

Abstract
Background
Accumulating evidence indicates that coronary microvascular dysfunction (CMD) caused by hypercholesterolemia can lead to myocardial ischemia, with or without obstructive atherosclerotic coronary artery disease. However, the molecular pathways associated with compromised coronary microvascular function before the development of myocardial ischemic injury remain poorly defined. In this study, we investigated the effects of hypercholesterolemia on the function and integrity of the coronary microcirculation in mice and the underlying mechanisms.
Methods and Results
Mice were fed a hypercholesterolemic Paigen's diet for 8 weeks. Echocardiography data showed that Paigen's diet caused CMD, characterized by significant reductions in coronary blood flow and coronary flow reserve, but did not affect cardiac remodeling or dysfunction. Immunofluorescence studies revealed that Paigen's diet–induced CMD was associated with activation of coronary arterioles inflammation and increased myocardial inflammatory cell infiltration. These pathological changes occurred in parallel with the upregulation of lysosomal signaling pathways in endothelial cells (ECs). Treating hypercholesterolemic mice with the cholesterol‐lowering drug ezetimibe significantly ameliorated Paigen's diet–induced adverse effects, including hypercholesterolemia, steatohepatitis, reduced coronary flow reserve, coronary endothelial cell inflammation, and myocardial inflammatory cell infiltration. In cultured mouse cardiac ECs, 7‐ketocholesterol increased mitochondrial reactive oxygen species and inflammatory responses. Meanwhile, 7‐ketocholesterol induced the activation of transcriptional factor EB and lysosomal signaling in mouse cardiac ECs, whereas the lysosome inhibitor bafilomycin A1 blocked 7‐ketocholesterol–induced transcriptional factor EB activation and exacerbated 7‐ketocholesterol–induced inflammation and cell death. Interestingly, ezetimibe synergistically enhanced 7‐ketocholesterol–induced transcriptional factor EB activation and attenuated 7‐ketocholesterol–induced mitochondrial reactive oxygen species and inflammatory responses in mouse cardiac ECs.
Conclusions
These results suggest that CMD can develop and precede detectable cardiac functional or structural changes in the setting of hypercholesterolemia and that upregulation of transcriptional factor EB–mediated lysosomal signaling in endothelial cells plays a protective role against CMD.
Keywords: coronary flow reserve, coronary microvascular dysfunction, endothelial dysfunction, hypercholesterolemia, lysosome
Subject Categories: Coronary Artery Disease, Metabolic Syndrome, Inflammatory Heart Disease, Echocardiography, Basic Science Research
Nonstandard Abbreviations and Acronyms
- CFR
coronary flow reserve
- CMD
coronary microvascular dysfunction
- EC
endothelial cell
- LAMP
lysosomal‐associated membrane protein
- LD
lipid droplet
- MCECs
mouse cardiac endothelial cells
- ND
normal diet
- NLRP3
NLR family pyrin domain containing 3
- PD
Paigen's diet
- PI
propidium iodide
- ROS
reactive oxygen species
- SMC
smooth muscle cell
- TFEB
transcriptional factor EB
- VCAM‐1
vascular cell adhesion molecule 1
Research Perspective.
What Is New?
We are the first to characterize coronary microvascular dysfunction (CMD) in the early stage of hypercholesterolemic mice.
Hypercholesterolemia‐induced CMD is associated with the activation of transcriptional factor EB–mediated lysosomal signaling, presenting a promising therapeutic approach to mitigate CMD by targeting the activation of this signaling pathway.
Ezetimibe, a Food and Drug Administration–approved cholesterol‐lowering drug, improves endothelial dysfunction and CMD by activating transcriptional factor EB–mediated lysosomal signaling.
What Question Should Be Addressed Next?
Future studies should evaluate whether statins alone or in combination with ezetimibe offer comparable or superior effects to ezetimibe alone on hypercholesterolemia‐induced CMD.
Future studies should investigate the role of lysosomal genes, such as transcriptional factor EB and lysosomal‐associated membrane proteins, by employing tissue‐specific knockout or knock‐in models in microvascular cells, including endothelial cells and pericytes, within the context of hypercholesterolemia‐induced CMD.
Coronary artery disease (CAD) is the most prevalent form of heart disease and a leading cause of death in the United States and other developed countries. 1 The coronary arterial system consists of distinct compartments, including large epicardial coronary arteries, arterioles, and capillaries. 2 The network of coronary arterioles and capillaries, known as the coronary microcirculation, plays a vital role in ensuring normal myocardial perfusion. 2 Functional or structural abnormalities in the coronary microcirculation can lead to impaired myocardial perfusion, resulting in a condition called coronary microvascular dysfunction (CMD), which can lead to myocardial ischemia. 2 , 3 CMD has a higher prevalence in individuals with suspected signs and symptoms of ischemia but no obstructive CAD, particularly in women. 4 CMD is associated with adverse outcomes and has important prognostic significance for various cardiovascular diseases, especially myocardial ischemia. 5 , 6 Coronary flow reserve (CFR) is often used as a clinical indicator to assess CMD in ischemia but no obstructive CAD. 7 , 8 To date, there is no specific treatment to prevent CMD, and therefore it is imperative to elucidate the molecular pathways associated with CMD.
The mechanisms leading to CMD are diverse and complex and are understudied compared with large coronary arteries. Coronary microvascular endothelial dysfunction is considered as an important mechanism in the initiation and progression of CMD. 9 , 10 Endothelial cells (ECs) play a crucial role in regulating vascular tone by producing and releasing vasoactive relaxing or constricting factors. Endothelium‐derived relaxing factors include NO, vasodilatory prostaglandins, and endothelium‐derived hyperpolarization factors. 11 , 12 Hydrogen peroxide is considered an important hyperpolarization factor in various vascular beds, including human coronary arteries. 13 ECs also produce vasoconstricting factors such as endothelin. In this regard, arterial endothelial dysfunction is often characterized by either an impaired vasodilatory response or an overactive vasoconstrictor response. Mechanistically, endothelial dysfunction is accompanied by increased oxidative stress, elevated production of reactive oxygen species (ROS) and vasoconstrictors, and a gradual decline in NO bioavailability. 14 In addition to impaired vasomotor responses, endothelial dysfunction involves a shift from a quiescent state to an activated, proinflammatory, and prothrombotic state, resulting in increased expression of inflammatory mediators and enhanced interaction with platelets and leukocytes. 15 Moreover, persistent and excessive endothelial dysfunction may lead to structural changes in coronary microcirculation including arterioles wall remodeling and microvascular rarefaction. 16 These CMD‐related pathological changes lead to abnormal myocardial hemodynamics and promote the development of myocardial ischemic injury.
Metabolic disorders, such as hypercholesterolemia, are recognized as important predisposing risk factors for CAD, with or without atherosclerotic obstruction and myocardial infarction. 17 , 18 , 19 Indeed, hypercholesterolemia is common in patients with ischemia but no obstructive CAD and is even higher in patients with obstructive CAD. 20 However, there are currently no studies using mouse models to characterize CMD and its associated pathological changes caused by hypercholesterolemia. The lack of mechanistic studies in this field has hampered the development of specific therapeutic interventions for CMD. The aim of this study was to determine the early effects of hypercholesterolemia on the coronary microcirculation and myocardial tissue in mice before the onset of cardiac remodeling and dysfunction. We used a murine hypercholesterolemia model established by high‐fat and high‐cholesterol Paigen's diet (PD) feeding, and CMD was characterized by reductions in coronary blood flow and CFR. We evaluated a series of cardiovascular‐related pathological changes in the coronary arterioles, capillaries, and myocardium, including arteriole wall thickening and lipid accumulation, microvascular rarefaction, thrombosis, and inflammation. We also investigated whether lysosomal signaling was changed in coronary ECs. In addition, we investigated the protective effects of the cholesterol‐lowering drug ezetimibe on CMD in hypercholesterolemic mice. We also confirmed the direct effects of ezetimibe on the activation of transcriptional factor EB (TFEB) lysosomal signaling and attenuation of EC inflammation and injury in a 7‐ketocholesterol–induced cellular model. Our findings provide new insights that activation of lysosome signaling may be a potential therapeutic target for treating CMD in metabolic disorders.
METHODS
Data Availability
All data and supporting materials have been provided with the published article and its online supplementary files.
Mice
All experimental protocols were reviewed and approved by the University of Houston Institutional Animal Care and Use Committee. Female wild‐type mice (B6.129 background) aged 6 to 12 months were used for all experiments. Mice were housed in a temperature‐controlled room with 12‐hour dark–light cycle and provided standard rodent chow and water ad libitum. Mice were randomly assigned into a normal chow diet (ND) group or a Paigen's diet (PD) (Research diet, D12336) group for 8 weeks. For experiments with ezetimibe administration, mice were divided into 3 groups: ND vehicle control, PD vehicle control, and PD with ezetimibe treatment. Ezetimibe (3 mg/kg) was administrated intraperitoneally every 2 days. Vehicle control groups received DMSO+PBS injection. Ezetimibe was dissolved in DMSO and diluted with PBS to keep DMSO concentration below 10% of the total injection volume. After 8 weeks, mice were euthanized, and blood, heart, liver, and aorta samples were collected and stored at −80 °C for further analysis.
Echocardiography
Transthoracic echocardiography was performed using a Vevo 3100 micro‐ultrasound imaging system with an MX550D‐0073 probe (VisualSonics Inc., Toronto, Ontario, Canada). Mice were anesthetized with 1.5% to 2% isoflurane mixed with 100% medical oxygen and maintained at 1% to 1.5% isoflurane to control the heart rate between 400 and 500 beats/min. Heart rate and ECG were monitored by limb electrodes. Body temperature was monitored and maintained between 36 and 37 °C. M‐mode images were obtained from the parasternal short‐axis view at the level of the papillary muscles after 8 weeks of diet feeding or ezetimibe administration and analyzed with Vevo LAB 2.1.0. To track the progression of CMD in the PD group over time, left anterior descending coronary artery flow velocity was measured at various time points (0, 2, 4, 6, and 8 weeks) using pulsed‐wave Doppler under baseline and hyperemic conditions induced by inhalation of 1.0% and 2.5% isoflurane, respectively. CFR was calculated as the ratio of peak blood flow velocity during hyperemia to baseline.
Antibodies and Reagents
Primary antibodies: CD41 (BD, 553847), fluorescein isothiocyanate–α‐smooth muscle actin (Sigma, F3777), CD45 (Abcam, ab25386), F4/80 (BD, 565411), von Willebrand factor (Abcam, ab11713), CD41 (BD, 553847), NG‐2 (Abcam ab275024), ZO‐1 (Thermo, 617 300), caspase‐1 (Adipogen, AG‐20B‐0044‐C100), high mobility group box 1 (Abcam, ab79823), vascular cell adhesion molecule 1 (VCAM‐1) (Abcam, ab134047), cathepsin B (Abcam, ab58802), lysosomal‐associated membrane protein (LAMP)‐1 (BD, 553792), LAMP‐2A (Abcam, ab18528), TFEB (Bethyl Laboratories, A303‐673A), transient receptor potential cation channel, mucolipin subfamily, member 1 (TRPML‐1; Thermo, PA1‐46474), perilipin 2 (Proteintech, 15 294‐1‐AP), microtubule‐associated proteins light chain 3‐I/II (CST, 12741S), CCL‐2 (Millipore, MABN712), β‐actin (CST, 3700S).
Secondary antibody for Western blot: IRDye 800CW anti‐mouse IgG (LICOR, 926‐32 212), IRDye 800CW anti‐Rabbit IgG (LICOR, 926‐32 213), anti‐mouse IgG, HRP (Thermo Fisher, A16011), stabilized peroxidase conjugated anti‐rabbit (Invitrogen, 32 460), anti‐rat IgG‐HRP (Fisher, 629 520). Secondary antibody for immunofluorescence: anti‐mouse IgG, Alexa Fluor 488 conjugate (Thermo Fisher, A‐21202), Alexa Fluor 488 conjugate anti‐rabbit IgG (Thermo Fisher, A21206), Alexa Fluor 555 conjugate anti‐mouse IgG (Thermo Fisher, A‐31570), Alexa Fluor 555 conjugate anti‐rabbit IgG (Thermo Fisher, A‐31572).
Reagents: Oil red (VWR, BT135140‐100G), Isolectin GS‐IB4 (Fisher, I21411), Enzychrom AF cholesterol Assay Kit (BioAssay, E2CH‐100), EnzyChrom Triglyceride Assay Kit (BioAssay, ETGA‐200), bafilomycin A1 from streptomyces griseus (Sigma, B1793), 7‐ketocholesterol (Sigma, C2394), Ezetimibe (Cayman, 16331), mitoSOX Red mitochondrial superoxide indicator (moleculae probes M36008), Green FLICA Caspase‐1 Assay Kit (Immunochemistry Technologies, No. 98), Cell‐Counting Kit 8 kit (APExBIO; K1018), Aurum Total RNA Mini Kits (Bio‐Rad, 732‐6820), iScript Reverse Transcription Supermix for RT‐qPCR (Bio‐Rad, 1708841), iTaq Universal SYBR Green supermix (Bio‐Rad, 1725121).
Immunofluorescence Staining
Immunofluorescence staining was performed using frozen tissues or cultured mouse cardiac endothelial cells (MCECs). For tissue staining, frozen section slides were fixed with 4% paraformaldehyde for 15 minutes at room temperature. For MCECs, ≈7.5×104 cells were seeded into 24‐well culture plates containing gelatin‐coated coverslips. After treatment with indicated stimuli, MCECs were fixed with 4% paraformaldehyde for 15 minutes at room temperature. After washing with PBS, frozen tissues or cells were blocked and permeabilized with 5% BSA+ 0.3% Triton X‐100 in PBS for 1 hour at room temperature and then incubated with primary antibodies overnight at 4 °C. After washing with PBST (PBS+0.05% Tween‐20), the slides were incubated with corresponding secondary antibodies conjugated to Alexa Fluor 488 or Alexa Fluor 555 (Invitrogen) for 1 hour at room temperature. After washing with PBST, slides were mounted by DAPI‐mounting solution and then analyzed by using Olympus IX73 imaging system. The Pearson's correlation for colocalization efficiency or mean fluorescence density was analyzed using the Image‐Pro Plus 6.0 software, as described previously. 21
Hematoxylin and Eosin Staining
Hematoxylin and eosin staining was performed using hematoxylin and eosin staining kit (Teomics HAE‐1). Heart or liver frozen section slides were fixed with 4% paraformaldehyde for 15 minutes at room temperature. After washing with distilled water, tissues were incubated with adequate hematoxylin (Mayer's) for 5 minutes, followed by 2 changes of distilled water to remove excess stain. Then bluing reagent was added to tissues for 10 to 15 seconds followed by 2 changes of distilled water. After dipping slides in absolute alcohol and wiping excess off, tissues were incubated with adequate Eosin Y solution for 2 to 3 minutes, followed with rinse using absolute alcohol. Then the slide was cleared and mounted in synthetic resin. Images were taken promptly using an Olympus IX73 imaging system.
