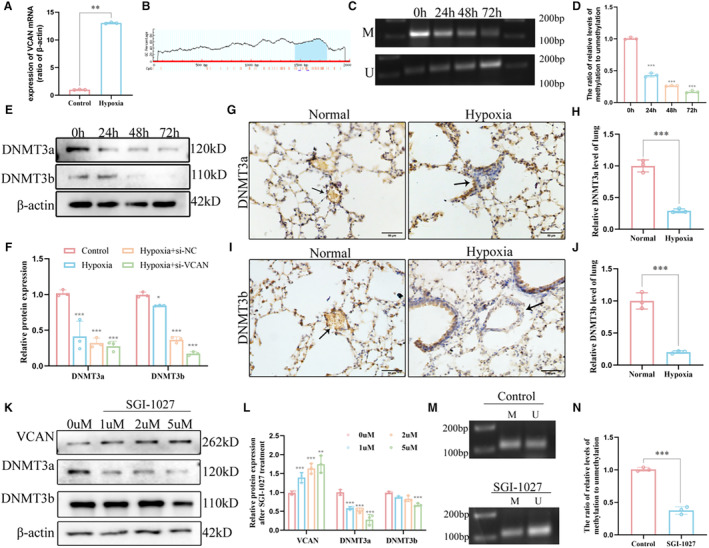
A, RT‐qPCR analysis of the mRNA level of VCAN from HPAECs (n=3). **P<0.01. (B) CpG sites of the promoter of VCAN gene. (C and D) DNA methylation levels of the VCAN promotor region in HPAECs and corresponding densitometric quantification as detected by MSP assay (n=3). ***P<0.001. (E and F) The protein levels and densitometric quantification of DNMT3a and DNMT3b were determined by Western blot analysis after the hypoxia treatment (n=3). *P<0.05, ***P<0.001. (G–J) Representative images of immunohistochemical staining confirmed that DNMT3a and DNMT3b were significantly decreased in HPH mice compared with the normal control group (n=3). Positive staining was indicated by a brown color and pointed out with black arrow. ×400, scale bar: 50 μm. ***P<0.001. (K and L) The protein levels and densitometric quantification of DNMTs and VCAN levels in SGI‐1027‐treated HPAECs (n=3). **P<0.01, ***P<0.001. (M and N) DNA methylation levels of the VCAN promotor region in 5 μM SGI‐1027‐treated HPAECs and corresponding densitometric quantification as detected by MSP assay (n=3). ***P<0.001. An unpaired 2‐tailed Student t test was performed for comparisons between 2 groups. CpG indicates Cytosine‐phosphate‐Guanine; DNMT, DNA methyltransferase; HPAECs, human pulmonary artery endothelial cells; HPH, hypoxia‐induced pulmonary hypertension; MSP, methylation‐specific polymerase chain reaction; RT‐qPCR, reverse transcription‐quantitative polymerase chain reaction; and VCAN, versican.
DISCUSSION
In this study, we conducted comprehensive validation by utilizing patient and mouse lung tissues, as well as cell models, to establish that versican, a novel indicator of EndMT, is involved in the development of HPH in a Twist1‐dependent manner. Additionally, our findings indicate that the upregulation of versican may be attributed to hypomethylation of CpG islands in the promoter region caused by a decline in DNMTs. Figure 7 depicts the research workflow and experimental procedures. Overall, our innovative discovery implies that versican has the potential to become therapeutic targets for addressing EndMT in HPH.
Figure 7. Schematic diagram summarizing how hypoxia‐induced promotor hypomethylation upregulates VCAN to promote EndMT in endothelial cells.
