

Abstract

The human CMP-sialic acid transporter (hCST) is a mammalian highly conserved type III antiporter that translocates CMP-sialic acid into the Golgi lumen, supporting sialylation. Although different works have focused on elucidating structure–function relationships in the hCST, this is the first study to address them in an alternatively spliced isoform. We have previously reported the expression of a functional human del177 isoform that has skipping of exon 6, resulting in a loss of 59 amino acids, without change in the open reading frame and conserving its C-terminal region. To elucidate structure–function relationships, we interrogated this isoform with a known pathogenic variant c.303C>T (p.Q101H) for the wt isoform, showing that its pathogenicity is significatively reduced in the mutated del177 isoform (del177Q101H). This is further explained by using a homology model based on previously reported mouse and maize crystal structures.
Introduction
Sialylation is terminal glycosylation where sialic acid (Sia) residues can be linked in an α2,3- or α2,6-glycosidic covalent bond to galactose (Gal) moieties, in an α2,6 bond to N-acetylgalactosamine (GalNAc) residues, and in an α2,9- or α2,8-bond to another Sia residue at the outer end of glycans. Sialylation plays a crucial role in multiple biological functions in both health and disease.1−4 Sialic acids are a family of negatively charged nonuloses with highly variable chemical structures and widely distributed in vertebrates; the N-acetylneuraminic acid (Neu5Ac) is the main form found in humans. Sia coupled to CMP (CMP-Sia) is the donor substrate of sialyltransferase enzymes necessary to carry out the sialylation of proteins, lipids, or RNA.5−8
The human CST (hCST) is a 337 aa, ten transmembrane domain (TMD) protein responsible for internalizing CMP-Sia into the Golgi lumen, and it is encoded by SLC35A1.9−11 Over the years, the functional mechanism of hCST has been described through several strategies, including punctual mutations and chimeras. A major advancement in the structural comprehension of the hCST was the elucidation of the crystallographic structure of the mouse transporter (mCST) reported in 2019, opening a window to explore different aspects of the hCST, including pathogenic variants and their impact on protein structure/function.9,12,13 Also, clinical and functional data have been obtained through studies in patients affected by pathogenic variants in SLC35A1 that cause the congenital disorder of glycosylation (CDG), SLC35A1-CDG. SLC35A1-CDG is mainly characterized by neurological and development delay, hypotonia, coagulation defects or macrothrombocytopenia, neutropenia, and a deficit of sialyl-Lewis-X antigen on the surface of neutrophils.14,15
Martínez-Duncker et al. reported the first patient affected by SLC35A1-CDG and described the presence of alternative splicing of the hCST including the del130 (227 amino acids), del244 (189 amino acids), del290 (65 amino acids), and del177 (278 amino acids) isoforms. Afterward, Salinas-Marín et al. showed that even though the human del177 (del177-hCST) isoform loses 177 bp corresponding to exon 6, related to TMD 7 (V208–G217 stretch),14,16,17 it remains functional, in contrast to the hamster del177 (haCST) isoform expressed in CHO mutant cells, namely, Lec2, characterized by deficient CMP-Sia transport (Cricetulus griseus; del177-haCST).18−20
Further SLC35A1-CDG cases have been reported, including a female with a homozygote c.303G>C (p.Q101H; NM_006416.5) missense mutation in SLC35A1 localized in TMD3 of the hCST that clinically presented with delayed psychomotor development, behavioral problems, microcephaly, dysmorphic facial features, reduction of some coagulation factors, and dying at the age of 22 years due to respiratory failure.21 The Q101H pathogenic variant did not affect Golgi localization, but its transporter activity was reduced by 50%.21
The overexpression of Q101H hCST in SLC35A1-deficient cells restored sialylation of O-glycans epitopes but not N-glycan sialylation.22 Dimerization analysis of the Q101H variant using the NanoBiT approach suggested that Q101H alters hCST dimerization. Interestingly, this variant partially rescued the sialylation defect in SLC35A1 knockout cells, suggesting the possibility that hCST works in monomeric and dimeric conformations because, apparently, dimerization may not be strictly required for its transport activity.23 Based on the crystal structure of mCST, homology modeling revealed that the Q101H variant loses interaction with the CMP-Sia substrate. Supporting this hypothesis, Li et al. proposed that the Q101 residue is positioned in the middle pockets of the transporter cavities, affecting its interaction with the phosphate group of CMP and consequently the internalization of the substrate.24
This work aimed to obtain novel structural information to explain the functionality of del177-hCST by assessing the impact of the p. Q101H (del177-hCSTQ101H) compared with the mutated full-length hCST (hCSTQ101H).
Materials and Methods
Cell Culture
The HEK293T wild-type (HEK293TWT) and knockdown (HEK293TKO) cell lines were obtained by Antje Banning and Ritva Tikkanen’s working group (University of Giessen faculty of medicine Giessen, Germany) and kindly donated to our laboratory.25 Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with high glucose supplemented with 2 mM l-glutamine, 1% penicillin/streptomycin, and 10% fetal bovine serum (FBS) (Thermo Fischer Scientific, Waltham, MA) at 37 °C in a humidified atmosphere containing 5% CO2.
Genotyping and Phenotyping of HEK293TKO Cells
HEK293TKO cells were analyzed by RT-PCR using Taq DNA polymerase (#EP0401, Thermo Fisher Scientific) to identify mutant SLC35A1 gene expression. Analysis was made using forward TRN-2s 5′-GAACCATGGCTGCCGAGAGAGAC-3′ and reverse NTR-4as 5′-GGAAAGGACAATGGCCGCTGCTGCTGC-3′ primers. PCR products were analyzed in melt agarose, and a lower size band than the wt of SLC35A1 was purified and Sanger sequenced (Figure S1). Furthermore, HEK293TKO cells and HEK293TWT (used as the control) were permeabilized with a Cytofix/Cytoperm kit (no. 554714, BD) following the manufacturer's protocol. Cells were washed with phosphate-buffered saline (PBS) and stained with anti-SLC35A1 antibody (#ab230196, Abcam) at 0.05 μg/mL. As the permeabilization control, we used an antitubulin DM1A antibody (#62204, Thermo Fisher Scientific) at 0.05 μg/mL and Alexa flour 647 goat antimouse as the secondary antibody. In addition, staining with biotinylated Sambucus nigra lectin (SNA, B-1305-2, Vector laboratories) was performed at 0.3 μg/mL, and cells stained were analyzed by flow (Figure S2).
Site-Directed Mutagenesis of c.303 G > C in SLC35A1
The vector Topo 3.3 TA (K8300–01, Life Technologies) was used to clone wt and del177 isoforms from SLC35A1 according to Salinas-Marín.16 Site-directed mutagenesis of c.303 G > C (Q101H) was made using Phusion Site-Directed enzyme (#F-541, Thermo Fisher Scientific). PCR amplicons were obtained using primers forward Mut-RSM5:5′-GTG TAT GCT GTT CAC AAC AAC ATG GCT T-3′ and reverse MUT-RSM3:5′-TAA CGA TGG CAC ACT TAA CTT CAA CAG TTC-3′, where the forward oligonucleotide carries punctual mutation. Thermocycling conditions of PCR were as follows: initial denaturation step at 98 °C for 30 s, 30 cycles of amplification at 98 °C for 10 s, 65 °C for 30 s, 72 °C for 5 min, and a final extension step at 72 °C for 10 min. PCR products (5 μL) were ligated with the T4 ligase enzyme (EL0012, Thermo Fisher Scientific).
