An official website of the United States government
Here's how you know
Official websites use .gov
A
.gov website belongs to an official
government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you've safely
connected to the .gov website. Share sensitive
information only on official, secure websites.
As a library, NLM provides access to scientific literature. Inclusion in an NLM database does not imply endorsement of, or agreement with,
the contents by NLM or the National Institutes of Health.
Learn more:
PMC Disclaimer
|
PMC Copyright Notice
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
Machine learning interatomic potentials, as a modern generation of classical force fields, take atomic environments as input and predict the corresponding atomic energies and forces. We challenge the commonly accepted assumption that the contribution of an atom can be learned from the short-range local environment of that atom. We employ density functional theory calculations to quantify the decay of the induced electron density and electrostatic potential in response to local perturbations throughout insulating, semiconducting and metallic samples of different dimensionalities. Molecules and thin layers are shown to fail keeping such disturbances localized. Therefore, the learnability of local atomic contributions, which guarantees scalability and transferability of a machine learning interatomic potential, is questionable in the case of molecules and low-dimensional samples. Similarly, the induced electrostatic effects due to substituted impurities or vacancy sites in a crystalline bulk are weakly damped and remain significant beyond several interatomic distances. However, geometric deformations in bulks are practically local within the first neighbors and induce a Yukawa-type electrostatic potential that exponentially vanishes. The practical importance of this finding is that it limits the application of the machine learning interatomic potentials to conformational search or thermal properties of bulk materials and so on, where only purely geometrical deformations are involved. Once chemically impactful defects like aliovalent impurities or vacancies are present, the interatomic potentials trained on local environments need to be corrected for long-range effects.
Keywords: Charge locality, Machine learning, Screening, Low-dimensional systems
Subject terms: Computational biophysics, Chemical physics, Condensed-matter physics, Information theory and computation
Introduction
Numerically solving the Schrödinger equation to determine the electronic structure of materials has already reached a prediction accuracy comparable to modern experimental methods1. As a major breakthrough in this field of research, machine learning (ML) has successfully proven as an economic alternative to the traditional paradigm in computational condensed matter physics and quantum chemistry2–13. For example, an artificial neural network (ANN) provides a highly efficient representation of the (ground state) wavefunction of many-particle systems3,4. An ANN is also flexible enough to learn from the samples the complicated structure-energy relations in atomic structures: once trained on a rich and diverse set of structure-energy pairs, the ANN can accurately regenerate the high-dimensional potential energy surface of large atomic structures at low computational cost14,15. The strategy that leads to such greatly reduction of computational cost of the ML structure-property mapping is the partitioning of the property in question into additive atomic contributions. Assuming that each atomic contribution can be learned locally and independently of the system size, the computational cost of evaluating the total property remains \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$${\mathcal {O}}(N)$$\end{document} with respect to the number of atoms N in the system. This resembles the advantageousness of the so-called linear scaling methods16. A second advantage of using the divide-and-conquer strategy is transferability of the locally learned atomic properties from one composition to a different composition.
Locality is generally accepted as a natural principle in chemistry which explains why, e.g., the chemical reactivity of a functional group is transferable between different molecules. This intuition is based on a physical concept known as “nearsightedness of electronic matter”, refereeing to the pioneering work by Kohn and Prodan17–19 who showed that the electron density at a given point for a many-electron system is practically insensitive to the perturbation of the external potential at distant points. Nearsightedness is different from classical screening of long-range electrostatic effects but it is a quantum mechanics effect due to Fermi-Dirac distribution and applies also to electrically neutral fermions at constant chemical potential. Generally, nearsightedness applies to electrons subject to a periodic external potential with a nearsightedness range of the lattice constant order18. On the other hand, nearsightedness, does not apply to few-electron systems while long-range electrostatic interactions are also long ranged and cause, e.g., the atomic charges in covalent molecules be affected beyond a few bond lengths15,20. The competition between nearsightedness and electrostatics determines the validity of locality in an atomic system and thus machine-learnability of an interatomic potential model for it. However, to the best of our knowledge, the locality radius is unknown in general. In essence, the aim of this study is to explore quantitatively the locality in different materials in response to three important types of perturbations. The change in the atomic charge, electron density and electrostatic potential as a function of the distance to the distortion position is obtained from a series of DFT calculations and compared across a variety of different types of samples with different dimensions. In this way, we are able to investigate the locality dependence on the type and dimensionality of the materials. We show that nearsightedness is questionable for samples with reduced bulkiness, i.e. small molecules or layered samples. In bulk samples, on the other hand, geometric deformations are nearsighted in contrast to chemical modifications like vacancies or aliovalent substitution. In particular, as long as ML interatomic potentials are employed for performing lattice vibrations (e.g. for thermal properties calculations) or molecular dynamics simulations without chemical reactions, the locality assumption is safely applicable. These results can be interpreted as evidence for certain limitations and applicability of ML interatomic potential models that are based on the local atomic contributions.
In the rest of this manuscript, we will first present the results of our first-principles calculations. We then compare the numerical values with analytic expressions for Green’s functions for screening. We finally draw our conclusions.
Results
Atomic charge
Inspired by previous work, we first consider the distribution of atomic charges in molecules with either a whole15 or partially20 chain-like structure. The results presented in the first three rows of Fig. 1 are meant to show how the type of a functional group X = OH, \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {O}^-$$\end{document} or SH which is positioned at one end of the linear molecule \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {XC}_7$$\end{document}\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {H}_7$$\end{document}O changes the partial charge on the terminal oxygen atoms (marked with a circle) at the other end. Similarly, the effect of protonation of one end of the carbon chain \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {C}_{{10}}$$\end{document}\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {H}_2$$\end{document} is shown in the next two rows. Finally, in the third set depicted in the two bottommost rows of Fig. 1, two energetically stable \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {C}_{{60}}$$\end{document} configurations with chain-cage structure are compared to verify that the charge distribution on the atomic chain depends on the geometry of the attached cage. (The atomic energy and density of states was also previously shown to be different for the \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {C}_{{60}}$$\end{document} configurations20).