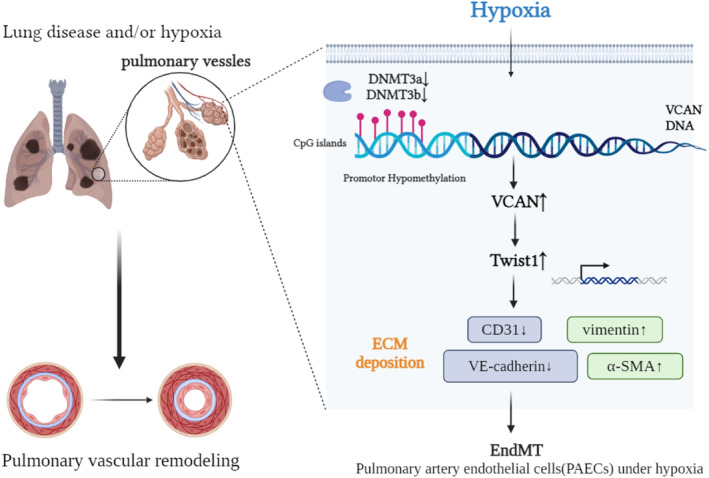
CpG indicates Cytosine‐phosphate‐Guanine; DNMT, DNA methyltransferase; ECM, extracellular matrix; EndMT, endothelial‐to‐mesenchymal transition; α‐SMA, α‐smooth muscle actin; VCAN, versican; and VE‐cadherin, vascular endothelial cadherin.
HPH is a condition characterized by abnormal growth of blood vessel cells, excessive deposition of ECM, and accumulation of inflammatory cells, resulting in irreversible remodeling of the pulmonary vasculature. 25 The process of EndMT is believed to be the trigger for HPH, leading to enhanced migration and invasion of endothelial cells. 26 During EndMT, there is a decrease in endothelial markers CD31 and vascular endothelial cadherin, and an increase in mesenchymal markers vimentin and α‐SMA as well as related TFs. 27 Our previous study has suggested that elevated levels of versican in endothelium of blood vessels may contribute to the progression of HPH through EndMT. 17 Versican is a major proteoglycan found in the ECM of human blood vessels and has been shown to affect the behavior of vascular cells, including endothelial cells. 28 , 29 However, existing studies were limited to animal or cell models and did not investigate the specific mechanism by which versican promotes EndMT in HPH. To further explore the role of versican in HPH, we collected lung tissues from patients with HPH and successfully established HPH mouse models and cell models. In our study, we observed colocalization of endothelial and mesenchymal markers in the endothelium of the pulmonary arteries, as well as significant proliferation and thickening of the middle layer and severe fibrosis surrounding the blood vessels (perivascular fibrosis). These findings suggest that EndMT and vascular remodeling occur in HPH and are accompanied by increased expression of versican at various levels, including the population, mice, and cellular levels.
As mentioned previously, we have successfully constructed HPH mouse and cell models, which have been recognized for their accuracy. Building upon this foundation, the increase in versican observed in the pulmonary endothelium in HPH is of significant importance. To validate the potential therapeutic effect of reducing versican expression on HPH, we conducted experiments both in vivo and in vitro. In the in vivo model, we knocked down versican and observed a notable attenuation of HPH progression in mouse models. This was demonstrated by a significant decrease in right ventricular systolic pressure, reduced arterial thickness, a decrease in vascular muscularization, and a reversal of EndMT transition. Furthermore, our in vitro experiments showed that knocking down versican prevented hypoxia‐induced EndMT in HPAECs. In conclusion, our study highlights the crucial role of versican in promoting EndMT and vascular remodeling in HPH. We are proud to be the first to confirm that knocking down versican exhibits promising therapeutic potential in mitigating the development and progression of HPH. These findings provide valuable insights for improving vascular remodeling in HPH.
Adequate research has confirmed that multiple TFs participate in the development of HPH by either promoting or inhibiting the transcription of marker genes involved in EndMT transition. 30 In our study, we investigated the role of versican and its interaction with Twist1, a key transcription factor associated with EndMT and known to be regulated by hypoxia‐related signaling pathways. 27 , 31 Through knockdown experiments targeting versican, we observed an influence on the expression of major TFs in endothelial cells, particularly Twist1. Previous studies have shown that Twist1 can repress the transcription of endothelial marker genes and promote EndMT. 32 , 33 Consistent with these findings, we observed increased Twist1 expression in patients with HPH, mice, and cell models. However, the mechanism underlying the interaction between versican and Twist1 in EndMT remains unknown. In our study, we demonstrated that upregulation or knockdown of versican affected Twist1 expression. Conversely, knocking down Twist1 did not significantly change versican expression. Furthermore, we identified the binding of Twist1 to versican protein, confirming the interaction between versican and downstream Twist1. Based on these findings, we propose that the versican/Twist1 pathway may partially regulate hypoxia‐induced EndMT, providing insights into the mechanism of versican in modulating EndMT and vascular remodeling in HPH.
