

Abstract
Objective
Asthma is a prevalent status attributing to lower respiratory tract chronic inflammation. Azithromycin (AZM) is known to be effective against asthma. Thus, this study delved into the mechanism of AZM repressing airway remodeling (AR) via the SAPK/JNK pathway in asthma.
Methods
Simulated asthmatic AR mouse model was developed by induction with ovalbumin (OVA) and intervened with AZM or dexamethasone (DEX) and anisomycin (JNK activator). Pathological changes in mouse lung tissues and AR were assessed by HE and Masson staining. The numbers of inflammatory cells, macrophages, eosinophils, neutrophils and lymphocytes in bronchoalveolar lavage fluid (BALF) were detected by Diff-Quik staining. Inflammatory factor levels (IL-6, TNF-α, IL-4) in BALF, and Collagen I, Collagen III, SAPK/JNK and p-SAPK/JNK protein levels in lung tissues were measured by ELISA and Western blot.
Results
The OVA-led asthmatic mouse model was successfully established. Relative to the OVA group, AZM and DEX treatment improved pulmonary smooth muscle thickening and bronchial epithelial fibrosis, reduced inflammatory cells, macrophages, eosinophils, neutrophils and lymphocytes in BALF, inhibited inflammatory factor TNF-α, IL-6, and IL-4 levels in BALF, and down-regulated Collagen I, Collagen III, and p-SAPK/JNK protein levels in lung tissues, with no prominent difference between the two regimens. JNK activator partially reversed the protective effect of AZM against OVA-induced asthma in mice.
Conclusion
AZM alleviated airway inflammation by inhibiting the SAPK/JNK pathway, thereby repressing AR in asthmatic mice. This study provided partial theoretical basis for clarifying asthma pathogenesis and new ideas for treating asthma.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13019-024-03193-w.
Keywords: Azithromycin, Asthma, Airway remodeling, Airway inflammation, Stress activated protein kinases/C-jun N-terminal kinases
Introduction
Asthma is a common disorder caused by chronic inflammation of the lower respiratory tract, affecting approximately 8% of the American adults and 4.7% Chinese adults [1, 2]. In clinical practice, asthma patients exhibit recurrent shortness of breath, wheezing, chest tightness, and coughing [3]. Notably, airway remodeling (AR) is a key pathological feature of asthma, closely tied up with the persistence of asthmatic symptoms and clinical adverse outcomes [4]. Airway structural changes in asthma consist of elevated smooth muscle mass, subepithelial fibrosis, neovascularization, epithelial alterations, and enlargement of glands [4]. Airway remodeling is often considered as a result of long-term airway inflammation [5]. Airway remodeling leads to thickening of the airway wall, thus causing airway stenosis, bronchial hyperresponsiveness, airway edema, and high secretion of mucus. However, due to the complexity and heterogeneity of asthma, the underlying pathogenesis is currently unclear. Furthermore, standardized management for asthma mainly show solicitude for the severity of the disease and selecting the appropriate medical treatment to decrease the risk of exacerbation and control symptoms, but irrespective of its severity and usually despite optimal medication, asthmatic patients may experience a loss of disease control and acute symptom exacerbations [6]. Hence, finding a treatment for asthma is essential.
The stress activated protein kinases/c-jun N-terminal kinases (SAPK/JNK) pathway is activated under different stimuli including pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α) and stress [7]. Accumulating evidence has suggested that the JNK/SAPK pathway is associated with inflammation in various diseases [8–10]. In addition, SAPK/JNK serves a momentous role in asthma pathogenes and is expressed by Th1 [interference gamma, interleukin (IL)-2] and Th2 cells (IL-25, IL-13, IL-9, IL-5, and IL-4) [11]. However, there is limited research on whether the SAPK/JNK pathway participates in airway inflammation and AR by regulating inflammatory cytokines. Azithromycin (AZM) is a macrolide antibiotic that has good effects against asthma attacks [12]. Research has shown that AZM improves AR and by allergic airway inflammation inhibiting apoptosis of rat lung epithelial cells [13]. What’s more, AZM improves AR induced by ovalbumin (OVA) in Balb/c mice by inhibiting epithelial mesenchymal transition [14]. However, it is currently unclear whether AZM can alleviate airway inflammation in asthmatic mice, thereby inhibiting AR by repressing SAPK/JNK pathway activation. This study aims to investigate the effect of AZM on airway inflammation and AR in asthmatic mice by curbing SAPK/JNK pathway activation, in an attempt to provide partial theoretical basis for clarifying the pathogenesis of asthma and novel ideas for treating asthma.
Materials and methods
Ethics statement
The study was authorized by the academic ethics committee of the Second Affiliated Hospital Zhejiang University School of Medicine (Approval number: 2023081). All procedures were abided by the Guide for the Care and Use of Laboratory Animals to minimize the pain of mice.
Experimental animals
Sixty Balb/c mice (30 male and 30 female, weighing 20 ± 2 g, 5–6 weeks old) from Vital River [SYXK (Beijing) 2022-0052, Beijing, China] were acclimated for 1 week under standard conditions of humidity (60% ± 10%), ambient temperature (22 °C ± 1 °C), and 12-h light/dark cycle (with light on at 8 am) with food and water ad libitum.
Animal model establishment and treatment
Sixty mice were arranged into the following 5 groups (n = 12): The Con group, the OVA group, the dexamethasone (DEX) group (positive control), the azithromycin (AZM) group and the AZM + Anisomycin group.
After 7 days of adaptive feeding, on the 0 and 14th days, mice were sensitized by 0.2 mL sensitizer [20 µg OVA (Sigma-Aldrich, St. Louis, MO, USA) and 20 mg aluminum hydroxide (Sigma-Aldrich) intraperitoneally, dissolved in normal saline] [14]. Mice in the Con group were intraperitoneally injected with the same volume of normal saline. Starting from the 21st day, mice in the Con group were nebulized and stimulated with OVA solution (1%) utilizing an ultrasound nebulizer (NE-U12; Omron, Tokyo, Japan). Mice in the Con group were stimulated with the same volume of normal saline (1 h each time, a total of 7 times). Starting from the 21st day, mice in the DEX and AZM groups were gavaged with 0.2 mL of normal saline containing 0.7 mg/kg DEX or 35 mg/kg AZM (equivalent to a clinical equivalent dose of 250 mg/day in humans) 1 h before each OVA nebulization stimulation [14]; the AZM + Anisomycin group mice were given 0.2 mL of normal saline containing 35 mg/kg AZM by gavage 1 h before each OVA nebulization stimulation, and injected with 5 mg/kg Anisomycin through tail vein [15]; the Con group mice were given an equal amount of normal saline by gavage in the same way. Among these, DEX tablets (national medicine permission number H44024276) are conventional drugs for treating asthma, purchased from SUCCHI Pharmaceutical (Zhongshan, Guangdong, China); AZM tablets (national medicine permission number H20056048) were purchased from Merro Pharmaceutical (Dalian, Liaoning, China); Anisomychin (HY-18982) is a JNK activator, purchased from MedChemExpress (Monmouth Junction, NJ, USA).