Comparison of charge nonlocality with five different partial charge assignment schemes, for the molecule \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {XC}_7$$\end{document}\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {H}_7$$\end{document}O with X = OH, \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {O}^-$$\end{document}, SH (top, three rows), \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {C}_{{10}}$$\end{document}\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {H}_2$$\end{document} and \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {C}_{{10}}$$\end{document}\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {H}^+_3$$\end{document} (middle, two rows) and two \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {C}_{{60}}$$\end{document} cage-tail structures (bottom, two rows). The color bars denote the net electric charge in units e.
Noting that atomic charge is not a physical observable and there is no unique and unambiguous definition for it, we generalize the results of previous studies and present in five columns of Fig. 1 the atomic charge from five different charge assignment schemes: real-space electron density decomposition via the smooth Voronoi20, Hirshfeld21 and Bader22 schemes, minimizing the electronic band energy with respect to the atomic charges within the DFTB framework23,24 and finally population analysis of the wavefunction to get the so-called Mulliken charge25 (see Methods). Not surprisingly, these schemes lead to scattered values of atomic charges. Nevertheless, in all test cases there exist occasions which show that the structural detail at one end of a linear conformation affects the atomic charge on the other end despite similar local environments. For example, the Hirshfeld and Mulliken charges detect a slight sensitivity of the charge of the terminal oxygen atom of the \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {XC}_7$$\end{document}\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {H}_7$$\end{document}O molecule on the functional group X at the other end of the molecule. More notably, the charge of the terminal atom in the 10-carbon chain (indicated by a circle) changes by \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sim 0.2$$\end{document} e upon protonation the opposite end of the chain according to the DFTB calculations in agreement with previously reported values from more accurate calculations15. Similarly, the DFTB, Hirshfeld and Voronoi methods indicate a clear dependence of the charges on the terminal atom of the tail of the two \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {C}_{{60}}$$\end{document} conformations on its cage structure.
The observed inconsistency between the charge values predicted by different atomic charge assignment schemes highlights the deficiency of atomic charge for an appropriate investigation of the issue of locality. We will exploit a rigorous method to quantify the nonlocality based on the electron density and electrostatic potential. Moreover, chemical modifications may hinder a quantitatively comparing of different atomic structures. Instead, one may consider randomly displacing a single atom by a given small amplitude or removing it to left a vacancy, which is identically applicable to any kind of samples.
Electron density and potential
In contrast to atomic charge, electron density is a smooth and continuous function of position and varies slowly in space. Moreover, this experimentally measurable field quantity, e.g. by X-ray or neutron diffraction, has a unique definition in all computational schemes in terms of the squared modulus of the N-electron wavefunction
where \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\textbf{r}_i$$\end{document} and \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$s_i$$\end{document} denote spatial and spin coordinates of electron i, respectively. In the spin-unpolarized Kohn-Sham version of the density functional theory (DFT)26, the electron density is approximated by the single-electron wavefunctions \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\Psi _i(\textbf{r})$$\end{document}. The corresponding Hartree potential satisfies the Poisson’s equation
where \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\epsilon _0$$\end{document} denotes the permittivity of vacuum while \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$e=1.602 \times 10^{-19}$$\end{document} C is the elementary charge. The latter equation lacks any quantum mechanical contribution from the Pauli exclusion principle and other correlations between the electrons, which are all lumped as the so-called exchange-correlation (XC) potential \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$V_\text {XC}(\textbf{r})$$\end{document} term in the DFT framework. The total electrostatic potential is then \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$V(\textbf{r})=V_\text {ext}(\textbf{r}) + V_\text {H}(\textbf{r})+V_\text {XC}(\textbf{r})$$\end{document}. For a system of \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$N_\alpha$$\end{document} atoms of atomic numbers \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$Z_\alpha$$\end{document} located at positions \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\textbf{R}_\alpha$$\end{document}, the external potential felt by the electrons reads \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$V_\text {ext}(\textbf{r}) =e \sum _{\alpha } Z_\alpha \delta (\textbf{r}-\textbf{R}_\alpha ).$$\end{document}
The induced electron density \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$${\Delta n}(\textbf{r}) = n(\textbf{r}) - n_0 (\textbf{r})$$\end{document} and electrostatic potential \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$${\Delta V}(\textbf{r}) = V(\textbf{r}) - V_0 (\textbf{r})$$\end{document} are calculated in every point \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\textbf{r}$$\end{document} in the space as the deviation from the unperturbed values, \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$n_0$$\end{document} and \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$V_0$$\end{document}, respectively, in response to applying a local modification (displacement, substitution or vacancy) into an atom in the structure. We map the three-dimensional fields into scalar functions of the radial distance x to the distortion center by averaging over successive thin shells. For linear molecules (one-dimensional systems) the shells are planar and perpendicular to the molecular axis at distance x from the displaced atom while for bulks (three-dimensional systems) the average is calculated on thin spherical shells of radius x centered at the original position of the displaced atom. For a sheet of atoms (a two-dimensional system), the shells are cylinders normal to the sheet with an axes passing through the displaced atom so that x is the cylinder radius. We calculate the induced electron density or electrostatic potential without relaxing the position of other atoms surrounding the perturbation center. This prevents too high peaks of induced electron density in the vicinity of the initial and final positions of the point-like atomic cores and thus allows for investigation of the perturbation applied to an individual atomic core.