Oil Red O Staining
Oil Red O stock solution: saturated oil Red O in isopropanol (0.3 g oil Red O in 100 mL isopropanol), using gentle heat from a water bath to dissolve. Oil Red O working solution (fresh): 30 mL of stock solution was mixed with 20 mL of distilled water and allowed to stand for 10 minutes, filtered twice with a syringe filter, and the working solution was used within 1 hour.
Staining procedure: Heart or liver frozen sections were prepared at a thickness of 8 μm, and air‐dried sections were placed onto slides, fixed in 4% paraformaldehyde for 15 minutes, and rinsed immediately in 3 changes of distilled water before being soaked for 20 minutes. The sections were then washed with 60% isopropanol for 1min to avoid water carryover. Sections were stained with freshly prepared Oil Red O working solution for 30 minutes, and then rinsed with 60% isopropanol for 2 minutes followed by 2 changes of distilled water. Nuclei were lightly stained with alum hematoxylin for 1 minute, then rinsed in running tap water for 10 minutes. Sections were mounted in mounting media and images were taken promptly with the Olympus IX73 imaging system.
Quantitative Real‐Time Polymerase Chain Reaction
Aurum Total RNA Mini Kits (Bio‐Rad, 732‐6820) were used to isolate total RNA from heart, aorta, and MCECs. iScript Reverse Transcription Supermix (Bio‐Rad, 1708841) was used to generate cDNA from isolated total RNA. Real‐time polymerase chain reaction was performed using the iTaq Universal SYBR Green supermix (Bio‐Rad, 1725121) on the Bio‐Rad CFX Connect Real‐Time System. Primers of NLR family pyrin domain containing 3 (NLRP3), NLRP‐1A, PYD and CARD domain containing, caspase‐1, interleukin‐1β, interleukin‐18, interleukin‐6, interleukin‐8, gasdermin D, tumor necrosis factor (TNF), intercellular adhesion molecule 1, VCAM‐1, P‐selectin, and E‐selectin were purchased from Bio‐Rad. Other primers used in the present study are listed in Table S1. The cycle threshold values were converted to relative gene expression levels using the 2ΔΔCt method. The data were normalized to internal control β‐actin or GAPDH. 22
Total Cholesterol and Triglyceride in Serum or Liver Homogenate
Total cholesterol and triglyceride levels in serum or liver homogenate were measured using a cholesterol assay kit (BioAssay, E2CH‐100) and a triglyceride assay kit (BioAssay, ETGA‐200), respectively. Briefly, for the cholesterol assay, 55 μL of assay buffer was mixed with 1 μL of enzyme mix and 1 μL of dye reagent before 50 μL of this working reagent was added to diluted 50‐μL standard and sample wells. For the triglyceride assay, working reagent was prepared by mixing 100 μL of assay buffer, 2 μL of enzyme mix, 5 μL of lipase, 1 μL of ATP, and 1 μL of dye reagent in each well; 100 μL of this working reagent mix was added to each diluted 10 μL of standard and sample well. The plates were tapped to mix and incubated at room temperature for 30 minutes before having the OD measured at 570 nM with a microplate reader (BMG Labtech). The cholesterol and triglyceride concentrations in the liver homogenate were normalized to protein concentration.
MCEC Culture
MCECs (Cedarlane, CLU510) were cultured in low glucose DMEM with 5% FBS, 1% penicillin/streptomycin, and 1 mmol/L of HEPES at 37 °C with 5% CO2.
Western Blotting
MCECs were lysed in Laemmli sample buffer (Bio‐Rad, 161‐0737) containing β‐mercaptoethanol (Sigma Aldrich, M3148) and boiled for 10 minutes at 95 °C before being placed in an ice‐cooled ultrasonic bath for 5 minutes. The prepared samples were separated by 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The proteins from these samples were then electrophoretically transferred onto a polyvinylidene fluoride membrane at 35 V at 4 °C overnight. The membranes were blocked with 5% BSA in Tris‐buffered saline with 0.05% Tween 20. After further washing, the membranes were incubated with primary antibodies as indicated according to the manufacturer's instructions. After washing with PBST, the membranes were then incubated with corresponding secondary antibodies for 1 hour at room temperature. Finally, the bands were washed with PBST, visualized, and analyzed by the LI‐COR Odyssey Fc System.
Mitochondrial ROS
Mitochondrial ROS production was measured using the MitoSOX Red mitochondrial superoxide indicator (molecular probes M36008). Approximately 7.5×104 MCECs were seeded on coverslips in 24‐well plates in DMEM with 5% FBS. After treatments, cells were incubated with 5 μmol/L of MitoSOX in culture medium for 10 minutes at 37 °C in the dark. Cells were washed with warmed PBS 3 times and incubated with Hoechst 33342 (0.5% v/v) for 5 minutes to stain nuclei. Coverslips were then mounted on slides for examination. Images were taken immediately using the Olympus IX73 imaging system.
Caspase‐1 Activity Assay by FAM‐FLICA Kit
Approximately 7.5×104 MCECs were seeded on coverslips in 24‐well plates for overnight, and the medium was replaced with DMEM containing 1% FBS. Cells were pretreated with or without 50 nmol/L of bafilomycin A1 for 1 hour, then cotreated with or without 40 μmol/L 7‐ketocholesterol for 24 hours. Subsequently, 1× FAM‐FLICA (diluted by culture media) was added to the cells on the coverslips for 1 hour. The medium on the coverslips was carefully removed and replaced with apoptosis wash buffer for 10 minutes. Wash buffer was then removed, and cells were stained with Hoechst 33342 at 0.5% v/v for 5 minutes in order to stain the nucleus. To identify dead cells, cells were incubated with propidium iodide (PI; 0.5% v/v) for 5 minutes. Cells were then fixed with a fixative solution at a v/v ratio of 1:10 in PBS for 15 minutes at room temperature. After fixation, coverslips were mounted on slides for examination. Images were taken immediately using the Olympus IX73 imaging system.
Cell Viability Assay
In brief, 1.0×104 MCECs were seeded into 96‐well plates overnight, and then the medium was replaced with DMEM containing 1% FBS. Cells were treated as described, and 10 μL Cell‐Counting Kit 8 solution was added to each well and incubated at 37 °C for 1 hour. The absorbance was measured at 450 nm using a microplate reader (BMG Labtech).
Calcein‐AM Labeled Monocyte Retention
Macrophage J774 cells were trypsinized and stained with 2 μmol/L fluorescent Calcein‐AM (Thermo Fisher, C3100MP) at 37 °C for 10 minutes. After treatment as indicated, 5×104 Calcein‐AM–labeled J774 cells were seeded onto MCECs for 10 to 30 minutes. Unbounded monocytes were discarded, and MCEC layers with attached monocytes were gently washed with PBS, followed by fixation with 4% paraformaldehyde for 10 minutes at room temperature. The nucleus was stained with DAPI for 15 minutes at room temperature and mounted with an antifluorescence quenching agent. Imaging was performed using the Olympus IX73 imaging system.
Statistical Analysis
Data are presented as mean±SEM. All normally distributed data sets were analyzed by the 1‐ or 2‐way ANOVA with different treatments as category factors, followed by Bonferroni's multiple comparisons test if applicable. Data sets that did not meet normality assumptions were analyzed using the Kruskal–Wallis test, followed by Dunn's multiple comparisons test. A Student's t test was used to detect significant difference between 2 groups for normally distributed data sets, while the Mann–Whitney test or Kolmogorov–Smirnov test was applied for nonnormally distributed data sets. Statistical analysis was carried out using GraphPad Prism 6.0 software (GraphPad Software, La Jolla, CA). P<0.05 was considered statistically significant.
RESULTS
Effects of PD on CFR, Cardiac Remodeling, and Heart Function
CMD was measured by noninvasive echocardiography and assessed by coronary blood flow and CFR. Representative images and summarized data (Figure 1A and 1B) indicate that PD treatment significantly decreased coronary blood flow velocity in hyperemic mice (2.5% isoflurane) at 6 and 8 weeks compared with 0 weeks but had no effects at the baseline (1.0% isoflurane), corresponding to a decrease in CFR. Next, we evaluated whether PD affected cardiac remodeling (Figure 1C) and heart function (Figure 1D). We found that PD for 8 weeks showed no significant effect on cardiac remodeling, as measured by diastolic left ventricular (LV) anterior wall, systolic LV anterior wall, diastolic LV posterior wall, systolic LV posterior wall, and calculated LV mass (Figure 1C). PD for 8 weeks had no significant effect on heart function, as measured by the diastolic left ventricular internal end, systolic left ventricular internal end, diastolic LV volume, systolic LV volume, and calculated LV ejection fraction and LV shortening fraction (Figure 1D). These data demonstrate that PD causes CMD without affecting cardiac remodeling or heart function.
Figure 1. Effects of PD on CFR, cardiac remodeling, and heart function.

A, Representative ultrasound images of isoflurane‐induced vasodilation of the left anterior descending coronary artery at basal (1.0% isoflurane) and hyperemia (2.5% isoflurane) levels in mice fed with ND and PD for 8 weeks. B, Summarized data of diastolic blood flow velocity peeks at 1.0% isoflurane, 2.5% isoflurane, and CFR (ratio of 2.5% isoflurane over 1.0% isoflurane) in mice fed with PD for 0, 2, 4, 6, and 8 weeks. C, Summarized data of echocardiographic parameters in cardiac remodeling: diastolic LVAW (d), systolic LVAW (s), diastolic LVPW (d), systolic LVPW (s), LV Mass, and corrected LV mass in mice fed with ND or PD for 8 weeks. D, Summarized data of echocardiographic parameters in heart function: diastolic LVID (d), systolic LVID (s), diastolic LV Vol (d), systolic LV Vol (s), LVEF, and LVFS in mice fed with ND or PD for 8 weeks. * vs PD 0 week or ND, P<0.05 (n=7–9). CFR indicates coronary flow reserve; LV, left ventricular; LVAW, left ventricular anterior wall; LVEF, LV ejection fraction; LVFS, LV fractional shortening; LVID, LV internal end; LVPW, left ventricular posterior wall; ND, normal diet; and PD, Paigen's diet.
Effects of PD on Coronary Arterioles Remodeling, Capillary Rarefaction, Atherothrombosis Events, and Arterioles and Cardiac Inflammation
Next, we characterized PD‐induced CMD by examining various cardiovascular‐related pathological changes. As shown in Figure 2A, hematoxylin and eosin staining showed that no neointima formation or changes in media thickness were observed in coronary arterioles in both ND and PD groups. These results indicate that PD for 8 weeks did not induce coronary arterioles remodeling. We also performed immunofluorescence analysis of myocardial capillary density by staining microvascular ECs with isolectin‐IB4 and pericytes with NG‐2. As shown in Figure 2B, PD did not affect IB4 or NG2 staining, nor did it alter the ratio of NG‐2 to IB4, suggesting that PD does not alter capillary density or pericyte coverage. Interestingly, Oil Red O staining in Figure 2C showed that lipid deposition/fatty streaks were detected in the coronary arterioles walls in 3 of 8 PD‐fed mice but not in the ND group. As shown in Figure 2D, immunofluorescence analysis detected CD41‐positive thrombus in some coronary arterioles in 2 of 8 PD‐fed mice, but not in the ND group. Furthermore, PD increased inflammasome activation in the endothelium of coronary arterioles, as evidenced by the increased colocalization (yellow color) of caspase‐1 (Figure 2E) or high mobility group box 1 (Figure 2F) with von Willebrand factor (EC marker). PD also increased the expression of VCAM‐1 in the endothelium of arterioles (Figure 2G), and increased the adhesion of CD45‐positive leukocytes (Figure 2H) and F4/80 positive macrophage (Figure 2I) in cardiac tissue. These results suggest that PD‐induced CMD is associated with increased activation of coronary arterioles inflammation and thrombosis and increased myocardial inflammatory cell infiltration, but not with coronary arterioles remodeling or capillary rarefaction.
Figure 2. Effects of PD on coronary vascular remodeling, capillary rarefaction, atherothrombosis events, and vascular and cardiac inflammation.
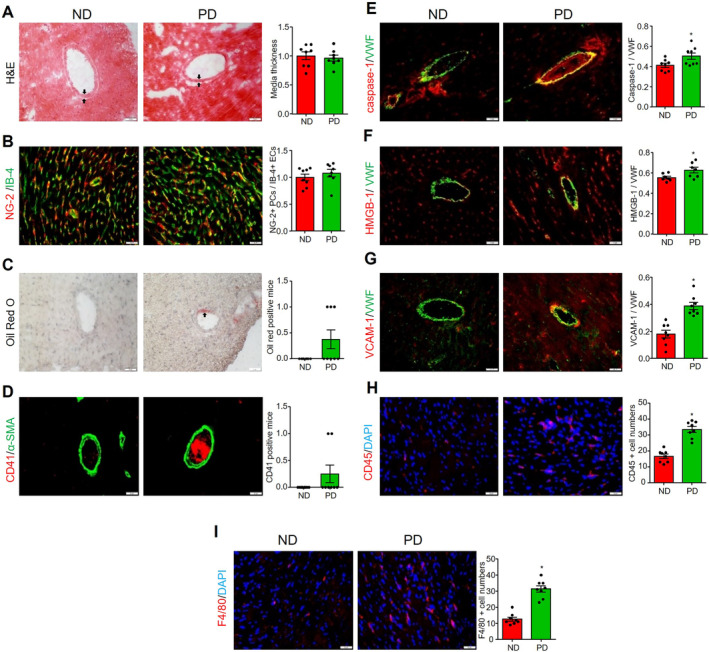
All heart samples were collected from mice fed with ND or PD for 8 weeks. A, Representative H&E staining and summary of coronary artery media thickness (black arrow). B, Representative capillary endothelial cell marker isolectin‐B‐4 staining plus pericyte marker NG‐2 staining, and summary of colocalization coefficient. C, Representative oil Red O staining for lipid deposition/fatty streaks (black arrow) in the coronary artery, and summary of oil Red O staining positive mice numbers. D, Representative CD41 staining for platelet activation, and summary of positive mice. α‐Smooth muscle actin staining was used to localize coronary arterioles. Representative images for caspase‐1/VWF (E), high mobility group box 1/VWF (F), and VCAM‐1/VWF (G) staining and their summarized colocalization coefficients. VWF staining was used to localize coronary arterioles endothelium. Cardiac inflammatory cells infiltration was indicated by positive staining of leukocyte marker CD45 (H) and macrophage marker F4/80 (I). Scale bar=20 μm. * vs ND, P<0.05 (n=6–8). H&E indicates hematoxylin and eosin; ND, normal diet; PD, Paigen's diet; and VWF, von Willebran factor.