Next, 24 h after administration, mice were euthanized by 1% pentobarbital sodium (PS) (100 mg/kg) intraperitoneally, with their lung tissue sample collected. The lung tissues of each group of mice were randomly assigned as follows: lung tissues of 6 mice were sliced and stained, and those of the other 6 mice were prepared into tissue homogenate for Western blot assay after bronchoalveolar lavage fluid (BALF) collection.
BALF collection
After euthanizing by an excessive intraperitoneal injection of PS (1%, 100 mg/kg), the mice were fixed on the operating table, with their trachea exposed. The neck skin was cut apart, the tissues were split, and the trachea was exposed. Afterwards, the mice were inserted with a sterile 22G indwelling needle, with their left lung ligated immediately, and slowly injected with 0.9% sodium chloride into the right lung, and then the fluid was slowly withdrawn, with the operation repeated 3 times. More than 1.2 mL of BALF was acquired and centrifuged. Supernatant was preserved at -20 °C for BALF inflammatory factor level assessment by enzyme-linked immunosorbent assay (ELISA) assay.
ELISA
BALF was collected, and inflammatory factor IL-6, TNF-α and IL-4 levels in BALF were measured by ELISA. IL-6 (PI326), TNF-α (PT512) and IL-4 (PI612) kits were all from Beyotime Biotechnology (Shanghai, China). All operations strictly followed the instructions of the reagent kits.
Histological staining
The tissues were cut into Sect. (4 μm thickness) and subjected to Masson trichrome staining and hematoxylin & eosin (HE; Sigma-Aldrich) staining to evaluate the collagen deposition degree and inflammation, separately. The lung tissue sections were magnified 200 times under the microscope, and three complete cross-sectional bronchial images with a diameter of 150–200 μm were selected on each section. ImagePro® Plus 6.0 software (Media Cybernetics, Bethesda, MD, USA) was used to measure airway wall thickness (Wat), bronchial basement membrane perimeter (Pbm), and bronchial smooth muscle thickness (Wam). Standardized by Pbm, the Wat/Pbm and Wam/Pbm ratios were utilized to express bronchial wall thickness and bronchial smooth muscle thickness, respectively. To quantify the fibrosis score, the bronchi with 150–200 μm diameter were selected randomly. The fibrosis degree was graded as 0–3 using the Masson-stained sections, with the detail methods referring to an existing study [14].
Analysis of inflammatory cells from BALF
The harvested BALF was subjected to centrifugation for 15 min at 3000 rpm. The resulting cell pellet was subsequently resuspended in phosphate-buffered saline (100 µL) at 4 °C. Next, the total cell count was determined using a hemocytometer. Cell smears were prepared via cytocentrifugation using a Cytospin 3 instrument (Thermo Scientific, Pittsburgh, PA, USA) and stained using the Diff-Quik staining kit (ZY-64664, Zeye Biotechnology, Shanghai, China). A minimum of 200 cells per slide were examined under a light microscope, and inflammatory cells (eosinophils, macrophages, lymphocytes, neutrophils) were identified and quantified based on their staining features and morphological criteria.
Western blot
Lung tissues were lysed and separated using protein lysis solution (R0278, Sigma-Aldrich), the protein content was analyzed with the help of the bicinoninic acid protein assay kit (Beyotime Biotechnology). Then, the protein was separated using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, followed by transfer onto the polyvinylidene fluoride (PVDF) membranes. Thereafter, membranes were blocked for 1 h in Tris-buffered saline with Tween-20 (TBST; 137 mM NaCl, 20 mM Tris, 0.1% Tween-20) containing 5% skim milk, followed by incubation with primary antibodies SAPK/JNK (1:1000, #67096, Cell Signaling Technology, Beverly, MA, USA), phospho-SAPK/JNK [1:1000, #9251, Cell Signaling Technology [16]], Collagen I (1:1000, ab260043, Abcam, Cambridge, UK), Collagen III (1:5000, ab7778, Abcam) and GAPDH (1:2500, ab9485, Abcam) at 4 °C overnight. After TBST washes, membranes were fostered with goat anti-rabbit horseradish peroxidase labeled secondary antibody (1:2000, ab6721, Abcam) at room temperature for 2 h, and ultimately, incubated with BeyoECL Plus (Beyotime Biotechnology). The grayscale of each band in Western blot images was quantified using ImageJ software (version 1.61; NIH Image, Bethesda, MD, USA), with GAPDH as the internal reference. Each experiment was repeated thrice.
Statistical analysis
All data were statistically analyzed and plotted using GraphPad Prism 8.01 (GraphPad, San Diego, CA, USA). The measurement data were listed in the form of mean ± standard deviation (SD). Independent sample t-test was employed for comparisons between 2 groups, one-way analysis of variance (ANOVA) analysis was applied for comparisons among groups, followed by Tukey’s test. P value was attained from a bilateral test, with P < 0.05 indicating a statistically significant difference.