Linear molecules
We first investigate the response of the electron density to random displacements of the terminal atoms. We illustrate in Fig. 2a the planar average of the induced electron density \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\overline{\Delta n}$$\end{document} along the linear molecules \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {HOC}_7$$\end{document}\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {H}_7$$\end{document}O and \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {C}_{{10}}$$\end{document}\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {H}^+_3$$\end{document} as a function of distance x to the reference terminal atom which is displaced in a random direction by 0.1 Å. For both molecules an initial bump, extended to \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$x\sim 2$$\end{document} Å, exists due to the displacement of the high-density electron cloud peaked over the displaced atom. Beyond the initial bump, \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\overline{\Delta n} (x)$$\end{document} decays as \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sim x^{-2}$$\end{document} within the molecule body but then develops as \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sim x^{-1}$$\end{document} into the vacuum region beyond \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sim 11$$\end{document} Å. On the other hand, Fig. 2b shows that the decay of the induced potential is similar to the decay of the induced electron density within the linear molecule body. The planar average of the induced Hartree potential \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\overline{\Delta V}_\text {H}(x)$$\end{document} decays as \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sim x^{-2}$$\end{document} throughout the molecules but tends to \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sim x^{-1}$$\end{document} in the vacuum. The dominant contribution to the total potential is the XC potential which exhibits \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$x^{-1}$$\end{document} decay within the molecular body. Surprisingly, \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\overline{\Delta V}_\text {XC}$$\end{document} tends to a constant in the vacuum while it, in principle, must vanish wherever the electron density is absent within the local density approximation (LDA) used in our calculations. It seems that the small induced XC potential in vacuum is related to the non-vanishing electron density in the void region between the periodic images of the molecule in the supercell method.
Induced electron density (a) and Hartree and XC potentials (b) in the two linear molecule test cases in response to small atomic displacements. The terminal oxygen atom of \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {HOC}_7$$\end{document}\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {H}_7$$\end{document}O or the terminal carbon atom in \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {C}_{{10}}$$\end{document}\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {H}^+_3$$\end{document} is moved in a random direction by 0.1 Å. The average of the induced electron density or potential is calculated over thin planar shells at distance x from the displaced atom along the molecular axis. The trends \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$x^{-1}$$\end{document} and \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$x^{-2}$$\end{document} are indicated by thin lines to help the eye.
Bulk samples
We now look at the locality of the induced electron density and electrostatic potential in extended materials. We consider three types of crystalline bulks as test cases: silicon in the diamond structure which is a covalent semiconductor, NaCl crystal as an ionic insulator and copper as a metallic crystal. Note that the model systems are periodic in three directions so that, in contrast to the molecules, all atomic sites are equivalent. For silicon and NaCl crystals, we construct \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$3\times 3$$\end{document} cubic supercells containing 216 atoms with lattice constant 16.3 and 17.0 Å, respectively. For copper, a cubic \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$4\times 4$$\end{document} supercell of lattice constant 14.5 Å is used which contains 256 Cu atoms. Although the supercells are not large enough to avoid finite-size effects as discussed later on, we are able to capture at least one order of magnitude reduction of the induced averageelectron density within one supercell in response to displacing a single atom by 0.1 Å.
As seen in Fig. 3, the decay of the shell-averaged induced electron density for bulk samples is not as smooth as for the molecules. For silicon, for example, the initial bump of the shift of the valence electrons of the displaced atomic core is followed by a few minor bumps that resemble the peaks of the radial distribution of atomic centers. Apart from the bumps, however, \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\overline{\Delta n} \sim x^{-3}$$\end{document} so that it is reduced by two orders of magnitude already at a distance \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sim 4$$\end{document} Å form the displaced atom. Similar inspection reveals a slower power-law decay, namely \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\overline{\Delta n} \sim x^{-2}$$\end{document} and \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sim x^{-1}$$\end{document} away from the distortion center for the ionic and metallic crystalline test cases, respectively. When a Cl ion is displaced, the first bump of width \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sim 2.5$$\end{document} Å is followed by a shallow one extended to 5 Å after which the decay is monotonic. However, the central bump around a displaced \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {Na}^+$$\end{document} ion is narrow because the \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {Na}^+$$\end{document} ion lacks a flexible electronic cloud. A few shallow and narrower bumps are present within one Na-Na distance (5.6 Å) which can be assigned to polarization of the eight electron-rich \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {Cl}^-$$\end{document} ions that surround the displaced Na core. Finally, the induced electron density in the metallic test case decays as \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sim x^{-1}$$\end{document} after a wide initial bump but it starts oscillating at distance \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sim 9$$\end{document} Å. The latter can be assigned to the periodic images of the displaced atom. In fact, the finite-size effect is clearly visible in the metallic sample because the decay of electron density perturbation is slow. This implies that locality is less fulfilled in a metallic bulk in comparison to covalent and ionic crystals.
Induced electron density (a) and the total electrostatic potential (b) in response to small atomic displacements in the three crystalline bulk test cases: diamond silicon, NaCl and copper perfect crystals. A single Si, Na, Cl or Si is displaced in a random direction by 0.1 Å. The average of the induced electron density or potential is calculated over thin spherical shells of radius x centered at the original position of the displaced atom. Note that all atoms are equivalent thanks to the translational symmetry of the crystal. The trends \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$x^{-1}$$\end{document} and \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$x^{-3}$$\end{document} and \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$x^{-1}\exp (-x/r_s)$$\end{document} are indicated by thin lines to help the eye. In (b), the screening length is arbitrarily set to \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$r_s=4$$\end{document} Å.
To verify the independence of the observed decay from the magnitude of the random displacements, we repeated selected tests by amplitudes 0.05, 0.1, 0.5 and 1 Å. The induced electron density increases by increasing the displacement amplitude but the x-dependence of \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\overline{\Delta n}$$\end{document} remains always the same. We also looked at the maximum value of the induced density within a shell which shows the same pattern as the shell-average value. Additionally, we verified the effect of the XC functional used in our DFT calculations. Note that locality of electron density is somehow an underlying assumption of the local density approximation (LDA) while the PBE27 functional is semi-local compared to the essentially non-local hybrid functional PBE0. Interestingly, our complementary tests indicate no appreciable difference between the monotonically-decaying induced electron densities calculated by the three different types of XC functionals for bulk silicon. For \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {C}_{{10}}$$\end{document}\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\hbox {H}_2$$\end{document} molecule and Cu bulk, however, the PBE and PBE0 calculations predict a slightly faster decay and a smaller asymptotic induced density.