Effect of PD on Transcriptional Levels of Lipogenesis, Thrombosis, and Inflammation‐Related Pathways
To confirm the pathological changes associated with CMD, we analyzed the expression levels of genes involved in lipid accumulation, thrombosis, and inflammation in the heart or aortas of mice fed with ND or PD for 8 weeks. We first examined genes related to lipid droplet (LD) biogenesis and formation, including glycerol‐3‐phosphate acyltransferase 4, 1‐acylglycerol‐3‐phosphate O‐acyltransferase 2, Lipin‐2, diacylglycerol O‐acyltransferase 1/2 (diacylglycerol O‐acyltransferase 1, diacylglycerol O‐acyltransferase 2), perilipins (perilipin1, 2, 3, 4, 5), and genes related to cholesterol biosynthesis, including acetyl‐CoA acetyltransferase 1/2 (ACAT1, ACAT2). As shown in Figure 3A, we found that PD had no significant effects on most of above‐mentioned genes in heart tissues except perilipin 1, a major perilipin isoform expressed in adipocytes. In contrast, PD significantly increased glycerol‐3‐phosphate acyltransferase 4, 1‐acylglycerol‐3‐phosphate O‐acyltransferase 2, diacylglycerol O‐acyltransferase 1/2, ACAT1/2, and perilipin 5 in aortas. These results suggest that PD treatment tends to increase lipogenesis and LD formation in the arterial wall.
Figure 3. Effect of PD on the transcriptional levels of lipogenesis, thrombosis, and inflammation pathways‐related genes.
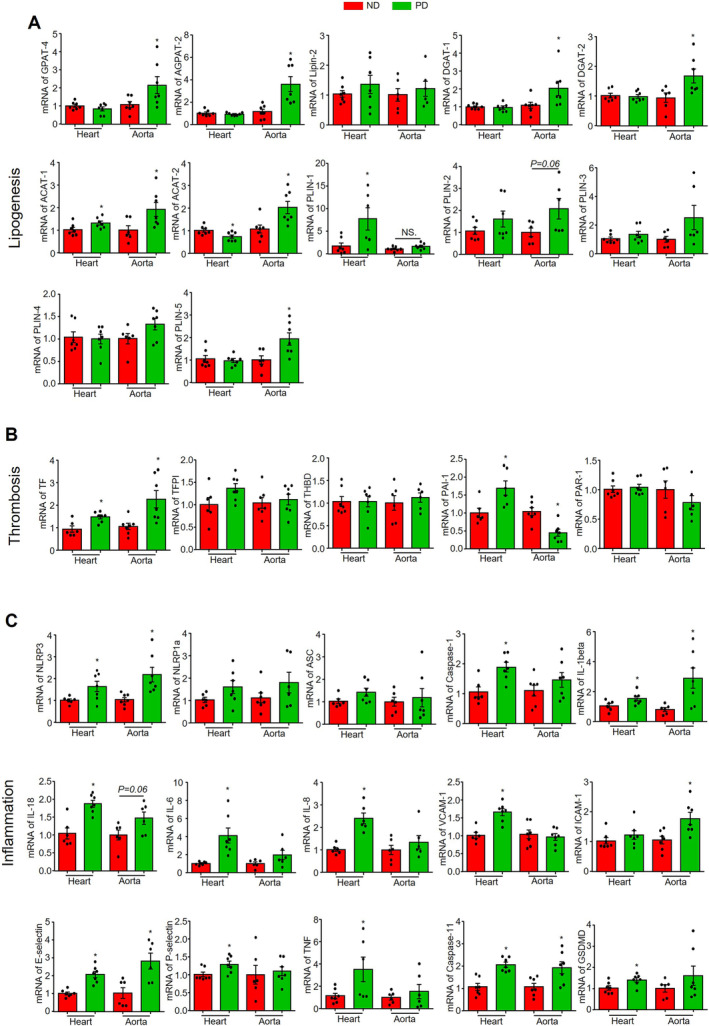
Heart or aorta tissue samples were collected from mice fed with ND or PD for 8 weeks. A, mRNA levels of lipogenesis‐related genes: GPAT‐4, AGPAT‐2, LIPIN‐2, DGAT‐1, DGAT‐2, ACAT‐1, ACAT‐2, PLIN‐1, PLIN‐2, PLIN‐3, PLIN‐4, and PLIN‐5. B, mRNA levels of thrombosis‐related genes: TF, TFPI, THBD, PAI‐1, and PAR‐1. C, mRNA levels of inflammation‐related genes: NLRP3, NLRP1a, ASC, caspase‐1, interleukin‐1β, interleukin‐18, interleukin‐6, interleukin‐8, VCAM‐1, intercellular adhesion molecule 1, E‐selectin, P‐selectin, TNF, caspase‐11, and gasdermin D. * vs ND, P<0.05 (n=6–7). ND indicates normal diet; and PD, Paigen's diet.
Then, we analyzed thrombosis‐related genes including tissue factor, tissue factor pathway inhibitor, thrombomodulin, proteinase‐activated receptor 1, and plasminogen activator inhibitor 1. As shown in Figure 3B, PD increased tissue factor in both hearts and aortas. Additionally, PD increased plasminogen activator inhibitor 1 in the heart but decreased it in the aorta. These results suggest that thrombotic pathways such as coagulation and fibrinolysis are altered in the arterial wall and myocardium.
As for inflammation, we analyzed genes of inflammasomes including NLRP3, NLRP1a, PYD and CARD domain containing, caspase‐1, caspase‐11, gasdermin D, interleukin‐1β, and interleukin‐18, adhesion molecules including VCAM‐1, intercellular adhesion molecule 1, E‐selectin, and P‐selectin, and inflammatory mediators including interleukin‐6, interleukin‐8, and TNF‐α. As shown in Figure 3C, PD significantly upregulated NLRP3, caspase‐1, caspase‐11, gasdermin D, interleukin‐1β, and interleukin‐18 in the heart, but only upregulated NLRP3, interleukin‐1β, and caspase‐11 in the aorta. These results suggest the NLRP3 inflammasome is activated in both the arterial wall and the myocardium, with more pronounced effects in the myocardium. PD also upregulated genes for various adhesion molecules, including VCAM‐1, E‐selectin, and P‐selectin in the heart and intercellular adhesion molecule 1 and E‐selectin in the aorta (Figure 3C). Interestingly, PD upregulated inflammatory mediators interleukin‐6, interleukin‐8, and TNF‐α only in the heart but not in the aorta (Figure 3C), which is consistent with the increased adhesion of CD45‐positive leukocytes and F4/80positive macrophage in cardiac tissue.
PD Upregulated Lysosomal Signaling Pathway in Coronary Arterioles
Recent studies have shown that lysosomal signaling pathways play a critical role in regulating endothelial homeostasis under various metabolic stresses, including hypercholesterolemia. 23 , 24 Here, we investigated whether hypercholesterolemia affects the lysosomal signaling in the coronary microcirculation by immunofluorescence studies. The expression of several lysosomal markers was significantly increased in the endothelium of coronary arterioles of mice fed with PD for 8 weeks, including transcription factor EB (TFEB) (Figure 4A), LAMP‐1 (Figure 4B), LAMP‐2A (Figure 4C), and transient receptor potential cation channel, mucolipin subfamily, member 1 (transient receptor potential cation channel, mucolipin subfamily, member 1 or mucolipin‐1) (Figure 4D). Furthermore, we quantified the mRNA levels of several lysosome/autophagy‐related genes in the heart and aorta tissues. As shown in Figure 4E, PD only upregulated the mRNA levels of LAMP‐2A and beclin‐1 in the heart, while almost all tested lysosome/autophagy‐related genes were significantly upregulated in the aorta, including LAMP‐1, transient receptor potential cation channel, mucolipin subfamily, member 1, sphingomyelin phosphodiesterase 1, microtubule‐associated proteins light chain 3, and p62/SQSTM1. Notably, PD did not affect TFEB mRNA levels in aorta and heart tissues (Figure 4E). Nonetheless, these data suggest that PD significantly activates the lysosome signaling pathway in coronary arterioles endothelium, and to a less extent, in the myocardium as well.
Figure 4. PD upregulated lysosome related signaling pathway in coronary arterioles.
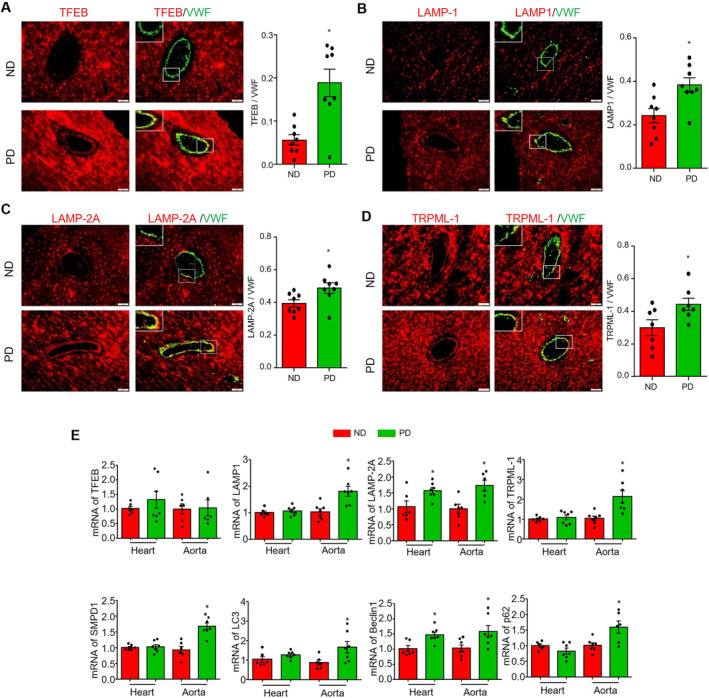
Heart or aorta tissue samples were collected from mice fed with ND or PD for 8 weeks. Representative images for staining of TFEB/VWF (A), LAMP‐1/VWF (B), LAMP‐2A/VWF (C), and TRPML‐1/VWF (D) and the summary of colocalization coefficients. VWF staining was used to localize coronary arterioles endothelium. E, mRNA levels of lysosome‐autophagy–related genes in heart or aorta tissues: TFEB, LAMP‐1, LAMP‐2A, TRPML‐1, SMPD‐1, LC3, beclin‐1, and p62/SQSTM1. scale bar=20 μm. * vs ND, P<0.05 (n=6–8). AOI indicates area of interest; ND, normal diet; PD, Paigen's diet; and VFW, von Willebrand factor.
Ezetimibe Alleviated PD‐Induced Steatohepatitis and Hypercholesterolemia
Ezetimibe is a Food and Drug Administration–approved drug for the treatment of hypercholesterolemia that works by inhibiting the absorption of dietary and biliary cholesterol from the small intestine. Here, we investigated whether ezetimibe could prevent the development of CMD in mice fed with PD for 8 weeks. We first examined the efficacy of ezetimibe administration by analyzing pathological features of the liver, including steatosis, inflammation, and serum cholesterol levels. As shown in Figure 5A, hematoxylin and eosin staining showed that ezetimibe prevented steatosis, which is confirmed by a decrease in oil Red O staining (Figure 5B and 5E) and a decrease in the expression of LD‐coating protein perilipin 2 (Figure 5C and 5F). Additionally, ezetimibe attenuated the PD‐induced increase in CD45‐positive inflammatory cells in the liver (Figure 5D and 5G). Next, we measured the total cholesterol and triglyceride levels in the serum and liver tissue. Consistent with the beneficial effects of ezetimibe on steatohepatitis, we found that PD induced an increase in serum and liver cholesterol levels, which were significantly attenuated by ezetimibe administration (Figure 5H and 5I). Notably, PD decreased serum triglycerides but did not affect liver triglycerides, whereas ezetimibe had no further effect (Figure 5J and 5K).
Figure 5. Ezetimibe alleviated PD‐induced liver steatohepatitis and hypercholesterolemia.
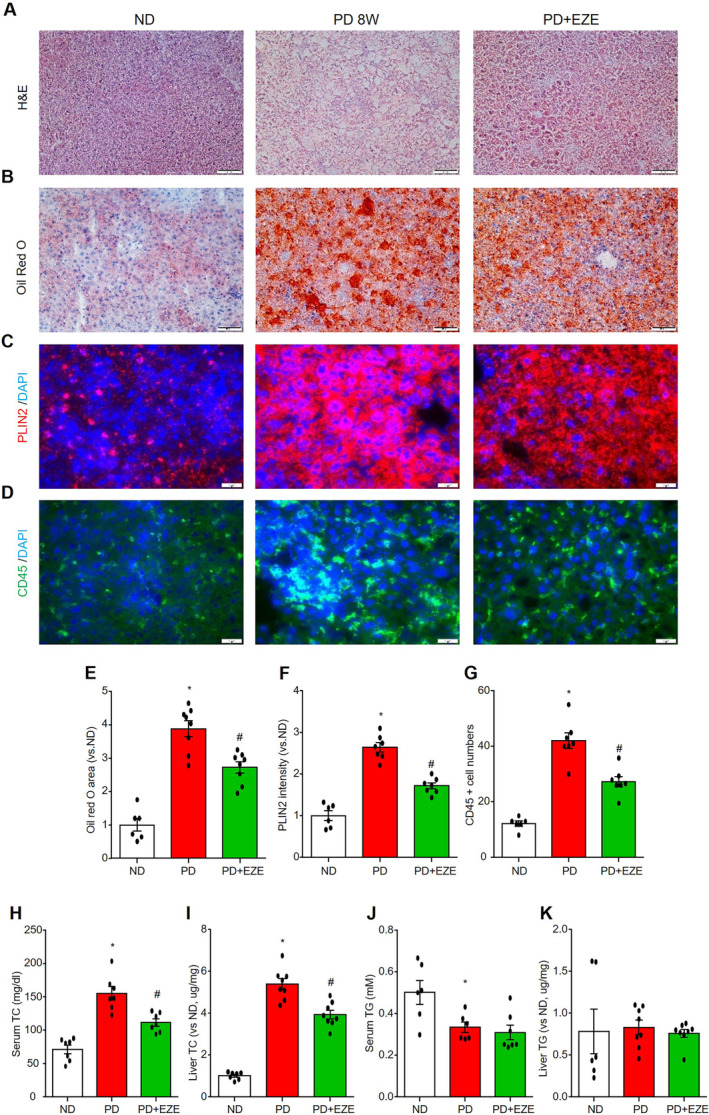
Liver sections, serum, and liver homogenate were collected from mice in the ND‐fed vehicle control mice group (PBS+DMSO), the 8‐weeks PD‐fed vehicle control group (PBS+DMSO) and the PD+EZE treatment group (I.P. 3 mg/kg per 2 days for 8 weeks). Liver histomorphology was measured using H&E staining (A). Liver lipid deposition was detected by oil Red O staining (B and E) and lipid droplet–associated protein perilipin 2 staining (C and F). Liver inflammatory cell infiltration was indicated by leukocyte marker CD45 staining (D and G). TC (H and I) and TG (J and K) levels in serum and liver homogenate were measured. Scale bar=20 μm. * vs ND, # vs PD, P<0.05 (n=6–8). EZE indicates ezetimibe; H&E, hematoxylin and eosin; ND, normal diet; PD, Paigen's diet; TC, total cholesterol; and TG, triglyceride.