Results
AZM suppressed OVA-stimulated AR in asthmatic mice
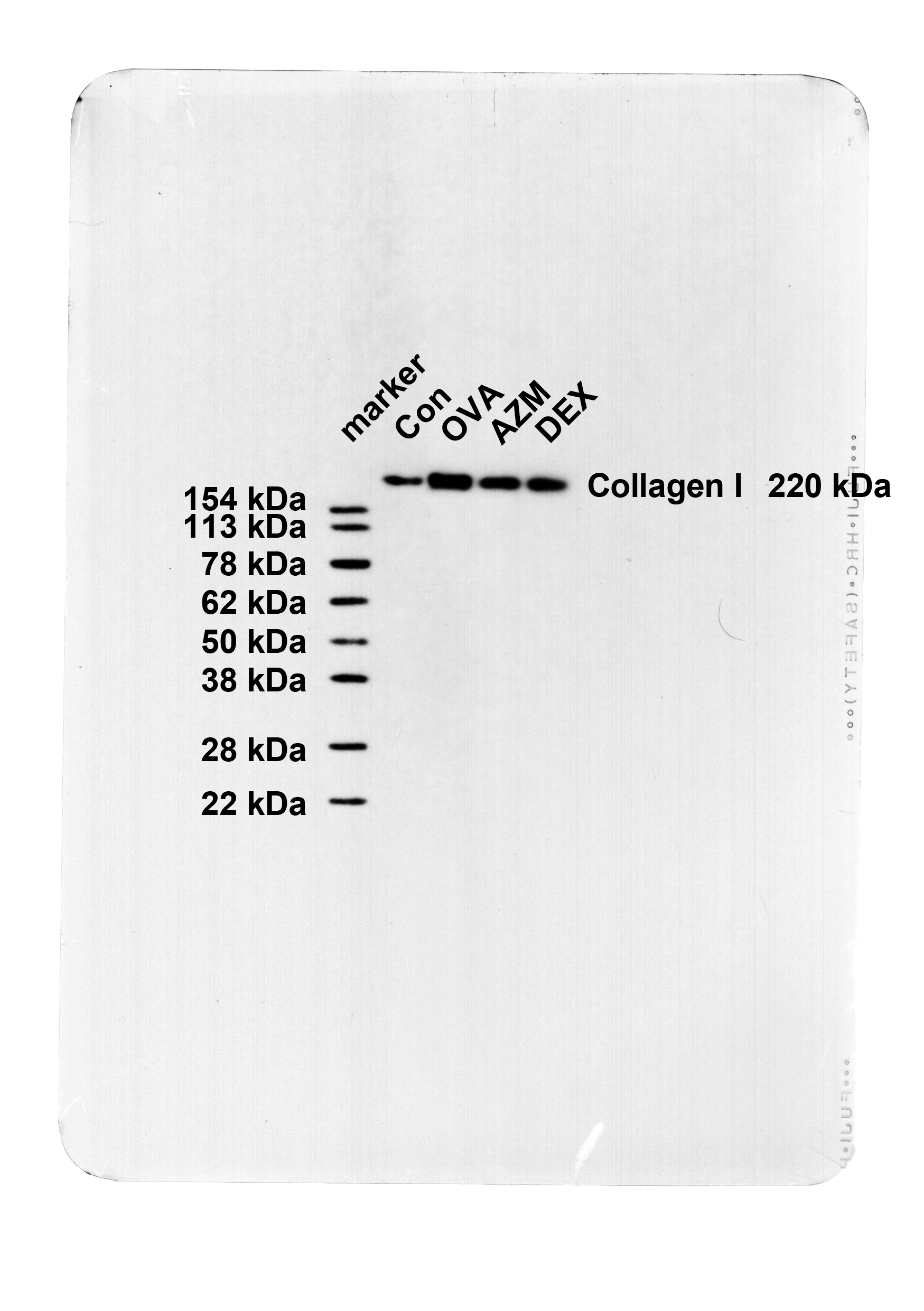
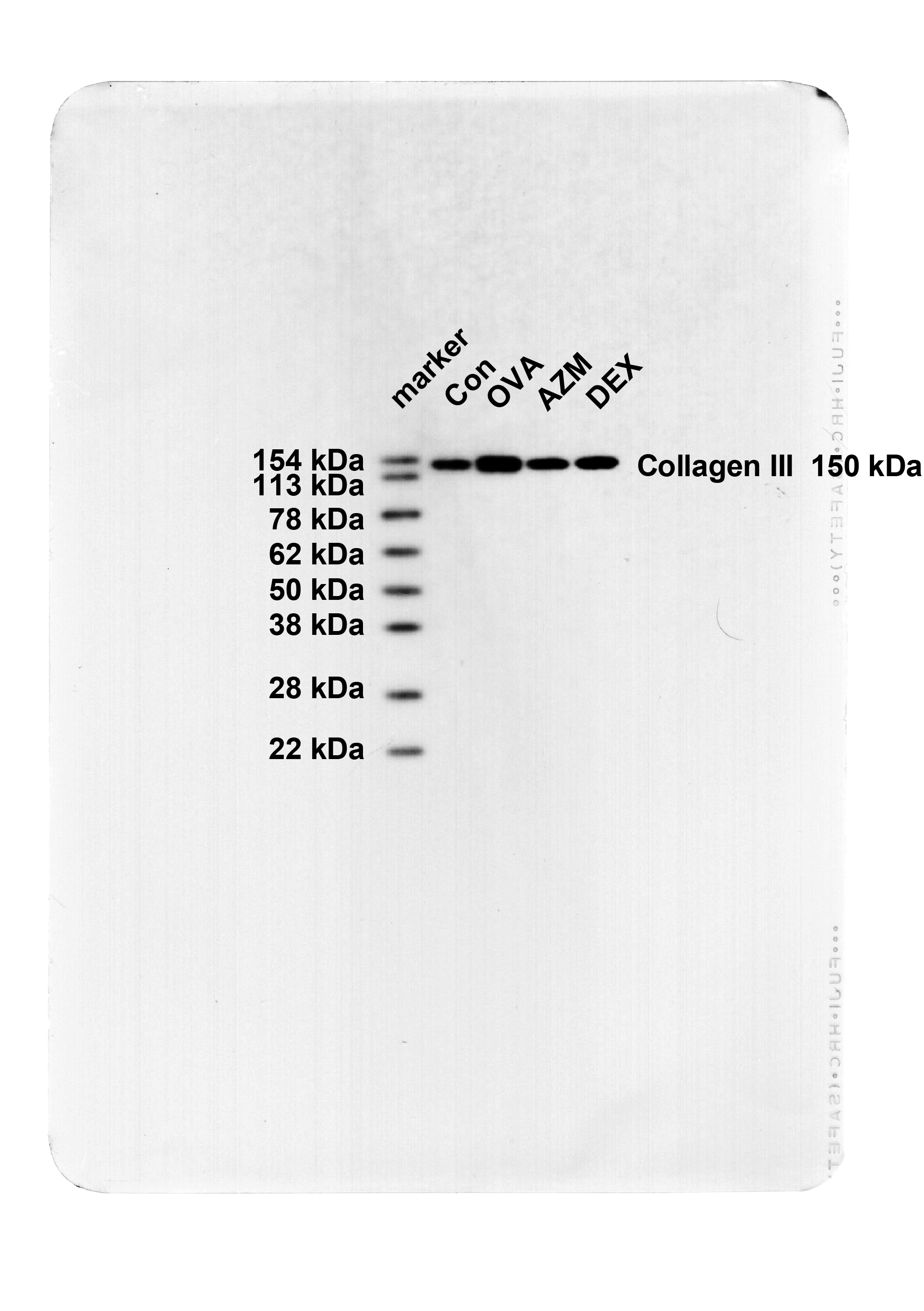
The pathological changes and bronchial epithelial fibrosis in the lung tissues of mice were observed by HE and Masson staining, which elicited that mice in the Con group exhibited intact bronchial mucosal epithelium and lung tissue structure, no obvious infiltration of inflammatory cells, and a small amount of collagen staining area around the airway. The bronchial wall and bronchial smooth muscle of the OVA group were memorably thickened, with a large number of inflammatory cells infiltrating around the bronchi, excessive collagen deposition, and an obvious increase in fibrotic area (Fig. 1A-B, all P < 0.001), whereas these symptoms were evidently attenuated after AZM or DEX treatment (Fig. 1A-B, P < 0.001), with no apparent difference between the two treatment regimens (Fig. 1A-B, all P > 0.05). Further determinations of Collagen I and Collagen III levels in lung tissues by Western blot unraveled that relative to the Con group, Collagen I and Collagen III levels in the lung tissues of the OVA group were in momentous upward inclination (Fig. 1C, P < 0.001), while after AZM or DEX treatment, the expression patterns were prominently down-regulated versus the OVA group (Fig. 1C, P < 0.01), with the two treatment regimens exhibiting no significant difference (Fig. 1C, all P > 0.05). Overall, the aforementioned results unveiled that AZM impeded OVA-led AR in asthmatic mice.