Chemicompetent, match-one clones (Escherichia coli) were transformed by heat shock and plated on selective LB agar plates. Positive clones were screened by conventional PCR with the oligonucleotides forward TRN-2s 5′-GAACCATGGCTGCCGAGAGAGAC-3′ and reverse NTR6-as 5′-GGAAAGGACAATGGCCGCTGCTGC-3′. Mutant constructions were sent for sequencing either on purified DNA and after confirmation by PCR-ARMS with the trioligonucleotide system forward 5′-GCTAGGAAAGCCATGTTGTTC-3′, normal reverse 5′-GCTAGGAAAGCCATGTTGTTC-3′, and the reverse oligonucleotide including punctual mutation 5′-GCTAGGAAAGCCATGTTGTTG-3′. Sanger sequencing results were analyzed by the Snap Gene Viewer obtaining WTQ101H and del177Q101H constructions.
Transfection of HEK293TKO Cells and Antibiotic Selection
The HEK293TKO cell is a model with a deficiency of sialic acid residues on the cell’s surface. 24 h before transfection, cells were seeded onto 24-well plates. Transfection of WT, del177, WTQ101H, and del177Q101H constructions was done using 3 μL of lipofectamine 2000 (#1668019, Thermo Fisher Scientific) and 2 μg of plasmid diluted with a DMEM medium free of serum and antibiotic in a 200 μL final volume. Thus, cells were incubated for 24 h, supplemented with 300 μL of DMEM medium with 10% FBS, and incubated for 48 h. Cell selection was performed by Geneticin G418 (#4727878001, Roche potency 892 μg/mg) at 2 mg/mL for 7 days. Then, total RNA was isolated using the Trizol reagent from the selected cells, and cDNA was synthesized from 2 μg of the total RNA. Finally, normal and mutated transcripts overexpressed were analyzed by PCR-ARMS, as explained above (Figure S3).
Cytotoxicity Assay by RCA-I Lectin
After 7 days of Geneticin selection, HEK293TKO transfected cells with WT, del177 constructions, and their mutated counterparts WTQ101H or del177Q101H were seeded into 96-well plates for 24 h and exposed to 10 ng/mL Ricinus communis Agglutinin lectin (RCA-I, Vector Laboratories, Inc.) for 48 h, and cell proliferation was measured through the MTT assay. For the MTT assay, 5 μg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide was diluted in PBS, and 10 μL of MTT solution was added to cells grown on 100 μL of DMEM medium and cells were incubated for 4 h under conditions described above. After that, we added 150 μL of MTT solvent (4 mM HCl/isopropanol, v/v) into each well, and the plates were incubated for 10 min at 37 °C under agitation. Then, the absorbance was measured on a Spectra Max M5 (Molecular devices) at 590 nm.
Lectin Staining
The HEK293TKO cells were transfected with the wt hCST isoform (KO-WT), the del177-hCST isoform (KO-del177), as well as the mutated Q101H wild-type variant (KO-WTQ101H) or the mutated Q101H del177 (KO-del177Q101H) isoform. These cells were fixed with 4% paraformaldehyde (PFA) and stained with 13 μg/mL SNA (FL-1301–2, Vector Laboratories) coupled to fluorescein isothiocyanate (FITC) diluted in a DMEM medium. Lectin-labeled samples were assessed by flow cytometry (Facs Canto II BD) and analyzed by Flow Jo V10 software.
Immunostaining Microscopy
Transfected and selected cells mentioned above were seeded in 8-well μ-slide microscopy chambers (80806, Ibidi) and permeabilized for 5 min with PBS supplemented with 0.05% triton X-100. After that, cells were fixed with 4% PFA for 30 min at 4 °C, three washes were performed with PBS, and blocking was made with 5% bovine serum albumin (BSA) for 1 h. Anti-TGN 46 antibody (ab2809, clone 2F7.1, Abcam) was used to label TGN using 2 μg/mL, and GM130 antibody (ab169276, Abcam) at 7 μg/mL was employed to stain Cis-Golgi incubating cells overnight at 4 °C. Secondary antibodies employed were antimouse IgG1-PE/CY7 (no. 406613, BioLegend) for TGN46 or antimouse IgG-Alexa 647 (ab150107, Abcam) for GM130 diluted in 5% BSA, and cells were incubated for 1 h at room temperature (RT). The hCST was marked with the anti-SLC35A1 antibody at 0.05 μg/mL for 2 h at RT and contra-stained with the antirabbit-Texas Red antibody (T-2767, Thermo Fisher Scientific) at 4 μg/mL for 1 h at RT. Confocal images were acquired with a Nikon A1R+ laser scanning confocal head coupled to an Eclipse Ti-E inverted microscope (Nikon Corporation, Tokyo, Japan) equipped with a motorized stage (TI-S-E, Nikon) and controlled through Nis Elements C v.5.00 software located at Unidad de Microscopía IIBO-UNAM, RRID:SCR_022204. Cells were analyzed under an Apo TIRF 100X Oil DIC N2 (N.A. 1.49); single plane images were sequentially captured using standard galvanometric scanners, excitation wavelengths of 405 (2 mW), 561 (2.2 mW), and 647 (17.1 mW) with AOTF modulation, the pinhole aperture set at 76.63 μm, and both standard and GaAsP detectors, including a transmitted light one, and analyzed by Fiji: ImageJ software.
Homology Models
Models were built in the AlphaFold 3 (AF3) server26 using the sequences for hCST and the del177-hCST variant from the UNIPROT databases P78382-1 and P78382-2, respectively.27 This generated structures for the open conformation, akin to the model of CST from maize (CSTZM), and in the partially closed conformation, 6OH3 was used for hCST, and the del177 variant was only retrieved in the open conformation. To obtain del177 models in a partially occluded conformation close to mCST, AF2 was used in conjunction with the mCST structure 6OH39 as a template. Given that AF3 and AF228 do not currently allow for arbitrary ligands in model building, coordinates for CMP-Sia were grafted onto each structure using the crystallographic structures of the mCST and CSTZM (PDB ID 6OH3(9) and 6I1R,29 respectively) as templates. As CSTZM is bound to CMP only, the orientation of Sia was manually corrected to avoid clashes with the transporter helices by rotating around the phosphate-sialic dihedral angle. Parameters for CMP-Sia were obtained in the Charmm-gui server30 with the ligand reader and the modeler tool31 and the Charmm36m32 force field. Mutants hCSTQ101H and del177Q101H were constructed in the same server using the mutation tool33 as a template for the AF2 and AF3 output models.
Structure Optimization
All structures (hCST, hCSTQ101H, del177-hCST, and del177-hCSTQ101H) in complex with CMP-Sia were submitted to the Charmm-gui server to add hydrogen atom coordinates and to generate the topology and parameter files needed for energy minimization. Energy minimization was performed in CHARMM48 software34 with the Charmm36m32 force field to remove steric clashes produced by grafting CMP-Sia onto the transporter structures. All atoms were kept fixed, except for those located 5.0 Å away from CMP-Sia. A first round of 100 steps for the hydrogen atoms only was carried out with the steepest descent, followed by 500 steps of conjugate gradient and by 500 steps of Adopted Basis Newton–Raphson (ABNR) minimization algorithms for all atoms in and around CMP-Sia. This was enough to achieve a convergence.