Ezetimibe Rescued Coronary Microvascular Function in Mice Fed With PD
As illustrated in Figure 6A and 6B, the administration of ezetimibe to PD‐fed mice significantly restored hyperemic coronary blood flow velocity and CFR compared with PD only group. In contrast, ezetimibe had no effect on cardiac remodeling or heart function (Figure 6C). As expected, ezetimibe inhibited the PD‐induced increase of VCAM‐1 (Figure 7A) and high mobility group box 1 (Figure 7B) expression in coronary arterioles and capillary ECs. Furthermore, ezetimibe prevented the PD‐induced infiltration of CD45‐positive inflammatory cells around coronary arterioles and capillaries (Figure 7C). These results suggest that ezetimibe prevents PD‐induced CMD and inflammatory responses in the coronary microcirculation.
Figure 6. Ezetimibe restored PD‐induced CFR decrease.
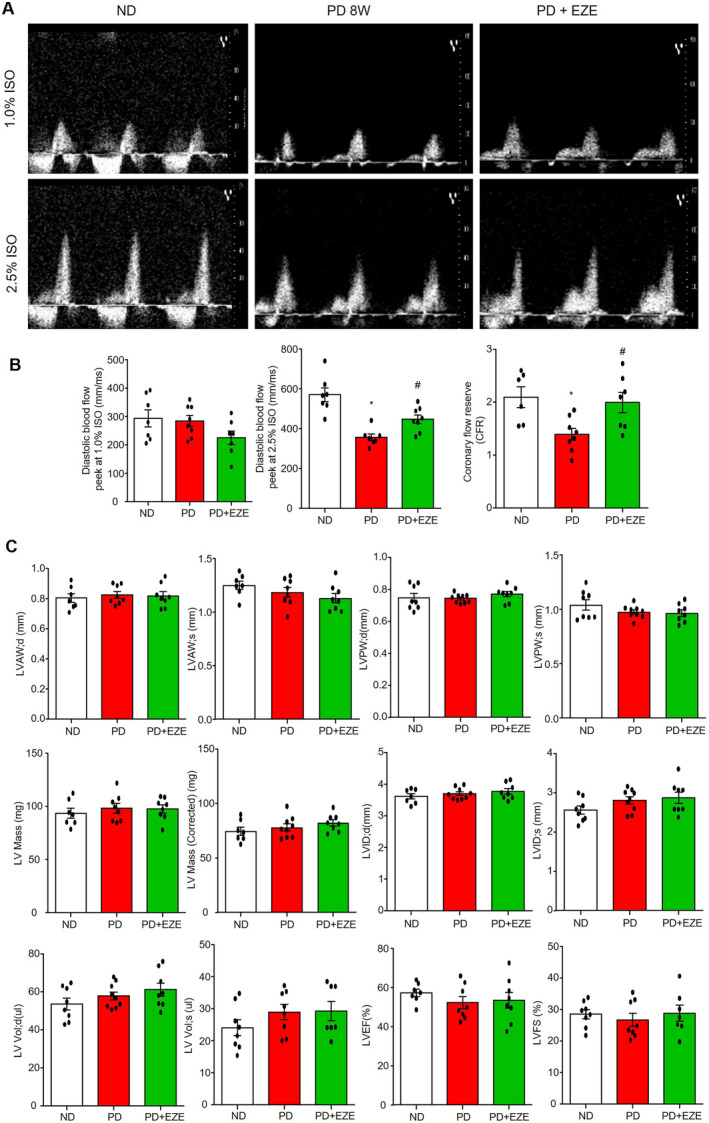
Echocardiographic parameters were measured in mice from the ND‐fed vehicle control mice group (PBS+DMSO), the 8‐weeks PD‐fed vehicle control group (PBS+DMSO) and the PD+EZE treatment group (I.P. 3 mg/kg per 2d for 8 weeks). A, Representative ultrasound images of isoflurane‐induced vasodilation of the left anterior descending coronary artery at basal (1.0% isoflurane) and hyperemia (2.5% isoflurane) levels. B, Summarized data of diastolic blood flow velocity peeks at 1.0% isoflurane, 2.5% isoflurane and CFR (ratio of 2.5% isoflurane over 1.0% isoflurane). C, Summarized data of echocardiographic parameters in cardiac remodeling and heart function: diastolic LVAW (d), systolic LVAW (s), diastolic LVPW (d), systolic LVPW (s), LV Mass, corrected LV mass, diastolic LVID (d), systolic LVID (s), diastolic LV Vol (d), systolic LV Vol (s), LVEF, and LVFS. * vs ND, # vs PD, P<0.05 (n=7–9). EZE indicates ezetimibe; LV, left ventricular; LVAW, left ventricular anterior wall; LVEF, LV ejection fraction; LVFS, LV fractional shortening; LVID, LV internal end; LVPW, left ventricular posterior wall; ND, normal diet; and PD, Paigen's diet.
Figure 7. Ezetimibe reduced PD‐induced inflammation and monocyte adhesion in both coronary arterioles and capillaries.
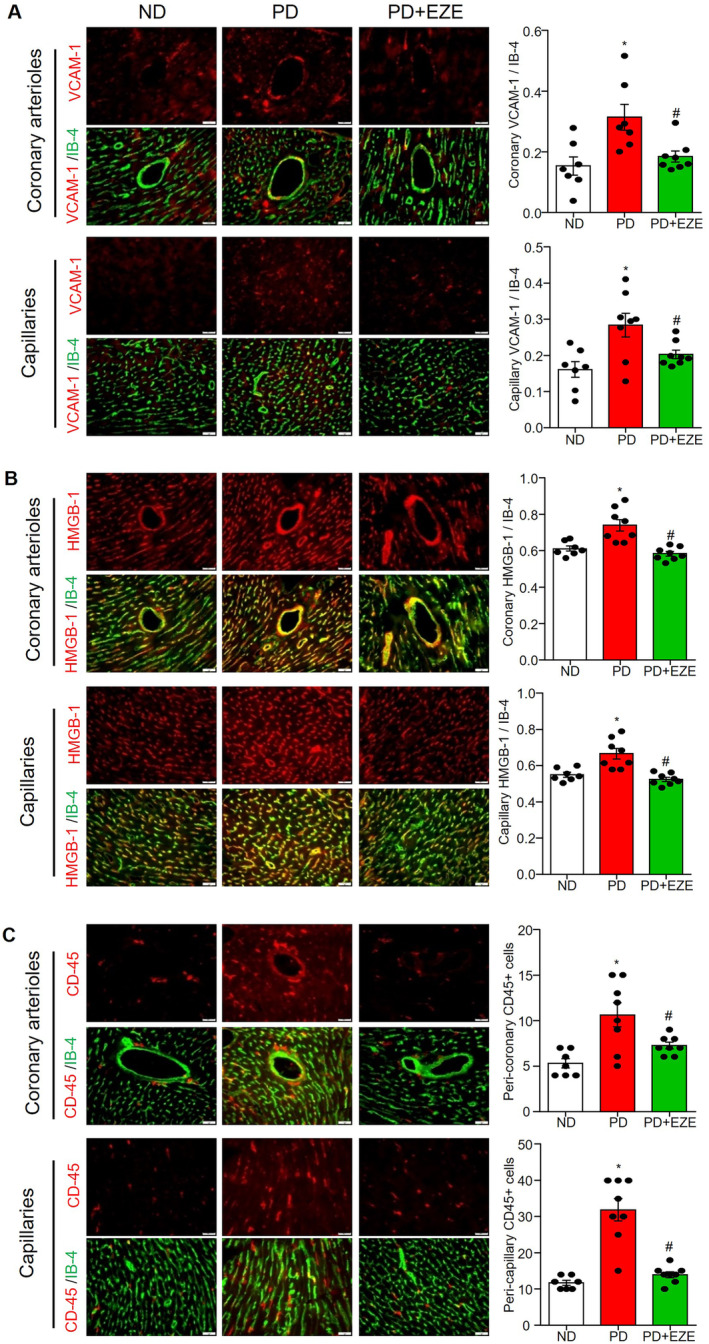
Heart sections collected were from mice in the ND‐fed vehicle control mice group (PBS+DMSO), the 8‐weeks PD‐fed vehicle control group (PBS+DMSO) and the PD+EZE treatment group (I.P. 3 mg/kg every 2 days for 8 weeks). The staining and summarized colocalization coefficients or expression of VCAM‐1/isolectin‐B4 (A), HMGB‐1/isolectin‐B4 (B), and CD45 (C) in coronary arterioles endothelium and capillary endothelial cells. Isolectin‐B4 staining was used to localize coronary arterioles endothelium and capillary endothelial cells. Scale bar=20 μm. * vs ND, # vs PD, P<0.05 (n=6–8). EZE, ezetimibe; HMGB‐1, high mobility group box 1; ND, normal diet; PD, Paigen's diet; and VCAM‐1, vascular cell adhesion molecule 1.
Mitochondrial ROS Production and Inflammation Is Associated With Upregulation of Lysosomal Signaling in Cultured MCECs
Next, we investigated the association between adverse effects and augmented lysosomal signaling in cultured MCECs. First, we confirmed that 7‐ketocholesterol dose‐dependently increased mitochondrial superoxide production as measured by mitoSOX Red staining (Figure 8A and 8D). 7‐Ketocholesterol treatment also significantly upregulated the expression of VCAM‐1 (Figure 8B and 8E) and C‐C motif chemokine ligand 2 (Figure 8C and 8F). These results suggest that 7‐ketocholesterol induces mitochondrial ROS production and inflammation in cultured MCECs, which mirrors the pathological changes in coronary ECs under hypercholesterolemic conditions in vivo. We then examined the effects of 7‐ketocholesterol on the lysosomal signaling pathway by analyzing the activation of TFEB, a master regulator of genes involved in autophagy and lysosomal signaling. 22 , 25 Once activated, TFEB translocates from the cytoplasm to the nucleus. As shown in Figure 8G, 7‐ketocholesterol dose‐dependently increased the nuclear translocation of TFEB in MCECs. Western blot analysis (Figure 8H) showed that 7‐ketocholesterol increased the protein expression of the autophagosome marker microtubule‐associated proteins light chain 3‐II. Furthermore, 7‐ketocholesterol significantly increased the mRNA levels of genes in autophagy and lysosomal signaling pathways, including LAMP‐2A, beclin‐1, microtubule‐associated proteins light chain 3 and p62/SQSTM1 (Figure 8I). In contrast, 7‐ketocholesterol had no effect on the LAMP‐1 and even suppressed the mRNA level of TFEB (Figure 8I). These results suggest that 7‐ketocholesterol activates TFEB‐mediated autophagy and lysosomal signaling in cultured MCECs.
Figure 8. Effect of 7‐ketochlesterol on mitochondrial superoxide, proinflammatory proteins and lysosome‐related signaling in cultured MCECs.
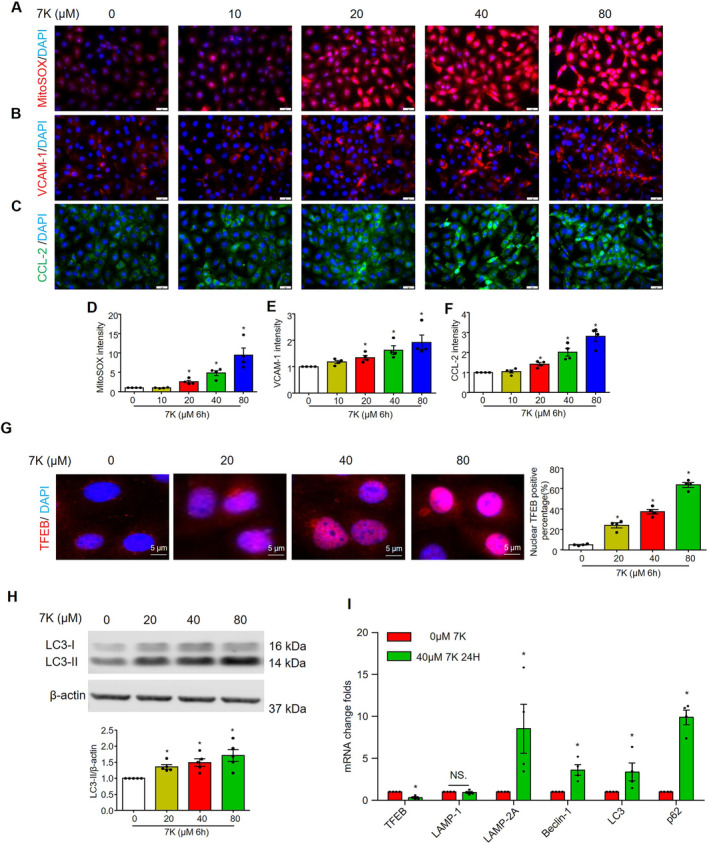
MCECs were cultured in low glucose DMEM with 5% FBS and treated with 7‐ketocholesterol for the indicated times. A and D, Representative immunofluorescence images and quantification show mitochondrial superoxide levels. Representative immunofluorescence images and quantification show the expression of proinflammatory proteins VCAM‐1 (B and E) and CCL2 (C and F). G, Representative immunofluorescence images and quantification show the nuclear TFEB positive percentage. Nuclei were stained with DAPI. H, Representative immunoblots and summarized data show the effects of 7‐ketocholesterol on the protein expression levels of microtubule‐associated proteins light chain 3‐II. I, Real‐time reverse transcription polymerase chain reaction analyses of TFEB, LAMP‐1, LAMP‐2A, beclin‐1, microtubule‐associated proteins light chain 3, and p62/SQSTM1 mRNA levels after treatment with 0 or 40 μM 7‐ketocholesterol for 24 hour. Scale bar=20 μm. * vs 0, P<0.05 (n=4–5). CCL2 indicates C‐C motif chemokine ligand 2; LAMP‐1, lysosomal‐associated membrane protein; MCECs, mouse cardiac endothelial cells; TFEB, transcriptional factor EB; and VCAM‐1, vascular cell adhesion molecule 1.