The distance-dependence of the induced potential for the same three bulk test cases, shown in Fig. 3b, is much smoother than the density. We do not represent the XC and Hartree terms separately because the XC contribution is still dominant and practically identical to the total electrostatic potential. The potential decay fits fairly well with \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sim x^{-1}\exp (- x/r_s)$$\end{document}. In other words, instead of the power-law \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$x^{-2}$$\end{document} in the one-dimensional molecules or \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$x^{-1}$$\end{document} in vacuum, the induced potential drops exponentially in bulks. With such a fast decay, the total induced potential reaches 1 mV already at distance \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$x\sim 10$$\end{document} Å in the bulk silicon or \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$x\sim 14$$\end{document} Å in the ionic and metallic samples. It will be shown in the following that distance-dependence of the electrostatic potential around a perturbation center in bulks follows a general and simple picture resembling charge screening and polarization.
Impurity and vacancy
Atomic displacement is the most frequent circumstance in molecular dynamics simulations. However, it is instructive to investigate the locality of bulks also in response to local chemical modifications. To this aim, we performed two additional sets of experiments by introducing an atom vacancy or an impurity substitution into the three bulk test cases. Shown on the left column of Fig. 4 is the spherically averaged induced electron density as a function of the radial distance to the position of the local disturbances. The significant oscillatory density which damps slowly away the vacancy or the substituted atom, is clearly visible even on the non-logarithmic scale (cf. Figs. 3 and 4). The corresponding distance-dependent induced potential, illustrated on the right column of Fig. 4, varies smoothly in contrast to the electron density and decays monotonically with radial distance. Note that when the type or the number of atoms is not the same in perturbed and unperturbed systems, apparent finite asymptotic value of the induced electrostatic potential is assigned to the arbitrary reference value of the electrostatic potential in DFT calculations. We set this asymptotic value to zero in order to align the potential between the perturbed and unperturbed systems.
Same as Fig. 3 but in non-logarithmic scale for comparison of two kinds of local perturbations, i.e. geometric (displacement by 0.1 Å, repeated from Fig. 3) and chemical (vacancy or impurity substitution). The impurity has the same number of valence electrons as the original atom when substituting Si by Ge, Cl by Br, Na by K, Cu by Ag while the number of valence electrons differ by one when substituting Si by P, Cl by S, Na by Mg, Cu by Zn.
We examined two types of impurity substitution. In one case, the impurity is an element in the same column of the periodic table as the original atom so that the number of valence electrons is preserved. In the second case, the substituted impurity is next to the original atom on the same row of the periodic table and thus has one less or one more valence electron than it. The latter leads to a significant local chemical structure change. As shown in the first row of Fig. 4, a germanium impurity introduced into the silicon crystal induces a rather localized electron density, fairly similar to the effect of an atomic displacement. In clear contrast, the phosphorus impurity with one more valence electron than silicon or a silicon atom vacancy both lead to a slowly damping and significant oscillations in the electron density. The right panel shows the same behavior for the induced potential: a silicon vacancy or a phosphorus substitution induces much larger electrostatic potential compared to geometric deformation or germanium substitution. The second and third rows of Fig. 4 correspond to the NaCl crystal with local disturbances introduced into a Cl or Na atomic site, respectively. Both isovalent substitution (Cl by Br, Na by K) and aliovalent substitution (Cl by S, Na by Mg) are examined. Interestingly, the behavior of NaCl bulk, as an ionic dielectric, looks similar to that of the silicon semiconducting bulk: In contrast to a pure geometric deformation or isovalent substitution, the Cl-vacancy or an S-substitution induce non-localized electron density and electric potential. Similarly, the electron density induced by randomly displacing a sodium atom or substituting it by potassium is much more localized than that by a sodium vacancy or substituting by magnesium (with different number of valence electrons). Note that S has one less electron than Cl while, e.g., P has one more valence electron than Si, hence the opposite sign of the corresponding induced electrostatic potential. A qualitatively similar behavior is observed for the metallic sample shown in the fourth row of Fig. 4. The pure geometric deformation by displacing a Cu atom induces a highly localized change in electron density which drops by three orders of magnitude within 3 Å. The corresponding electrostatic potential exponentially approaches zero. Dissimilarly, an impactful chemical modification by removing a Cu atom or substituting it by a Zn atom (which has one more valence electron than Cu) induces electron density fluctuations of large amplitude up to a distance a few Ångström. The induced electron density and electrostatic potential vanish faster if the Cu atom is replaced by a Ag atom which has the same number of valence electrons as Cu. Overall, chemically significant defects are not well screened in all the examined bulk crystals. This delimits the learnability of the interatomic interactions which are based on the locality assumption to purely geometric deformation or isovalent impurity substitution.