Fig. 1.

AZM suppressed OVA-induced AR in asthmatic mice. A: HE staining to observe the pathological changes of lung tissues (Wat: airway wall thickness; Pbm: bronchial basement membrane perimeter; Wam: bronchial smooth muscle thickness; Wat/Pbm: bronchial wall thickness; Wam/Pbm: bronchial smooth muscle thickness); B: Masson staining to observe bronchial epithelial fibrosis; C: Western blot to determine collagen I and Collagen III levels in lung tissues. n = 6. Data were expressed as mean ± SD and analyzed using one-way ANOVA, followed by post hoc testing using Tukey’s test. ** P < 0.01, *** P < 0.001
AZM alleviated OVA-led airway inflammation in asthmatic mice
The Diff-Quik staining findings indicated increments in the numbers of total inflammatory cells, eosinophils, neutrophil, lymphocytes and macrophages in the BALF of the OVA group compared to the Con group (Fig. 2A, P < 0.001). Following AZM or DEX treatment, decrements in the counts of total inflammatory cells, eosinophils, macrophages, lymphocytes, and neutrophils in the BALF of the asthmatic mice were observed compared to the mice in the OVA group (Fig. 2A, P < 0.01), with no significant difference between the two treatments (Fig. 2A, all P > 0.05). As reflected by ELISA results, in contrast to the Con group, IL-6, TNF-α and IL-4 levels in BALF of the OVA group mice measurably raised (Fig. 2B-D, all P < 0.001), whereas AZM or DEX treatment led to reductions in these levels (Fig. 2B-D, all P < 0.01), and the two treatment regimens didn’t vary in this effect (Fig. 2B-D, all P > 0.05). Taken together, the aforesaid results displayed that AZM mitigated OVA-induced airway inflammation in asthmatic mice.
Fig. 2.

AZM repressed airway inflammation induced by OVA in asthmatic mice. A: Results of Diff-Quik staining for BALF (total inflammatory cell, eosinophil, neutrophil, lymphocyte and macrophage counts); B-D: ELISA to measure inflammatory factor IL-6, TNF-α and IL-4 levels in BALF. n = 6. Data were expressed as mean ± SD and analyzed using one-way ANOVA, followed by post hoc testing using Tukey’s test. ** P < 0.01, *** P < 0.001
AZM subdued the SAPK/JNK pathway activation
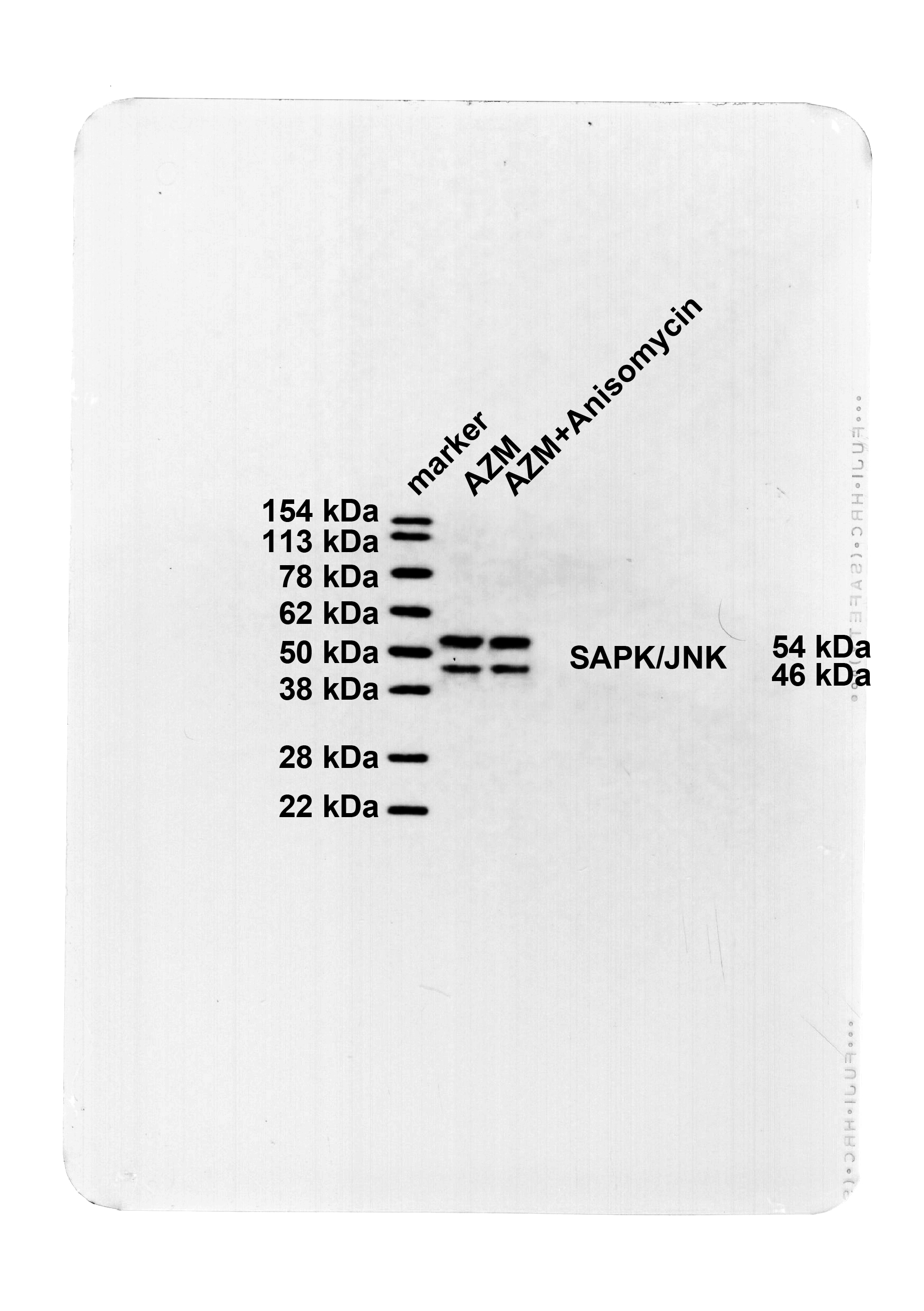
To further investigate whether AZM exerted a protective effect on mice with OVA-led asthma by curbing the SAPK/JNK pathway, p-SAPK/JNK and SAPK/JNK protein expression levels in the lung tissues of mice were measured by Western blot. p-SAPK/JNK was markedly boosted in the lung tissues of the OVA group mice versus the Con group (Fig. 3, P < 0.001) and no prominent difference was observed in SAPK/JNK protein level (Fig. 3, P > 0.05). p-SAPK/JNK protein was abated in the AZM group mice versus the OVA group (Fig. 3, P < 0.01), and there was no significant difference in the SAPK/JNK level (Fig. 3, P > 0.05). The results corroborated that AZM retarded SAPK/JNK pathway activation.