Ligand–protein interaction diagrams were analyzed with LigPlot+35 using the default parameters (maximum distance hydrogen–acceptor = 2.7 Å, and maximum distance donor–acceptor = 3.35 Å). Structure visualization and rendering were done in the VMD 1.9.3 software.36
Statistical Analysis
Flow cytometry data were analyzed using GraphPrism9 software, and statistical significance was determined by the one-way Brown–Forsythe ANOVA test and Tukey′s multiple comparisons test, using p < 0.05 as the significant value. The mean fluorescence intensities (MFIs) obtained from HEK293TWT and HEK293TKO cells were used as staining controls and were not considered in the multiple comparisons analysis to determine significance levels. The MFI was normalized by taking HEK293TWT cells as 100% in three independent experiments. For the MTT assay, we used n = 4 with three internal replicates per experiment and a one-way Brown–Forsythe ANOVA and Tukey′s multiple comparisons test, using p < 0.05 as the significant value.
Results
The Pathogenicity of the Q101H Variant Is Reduced in the del177 Isoform
To assess the pathogenicity of the Q101H variant, HEK293TKO cells were used. These cells have a defect in the transport of sialic acid compared to HEK293TWT cells and are characterized by a reduced sialophenotype previously reported.25 The glycophenotype of cells transfected with the full-length wild-type transcript (KO-WT), the del177 isoform (KO-del177), the full-length wild-type transcript bearing the Q101H variant (KO-WTQ101H), and the del177 isoforms bearing the Q101H variant (KO-del177Q101H) was assessed by flow cytometry by measuring the surface binding of the SNA lectin that recognizes α-2,6 Sia residues. As expected, the HEK293TKO cells showed a lower relative fluorescence (24.5% relative MFI with 11.4% positive cells) for SNA binding with respect to 100% MFI from HEK293TWT (95.3% positive cells). Furthermore, cells transfected with an empty plasmid (EP) were considered as a negative control and exhibited similar relative fluorescence (27.5% relative MFI, p < 0.00013, and 16.8% positive cells) for SNA reactivity as HEK293TKO cells (Figure 1).
Figure 1.

Analysis of SNA lectin cell surface binding by flow cytometry. (A) Graph of the relative fluorescence intensity corresponding to SNA lectin binding in transfected cells, normalized to levels of HEK293TWT cells. The relative fluorescence intensity from SNA reactivity was not significantly different between the KO-del177Q101H and KO-WT cells. Furthermore, KO-del177Q101H showed significantly improved SNA binding concerning KO-WTQ101H cells. (B) Representative cytometry histograms of percentages of SNA-positive cells. The horizontal axes show the fluorescence intensity of SNA lectin conjugated to FITC, while the vertical axes show the morphology in forward scatter. Figure S4 shows the percentage of cell positives for SNA lectin. n = 3, mean ± , ***p < 0.05. EP = HEK293TKO cells transfected with an EP.
KO-WT cells restored SNA binding to levels comparable to HEK293TWT cells (105.2% relative MFI and 91.9% positive cells normalized to HEK293TWT cells). In comparison, the KO-del177 cells showed improved SNA binding but to a lower degree (45.2% relative MFI, p < 0.015, and 74.5% positive cells) (Figure 1). The KO-WTQ101H cells were also able to partially restore sialylation (33.4% relative MFI, p < 0.0016; with 53.7% positive cells), consistent with previous findings21,23 (Figure 1). Interestingly, the KO-del177Q101H cells showed 67% relative MFI and 85.5% SNA+ binding, indicating that this variant is not as pathogenic in the del177 isoform.
To further corroborate the reduced pathogenicity of the del177Q101H isoform, HEK293TKO transfected cells were exposed to the RCA-I lectin, and cytotoxicity was measured through the MTT assay.37 This lectin has been previously used to isolate mutants from CHO cells lacking Sia because galactosylated epitopes for RCA-I are exposed. RCA-I binds to uncapped Gal or GalNAc residues of glycoconjugates attached to the cell membrane, resulting in a decreased expression of vascular endothelial growth factor receptor-2 and inhibition of glycopeptide synthesis, causing cell death.38,39 As expected, the RCA-I lectin was highly cytotoxic for HEK293TKO cells (39% survival rate) and EP cells (28%) compared to control HEK293TWT cells (92% survival), which were resistant. Interestingly, the KO-del177Q101H cells (80% survival rate) showed similar resistance as HEK293TWT cells (93%, p < 0.8780), in contrast to the KO-WTQ101H cells that presented a similar cytotoxicity (41%) as the HEK293TKO cell. By MTT results, the KO-del177Q101H cells showed a higher cell survival rate than the KO-del177 cells (65%, p < 0.6580), unlike KO-WTQ101H cells that were not resistant to RCA-I treatment (p < 0.0003), (Figure 2A). Hence, the Q101H mutation presents a less severe effect in the del177 isoform compared with the WT.
Figure 2.

Cytotoxicity assay with RCA-I lectin. (A) The absorbance was measured 48 h after exposition to RCA-I lectin, where KO-WTQ101H cells showed less proliferation compared to KO-WT and KO-del177Q101H cells. HEK293TWT cells were used as a positive control, HEK293TKO cells were used as a negative control, and HEK293TKO cells were transfected with an EP as a transfection control. For absorbance analysis, a one-way Brown–Forsythe ANOVA test and Tukey′s multiple comparisons test were used. n = 4, ****p < 0.05. (B) Micrographs were taken at 10X resolution, and cell morphology was contrasted with that of cells transfected without RCA-I (control) treatment.
Furthermore, micrographs of HEK293TWT cells exposed to RCA-I lectin showed a healthy morphology while HEK293TKO and EP cells showed stress like KO-WTQ101H cells. In contrast, the KO-del177Q101H cells showed a morphology like HEK293TWT and KO-WT cells (Figure 2B). Surprisingly, the KO-del177Q101H cells were observed to be healthier than the KO-del177 cells.
The del177Q101H Does Not Affect Golgi Localization or Structure
In previous reports, it was shown that Q101H hCST correctly localizes in the Golgi.24 To corroborate whether this also occurred with KO-del177Q101H, we examined its localization by confocal microscopy using the TGN46 antibody that labels the trans-Golgi network integral membrane protein 2 (TGN). We found that KO-del177Q101H cells properly colocalized with the TGN46 as did KO-WT, KO-del177Q101H, and KO-WTQ101H cells (Figure S5).
To further probe the status of the Golgi, we performed immunostaining of GM130 (red; cis-Golgi marker) in cells transfected with the different isoforms.40−43 Additionally, we analyzed the distribution of hCST (green) in wt and mutant cell lines. As expected, the variants did not colocalize with the cis-Golgi compartment. Following the micrographs, HEK293TKO cells showed a disaggregated and unlinked appearance of the GM130 protein and disorganized localization of the hCST compared with the compact composition and organized pattern of GM130 and hCST observed in HEK293TWT cells. Furthermore, KO-WT and KO-del177 cells displayed tight-packed GM130 situated close to the nucleus, restoration of disturbed Golgi, and organized localization of hCST as well as HEK293TWT cells. In addition, KO-del177Q101H cells exhibited noticeable compaction of GM130 and a restored hCST localization compared to KO-WTQ101H cells that displayed a slightly less rounded localization of GM130, and the hCST was loczalised in a kind of cluster that was less evident in KO-del177 cells (Figure 3).
Figure 3.