Inhibition of Lysosomal Function Exacerbated Inflammation and Cell Death in Cultured MCECs
To determine the role of lysosomal signaling activation in endothelial inflammation and injury, we inhibited lysosomal function in MCECs using bafilomycin A1, a selective inhibitor of vacuolar H+ ATPases on the lysosomal membrane. Bafilomycin A1 inhibits vacuolar H+ ases and leads to the impaired acidification of lysosomes. 26 Impaired lysosomal acidification may result in the inhibition of TRPML1, an upstream event of TFEB activation. 27 , 28 , 29 It was found that bafilomycin A1 significantly inhibited 7‐ketocholesterol–induced TFEB nuclear translocation (Figure 9A). We then examined the inflammatory and injury responses in MCECs by staining cells with VCAM‐1, FLICA (a green fluorescent probe for activated caspase‐1), 30 , 31 , 32 or PI (for detecting dead cells). We observed that bafilomycin A1 enhanced the 7‐ketocholesterol–induced increase in VCAM‐1 expression (Figure 9B), FLICA+ casapase‐1 activity (Figure 9C and 9D), and PI+ dead cells (Figure 9C and 9E). We also demonstrated that 7‐ketocholesterol increased the number of pyroptotic (FLICA+/PI+) (Figure 9F) and non‐pyroptotic (FLICA‐/PI+) (Figure 9G) dead cells, while bafilomycin A1 further increased the number of dead cells. By assaying cell viability, we confirmed that bafilomycin A1 exacerbated 7‐ketocholesterol–induced cell death (Figure 9H). Taken together, these results suggest that TFEB‐mediated lysosomal signaling protects against 7‐ketocholesterol–induced inflammatory responses and injury in cultured MCECs.
Figure 9. Inhibition of lysosome signaling by bafilomycin A1 exacerbates 7‐ketocholesterol –induced inflammation and cell death in cultured MCECs.
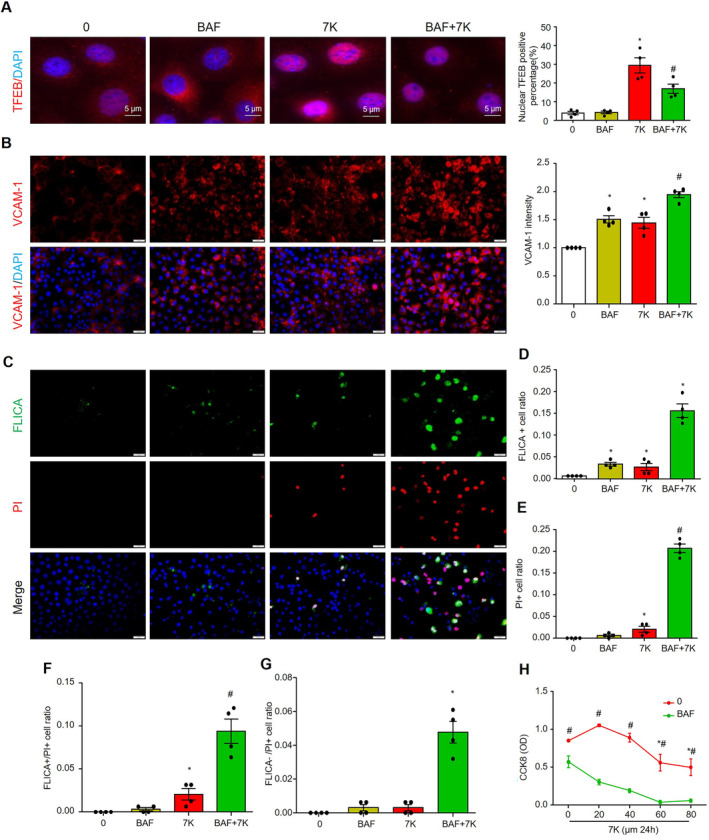
MCECs were cultured and treated in low glucose DMEM with 5% FBS, pretreated with or without 50 nM BAF for 1 hour, and then cotreated with or without 40 μM of 7‐ketocholesterol for 6 hour. A, Representative immunofluorescence images and quantification show the nuclear TFEB‐positive percentage. Nuclei were stained with DAPI. B through G, MCECs are treated in low‐glucose DMEM with 1% FBS for 2 hour before pretreatment with or without 50 nM of BAF for 1 hour, and then the cells are cotreated with or without 40 μM of 7‐ketocholesterol for 24 hour. B, Representative images of VCAM‐1 and summarized data. C through G, Representative images of FLICA/PI staining and summarized data. H, Cell numbers were detected by using CCK8 kit. Scale bar=20 μm. * vs 0, # vs BAF or 7‐ketocholesterol, P<0.05 (n=4). BAF indicates bafilomycin A1; CCK8, Cell‐Counting Kit 8; MCECs, mouse cardiac endothelial cells; PI, propidium iodide; TFEB, transcriptional factor EB; and VCAM‐1, vascular cell adhesion molecule 1.
Ezetimibe Activated TFEB and Attenuated Mitochondrial ROS and Inflammation in Cultured MCECs
Finally, we investigated whether ezetimibe could directly affect ECs in addition to its cholesterol‐lowering effects. We found that ezetimibe dose‐dependently increased TFEB nuclear translocation (Figure 10A) and further enhanced 7‐ketocholesterol–induced TFEB nuclear translocation (Figure 10B). Moreover, ezetimibe attenuated 7‐ketocholesterol–induced mitochondrial ROS production (Figure 10C), as well as the expression of VCAM‐1 (Figure 10D) and CCL‐2 (Figure 10E), and monocyte adhesion (Figure 10F). These results suggest that ezetimibe acts synergistically with 7‐ketocholesterol to increase TFEB‐mediated lysosomal signaling and prevent 7‐ketocholesterol–induced mitochondrial ROS and inflammation in cultured MCECs.
Figure 10. Ezetimibe attenuated 7‐ketocholesterol–induced mitochondrial superoxide, inflammation, and monocyte adhesion via increasing TFEB nuclear translocation in cultured MCECs.
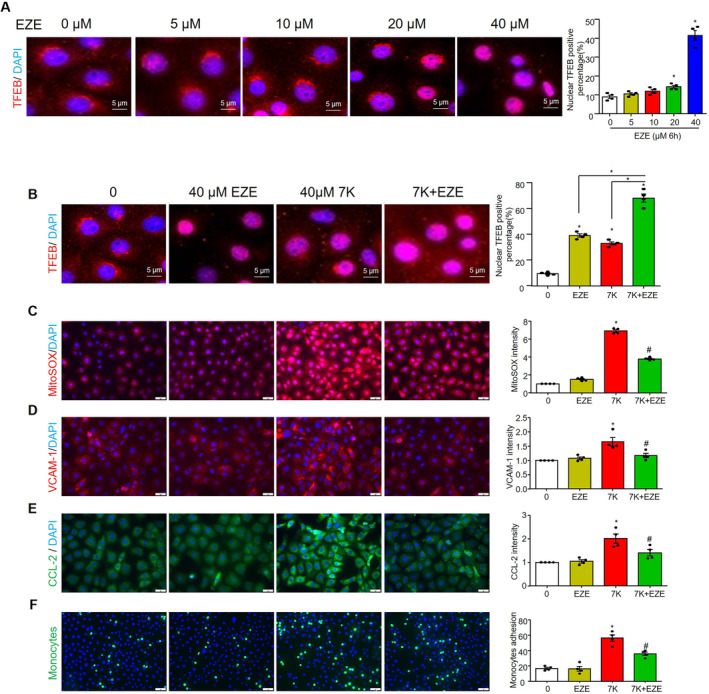
MCECs were cultured in low glucose DMEM with 5% FBS, then treated with EZE with or without 7‐ketocholesterol for the indicated time. A, Representative immunofluorescence images and quantification show the effect of ezetimibe on nuclear TFEB positive percentage. B, Representative immunofluorescence images and quantification show the effect of ezetimibe and 7‐ketocholesterol on nuclear TFEB positive percentage. Representative immunofluorescence images and quantification of mitochondrial superoxide (C), proinflammatory proteins vascular cell adhesion molecule 1 (VCAM‐1) (D) and CCL2 (E), and monocyte adhesion (F). Scale bar=20 μm. * vs 0, # vs 7‐keto, P<0.05 (n=4). CCL2 indicates C‐C motif chemokine ligand 2; EZE, ezetimibe; MCECs, mouse cardiac endothelial cells; TFEB, transcriptional factor EB; and VCAM‐1, vascular cell adhesion molecule 1.
DISCUSSION
An increasing number of studies have shown that the prevalence of CMD is high in patients diagnosed with hypercholesterolemia. 20 , 33 , 34 , 35 Even in patients with hypercholesterolemia without obstructive CAD or heart failure, CMD with reduced CFR has been observed. Despite the clinical significance of this disease, current management guidelines in the United States do not provide specific recommendations for the treatment of CMD caused by hypercholesterolemia. 4 , 34 One reason for this is that preclinical studies of hypercholesterolemia and CMD are significantly less extensive than those of obstructive vascular diseases such as atherosclerosis. A recent study reported that hypercholesterolemia resulted in reduced endomyocardial CFR and capillary density in a mini‐pig model without coronary stenosis. 36 , 37 Moreover, a high‐cholesterol diet has been shown to induce advanced occlusive coronary atherothrombosis and heart failure, leading to increased death, in scavenger receptor class B type 1 and low‐density lipoprotein receptor double knockout mice. 38 , 39 These findings have established a clear link between hypercholesterolemia and CMD in animal studies and their contribution to obstructive CAD. 38 , 39 , 40 , 41 , 42 However, the precise pathophysiological mechanisms underlying the initiation and progression of CMD caused by hypercholesterolemia remain unclear and understudied. In the present study, we established a hypercholesterolemia model in mice by feeding them a high‐fat and high‐cholesterol PD. Noninvasive imaging technique showed that PD decreased hyperemic coronary blood flow velocity and CFR as time increased (Figure 1). CFR began to decrease after 6 to 8 weeks of PD treatment, but no changes in cardiac remodeling or function were observed. Therefore, we used this model throughout this study to explore the early effects of hypercholesterolemia on the coronary microcirculation and myocardial tissue before the onset of cardiac remodeling and dysfunction.
Next, we characterized PD‐induced CMD by evaluating a range of cardiovascular‐related pathological changes in the coronary arterioles, capillaries, or myocardium, including arterial wall thickening and lipid accumulation, microvascular rarefaction, thrombosis, and inflammation. Histopathological analysis revealed that PD‐induced CMD was not associated with coronary arterioles wall remodeling that may cause vascular lumen reduction and insufficient blood perfusion (Figure 2A). Moreover, capillary density and pericyte coverage were not affected by PD (Figure 2B), suggesting that PD was insufficient to induce microvascular rarefaction and decrease blood perfusion. These data suggest that PD‐induced CMD may result from functional, rather than structural, changes in the coronary microcirculation.
Endothelial dysfunction is associated with increased permeability of plasma lipoproteins to the endothelium and their retention in the subendothelial area. 43 , 44 Increased retention of lipoproteins in the subendothelial area may lead to the uptake of lipoproteins by resident macrophages or nearby ECs or smooth muscle cells (SMCs). 43 , 44 The lipid content of the uptaken lipoproteins is then stored intracellularly as LDs. 44 , 45 These mechanisms may lead to lipid deposition in the vascular wall, forming fatty streaks at an early stage of occlusive vascular diseases. 44 , 45 Since hypercholesterolemia is a common risk factor for CMD and occlusive CAD, we analyzed lipid deposition in coronary arterioles by oil Red O staining. The results showed that the incidence of fatty streak formation in the coronary arterioles wall was increased, with fatty streaked arterioles observed in 3 of 8 mice examined (Figure 2C). Gene expression analysis further showed that PD treatment tends to increase lipogenesis and LD formation in the vessel wall (Figure 3A). The specific location of the lipid deposition is unclear, but it may accumulate in the subendothelium or in vascular wall cells, including macrophages, ECs, or SMCs. In particular, recent studies have shown that LDs in ECs promote atherosclerosis and hypertension in mice. 46 , 47 Therefore, it is possible that PD increases LD accumulation in coronary arterioles ECs, contributing to endothelial dysfunction and CMD, which deserves further investigation. Nonetheless, to our knowledge, these data for the first time demonstrate that PD‐induced CMD is associated with increased lipid deposition in the vessel wall of coronary arterioles in mice.
Recently, studies have reported that PD induced severe coronary atherothrombosis in scavenger receptor class B type 1 and low‐density lipoprotein receptor double knockout mice. 38 , 39 , 48 These knockout mice have plasma cholesterol levels of ≈750 mg/dL, an extremely high concentration not usually observed in patients with metabolic disorders. 48 In contrast, we and others have recently reported that wild‐type mice fed with PD developed a mild hypercholesterolemia as early as 2 weeks, with a 2‐fold increase in plasma cholesterol levels compared with ND‐fed controls, 25 , 49 which may better represent early‐stage metabolic disorders. Interestingly, the present study showed that CD41‐positive platelet aggregates were found in the lumen of coronary arterioles in 2 of 8 mice fed with PD, but not in ND‐fed controls (Figure 2D). The increased incidence of platelet aggregates was consistent with the upregulated gene expression of tissue factor in the heart or aortic tissues (Figure 3B). Therefore, these results suggest that mild hypercholesterolemia induced by PD promotes CMD, accompanied by a shift in coronary arterioles ECs from an antithrombotic to prothrombotic state.