Fig. 3.

AZM inactivated the SAPK/JNK pathway. Western blot to assess p-SAPK/JNK and SAPK/JNK protein levels in lung tissues. n = 6. Data were expressed as mean ± SD and analyzed using one-way ANOVA, followed by post hoc testing using Tukey’s test. ** P < 0.01, *** P < 0.001
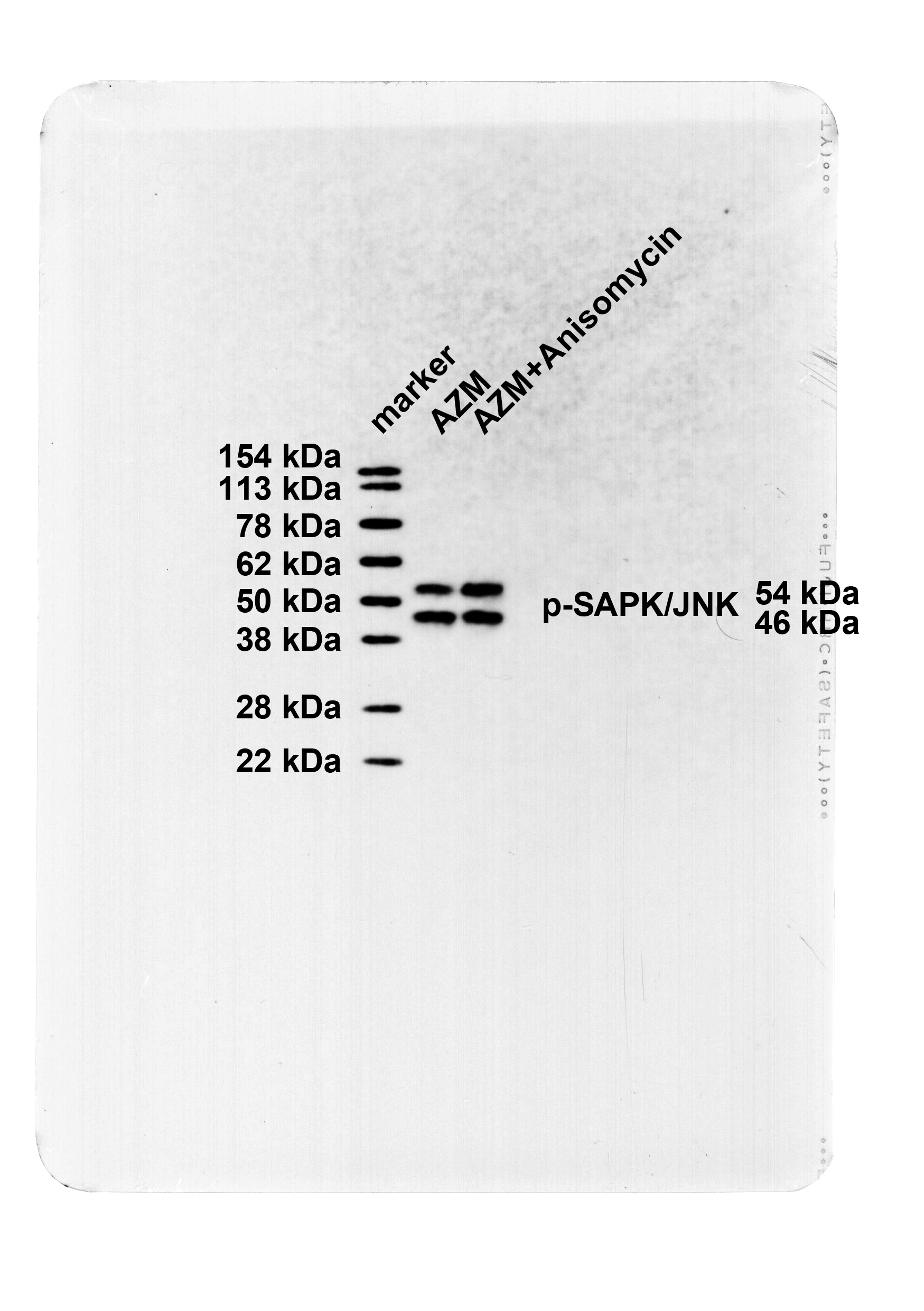
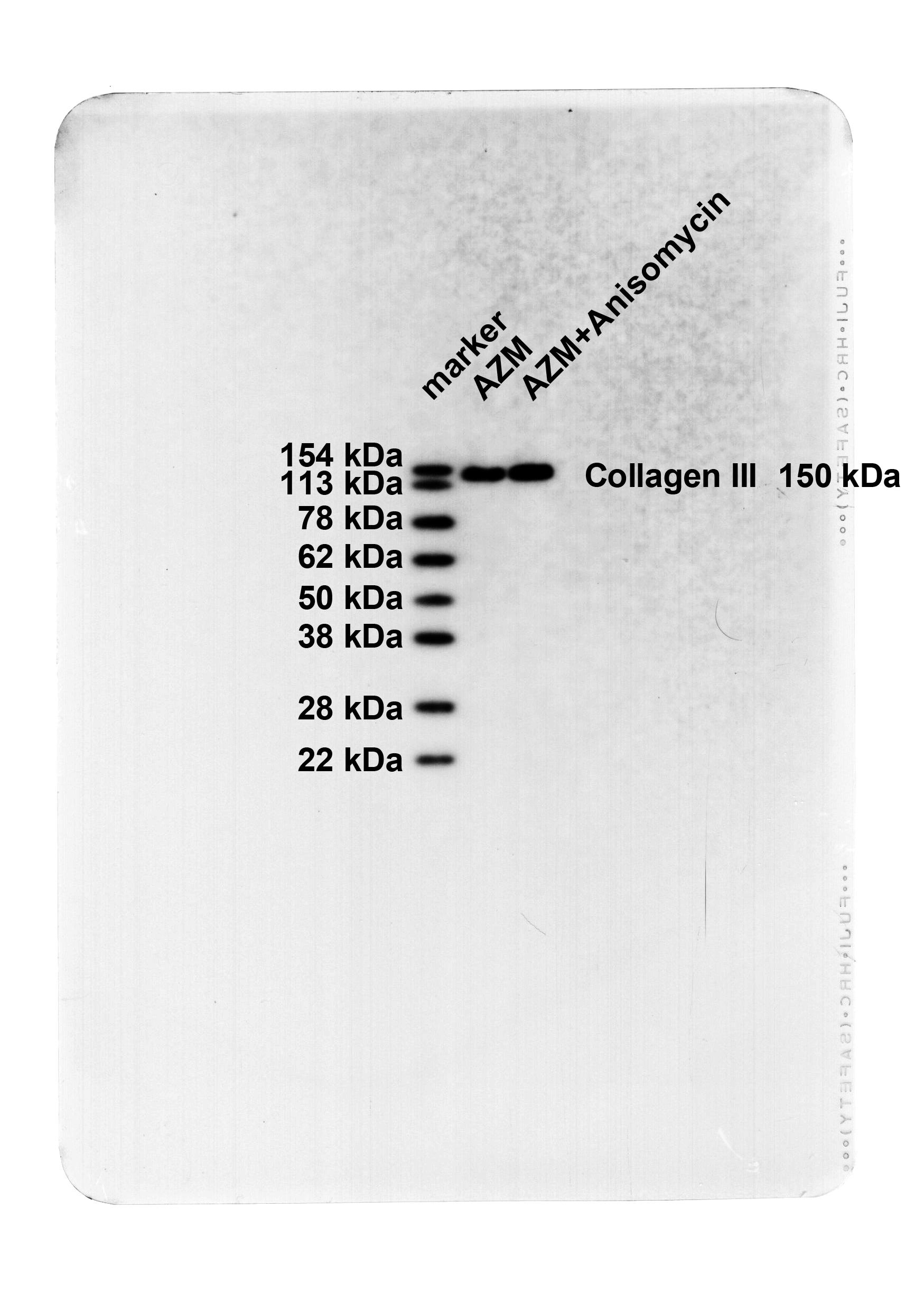
The SAPK/JNK pathway activation partially averted the protective effect of AZM on OVA-induced asthmatic mice
Asthmatic mice were injected with 5 mg/kg Anisomycin (JNK activator) via tail vein on top of AZM treatment. Western blot results manifested that in contrast to the AZM group, the p-SAPK/JNK protein level in the lung tissues of the AZM + Anisomycin group was apparently raised (Fig. 4A, P < 0.05), and SAPK/JNK protein levels were not significantly different (Fig. 4A, P > 0.05), indicating the successfully activated SAPK/JNK phosphorylation level. Subsequently, further assessments of pathological changes in mouse lung tissues, the numbers of total inflammatory cells,eosinophils, neutrophil, lymphocytes and macrophages in BALF, and bronchial epithelial fibrosis, lung tissue collagen Collagen I and Collagen III, and BALF inflammatory factor TNF-α, IL-6, and IL-4 levels revealed that relative to the AZM group, the AZM + Anisomychin group uncover saliently thickened bronchial wall and bronchial smooth muscle, with a large number of inflammatory cells infiltrating around the bronchus, excessive collagen deposition, markedly increased fibrotic area (Fig. 4B-C, all P < 0.05), increased numbers of total inflammatory cells, macrophages, eosinophils, neutrophils and lymphocytes in BALF (Fig. 4D, all P < 0.05), and hoisted levels of lung tissue Collagen I and Collagen III and BALF TNF-α, IL-6, and IL-4 (Fig. 4E-F, all P < 0.05). These results disclosed that activating the SAPK/JNK pathway partly abrogated the effect of AZM on protecting against OVA-induced asthma in mice.