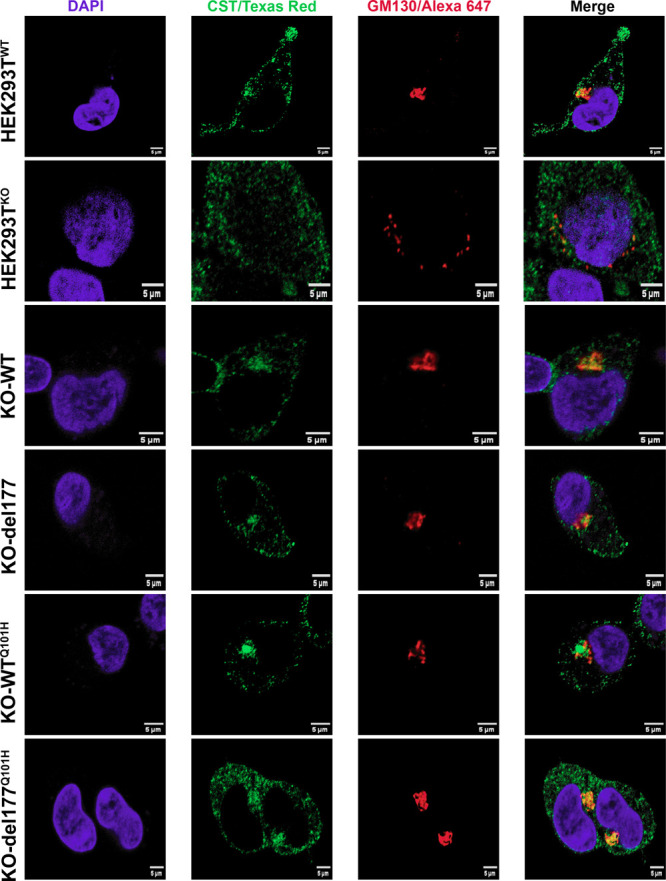
Confocal microscopy was employed to visualize GM130 in cells overexpressing the hCST isoforms in HEK293TKO cells. The cis-Golgi was specifically labeled using a GM130 antibody (red), while hCST was visualized with an anti-SLC35A1 antibody (green). Observations were made in KO-WT, KO-del177, KO-WTQ101H, and KO-del177Q101H cells, comparing the GM130 structure concerning HEK293TKO cells. In KO-WT and KO-del177Q101H cells, Golgi morphology restoration was evident compared to HEK293TKO cells. However, GM130 was observed disturbed in KO-WTQ101H cells, and hCST protein was detected as a cluster. Micrographs were obtained on a Nikon A1R+ STORM confocal microscope with a 100x objective. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole. Scale bar is 5 μm.
The del177-hCST Retains the Sialic Acid Cavity
A homology modeling was performed to explain how the CMP-Sia substrate might be interacting with binding cavities of the del177Q101H isoform and how these interactions could favor the increase of sialic acid in cells transfected with this variant. Inspection of the models produced by AF326 revealed two main conformations for the hCST. One structure corresponds to a closed conformation on both sides of the transporter, as in the mCST structures,9 while the other structure is in an outward-facing conformation, as in the CSTZM crystal structure.29 Both conformations differ in the conformation of the core helices and show a different position of the binding cavity toward the substrate, where a closed conformation recognizes the CMP-Sia with a cavity oriented in the middle. In contrast, the outward-facing conformation has an external cavity recognizing the CMP. The hCST and del177-hCST models were grafted using these two positions (Figure 4).
Figure 4.
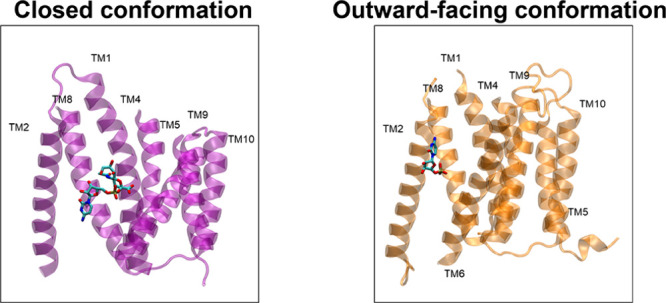
Different conformations of hCST transporters bound to CMP and the CMP-Sia substrate. Crystal structures for mCST (6OH3) and CSTZM (6I1R) are shown in purple and orange, respectively. TMDs 3, 6, and 7 are hidden for clarity. Models for the hCST and del177-hCST isoforms used these two structures as templates.
As previously described, the del177-hCST variant is functional despite losing 59 residues (192 aa-251 aa). AF3 model building of the del177 variant resulted in structures compatible with an outward-facing conformation as in CSTZM. Furthermore, the CMP-Sia substrate fits in the same interaction cavity as that in the full or hCST version. Since AF3 retrieved two different conformations for hCST, we built the partially occluded conformation of the del177-hCST isoform. As AF3 does not allow for template-target modeling, we used AF2 with the 6OH3 crystal structure as a template.
A comparison of the hCST and del177-hCST transporters reveals that the overall structure remains unchanged. Furthermore, truncation of hCST leads to the loss of the C-terminal fragment of TMD6 from residues 192 to 196, the entire TMD7 from residues 210 to 228, and the N-terminal fragment from residues 244 to 250 of TMD8 (Figure 5). These structural elements are close to the CMP-binding pocket, but the truncated transporter retains the sialic acid cavity. The interactions that ensure transport selectivity remain unchanged too, with specific residues reading both the cytosine and the sialic acid moieties.
Figure 5.

Missing region in the del177-hCST isoform. Front view of the mCST transporter; the residues lost by truncation are shown in yellow. A 90ο upward turn shows the bottom view of the transporter; each TMD is indicated. CMP-Sia is shown in a space-fill representation.
CMP-Sia Interacts with hCST-del177Q101H in an hCST-like Fashion
To investigate the effect of deletion and the effect of the point mutation Q101H on its interaction with the ligand, we constructed protein–ligand interaction maps for all complexes and mutants. Protein–ligand interaction maps were built in LigPlot+,35 using the CMP-Sia of the mCST transporter as a common reference. When CMP-Sia is in a mouse-like position in the hCST, it is stabilized by several interactions, all of them reported in previous publications as important for function.9,29,44 In both hCST and hCSTQ101H, N3 and C4-amine in the cytosine are recognized by Y214 and N210, conferring selectivity for cytosine over uracil. These residues are lost in the del177-hCST isoform, but their interactions could be compensated by a H-bond interaction between the C4-amine in the cytosine and the aromatic ring of Y259, reminiscent of a cation–pi interaction (Figure 6).
Figure 6.

Protein–ligand interaction maps of CMP-Sia in the mouse-like position. Each row shows the hydrogen bonds and van der Waals (VdW) interactions for the Q101 and H101 point mutations for the hCST and the del177-hCST isoform. On the bottom of each panel, the total number of H bonds and VdW interactions are shown as well as SNA lectin binding restoration percentages. For each map, the model engaging in more interactions is shown.