Epigenetic modifications, such as DNA methylation, play a crucial role in regulating the expression of cell type–specific genes, which is essential for cell differentiation and proliferation. 34 DNA methylation is a reversible modification of DNA that can impact gene expression without altering the underlying genome sequence. 35 It has been suggested to be involved in the development of lung diseases such as chronic obstructive pulmonary disease, pulmonary arterial hypertension, and asthma. 36 , 37 , 38 However, most studies have primarily focused on analyzing overall levels of DNA methylation in these lung diseases, with limited research investigating specific changes in the methylation levels of key genes during disease progression. 39 In a previous study by Hautefort et al, a bioinformatics analysis examining DNA methylation patterns was conducted on HPAECs isolated from the pulmonary vessels of patients with pulmonary arterial hypertension. This analysis identified 2 specific methylation signatures associated with pulmonary arterial hypertension, including hypomethylated and hypermethylated promoters. 40 To investigate whether the expression of versican (the gene of interest) was regulated by DNA methylation, we analyzed the versican sequence and predicted the presence of enriched CpG islands. Under hypoxic conditions mimicking the microenvironment of HPH, we observed a decrease in DNA methylation (hypomethylation) specifically at the versican locus.
Furthermore, we aimed to investigate the regulatory mechanism of versican methylation levels. Mammals possess 3 types of enzymes known as DNMTs‐DNMT1, DNMT3a, and DNMT3b, which are responsible for methyl transfer. 41 DNMT1's role is to maintain DNA methylation during DNA replication, preventing the passive loss of DNA methylation. On the other hand, DNMT3a and DNMT3b are involved in de novo DNA methylation, which regulates gene expression. 42 Previous studies have shown that the hypoxic microenvironment can influence the methylation levels of various genes by affecting the expression of DNMTs. 43 In our study, we examined the changes in the expression of upstream DNMTs and observed a decrease in DNMT3a and DNMT3b in pulmonary endothelial cells of HPH mice and hypoxia‐treated cell models. To further understand the role of DNMTs in versican regulation, we added the DNMTs inhibitor SGI‐1027 to HPAECs artificially. We observed the degradation of DNMT3a and DNMT3b, along with a decrease in versican methylation and an increase in versican mRNA expression. These findings suggest that the upregulation of versican under hypoxic conditions is largely regulated by DNA methylation. In conclusion, this discovery provides new insights into the epigenetic mechanisms involved in the upregulation of versican and identifies potential therapeutic targets for addressing pathological changes mediated by versican upregulation.
Our research has discovered that versican plays a crucial role in the development of HPH as a regulatory molecule for EndMT. It has been verified that versican acts as an upstream regulator of the transcription factor Twist1, thereby offering a novel therapeutic target for HPH to counteract abnormal pathological changes in the endothelium. However, there are still some limitations in our research. The molecular‐level interaction between versican and Twist1 remains unknown, and further investigation is required. Additionally, the specific mechanism through which DNMTs regulate versican needs to be explored in more depth, and the development of targeted treatment methods to upregulate versican methylation in vivo still requires further exploration. In conclusion, the research on HPH is an ongoing journey, and our experimental results only offer a limited perspective into the exploration of HPH.
Sources of Funding
This work was supported by the National Nature Science Foundation of China (82173472), General project of Wuxi Medical Center affiliated with Nanjing Medical University (WMCG202349), Wuxi Yanzhen Talent Project (2024‐YZ‐XKDTR‐WY‐2024), and Wuxi Science and Technology Development Fund Project (Y20222007).
Disclosures
The authors declare that they have no competing interests in this work.
Supporting information
Figures S1–S3
Acknowledgments
All authors made a significant contribution to the work reported, whether in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all of these areas. All authors took part in drafting, revising, or critically reviewing the article, gave final approval of the version to be published, have agreed on the journal to which the article has been submitted, and agree to be accountable for all aspects of the work.
This manuscript was sent to Julie K. Freed, MD, PhD, Associate Editor, for review by expert referees, editorial decision, and final disposition.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.124.036969
For Sources of Funding and Disclosures, see page 15.