Fig. 4.

The SAPK/JNK pathway activation partially annulled AZM-mediated protective effect against OVA-induced asthma in mice. A: Western blot to determine p-SAPK/JNK and SAPK/JNK protein levels in lung tissues; B: HE staining to observe the pathological changes of lung tissues (Wat: airway wall thickness; Pbm: bronchial basement membrane perimeter; Wam: bronchial smooth muscle thickness; Wat/Pbm: bronchial wall thickness; Wam/Pbm: bronchial smooth muscle thickness); C: Assessment of bronchial epithelial fibrosis by Masson staining; D: Results of Diff-Quik staining for BALF (total inflammatory cell, eosinophil, neutrophil, lymphocyte and macrophage counts); E: Western blot to measure Collagen I and Collagen III in lung tissues; F: ELISA to determine inflammatory factor IL-6, TNF-α and IL-4 levels in BALF. n = 6. Data were expressed as mean ± SD and tested using independent sample t-test. * P < 0.05, ** P < 0.01
Discussion
Asthma, a frequent disorder in the respiratory system, is estimated to impact up to 300 million population across the globe, with an obvious elevating prevalence [3, 17, 18]. An existing study has evidenced that AZM possesses the immunomodulatory properties, and benefits both asthmatic children and adults [19]. With the aim understanding asthma pathogenesis and providing novel ideas for the treatment of asthma, this study probed the specific role of AZM in modulating AR and airway inflammation via the SAPK/JNK pathway in asthmatic mice. Our findings highlighted that AZM mitigated AR and airway inflammation by curbing SAPK/JNK pathway activation in asthmatic mice.