Residues Q101 and Y121 read out two CMP-Sia regions, engaging one oxygen of the phosphate group and one from the sialic acid through H bonds. These interactions are reduced to only one hydrogen bond in the Q101H point mutant in the hCSTQ101H variant. To determine the importance of these interactions, SNA lectin binding restoration assays were carried out in hCST-deficient cells by complementation with different variants from hCST. Taking the rescue afforded by the wt hCST transporter as 100%, hCSTQ101H partially restores function with ∼33% hCST activity. Surprisingly, del177-hCSTQ101H is better at restoring SNA lectin binding, with ∼67% hCST activity, compared to del177-hCST with ∼45% activity. The higher activity of del177-hCSTQ101H could be explained in terms of H101 and Y121 creating a hydrogen-bond network that resembles the interactions seen in the hCST transporter, as shown in Figure 6. Despite having similar interactions, the reduction of the hydrogen-bond network leads to decreased activity (Figure 6). It is interesting to notice that not only the number of interactions is important but also the identity of these interactions plays a role in determining function. Our models suggest that reducing the number of hydrogen bonds to the phosphate group and sialic acid moieties is related to reduced SNA lectin binding restoration. This behavior could be explained by fewer interactions, which lead to the ligand not being read in the right position.
On the other hand, when we analyzed the interactions of CMP-Sia in a maize-like position, we noticed that the position of the cytosine moiety is pointing toward outside the transporter, and it is mainly stabilized by VdW interactions. Although residues N102, K55, Y214, K272, and Y98 are the only contacting shared residues between the two positions, these residues show H-bonding with different moieties compared to the mouse-like position (Figure 7).
Figure 7.

Protein–ligand interaction maps of CMP-Sia in the maize-like position. Each row shows the VdW and H-bond interactions between CMP-Sia and hCST and del177-hCST isoforms carrying the Q101H mutation.
All models with CMP-Sia in a maize-like conformation failed to engage in hCST-like interactions, as seen previously (Figure 6). This behavior might be explained by the upward position and the inversion of the ligand, which leads to a loss of H-bonding interactions. Also, residue Q101 and the point mutation Q101H are far from CMP-Sia in both conformations (partially occluded and outward-facing conformation). This suggests that the effect of the Q101H mutation happens only in the partially occluded and inward-facing conformations (Figure S6).
Discussion
The hCST functions as an NST for transporting CMP-Sia through an antiporter mechanism. It can also act as a uniporter, being temperature sensitive and behaving as a saturable system.29,45−47 Over the years, function–structure relationships have been proposed by site-directed mutagenesis, epitope insertion,12 and chimeras with UDP-Gal transporter, and recently, a mouse crystal model has been proposed.48−50 These experiments have provided important details about CST, including that it is highly specific for its substrate CMP-Sia/CMP with a higher affinity for CMP.9,51 However, this theory may be discarded because CST is also capable of recognizing CDP-Ribitol, suggesting an ability to translocate small molecules if they are attached to a nucleobase cytidine, forwarding the theory that substrate recognition is due to three small cavities that recognize sugar, nucleotide, and phosphate group where the nucleobase cavity is responsible for internalizing the substrate.24,44 Even though information on the structure and function of hCST has advanced due to these contributions, the structure–function relationships in its splice variants have not been completely explored.
In previous reports, we described the functionality of an alternative spliced isoform of hCST, named del177-hCST. Here, we explore the different functional effects of the c.303C>T (p.Q101H) pathogenic variant, previously reported in a patient affected by SLC35A1-CDG and that decreases 50% of the hCST activity.21 In this work, through SNA lectin binding assays and RCA-I lectin cytotoxicity assays on transfected HEK293TKO cells with normal and mutated (c.303C>T) wild-type and del177-hCST variants, we show that the deleterious effect of the p.Q101H WT variant is not observed in the p.Q101H del177 isoform, providing novel structural insights into CMP-sialic acid recognition.16
To determine if p.Q101H affected the Golgi localization of the del177 isoform, we analyzed its overlap with the TG46 Golgi marker. We did that because the inhibition of terminal Golgi glycosylation causes the accumulation of glycoconjugate proteins, leading to changes in the Golgi structure. Through the years, it has been shown that alterations in terminal fucosylation and sialylation disrupt the normal operation of Golgi, causing accumulation of glycoproteins because charge proteins in vesicular traffic are highly glycosylated, leading to an increase in Golgi size and loss of its function to export proteins, generating deficient traffic.40,52,53 Concerning Golgi localization experiments, all hCST variants showed overlap with the TGN46 Golgi marker. We observed that cells with sialic acid deficiency, such as HEK293TKO and the overexpressing KO-WTQ101H isoform, displayed disturbed distribution and widening stacks of TGN46 (Figure S5).
Furthermore, these changes were also accompanied by morphological modifications in GM130, which were restored in cells overexpressing the del177 isoform and its mutant del177Q101H, becoming similar in appearance to HEK293WT cells. Our findings confirmed Golgi fragmentation as a response to deficient sialylation induced by depletion in functional hCST. Considering GM130 morphology changes and its destabilization as a consequence of hCST deficiency, it could be proposed as a marker in the case of alterations in the SLC35A1, which compromise normal sialylation based on the fact that GM130 has already been proposed as a neurodegenerative marker in diseases such as amyotrophic lateral sclerosis, a neurodegenerative disease known to fragment Golgi due to cellular stress and protein transport dysfunction, similar to what occurs in hCST disfunction when the CST does not work correctly.41,54 Furthermore, a possible explanation could be that the collateral impact of sialylation depletion results in under-sialylation of the glycosyltransferases and enzymes that are required for the adequate and functional organization of Golgi.
In the homology models, it was observed that the del177-hCST model retains its substrate recognition cavity despite losing the C-terminal fragment of TMD6, the entire TMD7, and the N-terminal fragment of TMD8. In this cavity, the K55 residue interacts with the cytidine base in both the partially occluded and outward-facing conformations, like the mouse model.9
In the partially occluded model, the Y27 residue maintains its VdW interaction, which has been described as important for maintaining the cavity structure in the mouse model. However, in the outward-facing conformation in hCST and del177-hCST, Y27 forms a hydrogen bond with Sia, suggesting that once CMP-Sia is internalized into the cavity, Y27 helps stabilize the complex.
Additionally, other residues previously described in the literature as being important for CMP-Sia recognition lose their interaction in the del177-hCST model but are replaced by others. For example, Y214 in the hCST model recognizes cytosine. Still, in the del177-hCST model, this interaction is replaced by Y259 and Y98 through VdW interactions, providing the necessary stability to keep the substrate properly oriented. Therefore, upon comparison of the hCST and del177-hCST models, it is observed that the del177 isoform in both conformations presents a lower number of interactions in the middle cavity, and the nucleobase recognition site is in the occluded conformation. However, these are compensated by forming additional hydrogen bonds in the outward-facing conformation, which contributes to substrate stability once internalized into the cavity.9,24 This could correlate with the findings of Salinas-Marín et al., where the del177 isoform showed no significant variation compared to the wild-type isoform at 72 h post transfection.16
Focusing on the outward-facing conformation, we find that Q101 in the hCST belongs to the middle recognition cavity and interacts with phosphate and Sia groups. These interactions are recovered in the del177-hCSTQ101H and lost in the hCSTQ101H model. This is particularly significant as these interactions have been previously reported to be crucial for maintaining the stability and recognition of CMP-Sia. The loss of these interactions in hCSTQ101H could be one of the key reasons why this isoform loses 50% of its functionality. Furthermore, the hCSTQ101H model cannot be compensated because Q101 generates a steric hindrance affecting the formation of interactions that can stabilize the substrate; meanwhile, in the del177-hCSTQ101H model, the elimination of 59 amino acid residues leaves a more flexible cavity avoiding the Q residue steric hindrance, generating the recovery or compensation of certain interactions that help stabilize the substrate within the cavity.