Accumulating evidence suggests that the activation of the endothelial NLRP3 inflammasome serves as a critical initiating mechanism in the pathogenesis of vasculopathies associated with metabolic disorders, including hypercholesterolemia, hyperglycemia, and dyslipidemia, as well as other conditions such as smoking and sepsis. 21 , 31 , 50 , 51 , 52 Endothelial inflammasome activation leads to increased activity of caspase‐1, an inflammatory caspase, and production of proinflammatory cytokines such as interleukin‐1β and interleukin‐18. In addition, endothelial inflammasome activation may lead to caspase‐11 activation and gasdermin D–dependent pore formation in the plasma membrane, causing pyroptotic cell death. HMGB‐1 is being increasingly recognized as a key damage‐associated molecular pattern molecule actively released by immune cells or passively released from dying cells upon infection or sterile inflammation. 53 Our recent studies have shown that endothelial NLRP3 inflammasome activation promotes HMGB‐1 release from ECs in response to various cardiovascular risk factors. 54 , 55 , 56 HMGB‐1 can activate receptor for advanced glycation end products or Toll‐like 2/4 receptors to initiate various inflammatory responses or cell damage. 57 In particular, HMGB‐1 has been shown to be a potent vascular permeability factor that can lead to the loss of endothelial barrier function during metabolic disorders. 58 , 59 Upregulation of adhesion molecules, including VCAM‐1 and intercellular adhesion molecule 1, represents the initial inflammatory response of the ECs, which mediate the adhesion of leukocytes (eg, lymphocytes, monocytes, eosinophils, and basophils) to the vascular endothelium. 44 , 60 Recent studies have revealed that interleukin1β, interleukin‐18, and HMGB‐1 can upregulate the expression of these adhesion molecules in ECs. 61 PD has been shown to increase systemic inflammation in mice, 62 but its effect on inflammation in CMD has not been investigated. In the present study, we found that PD significantly increased the activation of the endothelial inflammasome and inflammatory responses in coronary arterioles, as indicated by elevated caspase‐1, HMGB‐1, and VCAM‐1 (Figure 2E‐G). In addition, there were more CD45‐positive immune cells in the myocardium of PD‐fed mice than ND‐fed controls (Figure 2H), suggesting that increased leukocyte adhesion and transendothelial migration contribute to cardiac inflammation. This increase in cardiac inflammation was also confirmed by our gene expression analysis of various inflammatory mediators such as interleukin‐6, interleukin‐8, and TNF‐α (Figure 3C). Interestingly, recent studies have shown that inflammatory cytokines such as TNF‐α promote LD formation in ECs. 63 In addition, these inflammatory cytokines increase the expression of chemokines, adhesion molecules, and thrombotic modulators such as tissue factor, which may also facilitate interactions with platelets and their activation and aggregation. 10 , 64 , 65 In this regard, the activation of the endothelial inflammasome and inflammatory responses may crosstalk with coronary lipid accumulation and thrombosis. These findings support the view that hypercholesterolemia upregulates the activation of the endothelial NLRP3 inflammasome in the coronary microcirculation and promotes the development of CMD, and that cardiac inflammation may be an early effect associated with CMD.
Another important finding of the present study is that PD‐induced CMD is associated with the upregulation of lysosomal signaling pathways. Specifically, we observed upregulation of lysosomal pathway proteins, including TFEB, LAMP‐1, LAMP‐2A, and TRPML‐1, in the endothelium of coronary arterioles (Figure 4). Lysosomal signaling pathways play a critical role in regulating endothelial homeostasis under various metabolic stresses. 66 , 67 For example, lysosomal function is integral to cholesterol homeostasis and is a key processing center for low‐density lipoprotein–derived cholesterol. 68 Within lysosomes, cholesterol esters are hydrolyzed by lysosomal acid lipase, and free cholesterols interact with Niemann‐Pick disease, type C (NPC) 1/2, and are transported to extralysosomal destinations. 69 Lysosomal membrane proteins LAMP‐1 and LAMP‐2A are essential for lysosomal function and LAMP‐1/2 deficiency causes embryonic lethality in mice, accompanied by the accumulation of cholesterol and autophagic vacuoles in various tissues, including ECs and Schwann cells. 70 Overexpression of LAMP‐2A, but not LAMP‐1, rescues cholesterol accumulation in LAMP‐1/2 double‐deficient fibroblasts. 70 In addition, genetic deletion of LAMP‐2 leads to luminal stenosis and medial thickening in coronary arteries. 71 In fact, LAMP‐2A‐dependent chaperone‐mediated autophagy plays a critical role in lipophagy, a lysosome‐dependent process of LD catabolism. 72 These previous findings highlight the more significant role of LAMP‐2 in lysosomal function and lipid handling compared with LAMP‐1. Furthermore, TRPML‐1 is a lysosomal Ca2+ channel, and mutations in TRPML‐1 cause mucolipidosis type IV, a severe lysosomal storage disorder. 73 Increased TRPML‐1 activity promotes lysosomal trafficking and fusion with intracellular organelles, including multivesicular bodies or autophagosomes, thereby facilitating lysosome‐dependent degradation of these organelles and their lipid and protein contents. 74 TRPML‐1–mediated lysosomal Ca2+ release also activates calcineurin, which in turn dephosphorylates TFEB, leading to its nuclear translocation. 75 TFEB is a master transcription factor that controls genes involved in lysosomal and autophagy pathways. TFEB has been shown to be involved in angiogenesis and EC survival. 76 We recently demonstrated that simvastatin induces TFEB‐mediated lysosomal biogenesis, thereby preventing lipotoxicity‐induced NLRP3 inflammasome activation and injury in ECs. 67 In addition to ECs, TFEB suppression also contributes to SMC dedifferentiation, whereas activation of TFEB‐lysosomal signaling promotes SMC differentiation and inhibits neointima formation under hypercholesterolemia. 22 , 25 Therefore, based on these previous observations, we speculate that hypercholesterolemia upregulates lysosome signaling as an early adaptive response to lipotoxicity, and alleviates lipotoxicity‐induced cellular stresses, including endoplasmic reticulum stress and mitochondrial stresses, thereby contributing to the maintenance of coronary microcirculatory homeostasis.
To date, there are no established therapeutic interventions that have been clearly proven to be effective in treating CMD. Current treatments are mainly aimed at alleviating symptoms and reducing the risk of adverse events. Commonly used drugs include aspirin, statins, angiotensin‐converting enzyme inhibitors, and angiotensin receptor blockers. 4 In addition, ezetimibe has been shown to improve atherogenic lipid profiles, insulin resistance, and liver dysfunction in patients with hypercholesterolemia. 77 , 78 In terms of its effect on CAD, ezetimibe, either alone or in combination with simvastatin, has demonstrated the potential to improve endothelial and microvascular function in patients with glycemic disorders and CAD. 79 , 80 Animal studies have also shown that ezetimibe reduces atherosclerosis and vascular inflammation. 81 , 82 Notably, ezetimibe showed a more pronounced inhibitory effect on total oxysterols, 7‐ketocholesterol, and 27‐hydroxycholesterol than on total cholesterol in mice with hypercholesterolemia. 80 In the present study, we demonstrated that ezetimibe administration significantly alleviated PD‐induced steatohepatitis and hypercholesterolemia (Figure 5), and prevented PD‐induced CMD and inflammatory responses in the coronary microcirculation (Figure 6 and Figure 7). Our results provide the first evidence that ezetimibe may be a promising candidate for the treatment and prevention of hypercholesterolemia‐induced CMD. Our data also provide insights into alternative therapeutic strategies for patients who cannot tolerate statins or who have inadequate response to statin therapy alone. Statins remain the cornerstone of treatment for hypercholesterolemia‐associated cardiovascular disease, and therefore future studies should evaluate whether statins alone or in combination with ezetimibe can provide comparable or better efficacy than ezetimibe alone in the treatment of hypercholesterolemia‐induced CMD.
Next, we further elucidated the functional significance of upregulated TFEB‐lysosomal signaling in endothelial inflammation and injury in the coronary microcirculation in vitro. A 7‐ketocholesterol–induced cell injury model was established in cultured MCECs. 7‐Ketocholesterol is the major oxidation product of cholesterol found in human atherosclerotic plaques and has shown greater atherogenic potential than cholesterol in animal studies. 83 Previous reports have revealed that 7‐ketocholesterol induces mitochondrial dysfunction in ECs, characterized by altered mitochondria morphology, decreased mitochondrial membrane potential, and elevated mitochondrial superoxide levels. 84 , 85 In addition, 7‐ketocholesterol has been shown to have significant proinflammatory effects both in vitro and in vivo. 86 , 87 We have also recently demonstrated that 7‐ketocholesterol causes endothelial dysfunction and injury associated with increased oxidative stress, NLRP3 inflammasome activation, and inflammation in cultured murine ECs isolated from large carotid arteries. 30 , 88 The function and integrity of lysosomes are essential for maintaining cellular homeostasis, 89 and lysosomal dysfunction and damage can trigger or promote inflammasome activation, endoplasmic reticulum stress, or mitochondrial injury in ECs under various risk factors, including free fatty acids, cholesterol crystals, and Lactobacillus casei wall components. 51 , 55 , 56 , 90 , 91 In this study, we demonstrated for the first time that 7‐ketocholesterol induced mitochondrial ROS production, inflammation, and cellular death in cultured MCECs (Figure 8). Moreover, consistent with our animal studies, we found that 7‐ketocholesterol also upregulated TFEB‐mediated lysosome signaling (Figure 8). Importantly, inhibition of lysosomal function with bafilomycin A1 suppressed TFEB activation and exacerbated 7‐ketocholesterol–induced inflammation and cell death (Figure 9). Therefore, our results support the view that upregulated TFEB‐mediated lysosomal signaling in ECs serves as an adaptive protective mechanism to mitigate the deleterious effects associated with lipotoxicity, including mitochondrial oxidative stress, inflammatory responses, and cellular damage. The present study did not attempt to decipher the underlying mechanisms of activation of TFEB‐lysosomal signaling under metabolic stress or lipotoxic conditions. Nuclear translocation of TFEB requires its dephosphorylation by inhibition of protein kinases, such as mammalian target of rapamycin complex 1, or activation of protein phosphatases such as calcineurin and protein phosphatase 2A. 92 , 93 Interestingly, sustained elevated ROS levels have been shown to inhibit mammalian target of rapamycin complex 1 and enhance calcineurin or PP2A activities. 92 , 94 We recently demonstrated that 7‐ketocholesterol triggers TRPML‐1–mediated lysosome‐Ca2+ release in SMCs. 95 The lysosomal Ca2+ release may activate calcineurin and in turn promote TFEB activation. 95 , 96 Therefore, intracellular ROS levels and TRPML‐1–Ca2+ release may be involved in TFEB activation in MCECs, which deserves further investigation in future studies.
Finally, another important finding of this study is that ezetimibe could synergize with 7‐ketocholesterol to increase TFEB–lysosomal signaling and prevent 7‐ketocholesterol–induced mitochondrial ROS and inflammation in cultured MCECs (Figure 10). Ezetimibe is a pharmacological inhibitor of NPC1L1, a cholesterol transporter that is highly expressed in the apical membrane of enterocytes and the canalicular membrane of hepatocytes. The main mechanism of ezetimibe's beneficial effects is inhibiting intestinal and hepatic cholesterol absorption through NPC1L1, thereby lowering cholesterol. 97 Interestingly, recent studies have shown that ezetimibe could inhibit oxidative stress, urokinase‐type plasminogen activator receptor expression, or interleukin‐6 secretion in human umbilical vein endothelial cells, and have direct vasodilatory effects on rat mesenteric resistance arteries. 98 , 99 , 100 These previous studies and the findings of this study support the view that ezetimibe can affect the cardiovascular system independently of its lipid‐lowering effects, and that upregulation of TFEB–lysosomal signaling may represent one of the non–lipid‐lowering effects of ezetimibe. As NPC1L1 is highly expressed in the liver and small intestine but relatively low in other sites, the mechanism of action of ezetimibe in the cardiovascular system may involve targets other than NPC1L1. Ezetimibe has recently been shown to activate TFEB in hepatocytes via stimulation of 5′ AMP–activated protein kinase. 101 The exact mechanism by which ezetimibe activates TFEB–lysosomal signaling and the possible role of 5′ AMP–activated protein kinase in this effect warrants further investigation.
Although we provided significant evidence that TFEB‐mediated lysosomal signaling activation can prevent hypercholesterolemia‐induced CMD, there are some limitations to our study. First, it is possible that lysosomal signaling could represent a secondary, broadly protective mechanism activated in ECs in response to various damage‐associated molecular patterns, including but not limited to hypercholesterolemia. Further studies are warranted to investigate lysosomal signaling induction under other damage‐associated molecular patterns known to activate ECs. Second, although we demonstrated that inhibition of lysosomal function via bafilomycin A1 exacerbated inflammation and cell death in cultured MCECs, the lack of direct manipulation of lysosomal signaling in the microcirculation system in vivo limits our ability to provide more definitive mechanistic insights. Future studies should investigate the role of lysosomal genes, such as TFEB and LAMPs, by employing tissue‐specific knockout or knock‐in models in microvascular cells, including endothelial cells and pericytes, within the context of hypercholesterolemia‐induced CMD.
In summary, the present study demonstrated that CMD develops in a mouse model of mild hypercholesterolemia before the onset of cardiac remodeling and dysfunction. The most notable pathological changes associated with CMD were enhanced inflammation in the coronary microcirculation and myocardium. Moreover, the progression of CMD was accompanied by adaptive upregulation of TFEB‐lysosomal signaling in coronary ECs, which may attenuate the inflammation and injury in ECs. Ezetimibe, a Food and Drug Administration–approved cholesterol‐lowering drug, effectively reversed these deleterious effects, which is thought to be achieved by activating TFEB–lysosomal signaling to exert its non–lipid‐lowering effect. Targeting TFEB–lysosomal signaling may be a promising therapeutic strategy to prevent hypercholesterolemia‐induced CMD.
Sources of Funding
This work was supported by grants from the National Institutes of Health (R01HL150007, R01HL122937).
Disclosures
None.
Supporting information
Table S1
Acknowledgments
Drs X. Li and Zhang designed the research and wrote the manuscript; Dr Y‐T. Wang wrote the manuscript; Drs Wang, Moura, and Zuo performed the research; Drs Wang, Moura, Zuo, and Zhou analyzed the data; Drs Z. Wang, Roudbari, Hu, and P‐L. Li edited the manuscript; and all authors read and approved the final manuscript.
This manuscript was sent to June‐Wha Rhee, MD, Associate Editor, for review by expert referees, editorial decision, and final disposition.
Preprint posted on BioRxiv July 12, 2024. doi: https://doi.org/10.1101/2024.07.10.603000.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.124.037460
For Sources of Funding and Disclosures, see page 22.
Contributor Information
Yang Zhang, Email: yzhan219@central.uh.edu.
Xiang Li, Email: xli61@central.uh.edu.