References
- 1. Bousseau S, Sobrano Fais R, Gu S, Frump A, Lahm T. Pathophysiology and new advances in pulmonary hypertension. BMJ Med. 2023;2:e000137. doi: 10.1136/bmjmed-2022-000137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, Carlsen J, Coats AJS, Escribano‐Subias P, Ferrari P, et al. 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;43:3618–3731. doi: 10.1093/eurheartj/ehac237 [DOI] [PubMed] [Google Scholar]
- 3. Mandras SA, Mehta HS, Vaidya A. Pulmonary hypertension: a brief guide for clinicians. Mayo Clin Proc. 2020;95:1978–1988. doi: 10.1016/j.mayocp.2020.04.039 [DOI] [PubMed] [Google Scholar]
- 4. Rose L, Prins KW, Archer SL, Pritzker M, Weir EK, Misialek JR, Thenappan T. Survival in pulmonary hypertension due to chronic lung disease: influence of low diffusion capacity of the lungs for carbon monoxide. J Heart Lung Transplant. 2019;38:145–155. doi: 10.1016/j.healun.2018.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Humbert M, Guignabert C, Bonnet S, Dorfmüller P, Klinger JR, Nicolls MR, Olschewski AJ, Pullamsetti SS, Schermuly RT, Stenmark KR, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. 2019;53:1801887. doi: 10.1183/13993003.01887-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cassady SJ, Ramani GV. Right heart failure in pulmonary hypertension. Cardiol Clin. 2020;38:243–255. doi: 10.1016/j.ccl.2020.02.001 [DOI] [PubMed] [Google Scholar]
- 7. Swisher JW, Weaver E. The evolving management and treatment options for patients with pulmonary hypertension: current evidence and challenges. Vasc Health Risk Manag. 2023;19:103–126. doi: 10.2147/VHRM.S321025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yun E, Kook Y, Yoo KH, Kim KI, Lee MS, Kim J, Lee A. Endothelial to mesenchymal transition in pulmonary vascular diseases. Biomedicine. 2020;8:639. doi: 10.3390/biomedicines8120639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Karamanos NK, Theocharis AD, Piperigkou Z, Manou D, Passi A, Skandalis SS, Vynios DH, Orian‐Rousseau V, Ricard‐Blum S, Schmelzer CEH, et al. A guide to the composition and functions of the extracellular matrix. FEBS J. 2021;288:6850–6912. doi: 10.1111/febs.15776 [DOI] [PubMed] [Google Scholar]
- 10. Zhou K, Tian KJ, Yan BJ, Gui DD, Luo W, Ren Z, Wei DH, Liu LS, Jiang ZS. A promising field: regulating imbalance of EndMT in cardiovascular diseases. Cell Cycle. 2021;20:1477–1486. doi: 10.1080/15384101.2021.1951939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Theocharis AD, Manou D, Karamanos NK. The extracellular matrix as a multitasking player in disease. FEBS J. 2019;286:2830–2869. doi: 10.1111/febs.14818 [DOI] [PubMed] [Google Scholar]
- 12. Peng Q, Shan D, Cui K, Li K, Zhu B, Wu H, Wang B, Wong S, Norton V, Dong Y, et al. The role of endothelial‐to‐mesenchymal transition in cardiovascular disease. Cells. 2022;11:1834. doi: 10.3390/cells11111834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gaikwad AV, Lu W, Dey S, Bhattarai P, Haug G, Larby J, Chia C, Jaffar J, Westall G, Singhera GK, et al. Endothelial‐to‐mesenchymal transition: a precursor to pulmonary arterial remodelling in patients with idiopathic pulmonary fibrosis. ERJ Open Res. 2023;9:487. doi: 10.1183/23120541.00487-2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andersson‐Sjöland A, Hallgren O, Rolandsson S, Weitoft M, Tykesson E, Larsson‐Callerfelt AK, Rydell‐Törmänen K, Bjermer L, Malmström A, Karlsson JC, et al. Versican in inflammation and tissue remodeling: the impact on lung disorders. Glycobiology. 2015;25:243–251. doi: 10.1093/glycob/cwu120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Islam S, Watanabe H. Versican: a dynamic regulator of the extracellular matrix. J Histochem Cytochem. 