Conclusions
In this study, we confirm that the pathogenic variant c.303C>T (p.Q101H) of hCST, previously reported to decrease transporter activity by 50% in an SLC35A1-CDG patient, shows significantly reduced pathogenicity in the mutated del177 isoform (del177Q101H) when it is overexpressed in HEK293TKO cells. Furthermore, we contribute to the understanding of the structure and function of the del177 variant, an isoform that challenges the paradigms of membrane protein studies by remaining functional even when it loses 59 aa. This is because the recognition cavity retains a structure similar to that of the WT isoform. Thus, the remarkable similar ability to restore sialylation between del177Q101H and the wild-type isoform opens a window for the del177 variant as a potential genetic therapy in SLC35A1-CDG caused by the Q101H variant.
Previously, we reported the oligonucleotide morpholino, which allows blocking of the inclusion of exon 6 in pre-RNAm from the SLC35A1 gene. This alternative could be considered in these patients to redirect the protein translation process toward the del177Q101H variant instead of the WTQ101H mutant. This approach holds promise for therapeutic assistance to individuals affected by this CDG (Figure 8).
Figure 8.

Gene therapy by morpholino oligos in vivo. The backbone of these oligos consists of morpholine rings, distinctively connected to nitrogen atoms instead of the conventional oxygen atoms found in DNA or RNA. This atypical structure confers increased stability and resistance to nucleases. Upon binding to its target RNA sequence, the morpholino oligo disrupts normal cellular processes associated with the recognition and binding of RNA by proteins or other nucleic acids. This steric hindrance interferes with the proper functioning of the target RNA, often resulting in the inhibition of translation or the induction of exon skipping in pre-mRNA. In SLC35A1-CDG patients carrying the c.303C>T punctual mutation in the SLC35A1 gene, exon 6 would be blocked, causing a mature mRNA, which subsequently could be translated into the del177Q101H protein. This strategic manipulation of gene expression demonstrates the potential of morpholino oligomers in modulating splicing events for therapeutic purposes.
The use of oligonucleotides as biotherapeutics has increased in recent years, with successful applications in treating infectious diseases, such as vaccines against hepatitis B or SARS-CoV-2, and chronic diseases, such as muscular dystrophy.55−57 However, its potential for treating rare diseases has not yet been fully explored. Research into the use of oligo morpholinos in treating rare diseases is ongoing, and initial results are promising. These drugs have the potential to offer new hope for patients with rare diseases to allow more treatment options.
Acknowledgments
The authors are grateful to Dr. Antje Banning and Ritva Tikkanen for donating HEK293TWT and HEK293TKO cells. The authors also acknowledge the technical support received from Ph.D. Miguel Tapia-Rodriguez in the Microscopy Unit of IIBO-UNAM. Finally, the authors also thank Leticia Olvera, Maricela Olvera, and Erika Melchy Pérez for technical support in molecular biology and for providing facilities to use cytometry from the Biotechnology Institute, UNAM.
Glossary
Abbreviations
- aa
amino acids
- A.U.
absorbance units
- BSA
bovine serum albumin
- CDG
congenital disorder of glycosylation
- CMP-Sia
cytidine monophosphate N-acetylneuraminic acid
- EP
empty plasmid
- CST
CMP-Sia transporter
- hCST
human CST
- haCST
hamster CMP-Sia transporter
- mCST
mouse CMP-Sia transporter
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- Gal
galactose
- GalNAc
N-acetyl galactosamine
- MFI
mean fluorescence intensity
- MTT
(3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide
- Neu5Ac
N-acetylneuraminic acid
- NST
nucleotide sugar transporter
- PBS
phosphate-buffered saline
- PFA
paraformaldehyde
- PI
propidium iodide
- RCA-I
Ricinus communis agglutinin lectin
- RT
room temperature
- Sia
sialic acid
- Siglecs
sialic acid-binding immunoglobulin-type lectins
- SNA
Sambucus nigra lectin
- STs
sialyltransferases
- TMDs
transmembrane domains
- TGN
trans-Golgi network
- WGA
wheat germ agglutinin
- wt
wild-type
Data Availability Statement
All software described in the modeling section are free for use and available for download from their respective Web sites after prior registration: LigPlot+ version 2.2 (https://www.ebi.ac.uk/thornton-srv/software/LigPlus/download2.html), CHARMM48 (https://academiccharmm.org/program), and VMD 1.9.3 (https://www.ks.uiuc.edu/Development/) were used for visualization and are available as free software. Web servers such as AlphaFold 2, AlphaFold 3, and charmm-gui can be found at https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb, https://alphafoldserver.com/, and https://charmm-gui.org/, respectively. Finally, the UNIPROT and PDB databases are available at https://www.uniprot.org/ and https://www.rcsb.org/. The final AlphaFold models were selected after grafting the CMP-sialic acid onto each model and searching for steric clashes. The selected models were then minimized and analyzed for their ligand–protein interactions. All mutants Q101H were generated using the previously selected models. The models described previously as well as the input for CHARMM48 minimization and the input parameters are available for download as a.rar compressed archive. GraphPad Prism 9 was used for calculating IC50 and CC50 values (https://www.graphpad.com/). Origin 8.5 and Adobe Photoshop 2020 were used for data visualization (https://www.adobe.com/mx/).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c08466.
Figure S1, analysis of genotype from HEK293TKO cells; Figure S2, analysis of glycophenotype from HEK293TKO cells; Figure S3, PCR-ARMS to assess c.303G>C point mutation in transfected HEK293TKO cells; Figure S4, percentage of SNA-positive cells by flow cytometry; Figure S5, analysis of TGN and hCST localization by confocal microscopy; and Figure S6, interaction of the outward-facing and partially occluded conformations of the hCST and del177-hCST models (PDF)
Author Contributions
Conceptualization: B.I.V.-D., M.A.R.-M., N.P., and R.S.-M.; methodology: B.I.V.-D., M.A.R.-M., and N.P.; software: B.I.-V.D., M.A.-R.M., and N.P.; validation: B.I.V.-D., M.A.R.-M., N.P., and R.S.-M.; formal analysis: B.I.V.-D., M.A.R.-M., N.P.; B.D.-M., H.M.M.-M., Y.P.–C., I.M.-D., and R.S.-M.; investigation: B.I.V.-D., M.A.R.-M., N.P, I.M.-D., and R.S.-M.; resources: B.I.V.-D., M.A.R.-M., N.P.; B.D.-M., H.M.M.-M., Y.P.-C., I.M.-D., and R.S.-M.; data curation: B.I.V.-D., M.A.R.-M., N.P., I.M.-D., and R.S.-M.; writing—original draft preparation: B.I.V.-D., M.A.R.-M., N.P., I.M.-D., and R.S.-M.; writing—review and editing: B.I.V.-D., M.A.R.-M., N.P.; B.D.-M., H.M.M.-M., Y.P.-C., I.M.-D., and R.S.-M.; visualization: B.I.V.-D., M.A.R.-M., N.P., I.M.-D., and R.S.-M.; supervision: N.P., I.M.-D., and R.-S.M.; project administration: B.I.V.-D. and R.S.-M.; funding: R.S.-M.
The work carried out in our laboratory was supported by CONACYT grant number A1-S-27518, and BIVD was supported by CONACYT scholarship number 858568.
The authors declare no competing financial interest.