Discussion
Screened Green’s function
The induced electrostatic potential from our first-principles calculations for all cases presented in Figs. 2, 3 and 4, is an essentially monotonic and smooth function of distance x to the disturbance center. In particular, the data points for bulk samples in Fig. 3 fit well with the exponentially decaying function, \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$x^{-1}\exp (-x/r_s)$$\end{document}, which implies that a geometry deformation is electrically shielded over a distance \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sim r_s$$\end{document}. This type of exponentially decaying electrostatic potential resemble the well-known screened Coulomb potential. In fact, the same form of this potential can be derived from different approaches with different physical assumptions, e.g. the Debye-Hückel model28 for classical plasmas or electrolyte liquids and the Thomas-Fermi model29 for metals. Both approaches share the assumption of proportionality of the induced charge density to the electrostatic potential at the same point in space \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\Delta \rho (\textbf{r}) \propto \rho (\textbf{r}) V(\textbf{r})$$\end{document}, where \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\rho (\textbf{r})$$\end{document} denotes the free charge density (not including the induced one \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\Delta \rho$$\end{document}). This converts the Poisson’s equation \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\nabla ^2 V(\textbf{r}) = -(\rho +\Delta \rho )/\epsilon _0$$\end{document}, where \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\epsilon _0$$\end{document} is the permittivity of vacuum, into the screened Poisson’s equation
which implies that the electrostatic potential developed by a point charge embedded in an electron gas or electrolyte liquid falls off faster than as \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$|\mathbf{r-r'}|^{-1}$$\end{document} in vacuum. The potential \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$V(\textbf{r})= \frac{1}{4\pi \epsilon _0} \int \textrm{d} \mathbf{r'} \rho (\mathbf{r'}) G(\mathbf{r, r'} )$$\end{document} solves the screened Poisson’s Eq. (3). But as the screening length \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\kappa _s^{-1}$$\end{document} becomes very large, the unscreened Coulomb potential \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$V(\textbf{r})= Q/4\pi \epsilon _0 |\textbf{r} -\mathbf{r'}|$$\end{document} is recovered with \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\rho (\textbf{r}) = Q\delta (\mathbf{r-r'})$$\end{document} for a point charge Q at position \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\mathbf{r'}$$\end{document}.
Note that Eq. (4) is identical to the function \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$x^{-1}\exp (-x/r_s)$$\end{document} used for fitting with \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$r_s=\kappa _s^{-1}$$\end{document} being the screening length while \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$x=|\mathbf{r-r'}|$$\end{document} is the radial distance to the displaced atom position rather than to an external point charge. Interestingly, the screened Green’s function (4) fits well to the data points of the Si sample in Fig. 3b with a screening length 3.1 Å. Displacing a Na or Cl core in the NaCl crystal leads to practically the same screening effect and the damping electrostatic potential obeys Eq. (4). The screening lengths fitted to the data points in Fig. 3b corresponding to displacing a Na or Cl core are 3.5 or 3.7 Å, respectively. Finally, for Cu, \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$k_s^{-1} = 2.8$$\end{document} Å. Therefore, it seems that the first atomic layer surrounding a displaced atom shields it from the next atomic layers in all cases. It is surprising that metallic, ionic and covalent samples all show the same damping behavior despite the underlying mechanism should be very different in different materials.
Spatial distribution of the induced electron density and electrostatic potential around an impurity, vacancy or displaced atom in materials is an old problem. In the simplest picture, consider a positive point charge embedded in an initially homogeneous electron gas. From a classical electrodynamics viewpoint, electron concentration around the charge increases in response to the electrostatic attraction with the positive charge. The charge is thus screened by the accumulated electrons. Similarly, a point charge embedded in plasma or electrolyte liquid is screened by the attracted ions of opposite charge. Moreover, the point charge in a dielectric medium polarizes the surrounding atoms and the static dielectric constant (also known as relative permittivity) in enhanced. All these tend to render the electrostatic interactions short range.
From a quantum mechanical perspective, on the other hand, a local disturbance is a scatterer of the unlocalized electrons in the medium. The Linhard approach predicts the so-called Friedel oscillations of the electron density around an external point charge which damp as the inverse cube of distance \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\Delta n(\textbf{r}) \propto |\mathbf{r-r'}|^{-3} \cos (2 k_F|\mathbf{r-r'}|)$$\end{document}29. With the Fermi wavevectors of metallic elements, \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$k_F \sim 0.6-1.8~\text {\text{\AA }}^{-1}$$\end{document}, the expected spatial period of the oscillations accounts to \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sim 2-5$$\end{document} Å, which is slightly longer than that of damped oscillations around vacancies and impurities in Fig. 4a. Note that the oscillatory electron density originates from the fermion character of electron and fundamentally independent of its electric charge and thus of the electric polarization.
In contrast, geometric deformations always lead to exponentially damping potential, Eq. (4), as discussed earlier. Such a static screening with exponential damping is predicted within the Thomas-Fermi approximation in the limit of very small wavevectors \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\ll k_F \sim 1~\text {\text{\AA }}^{-1}$$\end{document}. In that case, the Thomas-Fermi screening length decreases very slowly when the electron concentration increases as \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\kappa _s^{-1} \simeq \frac{1}{2} \sqrt{a_0n^{-1/3}}$$\end{document}, where the Bohr radius \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$a_0=4\pi \epsilon _0 \hslash ^2/me^2 \simeq 0.529$$\end{document} Å. Let us exemplify two simple and naïve cases. In vacuum, i.e. as n approaches zero, \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\kappa _s^{-1} \rightarrow \infty$$\end{document} and screening vanishes. In this limit, Eq. (4) reduces to the Green’s function of vacuum \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$G(\mathbf{r,r'})=|\mathbf{r-r'}|^{-1}$$\end{document}. On the other hand, for a homogeneous electron gas with density \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$n= 8.45 \times 10^{22}~\text {cm}^{-3}$$\end{document} of copper30 the screening length is \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\kappa _s^{-1} = 0.55$$\end{document} Å. It is obvious that this value is unrealistically short noting that for an ordinary metal a local disturbance is expected to be screened on distances comparable to interatomic distances29. Once the homogeneous gas is replaced by a realistic one is in our DFT computations, we get \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$r_s= 2.8$$\end{document} Å from fitting Eq. (4) to the data points of our first-principles for bulk copper, presented in Fig. 3b.