2020;68:763–775. doi: 10.1369/0022155420953922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang L, Wang L, Yang Z, Jin H, Zou Q, Zhan Q, Tang Y, Tao Y, Lei L, Jing Y, et al. Up‐regulation of EMT‐related gene VCAN by NPM1 mutant‐driven TGF‐β/cPML signalling promotes leukemia cell invasion. J Cancer. 2019;10:6570–6583. doi: 10.7150/jca.30223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yu J, Huang S, Shen W, Zhang Z, Ye S, Chen Y, Yang Y, Bian T, Wu Y. Expression profiles of circRNAs and identification of hsa_circ_0007608 and hsa_circ_0064656 as potential biomarkers for COPD‐PH patients. Int J Chron Obstruct Pulmon Dis. 2023;18:2457–2471. doi: 10.2147/COPD.S424712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Y, Zou X, Qian W, Weng X, Zhang L, Zhang L, Wang S, Cao X, Ma L, Wei G, et al. Enhanced PAPSS2/VCAN sulfation axis is essential for snail‐mediated breast cancer cell migration and metastasis. Cell Death Differ. 2019;26:565–579. doi: 10.1038/s41418-018-0147-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gu S, Goel K, Forbes LM, Kheyfets VO, Yu YA, Tuder RM, Stenmark KR. Tensions in taxonomies: current understanding and future directions in the pathobiologic basis and treatment of group 1 and group 3 pulmonary hypertension. Compr Physiol. 2023;13:4295–4319. doi: 10.1002/cphy.c220010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ho L, Hossen N, Nguyen T, Vo A, Ahsan F. Epigenetic mechanisms as emerging therapeutic targets and microfluidic chips application in pulmonary arterial hypertension. Biomedicine. 2022;10:170. doi: 10.3390/biomedicines10010170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheng X, Wang Y, Du L. Epigenetic modulation in the initiation and progression of pulmonary hypertension. Hypertension. 2019;74(4):733–739. doi: 10.1161/HYPERTENSIONAHA.119.13458 [DOI] [PubMed] [Google Scholar]
- 22. Nanduri J, Semenza GL, Prabhakar NR. Epigenetic changes by DNA methylation in chronic and intermittent hypoxia. Am J Physiol Lung Cell Mol Physiol. 2017;313:L1096–L1100. doi: 10.1152/ajplung.00325.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu XQ, Chen M, Dasgupta C, Xiao D, Huang X, Yang S, Zhang L. Chronic hypoxia upregulates DNA methyltransferase and represses large conductance Ca2+−activated K+ channel function in ovine uterine arteries. Biol Reprod. 2017;96:424–434. doi: 10.1095/biolreprod.116.145946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luo HL, Chang YL, Liu HY, Wu YT, Sung MT, Su YL, Huang CC, Wang PC, Peng JM. VCAN Hypomethylation and expression as predictive biomarkers of drug sensitivity in upper urinary tract urothelial carcinoma. Int J Mol Sci. 2023;24:7486. doi: 10.3390/ijms24087486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ye Y, Xu Q, Wuren T. Inflammation and immunity in the pathogenesis of hypoxic pulmonary hypertension. Front Immunol. 2023;14:1162556. doi: 10.3389/fimmu.2023.1162556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gorelova A, Berman M, Al Ghouleh I. Endothelial‐to‐mesenchymal transition in pulmonary arterial hypertension. Antioxid Redox Signal. 2021;34:891–914. doi: 10.1089/ars.2020.8169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mammoto T, Muyleart M, Konduri GG, Mammoto A. Twist1 in hypoxia‐induced pulmonary hypertension through transforming growth factor‐β‐Smad signaling. Am J Respir Cell Mol Biol. 2018;58:194–207. doi: 10.1165/rcmb.2016-0323OC [DOI] [PubMed] [Google Scholar]