Supplementary Material
References
- Varki A. Sialic acids in human health and disease. Trends Mol. Med. 2008, 14 (8), 351–360. 10.1016/j.molmed.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobie C.; Skropeta D. Insights into the role of sialylation in cancer progression and metastasis. Br. J. Cancer 2021, 124 (1), 76–90. 10.1038/s41416-020-01126-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traving C.; Schauer R. Structure, Function and Metabolism of Sialic Acids. CMLS, Cell. Mol. Life Sci. 1998, 54, 1330–1349. 10.1007/s000180050258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer R. Sialic acids as regulators of molecular and cellular interactions. Curr. Opin Struct Biol. 2009, 19 (5), 507–514. 10.1016/j.sbi.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A; Schauer R.. Sialic Acids. In Laboratory Guide to the Methods in Biochemical Genetics; Springer, 2009; pp. 335–349. [Google Scholar]
- Varki A. Biological roles of glycans. Glycobiology. 2017, 27 (1), 3. 10.1093/glycob/cww086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer R; Kamerling J. P.. Exploration of the Sialic Acid World. In Advances in Carbohydrate Chemistry and Biochemistry; Academic Press Inc., 2018; Vol. 75, pp.1–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis A. L.; Chen X.; Schnaar R. L.; Varki A.. Sialic Acids and Other Nonulosonic Acids. In Essentials of Glycobiology; Cold Spring Harbor Laboratory Press, 2022. [PubMed] [Google Scholar]
- Ahuja S.; Whorton M. R. Structural basis for mammalian nucleotide sugar transport. Elife. 2019, 8, 1–27. 10.7554/eLife.45221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W; Colley K. J.. Nucleotide sugar transporters of the Golgi apparatus. In The Golgi Apparatus: State of the Art 110 Years after Camillo Golgi’s Discovery; Springer: Vienna; 2008; pp. 190–206. [Google Scholar]
- Zhao W.; Chen T. L. L.; Vertel B. M.; Colley K. J. The CMP-sialic acid transporter is localized in the medial-trans Golgi and possesses two specific endoplasmic reticulum export motifs in its carboxyl-terminal cytoplasmic tail. J. Biol. Chem. 2006, 281 (41), 31106–31118. 10.1074/jbc.M605564200. [DOI] [PubMed] [Google Scholar]
- Eckhardt M.; Gotza B.; Gerardy-Schahn R. Membrane topology of the mammalian CMP-sialic acid transporter. J. Biol. Chem. 1999, 274 (13), 8779–8787. 10.1074/jbc.274.13.8779. [DOI] [PubMed] [Google Scholar]
- Eckhardt M.; Mühlenhoff M.; Bethe A.; Gerardy-Schahn R. Expression cloning of the Golgi CMP-sialic acid transporter. Proceedings of the National Academy of Sciences. 1996, 93 (15), 7572–7576. 10.1073/pnas.93.15.7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willig T. N.; Breton-Gorius J.; Elbim C.; et al. Macrothrombocytopenia with abnormal demarcation membranes in megakaryocytes and neutropenia with a complete lack of sialyl-Lewis-X antigen in leukocytes - A new syndrome?. Blood. 2001, 97 (3), 826–828. 10.1182/blood.V97.3.826. [DOI] [PubMed] [Google Scholar]
- Kauskot A.; Pascreau T.; Adam F.; et al. A mutation in the gene coding for the sialic acid transporter SLC35A1 is required for platelet life span but not proplatelet formation. Haematologica. 2018, 103 (12), e613–e617. 10.3324/haematol.2018.198028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas-Marín R.; Mollicone R.; Martínez-Duncker I. A functional splice variant of the human Golgi CMP-sialic acid transporter. Glycoconj J. 2016, 33 (6), 897–906. 10.1007/s10719-016-9697-8. [DOI] [PubMed] [Google Scholar]
- Martinez-Duncker I.; Dupré T.; Piller V.; et al. Genetic complementation reveals a novel human congenital disorder of glycosylation of type II, due to inactivation of the Golgi CMP-sialic acid transporter. Blood. 2005, 105 (7), 2671–2676. 10.1182/blood-2004-09-3509. [DOI] [PubMed] [Google Scholar]
- Eckhardt M.; Gotza B.; Gerardy-Schahn R. Mutants of the CMP-sialic acid transporter causing the Lec2 phenotype. J. Biol. Chem. 1998, 273 (32), 20189–20195. 10.1074/jbc.273.32.20189. [DOI] [PubMed] [Google Scholar]
- Eckhardt M.; Gerardy-Schahn R. Molecular cloning of the hamster CMP-sialic acid transporter. Eur. J. Biochem. 1997, 248 (1), 187–192. 10.1111/j.1432-1033.1997.00187.x. [DOI] [PubMed] [Google Scholar]
- Deutscher S. L.; Nuwayhid N.; Stanley P.; Briles E. I. B.; Hirschberg C. B. Translocation across golgi vesicle membranes: A CHO glycosylation mutant deficient in CMP-sialic acid transport. Cell. 1984, 39 (2), 295–299. 10.1016/0092-8674(84)90007-2. [DOI] [PubMed] [Google Scholar]
- Mohamed M.; Ashikov A.; Guillard M.; et al. Intellectual disability and bleeding diathesis due to deficient CMP-sialic acid transport. Neurology. 2013, 81 (7), 681–687. 10.1212/WNL.0b013e3182a08f53. [DOI] [PubMed] [Google Scholar]
- Ng B. G.; Asteggiano C. G.; Kircher M.; et al. Encephalopathy caused by Novel Mutations in the CMP-Sialic Acid Transporter, SLC35A1. Am. J. Med. Genet A 2017, 173 (11), 2906. 10.1002/ajmg.a.38412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulc B.; Zadorozhna Y.; Olczak M.; Maszczak-Seneczko D.; Wiertelak W. Novel insights into selected disease-causing mutations within the slc35a1 gene encoding the cmp-sialic acid transporter. Int. J. Mol. Sci. 2021, 22 (1), 304–324. 10.3390/ijms22010304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.; Mukhopadhyay S. A three-pocket model for substrate coordination and selectivity by the nucleotide sugar transporters SLC35A1 and SLC35A2. J. Biol. Chem. 2021, 297 (3), 101069 10.1016/j.jbc.2021.101069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banning A.; Zakrzewicz A.; Chen X.; Gray S. J.; Tikkanen R. Knockout of the cmp–sialic acid transporter slc35a1 in human cell lines increases transduction efficiency of adeno-associated virus 9: Implications for gene therapy potency assays. Cells 2021, 10 (5), 1259. 10.3390/CELLS10051259/S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramson J.; Adler J.; Dunger J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. Published online 2024, 630, 493. 10.1038/s41586-024-07487-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. 10.1093/nar/gkac1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirdita M.; Schütze K.; Moriwaki Y.; Heo L.; Ovchinnikov S.; Steinegger M. ColabFold: making protein folding accessible to all. Nat. Methods. 2022, 19 (6), 679–682. 10.1038/s41592-022-01488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nji E.; Gulati A.; Qureshi A. A.; Coincon M.; Drew D. Structural basis for the delivery of activated sialic acid into Golgi for sialyation. Nat. Struct Mol. Biol. 2019, 26 (6), 415–423. 10.1038/s41594-019-0225-y. [DOI] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29 (11), 1859–1865. 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Kim S.; Lee J.; Jo S.; Brooks C. L.; Lee H. S.; Im W. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem. 2017, 38 (21), 1879–1886. 