Dimensionality
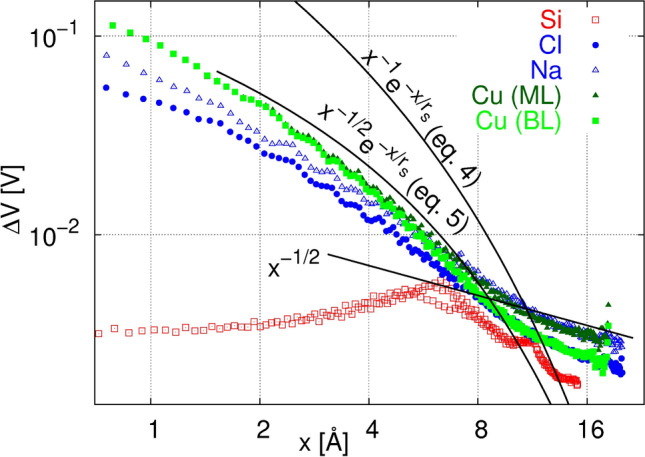
We have shown that locality, quantified as the declining behavior of induced electron density and electrostatic potential, is not satisfied in linear molecules in contrast to extended covalent, ionic and metallic crystalline samples. It is thus interesting to investigate the role of dimensionality on the locality. In the following, we present the result of our tests for the two-dimensional variants of the same bulk test cases, namely: (i) buckled silicene (silicon monolayer), (ii) NaCl (001) bilayer film, and (iii) copper monolayer and bilayer. In contrast to silicene and NaCl films, a single or two layers of Cu(111) planes might be structurally unstable which has no importance for the present test. The averages of the induced electrostatic potential is calculated on cylindrical shells around the perturbation center. The axis of the cylinder is normal to the plane of the two-dimensional sample. The shell-average of the total electrostatic potential calculated as a function of the radial distance to the position of the displaced atom, (cylinder radius) is shown in Fig. 5. It is clearly seen that upon reducing the sample dimension from three to two, the decay of the electrostatic potential slows down. While the bulk samples all obey \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$x^{-1}\exp (-x/r_s)$$\end{document}, the two-dimensional variants of the same samples show a \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$x^{-1/2}\exp (-x/r_s)$$\end{document} decaying regime up to about 10 Å which is then followed by a \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$x^{-1/2}$$\end{document} regime. The case of silicene is different within the first neighbours of the displaced atom. The difference is seemingly due to the very sparse structure of this buckled hexagonal sheet.
Decay of total electrostatic potential (\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$V_\text {XC}+V_\text {H}$$\end{document}) in two-dimensional samples in response to local geometric deformation i.e. displacement of a single atom, averaged on thin cylindrical shells of radius x centered at the position of the displaced atom. Thin lines show the decay trends, including Eq. (4) repeated from Fig. 3b, for comparison with the declining exponential behavior in corresponding bulks. The screening length is arbitrarily set to \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$r_s=4$$\end{document} Å.
The Green’s function corresponding to the two-dimensional screened Poisson’s Eq. (3) is
where \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$K_0(x)$$\end{document} denotes the zeroth order modified Bessel function of the second kind. The approximate Green’s function on the right hand side is obtained using the large-x asymptotic approximation of \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$K_0(x)\rightarrow \sqrt{\frac{\pi }{2x}}e^{-x}\big [1+0\big (\frac{1}{x}\big )\big ]$$\end{document}31. Neglecting the odd case of silicene, Eq. (5) fits fairly well with the data-points of the induced electrostatic potential in thin layers illustrated in Fig. 5 within the range \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$x \lesssim 10$$\end{document} Å. For larger distances that are comparable to the supercell box size, however, the electrostatic potential deviates from Eq. (5) and the decay is no longer exponential but becomes power-law (\documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sim x^{-1/2}$$\end{document}). Similar to linear molecules shown in Fig. 2b, the magnitude of the induced electrostatic potential due to displacing an atom by 0.1 Å remains within a few mV at such distances. We conclude that the electrostatic potential does not vanish even beyond a few interatomic distances away the geometric deformation center in one- and two-dimensional samples. We consider the latter as an obvious manifestation of nonlocality in low-dimensional materials. Moreover, the static screening in (quasi-) two-dimensional samples is weak32,33 consistent with our first-principles calculations presented in Fig. 5. Finally, screening of impurities in a semiconductor nanocrystal has also been shown to depend on its size34.
Concluding remarks
“Nearsightedness in electronic matter” was introduced by Kohn in 199617 to refer to the limited dependence of the electron density at a given point on the variation of external potential at large distances. The other way round, screening is also known to greatly suppress the sensitivity of the electrostatic potential at a given point to the change of electron density at large distances. The mechanism behind the two phenomena is not necessarily the same. Our first principles calculations reveal that the electron density around a vacancy or impurity in a bulk sample shows Friedel-like oscillations. The corresponding electrostatic potential decays slowly. We consider this as a nonlocal effect (see Fig. 4).
On the other hand, if an atom is displaced from its equilibrium position in bulk even by an Å, the deformation remains localized and is screened so that the electrostatic potential change drops exponentially away the disturbance center. the underlying assumption of almost all interatomic potentials developed for large-scale materials simulations is locality, based on the concepts like nearsightedness and screening. We verified in this study how factors like bulkiness and structure of the sample or the type of the local disturbance determines the locality range in practice. Our generally applicable technique makes it possible to compare locality in different kinds of samples by quantifying the decay of the induced shell-averaged electrostatic potential away the local disturbance. The disturbance might be an impurity substitution, vacancy or atom displacement with an arbitrary amplitude and direction. One of advantage over using the induced atomic charges previously suggested for such tests is that the induced density or potential is a continuous and smoothly varying function of distance.