- 28. Wight TN. A role for proteoglycans in vascular disease. Matrix Biol. 2018;71–72:396–420. doi: 10.1016/j.matbio.2018.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nandadasa S, O'Donnell A, Murao A, Yamaguchi Y, Midura RJ, Olson L, Apte SS. The versican‐hyaluronan complex provides an essential extracellular matrix niche for Flk1+ hematoendothelial progenitors. Matrix Biol. 2021;97:40–57. doi: 10.1016/j.matbio.2021.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Piera‐Velazquez S, Jimenez SA. Endothelial to mesenchymal transition: role in physiology and in the pathogenesis of human diseases. Physiol Rev. 2019;99:1281–1324. doi: 10.1152/physrev.00021.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang B, Niu W, Dong HY, Liu ML, Luo Y, Li ZC. Hypoxia induces endothelial‐mesenchymal transition in pulmonary vascular remodeling. Int J Mol Med. 2018;42:270–278. doi: 10.3892/ijmm.2018.3584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li Z, Chen B, Dong W, Kong M, Fan Z, Yu L, Wu D, Lu J, Xu Y. MKL1 promotes endothelial‐to‐mesenchymal transition and liver fibrosis by activating TWIST1 transcription. Cell Death Dis. 2019;10:899. doi: 10.1038/s41419-019-2101-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang EL, Zhang JJ, Luo FM, Fu MY, Li D, Peng J, Liu B. Cerebellin‐2 promotes endothelial‐mesenchymal transition in hypoxic pulmonary hypertension rats by activating NF‐κB/HIF‐1α/Twist1 pathway. Life Sci. 2023;328:121879. doi: 10.1016/j.lfs.2023.121879 [DOI] [PubMed] [Google Scholar]
- 34. Li S, Tollefsbol TO. DNA methylation methods: global DNA methylation and methylomic analyses. Methods. 2021;187:28–43. doi: 10.1016/j.ymeth.2020.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Villicaña S, Bell JT. Genetic impacts on DNA methylation: research findings and future perspectives. Genome Biol. 2021;22:127. doi: 10.1186/s13059-021-02347-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sundar IK, Yin Q, Baier BS, Yan L, Mazur W, Li D, Susiarjo M, Rahman I. DNA methylation profiling in peripheral lung tissues of smokers and patients with COPD. Clin Epigenetics. 2017;9:38. doi: 10.1186/s13148-017-0335-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clifford RL, Patel J, MacIsaac JL, McEwen LM, Johnson SR, Shaw D, Knox AJ, Hackett TL, Kobor MS. Airway epithelial cell isolation techniques affect DNA methylation profiles with consequences for analysis of asthma related perturbations to DNA methylation. Sci Rep. 2019;9:14409. doi: 10.1038/s41598-019-50873-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Duan J, Zhong B, Fan Z, Zhang H, Xu M, Zhang X, Sanders YY. DNA methylation in pulmonary fibrosis and lung cancer. Expert Rev Respir Med. 2022;16:519–528. doi: 10.1080/17476348.2022.2085091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yan Y, He YY, Jiang X, Wang Y, Chen JW, Zhao JH, Ye J, Lian TY, Zhang X, Zhang RJ, et al. DNA methyltransferase 3B deficiency unveils a new pathological mechanism of pulmonary hypertension. Sci Adv. 2020;6:eaba2470. doi: 10.1126/sciadv.aba2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hautefort A, Chesné J, Preussner J, Pullamsetti SS, Tost J, Looso M, Antigny F, Girerd B, Riou M, Eddahibi S, et al. Pulmonary endothelial cell DNA methylation signature in pulmonary arterial hypertension. Oncotarget. 2017;8:52995–53016. doi: 10.18632/oncotarget.18031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Castillo‐Aguilera O, Depreux P, Halby L, Arimondo PB, Goossens L. DNA methylation targeting: the DNMT/HMT crosstalk challenge. Biomol Ther. 2017;7:3. doi: 10.3390/biom7010003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dan J, Chen T. Genetic studies on mammalian DNA Methyltransferases. Adv Exp Med Biol. 2022;1389:111–136. doi: 10.1007/978-3-031-11454-0_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ali MM, Phillips SA, Mahmoud AM. HIF1α/TET1 pathway mediates hypoxia‐induced adipocytokine promoter Hypomethylation in human adipocytes. Cells. 2020;9:134. doi: 10.3390/cells9010134 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S3
Data Availability Statement
All data and supporting materials have been provided with the published article.