10.1002/jcc.24829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Rauscher S.; Nawrocki G.; et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods. 2017, 14 (1), 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. J.; Kern N.; Brown T.; Lee J.; Im W. CHARMM-GUI PDB Manipulator: Various PDB Structural Modifications for Biomolecular Modeling and Simulation. J. Mol. Biol. 2023, 435 (14), 167995 10.1016/j.jmb.2023.167995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks C. L.; Mackerell A. D.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30 (10), 1545–1614. 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman A. L.; Mark B. S. LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14 (1), 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Ghasemi M.; Turnbull T.; Sebastian S.; Kempson I. The mtt assay: Utility, limitations, pitfalls, and interpretation in bulk and single-cell analysis. Int. J. Mol. Sci. 2021, 22 (23), 12827. 10.3390/IJMS222312827/S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J. H.; Singh T.; Herp A.; Wu A. M. Carbohydrate recognition factors of the lectin domains present in the Ricinus communis toxic protein (ricin). Biochimie. 2006, 88 (2), 201–217. 10.1016/j.biochi.2005.07.007. [DOI] [PubMed] [Google Scholar]
- You W. K.; Kasman I.; Hu-Lowe D. D.; McDonald D. M. Ricinus communis Agglutinin I Leads to Rapid Down-Regulation of VEGFR-2 and Endothelial Cell Apoptosis in Tumor Blood Vessels. Am. J. Pathol. 2010, 176 (4), 1927. 10.2353/ajpath.2010.090561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg-Lerner A.; Benyair R.; Hizkiahou N.; et al. Golgi organization is regulated by proteasomal degradation. Nat. Commun. 2020, 11 (1), 1–14. 10.1038/s41467-019-14038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B.; Li X.; Zhu X. The Role of GM130 in Nervous System Diseases. Front. Neurol. 2021, 12, 743787 10.3389/fneur.2021.743787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N.; Rabouille C.; Watson R.; et al. Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 1995, 131 (6 Pt 2), 1715–1726. 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N. Emerging new roles of GM130, a cis-Golgi matrix protein, in higher order cell functions. J. Pharmacol Sci. 2010, 112 (3), 255–264. 10.1254/jphs.09R03CR. [DOI] [PubMed] [Google Scholar]
- Ury B.; Potelle S.; Caligiore F.; Whorton M. R.; Bommer G. T. The promiscuous binding pocket of SLC35A1 ensures redundant transport of CDP-ribitol to the Golgi. J. Biol. Chem. 2021, 296, 100789 10.1016/j.jbc.2021.100789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berninsone P.; Eckhardt M.; Gerardy-Schahn R.; Hirschberg C. B. Functional expression of the murine Golgi CMP-sialic acid transporter in Saccharomyces cerevisiae. J. Biol. Chem. 1997, 272 (19), 12616–12619. 10.1074/jbc.272.19.12616. [DOI] [PubMed] [Google Scholar]
- Capasso J. M.; Hirschberg C. B. Mechanisms of glycosylation and sulfation in the Golgi apparatus: evidence for nucleotide sugar/nucleoside monophosphate and nucleotide sulfate/nucleoside monophosphate antiports in the Golgi apparatus membrane. Proc. Natl. Acad. Sci. U. S. A. 1984, 81 (22), 7051. 10.1073/pnas.81.22.7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaramonte M.; Koviach J. L.; Moore C.; et al. Inhibition of CMP-Sialic Acid Transport into Golgi Vesicles by Nucleoside Monophosphates†. Biochemistry. 2001, 40 (47), 14260–14267. 10.1021/bi011262w. [DOI] [PubMed] [Google Scholar]
- Aoki K.; Ishida N.; Kawakita M. Substrate Recognition by UDP-galactose and CMP-sialic Acid Transporters. J. Biol. Chem. 2001, 276 (24), 21555–21561. 10.1074/jbc.M101462200. [DOI] [PubMed] [Google Scholar]
- Aoki K.; Ishida N.; Kawakita M. Substrate Recognition by Nucleotide Sugar Transporters. J. Biol. Chem. 2003, 278 (25), 22887–22893. 10.1074/jbc.M302620200. [DOI] [PubMed] [Google Scholar]
- Takeshima-Futagami T.; Sakaguchi M.; Uehara E.; et al. Amino acid residues important for CMP-sialic acid recognition by the CMP-sialic acid transporter: analysis of the substrate specificity of UDP-galactose/CMP-sialic acid transporter chimeras. Glycobiology. 2012, 22 (12), 1731–1740. 10.1093/glycob/cws116. [DOI] [PubMed] [Google Scholar]
- Ahuja S.; Cahill J.; Hartfield K.; Whorton M. R. Inhibition of CMP-sialic acid transport by endogenous 5-methyl CMP. PLoS One. 2021, 16 (6), e0249905 10.1371/journal.pone.0249905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y.; Zhang X.; Nix D. B.; et al. Regulation of protein glycosylation and sorting by the Golgi matrix proteins GRASP55/65. Nat. Commun. 2013, 4, 4. 10.1038/ncomms2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Wang Y. Glycosylation quality control by the Golgi structure. J. Mol. Biol. 2016, 428 (16), 3183–3193. 10.1016/j.jmb.2016.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bommel D. M.; Toonen R. F.; Verhage M. Mapping localization of 21 endogenous proteins in the Golgi apparatus of rodent neurons. Scientific Reports. 2023, 13 (1), 1–9. 10.1038/s41598-023-29998-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.; Adams A. M.; Johnsen R. D.; Fletcher S.; Wilton S. D. Morpholino Oligomer-Induced Dystrophin Isoforms to Map the Functional Domains in the Dystrophin Protein. Mol. Ther Nucleic Acids. 2020, 22, 263–272. 10.1016/j.omtn.2020.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S.; Sharma S. N.; Kundu J.; Pattanayak S.; Sinha S. Morpholino oligonucleotide-mediated exon skipping for DMD treatment: Past insights, present challenges and future perspectives. J. Biosci. 2023, 48 (4), 38. 10.1007/s12038-023-00365-z. [DOI] [PubMed] [Google Scholar]
- Egli M.; Manoharan M. Chemistry, structure and function of approved oligonucleotide therapeutics. Nucleic Acids Res. 2023, 51 (6), 2529–2573. 10.1093/nar/gkad067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All software described in the modeling section are free for use and available for download from their respective Web sites after prior registration: LigPlot+ version 2.2 (https://www.ebi.ac.uk/thornton-srv/software/LigPlus/download2.html), CHARMM48 (https://academiccharmm.org/program), and VMD 1.9.3 (https://www.ks.uiuc.edu/Development/) were used for visualization and are available as free software. Web servers such as AlphaFold 2, AlphaFold 3, and charmm-gui can be found at https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb, https://alphafoldserver.com/, and https://charmm-gui.org/, respectively. Finally, the UNIPROT and PDB databases are available at https://www.uniprot.org/ and https://www.rcsb.org/. The final AlphaFold models were selected after grafting the CMP-sialic acid onto each model and searching for steric clashes. The selected models were then minimized and analyzed for their ligand–protein interactions. All mutants Q101H were generated using the previously selected models. The models described previously as well as the input for CHARMM48 minimization and the input parameters are available for download as a.rar compressed archive. GraphPad Prism 9 was used for calculating IC50 and CC50 values (https://www.graphpad.com/). Origin 8.5 and Adobe Photoshop 2020 were used for data visualization (https://www.adobe.com/mx/).