For a geometric deformation in bulk materials, the potential mimics the behavior predicted by the screened Poisson’s Eq. (3). In other words, the electric consequences of geometric deformations are screened in materials extended in three dimensions, just as a point charge is screened in an electrolyte liquid or fermion gas. In contrast, chemically impactful defects like aliovalent substation or vacancies show significant long-range effects. Moreover, structures composed of sheets or chains of atoms, also fail to screen chemical modifications or geometric deformations. In such cases, long-range electrostatic interactions must be taken into account when atomic contributions to interatomic potentials are trained. An effective solution to this issue is to enforce the atomic system to satisfy the charge equilibration requirement throughout the system. It can be realized, e.g., via training atomic electronegativity35,36. The resultant atomic ML atomic contributions to the interatomic potentials are equipped with non-local electrostatic interactions throughout the whole structure in addition to the local chemical environment of the corresponding atom. For liquid water, it has been shown that adding the long-range electrostatics, from fixed point charges37 or trained directly by an ANN38, outperforms the ML atomistic models. Grisafi and Ceriotti39 proposed a long distance equivarient representation to incorporate long-range electrostatics into the ML potentials which can generally capture any nonlocal physics. Finally, it should be noted that the DFT calculations data set used in this work is known to suffer from missing long-range electrostatics. It would be interesting to see whether the same shortcoming is present if the interatomic potentials are trained on more accurate ab initio reference data.
Nevertheless, we showed that the induced electrostatic potential of a geometric distortion is very localized inside a dense piece of material. The physics of the localization in different types of materials remains to be explored in future work. Our numeric results showed that bulk samples, in particular the covalent crystals, screen out the geometric disturbances beyond the first atomic neighbours. The impact of this finding is that lattice dynamics computations and molecular dynamics simulations can be safely performed using the ML interatomic potentials that split the energy and forces into atomic contributions. In fact, transferability and scalability of a ML interatomic potential critically depends on how accurate is the training of an atomic contribution from a limited local environment. This requirement is not satisfied in low dimensional systems, namely two-dimensional sheets or clusters and molecules.
Methods
The optimized atomic structures are determined with DFT calculations using the Quantum ESPRESSO code package40–42. A cutoff of 30 Ry is used to expand the wavefunction of the valence electrons in terms of plane waves. The PBE27 scheme of the generalized gradient approximation is used to describe the XC functional. For selected cases, the electron density is reported also from the calculations with the LDA and PBE0 XC functionals. The DFTB self-consistent atomic charges are calculated by the DFTB+ code23,24. The number density of electrons in real space \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$n (\textbf{r})$$\end{document} is integrated over the whole space to assign to an atom \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\alpha$$\end{document} a (real) number of electrons
according to associated weight functions \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$w_\alpha (\textbf{r})$$\end{document}. The weights are usually maximized at the position of the corresponding atomic centers \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\textbf{R}_\alpha$$\end{document}. For example, in the Hirshfeld method21
where \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$n^0_\alpha (\textbf{r})$$\end{document} is the electron density of isolated neutral atoms. Moreover, the space might be partitioned into the non-overlapping atomic basins with the condition \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$w_\alpha (\textbf{r})=1$$\end{document} if \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\textbf{r}$$\end{document} is within the basin \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\alpha$$\end{document} and \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$w_\alpha (\textbf{r})=0$$\end{document} elsewhere. The Bader analysis22 of the electron density determines the zero-flux surfaces of the electron density as the dividing border between the atomic basins while the strictly geometric Voronoi decomposition exploits the Wigner-Seitz cells. We also consider the smooth Voronoi decomposition scheme20 by replacing \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$n^0_\alpha (\textbf{r}) \rightarrow \exp \left( -|\mathbf{r-R}_\alpha |^2/\sigma ^2_\alpha \right. )$$\end{document} in Eq. (7). This intuitive approach combines the geometric information of the atomic configuration with the atomic radius \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sigma _\alpha$$\end{document}. The strictly geometric Voronoi decomposition is recovered in the limit of \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sigma _\alpha \rightarrow 0$$\end{document}, but we set \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sigma _\alpha$$\end{document} to the atomic covalent radius to mimic a simplified Hirshfeld scheme with a single Gaussian-type orbital. Note that, in contrast to Mulliken and DFTB atomic charges, \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sum _\alpha w_\alpha (\textbf{r})=1$$\end{document} at every point \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\textbf{r}$$\end{document} of the real space so that Eq. (6) leads to atomic charges that preserve the total number of electrons in the system \documentclass[12pt]{minimal}
\usepackage{amsmath}
\usepackage{wasysym}
\usepackage{amsfonts}
\usepackage{amssymb}
\usepackage{amsbsy}
\usepackage{mathrsfs}
\usepackage{upgreek}
\setlength{\oddsidemargin}{-69pt}
\begin{document}$$\sum _\alpha N_\alpha =N$$\end{document}.
Data availibility
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
1.Lejaeghere, K. et al. Reproducibility in density functional theory calculations of solids. Science351, aad3000 (2016). [DOI] [PubMed]
2.Bogojeski, M., Vogt-Maranto, L., Tuckerman, M. E., Müller, K.-R. & Burke, K. Quantum chemical accuracy from density functional approximations via machine learning. Nat. Comm.11, 5223 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
3.Carleo, G. & Troyer, M. Solving the quantum many-body problem with artificial neural networks. Science355, 602–606 (2017).
[DOI] [PubMed] [Google Scholar]
4.Melko, R. G., Carleo, G., Carrasquilla, J. & Cirac, J. I. Restricted Boltzmann machines in quantum physics. Nat. Phys.15, 887–892 (2019). [Google Scholar]
6.Brockherde, F. et al. Bypassing the Kohn-Sham equations with machine learning. Nat. Commun.8, 872 (2017).
[DOI] [PMC free article] [PubMed] [Google Scholar]
7.Schmidt, J., Marques, M. R., Botti, S. & Marques, M. A. Recent advances and applications of machine learning in solid-state materials science. npj Comput. Mater.5, 1–36 (2019). [Google Scholar]
8.Fiedler, L., Shah, K., Bussmann, M. & Cangi, A. Deep dive into machine learning density functional theory for materials science and chemistry. Phys. Rev. Mater.6, 040301 (2022). [Google Scholar]
9.Xia, R. & Kais, S. Quantum machine learning for electronic structure calculations. Nat. Commun.9, 4195 (2018).