References
- 1. Guilbert JJ. The world health report 2002—reducing risks, promoting healthy life. Educ Health (Abingdon). 2003;16:230. doi: 10.1080/1357628031000116808 [DOI] [PubMed] [Google Scholar]
- 2. Camici PG, d'Amati G, Rimoldi O. Coronary microvascular dysfunction: mechanisms and functional assessment. Nat Rev Cardiol. 2015;12:48–62. doi: 10.1038/nrcardio.2014.160 [DOI] [PubMed] [Google Scholar]
- 3. Godo S, Suda A, Takahashi J, Yasuda S, Shimokawa H. Coronary microvascular dysfunction. Arterioscler Thromb Vasc Biol. 2021;41:1625–1637. doi: 10.1161/ATVBAHA.121.316025 [DOI] [PubMed] [Google Scholar]
- 4. Marano P, Wei J, Merz CNB. Coronary microvascular dysfunction: what clinicians and investigators should know. Curr Atheroscler Rep. 2023;25:435–446. doi: 10.1007/s11883-023-01116-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brainin P, Frestad D, Prescott E. The prognostic value of coronary endothelial and microvascular dysfunction in subjects with normal or non‐obstructive coronary artery disease: a systematic review and meta‐analysis. Int J Cardiol. 2018;254:1–9. doi: 10.1016/j.ijcard.2017.10.052 [DOI] [PubMed] [Google Scholar]
- 6. Chaudhary R, Bashline M, Novelli EM, Bliden KP, Tantry US, Olafiranye O, Rahman A, Gurbel PA, Pacella JJ. Sex‐related differences in clinical outcomes among patients with myocardial infarction with nonobstructive coronary artery disease: a systematic review and meta‐analysis. Int J Cardiol. 2022;369:1–4. doi: 10.1016/j.ijcard.2022.07.050 [DOI] [PubMed] [Google Scholar]
- 7. Schindler TH, Dilsizian V. Coronary microvascular dysfunction: clinical considerations and noninvasive diagnosis. JACC Cardiovasc Imaging. 2020;13:140–155. doi: 10.1016/j.jcmg.2018.11.036 [DOI] [PubMed] [Google Scholar]
- 8. Marinescu MA, Loffler AI, Ouellette M, Smith L, Kramer CM, Bourque JM. Coronary microvascular dysfunction, microvascular angina, and treatment strategies. JACC Cardiovasc Imaging. 2015;8:210–220. doi: 10.1016/j.jcmg.2014.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ong P, Athanasiadis A, Borgulya G, Mahrholdt H, Kaski JC, Sechtem U. High prevalence of a pathological response to acetylcholine testing in patients with stable angina pectoris and unobstructed coronary arteries. The ACOVA study (abnormal COronary VAsomotion in patients with stable angina and unobstructed coronary arteries). J Am Coll Cardiol. 2012;59:655–662. doi: 10.1016/j.jacc.2011.11.015 [DOI] [PubMed] [Google Scholar]
- 10. Huang AL, Vita JA. Effects of systemic inflammation on endothelium‐dependent vasodilation. Trends Cardiovasc Med. 2006;16:15–20. doi: 10.1016/j.tcm.2005.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vanhoutte PM, Shimokawa H, Feletou M, Tang EH. Endothelial dysfunction and vascular disease—a 30th anniversary update. Acta Physiol (Oxf). 2017;219:22–96. doi: 10.1111/apha.12646 [DOI] [PubMed] [Google Scholar]
- 12. Godo S, Takahashi J, Yasuda S, Shimokawa H. Endothelium in coronary macrovascular and microvascular diseases. J Cardiovasc Pharmacol. 2021;78:S19–S29. doi: 10.1097/FJC.0000000000001089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shimokawa H. Hydrogen peroxide as an endothelium‐derived hyperpolarizing factor. Pflugers Arch. 2010;459:915–922. doi: 10.1007/s00424-010-0790-8 [DOI] [PubMed] [Google Scholar]
- 14. Incalza MA, D'Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc Pharmacol. 2018;100:1–19. doi: 10.1016/j.vph.2017.05.005 [DOI] [PubMed] [Google Scholar]
- 15. Rossaint J, Margraf A, Zarbock A. Role of platelets in leukocyte recruitment and resolution of inflammation. Front Immunol. 2018;9:2712. doi: 10.3389/fimmu.2018.02712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Del Buono MG, Montone RA, Camilli M, Carbone S, Narula J, Lavie CJ, Niccoli G, Crea F. Coronary microvascular dysfunction across the spectrum of cardiovascular diseases: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2021;78:1352–1371. doi: 10.1016/j.jacc.2021.07.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bergstrand L, Olsson AG, Erikson U, Holme I, Johansson J, Kaijser L, Lassvik C, Molgaard J, Nilsson S, Stenport G, et al. The relation of coronary and peripheral arterial disease to the severity of femoral atherosclerosis in hypercholesterolaemia. J Intern Med. 1994;236:367–375. doi: 10.1111/j.1365-2796.1994.tb00812.x [DOI] [PubMed] [Google Scholar]
- 18. Lettino M. Management of hypercholesterolemia in patients with acute coronary syndrome: current mechanisms and future perspectives. G Ital Cardiol (Rome). 2016;17:31S–37S. doi: 10.1714/2254.24282 [DOI] [PubMed] [Google Scholar]
- 19. Zhang S, Picard MH, Vasile E, Zhu Y, Raffai RL, Weisgraber KH, Krieger M. Diet‐induced occlusive coronary atherosclerosis, myocardial infarction, cardiac dysfunction, and premature death in scavenger receptor class B type I‐deficient, hypomorphic apolipoprotein ER61 mice. Circulation. 2005;111:3457–3464. doi: 10.1161/CIRCULATIONAHA.104.523563 [DOI] [PubMed] [Google Scholar]
- 20. van Schalkwijk DL, Widdershoven J, Magro M, Smaardijk V, Bekendam M, Vermeltfoort I, Mommersteeg P. Clinical and psychological characteristics of patients with ischemia and non‐obstructive coronary arteries (INOCA) and obstructive coronary artery disease. Am Heart J Plus. 2023;27:100282. doi: 10.1016/j.ahjo.2023.100282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xia M, Boini KM, Abais JM, Xu M, Zhang Y, Li PL. Endothelial NLRP3 inflammasome activation and enhanced neointima formation in mice by adipokine visfatin. Am J Pathol. 2014;184:1617–1628. doi: 10.1016/j.ajpath.2014.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang YT, Chen J, Li X, Umetani M, Chen Y, Li PL, Zhang Y. Contribution of transcription factor EB to adipoRon‐induced inhibition of arterial smooth muscle cell proliferation and migration. Am J Physiol Cell Physiol. 2019;317:C1034–C1047. doi: 10.1152/ajpcell.00294.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vion AC, Kheloufi M, Hammoutene A, Poisson J, Lasselin J, Devue C, Pic I, Dupont N, Busse J, Stark K, et al. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc Natl Acad Sci USA. 2017;114:E8675–E8684. doi: 10.1073/pnas.1702223114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang J, Yu J, Li D, Yu S, Ke J, Wang L, Wang Y, Qiu Y, Gao X, Zhang J, et al. Store‐operated calcium entry‐activated autophagy protects EPC proliferation via the CAMKK2‐MTOR pathway in ox‐LDL exposure. Autophagy. 2017;13:82–98. doi: 10.1080/15548627.2016.1245261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang YT, Li X, Chen J, McConnell BK, Chen L, Li PL, Chen Y, Zhang Y. Activation of TFEB ameliorates dedifferentiation of arterial smooth muscle cells and neointima formation in mice with high‐fat diet. Cell Death Dis. 2019;10:676. doi: 10.1038/s41419-019-1931-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mauvezin C, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V‐ATPase‐dependent acidification and Ca‐P60A/SERCA‐dependent autophagosome‐lysosome fusion. Autophagy. 2015;11:1437–1438. doi: 10.1080/15548627.2015.1066957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee JH, McBrayer MK, Wolfe DM, Haslett LJ, Kumar A, Sato Y, Lie PP, Mohan P, Coffey EE, Kompella U, et al. Presenilin 1 maintains lysosomal Ca(2+) homeostasis via TRPML1 by regulating vATPase‐mediated lysosome acidification. Cell Rep. 2015;12:1430–1444. doi: 10.1016/j.celrep.2015.07.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang W, Gao Q, Yang M, Zhang X, Yu L, Lawas M, Li X, Bryant‐Genevier M, Southall NT, Marugan J, et al. Up‐regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc Natl Acad Sci USA. 2015;112:E1373–E1381. doi: 10.1073/pnas.1419669112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Di Paola S, Medina DL. TRPML1‐/TFEB‐dependent regulation of lysosomal exocytosis. Methods Mol Biol. 2019;1925:143–144. doi: 10.1007/978-1-4939-9018-4_12 [DOI] [PubMed] [Google Scholar]
- 30. Yuan X, Bhat OM, Zou Y, Li X, Zhang Y, Li PL. Endothelial acid sphingomyelinase promotes NLRP3 inflammasome and neointima formation during hypercholesterolemia. J Lipid Res. 2022;63:100298. doi: 10.1016/j.jlr.2022.100298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen Y, Pitzer AL, Li X, Li PL, Wang L, Zhang Y. Instigation of endothelial Nlrp3 inflammasome by adipokine visfatin promotes inter‐endothelial junction disruption: role of HMGB1. J Cell Mol Med. 2015;19:2715–2727. doi: 10.1111/jcmm.12657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Y, Li X, Pitzer AL, Chen Y, Wang L, Li PL. Coronary endothelial dysfunction induced by nucleotide oligomerization domain‐like receptor protein with pyrin domain containing 3 inflammasome activation during hypercholesterolemia: beyond inflammation. Antioxid Redox Signal. 2015;22:1084–1096. doi: 10.1089/ars.2014.5978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yokoyama I, Ohtake T, Momomura S, Nishikawa J, Sasaki Y, Omata M. Reduced coronary flow reserve in hypercholesterolemic patients without overt coronary stenosis. Circulation. 1996;94:3232–3238. doi: 10.1161/01.cir.94.12.3232 [DOI] [PubMed] [Google Scholar]
- 34. Vuorio A, Kovanen PT, Raal FJ. Coronary microcirculatory dysfunction in hypercholesterolemic patients with COVID‐19: potential benefit from cholesterol‐lowering treatment. Ann Med. 2023;55:2199218. doi: 10.1080/07853890.2023.2199218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Galderisi M, de Simone G, Cicala S, Parisi M, D'Errico A, Innelli P, de Divitiis M, Mondillo S, de Divitiis O. Coronary flow reserve in hypertensive patients with hypercholesterolemia and without coronary heart disease. Am J Hypertens. 2007;20:177–183. doi: 10.1016/j.amjhyper.2006.06.017 [DOI] [PubMed] [Google Scholar]
- 36. Sorop O, van den Heuvel M, van Ditzhuijzen NS, de Beer VJ, Heinonen I, van Duin RW, Zhou Z, Koopmans SJ, Merkus D, van der Giessen WJ, et al. Coronary microvascular dysfunction after long‐term diabetes and hypercholesterolemia. Am J Physiol Heart Circ Physiol. 2016;311:H1339–H1351. doi: 10.1152/ajpheart.00458.2015 [DOI] [PubMed] [Google Scholar]
- 37. Theilmeier G, Verhamme P, Dymarkowski S, Beck H, Bernar H, Lox M, Janssens S, Herregods MC, Verbeken E, Collen D, et al. Hypercholesterolemia in minipigs impairs left ventricular response to stress: association with decreased coronary flow reserve and reduced capillary density. Circulation. 2002;106:1140–1146. doi: 10.1161/01.cir.0000026805.41747.54 [DOI] [PubMed] [Google Scholar]
- 38. Fuller M, Dadoo O, Serkis V, Abutouk D, MacDonald M, Dhingani N, Macri J, Igdoura SA, Trigatti BL. The effects of diet on occlusive coronary artery atherosclerosis and myocardial infarction in scavenger receptor class B, type 1/low‐density lipoprotein receptor double knockout mice. Arterioscler Thromb Vasc Biol. 2014;34:2394–2403. doi: 10.1161/ATVBAHA.114.304200 [DOI] [PubMed] [Google Scholar]
- 39. Liao J, Guo X, Wang M, Dong C, Gao M, Wang H, Kayoumu A, Shen Q, Wang Y, Wang F, et al. Scavenger receptor class B type 1 deletion led to coronary atherosclerosis and ischemic heart disease in low‐density lipoprotein receptor knockout mice on modified Western‐type diet. J Atheroscler Thromb. 2017;24:133–146. doi: 10.5551/jat.33019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pearson JT, Yoshimoto M, Chen YC, Sultani R, Edgley AJ, Nakaoka H, Nishida M, Umetani K, Waddingham MT, Jin HL, et al. Widespread coronary dysfunction in the absence of HDL receptor SR‐B1 in an ischemic cardiomyopathy mouse model. Sci Rep. 2017;7:18108. doi: 10.1038/s41598-017-18485-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gonzalez L, MacDonald ME, Deng YD, Trigatti BL. Hyperglycemia aggravates diet‐induced coronary artery disease and myocardial infarction in SR‐B1‐knockout/ApoE‐Hypomorphic mice. Front Physiol. 2018;9:1398. doi: 10.3389/fphys.2018.01398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nakaoka H, Nakagawa‐Toyama Y, Nishida M, Okada T, Kawase R, Yamashita T, Yuasa‐Kawase M, Nakatani K, Masuda D, Ohama T, et al. Establishment of a novel murine model of ischemic cardiomyopathy with multiple diffuse coronary lesions. PLoS One. 2013;8:e70755. doi: 10.1371/journal.pone.0070755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tabas I, Garcia‐Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209:13–22. doi: 10.1083/jcb.201412052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thayse K, Kindt N, Laurent S, Carlier S. VCAM‐1 target in non‐invasive imaging for the detection of atherosclerotic plaques. Biology (Basel). 2020;9:368. doi: 10.3390/biology9110368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Plakkal Ayyappan J, Paul A, Goo YH. Lipid droplet‐associated proteins in atherosclerosis (review). Mol Med Rep. 2016;13:4527–4534. doi: 10.3892/mmr.2016.5099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim B, Zhao W, Tang SY, Levin MG, Ibrahim A, Yang Y, Roberts E, Lai L, Li J, Assoian RK, et al. Endothelial lipid droplets suppress eNOS to link high fat consumption to blood pressure elevation. J Clin Invest. 2023;133:e173160. doi: 10.1172/JCI173160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu S, Offermanns S. Endothelial lipid droplets drive atherosclerosis and arterial hypertension. Trends Endocrinol Metab. 2024;35:453–455. doi: 10.1016/j.tem.2024.02.014 [DOI] [PubMed] [Google Scholar]
- 48. Medical Advisory S . Low‐density lipoprotein apheresis: an evidence‐based analysis. Ont Health Technol Assess Ser. 2007;7:1–101. [PMC free article] [PubMed] [Google Scholar]