Recently, an emerging concept supports that AR as a major asthmatic pathological feature, is of vital importance in the pathogenesis of asthma, where structural alterations linked to asthma consist of subepithelial fibrosis, disrupted epithelial integrity, smooth muscle hyperplasia/hypertrophy, goblet cell metaplasia/hyperplasia, and enhanced vascularity; these changes are hypothesized to lead to airflow limitation, airway obstruction, airway hyperresponsiveness, and progressive impact on lung function in the asthmatic individuals [20]. On the other hand, as a chronic condition of the airways, asthma has long been considered primarily as an inflammatory condition, and as a result, resolving inflammation is the main focus of current therapeutic interventions [20]. Notably, AZM is indicated for dermal, urogenital, respiratory and other bacterial infections, which plays immunomodulatory roles in various chronic inflammatory diseases, such as rosacea, diffuse panbronchiolitis and post-transplant bronchiolitis, with regulation of host responses stimulating its long-term therapeutic benefit in non-eosinophilic asthma, exacerbations of chronic obstructive pulmonary disease, non-cystic fibrosis bronchiectasis and cystic fibrosis [21]. Unsurprisingly, our findings also elicited increased bronchial wall and bronchial smooth muscle thickness, expanded fibrotic area, elevated Collagen I and Collagen III protein levels, excessive collagen deposition, and inflammatory infiltration around the bronchi, as well as elevated IL-4, TNF-α and IL-6 levels in BALF of OVA-induced asthmatic mice, while AZM or DEX treatment brought about the opposite trends. In a previous study, AZM has been reported to limit the elevation in Collagen I mRNA level and total collagen accumulation in culture media in cyclosporine A-treated renal transplant fibroblasts, suggesting its property to ameliorate cyclosporine A-induced gingival overgrowth [22]. More importantly, AZM can markedly reduce both restrictive lung function pattern and pulmonary fibrosis [23]. AZM reduces epithelial thickening, the thickness of peribronchial smooth muscle layer, deposition of collage fiber, and effectively represses apoptotic index of airway epithelium, thus ameliorating AR, and also leads to reduced inflammatory scores [13, 14]. Also, AZM suppresses the OVA-dependent airway inflammation, leading to a reduction in cytokine expressions, for instance, IL-13 and IL-5 in BALF, suggesting its beneficial actions on treating noninfectious airway inflammatory disorders, such as asthma [24]. Another study has also drawn the conclusion that long-term AZN treatment improves not only AR but also airway inflammation in a chronic asthmatic murine model [25]. To conclude, AZM curbed OVA-induced AR and airway inflammation in asthmatic mice.
SAPK/JNK, expressed by both Th1 and Th2 cells, plays an essential role in asthma pathogenesis, with the JNK encoding gene inhibitor decreasing airway smooth muscle proliferation and bronchial eosinophil infiltration [11]. It has been documented that AZM regulates the expression levels of asthma-related chemokines via the JNK pathway [26]. Furthermore, AZM curbs the SAPK phosphorylation in animals [27]. Consistently, our results uncovered raised p-SAPK/JNK level in OVA-induced asthmatic mice, while diminished p-SAPK/JNK level after AZM treatment. Moreover, our study demonstrated the inhibitory effect of AZM on the SAPK/JNK pathway in an animal model of asthma in vivo for the first time. Further, to confirm this, we injected JNK activator into asthmatic mice on the basis of AZM treatment, and discovered that bronchial epithelial fibrosis and pathological changes in lung tissues were aggravated, and total inflammatory cell, macrophage, eosinophil, neutrophil, and lymphocyte counts and inflammatory cytokine levels in BALF and collagen levels in lung tissues were elevated in asthmatic mice, suggesting for the first time that activating the SAPK/JNK pathway partly abrogated the protective role of AZM on OVA-induced asthmatic mice.
The present study demonstrated that p-SAPK/JNK was up-regulated in the lung tissues of mice in the OVA group compared to the Con group. Relative to the OVA group, p-SAPK/JNK was down-regulated in the lung tissues of mice in the AZM group, but the level in the AZM group was still higher than that in the Con group, and did not restore to the same level as the Con group, which indicated that AZM might exert its effects through other pathways. It has been documented that AZM reduces IL-6 and IL-2 levels, elevates IL-10 level, and attenuates asthma triggered by lung inflammation by suppressing EZH2-mediated NF-κB p65-associated hypermethylation of H3K27me3 [28]. AZM exerts direct impacts on various cytokines, leading to the inhibition of inflammation and decrease of airway reactivity, which also plays a protective role against airway remodeling by modulating the phosphoinositide 3-kinase/Akt/mechanistic target of the rapamycin kinase/hypoxia-inducible factor 1α/vascular endothelial growth factor pathway [29]. Additionally, AZM facilitates the proliferation of airway smooth muscle cells and restores asthma airway remodeling through the mitochondrial pathway [30]. Therefore, AZM may also attenuate inflammatory responses and pathological changes via other pathways.
Collectively, this study highlighted that AZM attenuated airway inflammation and AR by repressing SAPK/JNK pathway activation in asthmatic mice, which provided some theoretical basis for understanding the pathogenesis of asthma and also provided new ideas for treating asthma. Nevertheless, this article only developed an asthmatic mouse model through OVA induction, and preliminarily explored how AZM could mitigate airway inflammation in asthmatic mice by inhibiting the JNK/SAPK pathway, thereby inhibiting AR in asthmatic mice. Whether AZM alleviated lung function in asthmatic mice has not been studied. Meanwhile, this mechanism is currently not confirmed in cell experiments, with deeper molecular mechanisms unexplored. These are the direction of our study in the future.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
Not applicable.
Author contributions
Author Contribution StatementAll authors contributed to the study conception and design, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript. D.M. was responsible for designing the review protocol, writing the protocol and report, conducting the search, screening potentially eligible studies, extracting and analysing data, interpreting results, updating reference lists. H.D. was responsible for designing the review protocol and screening potentially eligible studies. Y.H. and A.P. contributed to writing the report, extracting and analysing data, interpreting results. D.M. and L.G. provided feedback on the report.
Funding
Not applicable.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Declarations
Ethics approval and consent to participate
The study was authorized by the academic ethics committee of the Second Affiliated Hospital Zhejiang University School of Medicine (Approval number: 2023081). All procedures were strictly implemented by the Guide for the Care and Use of Laboratory Animals. All the laboratory procedures were used to minimize the pain of mice.
Consent for publication
Not applicable.
Disclosure
The authors report there are no competing interests to declare.