[DOI] [PMC free article] [PubMed] [Google Scholar]
10.Dral, P. O. & Barbatti, M. Molecular excited states through a machine learning lens. Nat. Rev. Chem.5, 388–405 (2021).
[DOI] [PubMed] [Google Scholar]
11.Fedik, N. et al. Extending machine learning beyond interatomic potentials for predicting molecular properties. Nat. Rev. Chem.6, 653–672 (2022).
[DOI] [PubMed] [Google Scholar]
12.Unke, O. T. et al. Biomolecular dynamics with machine-learned quantum-mechanical force fields trained on diverse chemical fragments. Sci. Adv.10, eadn4397 (2024).
[DOI] [PubMed] [Google Scholar]
13.Burrill, D. et al. MLTB: Enhancing transferability and extensibility of density functional tight binding theory with multi-body interaction corrections. ChemRxiv 0n3q0 (2024).
14.Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett.98, 146401 (2007).
[DOI] [PubMed] [Google Scholar]
15.Ko, T. W., Finkler, J. A., Goedecker, S. & Behler, J. A fourth-generation high-dimensional neural network potential with accurate electrostatics including non-local charge transfer. Nat. Commun.12, 1–11 (2021).
[DOI] [PMC free article] [PubMed] [Google Scholar]
16.Goedecker, S. Linear scaling electronic structure methods. Rev. Mod. Phys.71, 1085 (1999). [Google Scholar]
17.Kohn, W. Density functional and density matrix method scaling linearly with the number of atoms. Phy. Rev. Lett.76, 3168 (1996). [DOI] [PubMed] [Google Scholar]
18.Prodan, E. & Kohn, W. Nearsightedness of electronic matter. Proc. Natl. Acad. Sci.102, 11635–11638 (2005).
[DOI] [PMC free article] [PubMed] [Google Scholar]
19.Prodan, E. Nearsightedness of electronic matter in one dimension. Phy. Rev. B73, 085108 (2006). [Google Scholar]
20.Parsaeifard, B., De, D. S., Finkler, J. A. & Goedecker, S. Fingerprint-based detection of non-local effects in the electronic structure of a simple single component covalent system. Cond. Mat.6, 9 (2021). [Google Scholar]
21.Hirshfeld, F. L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta44, 129–138 (1977). [Google Scholar]
22.Bader, R. Atoms in Molecules: A Quantum Theory Clarendon Press (Oxford New York, 1990).
23.Elstner, M. et al. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B58, 7260 (1998). [Google Scholar]
24.Aradi, B., Hourahine, B. & Frauenheim, T. DFTB+, a sparse matrix-based implementation of the DFTB method. J. Phys. Chem. A111, 5678–5684 (2007).
[DOI] [PubMed] [Google Scholar]
25.Mulliken, R. S. Electronic population analysis on LCAO-MO molecular wave functions. I. J. Chem. Phys.23, 1833–1840 (1955). [Google Scholar]
26.Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev.140, A1133 (1965). [Google Scholar]
27.Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett.77, 3865 (1996).
[DOI] [PubMed] [Google Scholar]
28.Huckel, E. & Debye, P. Zur Theorie der Elektrolyte. I. Gefrierpunktserniedrigung und verwandte Erscheinungen. Phys. Z24, 185–206 (1923). [Google Scholar]
29.Ashcroft, N. W. & Mermin, N. D. Solid State Physics, chap. 17 (Cengage Learning, 2022).
30.Kittel, C. Introduction to Solid State Physics, chap. 14 (2021), 8th edn.
31.Jackson, J. D. Classical Electrodynamics, chap. 3 (Wiley, New York, NY, 1999), 3rd edn.
32.Xiao, K., Kan, C. & Cui, X. Coulomb potential screening via charged carriers and charge-neutral dipoles/excitons in two-dimensional case. arXiv:2309.14101 (2023).
33.Ataei, S. S. & Sadeghi, A. Competitive screening and band gap renormalization in n-type monolayer transition metal dichalcogenides. Phys. Rev. B104, 155301 (2021). [Google Scholar]
34.Trani, F. et al. Screening in semiconductor nanocrystals: Ab initio results and Thomas-Fermi theory. Phys. Rev. B.73, 245430 (2006). [Google Scholar]
35.Ghasemi, S. A., Hofstetter, A., Saha, S. & Goedecker, S. Interatomic potentials for ionic systems with density functional accuracy based on charge densities obtained by a neural network. Phys. Rev. B92, 045131 (2015). [Google Scholar]
36.Faraji, S. et al. High accuracy and transferability of a neural network potential through charge equilibration for calcium fluoride. Phys. Rev. B95, 104105 (2017). [Google Scholar]
37.Yue, S. et al. When do short-range atomistic machine-learning models fall short? J. Chem. Phys.154, 034111. 10.1063/5.0031215 (2021). [DOI] [PubMed]
38.Gao, A. & Remsing, R. C. Self-consistent determination of long-range electrostatics in neural network potentials. Nat. Commun.13, 1572. 10.1038/s41467-022-29243-2 (2022).
[DOI] [PMC free article] [PubMed] [Google Scholar]
39.Grisafi, A. & Ceriotti, M. Incorporating long-range physics in atomic-scale machine learning. J. Chem. Phys.151, 204105. 10.1063/1.5128375 (2019). [DOI] [PubMed]
40.Giannozzi, P. et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Cond. Mat.21, 395502 (2009). [DOI] [PubMed] [Google Scholar]
41.Giannozzi, P. et al. Advanced capabilities for materials modelling with QUANTUM ESPRESSO. J. Phys. Cond. Mat.29, 465901 (2017). [DOI] [PubMed] [Google Scholar]
42.Giannozzi, P. et al. Quantum ESPRESSO toward the exascale. J. Chem. Phys.152, 154105 (2020).
[DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.