- 49. Zhu M, Ji G, Jin G, Yuan Z. Different responsiveness to a high‐fat/cholesterol diet in two inbred mice and underlying genetic factors: a whole genome microarray analysis. Nutr Metab (Lond). 2009;6:43. doi: 10.1186/1743-7075-6-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou X, Wu Y, Ye L, Wang Y, Zhang K, Wang L, Huang Y, Wang L, Xian S, Zhang Y, et al. Aspirin alleviates endothelial gap junction dysfunction through inhibition of NLRP3 inflammasome activation in LPS‐induced vascular injury. Acta Pharm Sin B. 2019;9:711–723. doi: 10.1016/j.apsb.2019.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang Y, Chen Y, Zhang Y, Li PL, Li X. Contribution of cathepsin B‐dependent Nlrp3 inflammasome activation to nicotine‐induced endothelial barrier dysfunction. Eur J Pharmacol. 2019;865:172795. doi: 10.1016/j.ejphar.2019.172795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen Y, Li X, Boini KM, Pitzer AL, Gulbins E, Zhang Y, Li PL. Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim Biophys Acta. 2015;1853:396–408. doi: 10.1016/j.bbamcr.2014.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen Y, Wang L, Pitzer AL, Li X, Li PL, Zhang Y. Contribution of redox‐dependent activation of endothelial Nlrp3 inflammasomes to hyperglycemia‐induced endothelial dysfunction. J Mol Med (Berl). 2016;94:1335–1347. doi: 10.1007/s00109-016-1481-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jia C, Zhang J, Chen H, Zhuge Y, Chen H, Qian F, Zhou K, Niu C, Wang F, Qiu H, et al. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis. 2019;10:778. doi: 10.1038/s41419-019-2021-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang L, Chen Y, Li X, Zhang Y, Gulbins E, Zhang Y. Enhancement of endothelial permeability by free fatty acid through lysosomal cathepsin B‐mediated Nlrp3 inflammasome activation. Oncotarget. 2016;7:73229–73241. doi: 10.18632/oncotarget.12302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Andersson U, Yang H. HMGB1 is a critical molecule in the pathogenesis of gram‐negative sepsis. J Intensive Med. 2022;2:156–166. doi: 10.1016/j.jointm.2022.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhao MJ, Jiang HR, Sun JW, Wang ZA, Hu B, Zhu CR, Yin XH, Chen MM, Ma XC, Zhao WD, et al. Roles of RAGE/ROCK1 pathway in HMGB1‐induced early changes in barrier permeability of human pulmonary microvascular endothelial cell. Front Immunol. 2021;12:697071. doi: 10.3389/fimmu.2021.697071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huang W, Liu Y, Li L, Zhang R, Liu W, Wu J, Mao E, Tang Y. HMGB1 increases permeability of the endothelial cell monolayer via RAGE and Src family tyrosine kinase pathways. Inflammation. 2012;35:350–362. doi: 10.1007/s10753-011-9325-5 [DOI] [PubMed] [Google Scholar]
- 60. Cook‐Mills JM, Marchese ME, Abdala‐Valencia H. Vascular cell adhesion molecule‐1 expression and signaling during disease: regulation by reactive oxygen species and antioxidants. Antioxid Redox Signal. 2011;15:1607–1638. doi: 10.1089/ars.2010.3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, Suffredini AF. Inflammation‐promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–2660. doi: 10.1182/blood-2002-05-1300 [DOI] [PubMed] [Google Scholar]
- 62. Pindjakova J, Sartini C, Lo Re O, Rappa F, Coupe B, Lelouvier B, Pazienza V, Vinciguerra M. Gut Dysbiosis and adaptive immune response in diet‐induced obesity vs. systemic inflammation. Front Microbiol. 2017;8:1157. doi: 10.3389/fmicb.2017.01157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jung HS, Shimizu‐Albergine M, Shen X, Kramer F, Shao D, Vivekanandan‐Giri A, Pennathur S, Tian R, Kanter JE, Bornfeldt KE. TNF‐alpha induces acyl‐CoA synthetase 3 to promote lipid droplet formation in human endothelial cells. J Lipid Res. 2020;61:33–44. doi: 10.1194/jlr.RA119000256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263–271. doi: 10.1016/j.jacc.2013.02.092 [DOI] [PubMed] [Google Scholar]
- 65. Godo S, Takahashi J, Yasuda S, Shimokawa H. Role of inflammation in coronary epicardial and microvascular dysfunction. Eur Cardiol. 2021;16:e13. doi: 10.15420/ecr.2020.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fischer AW, Jaeckstein MY, Gottschling K, Heine M, Sass F, Mangels N, Schlein C, Worthmann A, Bruns OT, Yuan Y, et al. Lysosomal lipoprotein processing in endothelial cells stimulates adipose tissue thermogenic adaptation. Cell Metab. 2021;33:547–564.e7. doi: 10.1016/j.cmet.2020.12.001 [DOI] [PubMed] [Google Scholar]
- 67. Zhang Y, Wang YT, Koka S, Zhang Y, Hussain T, Li X. Simvastatin improves lysosome function via enhancing lysosome biogenesis in endothelial cells. Front Biosci (Landmark Ed). 2020;25:283–298. doi: 10.2741/4807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Thelen AM, Zoncu R. Emerging roles for the lysosome in lipid metabolism. Trends Cell Biol. 2017;27:833–850. doi: 10.1016/j.tcb.2017.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Meng Y, Heybrock S, Neculai D, Saftig P. Cholesterol handling in lysosomes and beyond. Trends Cell Biol. 2020;30:452–466. doi: 10.1016/j.tcb.2020.02.007 [DOI] [PubMed] [Google Scholar]
- 70. Eskelinen EL, Schmidt CK, Neu S, Willenborg M, Fuertes G, Salvador N, Tanaka Y, Lullmann‐Rauch R, Hartmann D, Heeren J, et al. Disturbed cholesterol traffic but normal proteolytic function in LAMP‐1/LAMP‐2 double‐deficient fibroblasts. Mol Biol Cell. 2004;15:3132–3145. doi: 10.1091/mbc.e04-02-0103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nguyen HT, Noguchi S, Sugie K, Matsuo Y, Nguyen CTH, Koito H, Shiojima I, Nishino I, Tsukaguchi H. Small‐vessel vasculopathy due to aberrant autophagy in LAMP‐2 deficiency. Sci Rep. 2018;8:3326. doi: 10.1038/s41598-018-21602-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Alfaro IE, Albornoz A, Molina A, Moreno J, Cordero K, Criollo A, Budini M. Chaperone mediated autophagy in the crosstalk of neurodegenerative diseases and metabolic disorders. Front Endocrinol (Lausanne). 2018;9:778. doi: 10.3389/fendo.2018.00778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang Z, El Zowalaty AE, Li Y, Andersen CL, Ye X. Association of luteal cell degeneration and progesterone deficiency with lysosomal storage disorder mucolipidosis type IV in Mcoln1−/− mouse modeldagger. Biol Reprod. 2019;101:782–790. doi: 10.1093/biolre/ioz126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Li G, Li PL. Lysosomal TRPML1 channel: implications in cardiovascular and kidney diseases. Adv Exp Med Biol. 2021;1349:275–301. doi: 10.1007/978-981-16-4254-8_13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Scotto Rosato A, Montefusco S, Soldati C, Di Paola S, Capuozzo A, Monfregola J, Polishchuk E, Amabile A, Grimm C, Lombardo A, et al. TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKbeta/VPS34 pathway. Nat Commun. 2019;10:5630. doi: 10.1038/s41467-019-13572-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fan Y, Lu H, Liang W, Garcia‐Barrio MT, Guo Y, Zhang J, Zhu T, Hao Y, Zhang J, Chen YE. Endothelial TFEB (transcription factor EB) positively regulates postischemic angiogenesis. Circ Res. 2018;122:945–957. doi: 10.1161/CIRCRESAHA.118.312672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tamaki N, Ueno H, Morinaga Y, Shiiya T, Nakazato M. Ezetimibe ameliorates atherosclerotic and inflammatory markers, atherogenic lipid profiles, insulin sensitivity, and liver dysfunction in Japanese patients with hypercholesterolemia. J Atheroscler Thromb. 2012;19:532–538. doi: 10.5551/jat.10835 [DOI] [PubMed] [Google Scholar]
- 78. Park H, Shima T, Yamaguchi K, Mitsuyoshi H, Minami M, Yasui K, Itoh Y, Yoshikawa T, Fukui M, Hasegawa G, et al. Efficacy of long‐term ezetimibe therapy in patients with nonalcoholic fatty liver disease. J Gastroenterol. 2011;46:101–107. doi: 10.1007/s00535-010-0291-8 [DOI] [PubMed] [Google Scholar]
- 79. Settergren M, Bohm F, Ryden L, Pernow J, Kalani M. Lipid lowering versus pleiotropic effects of statins on skin microvascular function in patients with dysglycaemia and coronary artery disease. J Intern Med. 2009;266:492–498. doi: 10.1111/j.1365-2796.2009.02128.x [DOI] [PubMed] [Google Scholar]
- 80. Honda K, Matoba T, Antoku Y, Koga JI, Ichi I, Nakano K, Tsutsui H, Egashira K. Lipid‐lowering therapy with ezetimibe decreases spontaneous atherothrombotic occlusions in a rabbit model of plaque erosion: a role of serum oxysterols. Arterioscler Thromb Vasc Biol. 2018;38:757–771. doi: 10.1161/ATVBAHA.117.310244 [DOI] [PubMed] [Google Scholar]
- 81. Kuhlencordt PJ, Padmapriya P, Rutzel S, Schodel J, Hu K, Schafer A, Huang PL, Ertl G, Bauersachs J. Ezetimibe potently reduces vascular inflammation and arteriosclerosis in eNOS‐deficient ApoE ko mice. Atherosclerosis. 2009;202:48–57. doi: 10.1016/j.atherosclerosis.2008.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Davis HR Jr, Compton DS, Hoos L, Tetzloff G. Ezetimibe, a potent cholesterol absorption inhibitor, inhibits the development of atherosclerosis in ApoE knockout mice. Arterioscler Thromb Vasc Biol. 2001;21:2032–2038. doi: 10.1161/hq1201.100260 [DOI] [PubMed] [Google Scholar]
- 83. Lyons MA, Brown AJ. 7‐Ketocholesterol. Int J Biochem Cell Biol. 1999;31:369–375. doi: 10.1016/s1357-2725(98)00123-x [DOI] [PubMed] [Google Scholar]
- 84. Dasari B, Prasanthi JR, Meiers C, Singh BB, Ghribi O. Differential effects of the estrogen receptor agonist estradiol on toxicity induced by enzymatically‐derived or autoxidation‐derived oxysterols in human ARPE‐19 cells. Curr Eye Res. 2013;38:1159–1171. doi: 10.3109/02713683.2013.811257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Fu X, Huang X, Li P, Chen W, Xia M. 7‐Ketocholesterol inhibits isocitrate dehydrogenase 2 expression and impairs endothelial function via microRNA‐144. Free Radic Biol Med. 2014;71:1–15. doi: 10.1016/j.freeradbiomed.2014.03.010 [DOI] [PubMed] [Google Scholar]
- 86. Amaral J, Lee JW, Chou J, Campos MM, Rodriguez IR. 7‐Ketocholesterol induces inflammation and angiogenesis in vivo: a novel rat model. PLoS One. 2013;8:e56099. doi: 10.1371/journal.pone.0056099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Tani M, Kamata Y, Deushi M, Osaka M, Yoshida M. 7‐Ketocholesterol enhances leukocyte adhesion to endothelial cells via p38MAPK pathway. PLoS One. 2018;13:e0200499. doi: 10.1371/journal.pone.0200499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Koka S, Xia M, Chen Y, Bhat OM, Yuan X, Boini KM, Li PL. Endothelial NLRP3 inflammasome activation and arterial neointima formation associated with acid sphingomyelinase during hypercholesterolemia. Redox Biol. 2017;13:336–344. doi: 10.1016/j.redox.2017.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lakpa KL, Khan N, Afghah Z, Chen X, Geiger JD. Lysosomal stress response (LSR): physiological importance and pathological relevance. J Neuroimmune Pharmacol. 2021;16:219–237. doi: 10.1007/s11481-021-09990-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wang T, Xiao G, Lu Q, Zhou Y, Wang S, Liang X, Song Y, Xu M, Zhu Y, Li N. Synergistic lysosomal impairment and ER stress activation for boosted autophagy dysfunction based on Te double‐headed Nano‐bullets. Small. 2022;18:e2201585. doi: 10.1002/smll.202201585 [DOI] [PubMed] [Google Scholar]
- 91. de la Mata M, Cotan D, Villanueva‐Paz M, de Lavera I, Alvarez‐Cordoba M, Luzon‐Hidalgo R, Suarez‐Rivero JM, Tiscornia G, Oropesa‐Avila M. Mitochondrial dysfunction in lysosomal storage disorders. Diseases. 2016;4:31. doi: 10.3390/diseases4040031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hasegawa J, Tokuda E, Yao Y, Sasaki T, Inoki K, Weisman LS. PP2A‐dependent TFEB activation is blocked by PIKfyve‐induced mTORC1 activity. Mol Biol Cell. 2022;33:ar26. doi: 10.1091/mbc.E21-06-0309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–914. doi: 10.4161/auto.19653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hambright HG, Meng P, Kumar AP, Ghosh R. Inhibition of PI3K/AKT/mTOR axis disrupts oxidative stress‐mediated survival of melanoma cells. Oncotarget. 2015;6:7195–7208. doi: 10.18632/oncotarget.3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Xu M, Zhang Q, Li PL, Nguyen T, Li X, Zhang Y. Regulation of dynein‐mediated autophagosomes trafficking by ASM in CASMCs. Front Biosci (Landmark Ed). 2016;21:696–706. doi: 10.2741/4415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sul OJ, Li G, Kim JE, Kim ES, Choi HS. 7‐ketocholesterol enhances autophagy via the ROS‐TFEB signaling pathway in osteoclasts. J Nutr Biochem. 2021;96:108783. doi: 10.1016/j.jnutbio.2021.108783 [DOI] [PubMed] [Google Scholar]
- 97. Ikeda S, Maemura K. Ezetimibe and vascular endothelial function. Curr Vasc Pharmacol. 2011;9:87–98. doi: 10.2174/157016111793744797 [DOI] [PubMed] [Google Scholar]
- 98. Qin J, Wang LL, Liu ZY, Zou YL, Fei YJ, Liu ZX. Ezetimibe protects endothelial cells against oxidative stress through Akt/GSK‐3beta pathway. Curr Med Sci. 2018;38:398–404. doi: 10.1007/s11596-018-1892-3 [DOI] [PubMed] [Google Scholar]
- 99. Becher T, Schulze TJ, Schmitt M, Trinkmann F, El‐Battrawy I, Akin I, Kalsch T, Borggrefe M, Stach K. Ezetimibe inhibits platelet activation and uPAR expression on endothelial cells. Int J Cardiol. 2017;227:858–862. doi: 10.1016/j.ijcard.2016.09.122 [DOI] [PubMed] [Google Scholar]
- 100. Esmailpourmoghadam E, Salehi H, Moshtaghi N. Differential gene expression responses to salt and drought stress in tall fescue (Festuca arundinacea Schreb.). Mol Biotechnol. 2023;66:2481–2496. doi: 10.1007/s12033-023-00888-8 [DOI] [PubMed] [Google Scholar]
- 101. Kim SH, Kim G, Han DH, Lee M, Kim I, Kim B, Kim KH, Song YM, Yoo JE, Wang HJ, et al. Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase‐TFEB‐mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy. 2017;13:1767–1781. doi: 10.1080/15548627.2017.1356977 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Data Availability Statement
All data and supporting materials have been provided with the published article and its online supplementary files.