Submission declaration and verification
The work described has not been published previously, that it is not under consideration for publication elsewhere, that its publication is approved by all authors and tacitly or explicitly by the responsible authorities where the work was carried out, and that, if accepted, it will not be published elsewhere in the same form, in English or in any other language, including electronically without the written consent of the copyright-holder.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Wu TD, Brigham EP, McCormack MC. Asthma in the primary care setting. Med Clin North Am. 2019;103(3):435–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cao C, Wang Y, Peng L, Wu W, Yang H, Li Z. Asthma and other respiratory diseases of children in relation to Personal Behavior, Household, parental and environmental factors in West China. Toxics. 2023;11(12). [DOI] [PMC free article] [PubMed]
- 3.Mims JW. Asthma: definitions and pathophysiology. Int Forum Allergy Rhinol. 2015;5(Suppl 1):S2–6. [DOI] [PubMed] [Google Scholar]
- 4.Bergeron C, Al-Ramli W, Hamid Q. Remodeling in asthma. Proc Am Thorac Soc. 2009;6(3):301–5. [DOI] [PubMed] [Google Scholar]
- 5.Hirota N, Martin JG. Mechanisms of airway remodeling. Chest. 2013;144(3):1026–32. [DOI] [PubMed] [Google Scholar]
- 6.Castillo JR, Peters SP, Busse WW. Asthma exacerbations: Pathogenesis, Prevention, and treatment. J Allergy Clin Immunol Pract. 2017;5(4):918–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nishina H, Wada T, Katada T. Physiological roles of SAPK/JNK signaling pathway. J Biochem. 2004;136(2):123–6. [DOI] [PubMed] [Google Scholar]
- 8.Lu TC, Wu YH, Chen WY, Hung YC. Targeting oxidative stress and endothelial dysfunction using tanshinone IIA for the Treatment of Tissue Inflammation and fibrosis. Oxid Med Cell Longev. 2022;2022:2811789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Papaconstantinou J. The role of signaling pathways of inflammation and oxidative stress in Development of Senescence and Aging Phenotypes in Cardiovascular Disease. Cells. 2019;8(11). [DOI] [PMC free article] [PubMed]
- 10.Wronka M, Krzeminska J, Mlynarska E, Rysz J, Franczyk B. The influence of lifestyle and treatment on oxidative stress and inflammation in diabetes. Int J Mol Sci. 2022;23(24). [DOI] [PMC free article] [PubMed]
- 11.Dandekar RD, Khan MM. Involvement of histamine receptors in SAPK/JNK phosphorylation. Int Immunopharmacol. 2012;13(2):190–6. [DOI] [PubMed] [Google Scholar]
- 12.Brusselle G, Pavord I. Azithromycin in uncontrolled asthma. Lancet. 2017;390(10095):629–30. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y, Pu Y, Li D, Zhou L, Wan L. Azithromycin ameliorates airway remodeling via inhibiting airway epithelium apoptosis. Life Sci. 2017;170:1–8. [DOI] [PubMed] [Google Scholar]
- 14.Pu Y, Liu Y, Liao S, Miao S, Zhou L, Wan L. Azithromycin ameliorates OVA-induced airway remodeling in Balb/c mice via suppression of epithelial-to-mesenchymal transition. Int Immunopharmacol. 2018;58:87–93. [DOI] [PubMed] [Google Scholar]
- 15.Yin N, Wu C, Qiu J, Zhang Y, Bo L, Xu Y, et al. Protective properties of heme oxygenase-1 expressed in umbilical cord mesenchymal stem cells help restore the ovarian function of premature ovarian failure mice through activating the JNK/Bcl-2 signal pathway-regulated autophagy and upregulating the circulating of CD8(+)CD28(-) T cells. Stem Cell Res Ther. 2020;11(1):49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Y, Lin Z, Han Z, Wu Z, Hua J, Zhong R, et al. miR-539 activates the SAPK/JNK signaling pathway to promote ferropotosis in colorectal cancer by directly targeting TIPE. Cell Death Discov. 2021;7(1):272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartel S, La Grutta S, Cilluffo G, Perconti G, Bongiovanni A, Giallongo A, et al. Human airway epithelial extracellular vesicle miRNA signature is altered upon asthma development. Allergy. 2020;75(2):346–56. [DOI] [PubMed] [Google Scholar]
- 18.Kaur R, Chupp G. Phenotypes and endotypes of adult asthma: moving toward precision medicine. J Allergy Clin Immunol. 2019;144(1):1–12. [DOI] [PubMed] [Google Scholar]
- 19.Ghimire JJ, Jat KR, Sankar J, Lodha R, Iyer VK, Gautam H, et al. Azithromycin for poorly controlled asthma in children: a Randomized Controlled Trial. Chest. 2022;161(6):1456–64. [DOI] [PubMed] [Google Scholar]
- 20.Banno A, Reddy AT, Lakshmi SP, Reddy RC. Bidirectional interaction of airway epithelial remodeling and inflammation in asthma. Clin Sci (Lond). 2020;134(9):1063–79. [DOI] [PubMed] [Google Scholar]
- 21.Parnham MJ, Erakovic Haber V, Giamarellos-Bourboulis EJ, Perletti G, Verleden GM, Vos R. Azithromycin: mechanisms of action and their relevance for clinical applications. Pharmacol Ther. 2014;143(2):225–45. [DOI] [PubMed] [Google Scholar]
- 22.Kim JY, Park SH, Cho KS, Kim HJ, Lee CK, Park KK, et al. Mechanism of azithromycin treatment on gingival overgrowth. J Dent Res. 2008;87(11):1075–9. [DOI] [PubMed] [Google Scholar]
- 23.Wuyts WA, Willems S, Vos R, Vanaudenaerde BM, De Vleeschauwer SI, Rinaldi M, et al. Azithromycin reduces pulmonary fibrosis in a bleomycin mouse model. Exp Lung Res. 2010;36(10):602–14. [DOI] [PubMed] [Google Scholar]
- 24.Beigelman A, Gunsten S, Mikols CL, Vidavsky I, Cannon CL, Brody SL, et al. Azithromycin attenuates airway inflammation in a noninfectious mouse model of allergic asthma. Chest. 2009;136(2):498–506. [DOI] [PubMed] [Google Scholar]
- 25.Kang JY, Jo MR, Kang HH, Kim SK, Kim MS, Kim YH, et al. Long-term azithromycin ameliorates not only airway inflammation but also remodeling in a murine model of chronic asthma. Pulm Pharmacol Ther. 2016;36:37–45. [DOI] [PubMed] [Google Scholar]
- 26.Kuo CH, Lee MS, Kuo HF, Lin YC, Hung CH. Azithromycin suppresses Th1- and Th2-related chemokines IP-10/MDC in human monocytic cell line. J Microbiol Immunol Infect. 2019;52(6):872–9. [DOI] [PubMed] [Google Scholar]
- 27.Gao L, Tang Z, Li T, Wang J. Combination of kaempferol and azithromycin attenuates Staphylococcus aureus-induced osteomyelitis via anti-biofilm effects and by inhibiting the phosphorylation of ERK1/2 and SAPK. Pathog Dis. 2021;79(8). [DOI] [PubMed]
- 28.Wu S, Tian X, Mao Q, Peng C. Azithromycin attenuates wheezing after pulmonary inflammation through inhibiting histone H3K27me3 hypermethylation mediated by EZH2. Clin Epigenetics. 2023;15(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao X, Yu FQ, Huang XJ, Xu BY, Li YL, Zhao XY, et al. Azithromycin influences airway remodeling in asthma via the PI3K/Akt/MTOR/HIF-1alpha/VEGF pathway. J Biol Regul Homeost Agents. 2018;32(5):1079–88. [PubMed] [Google Scholar]
- 30.Wu L, Yin J, Zhang Q, Wang M, Dai W, Zhou J, et al. Azithromycin induces apoptosis in airway smooth muscle cells through mitochondrial pathway in a rat asthma model. Ann Transl Med. 2021;9(14):1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.