

Abstract
High-grade serous ovarian cancer (HGSOC) is one of the deadliest cancers for women, with a low survival rate, no early detection biomarkers, a high rate of recurrence, and few therapeutic options. Forskolin, an activator of cyclic AMP signaling, has several anticancer activities, including against HGSOC, but has limited use in vivo. Its water-soluble derivative, colforsin daropate, has the same mechanism of action as forskolin and is used to treat acute heart failure. Here, we investigated the potential of colforsin daropate as a treatment for HGSOC. We found that colforsin daropate induced cell cycle arrest and apoptosis in cultured HGSOC cells and spheroids but had negligible cytotoxicity in immortalized, nontumorigenic fallopian tube secretory cells and ovarian surface epithelial cells. Colforsin daropate also prevented HGSOC cells from invading ovarian surface epithelial cell layers in culture. In vivo, colforsin daropate reduced tumor growth, synergized with cisplatin (a standard chemotherapy in ovarian cancer care), and improved host survival in subcutaneous and intraperitoneal xenograft models. These anti-tumor effects of colforsin daropate were mediated in part by its reduction in the abundance and transcriptional activity of the oncoprotein c-MYC, which is often increased in HGSOC. Our findings demonstrate that colforsin daropate may be a promising therapeutic that could be combined with conventional therapies to treat HGSOC.
Introduction
Ovarian cancer is currently the 2nd deadliest gynecological malignancy worldwide and is increasing in incidence and mortality, with well over 300,000 new cases and over 200,000 deaths as of the latest statistics1. Epithelial high-grade serous ovarian carcinoma (HGSOC) is the most common and deadly subtype of ovarian cancer, accounting for approximately 70% of cases diagnosed and 75% of ovarian cancer deaths 2–4. Its high mortality rate is due to the absence of early symptoms, resulting in 80% of patients being diagnosed at later stages often after metastatic progression throughout the peritoneal cavity. Around 80% of tumors or metastases will recur within 5 years despite initially responding to standard platinum/taxane therapy. Patient with recurrent and chemoresistant tumors have limited treatment options. Currently, the only adjuvant therapies that are approved by the Food and Drug Administration (FDA) and are regularly used for HGSOC treatment are PARP inhibitors (which generally only benefits the sub-population of patients with well-defined defects in DNA damage response (DDR) pathways, such as BRCA1/2 mutation) and anti-angiogenic therapy (which produces improvements in progression-free survival but not overall survival)5–12. Thus, there is a critical need for therapies that can limit recurrence and improve survival outcomes.
Forskolin, the well-established small-molecule activator of intracellular cyclic AMP (cAMP) signaling, has been shown to have a variety of clinical uses, including the treatment of cancer13. Forskolin is a labdane diterpene derived from the roots of the Indian Coleus forskohlii plant, an Ayurvedic herbal medicine long used to treat a variety of disorders, such as angina, hypertension, and asthma14–16. The canonical function of forskolin is to act as a potent, reversible stimulator of adenylyl cyclase (AC) through direct interaction with the AC catalytic subunit. This generates a rapid increase of intracellular cAMP levels and activates cAMP signaling pathways through effector proteins, such as cAMP-dependent protein kinase (PKA) and exchange protein directly activated by cAMP (EPAC)16–18. Cyclic AMP acts as a second messenger and binds to the regulatory subunits of PKA causing the release of the catalytic PKA subunits which can then phosphorylate downstream targets that include cAMP response element binding protein (CREB)19. Phosphorylated CREB then translocates into the nucleus and binds to cAMP response elements to activate gene expression programs20. EPAC is also activated by direct binding of cAMP and facilitates signaling changes involved in cell adhesion and proliferation21.
With respect to cancer, forskolin can have several anti-tumorigenic effects (typically through canonical activation of cAMP signaling), such as inhibition of cell growth and induction of cell death in gastric, lymphoid, and colon cancers22–24. Forskolin has also been shown to inhibit cell migration and invasion, and metastatic colonization by multiple cancer types25–28. In addition, combination therapy of forskolin with other anti-cancer drugs has been shown to be synergistic in treating chemoresistant colon cancer cells in culture. Finally, evidence has also emerged that forskolin may be a valuable tool for targeting tumor-initiating cells by causing mesenchymal, stem-like cancer cells to transition into less aggressive epithelial-differentiated states29. The effects of forskolin on ovarian cancer biology remain poorly defined and only a few studies have examined the direct effects of forskolin treatment on ovarian cancer cells. Mann et al. have reported that combination treatment of forskolin with cisplatin enhanced cisplatin accumulation within ovarian cancer cells30, but other studies reported that forskolin treatment was inhibitory31. Forskolin stimulation of cAMP signaling has been reported to cause phosphorylation of claudin-3 to alter tight junctions and stimulate production of granulin-epithelin precursor (GEP) in ovarian cancer cells32,33. However, these studies did not examine whether forskolin caused any phenotypic changes in the ovarian cancer cells. An additional study by Warrenfeltz and colleagues showed that forskolin treatment could inhibit the migration of SKOV3 cells in culture 31. There are conflicting reports as to whether cAMP signaling itself is pro- or anti-tumorigenic with respect to ovarian cancer. However, most of the data in the literature support an anti-tumorigenic role for activated cAMP signaling through mechanisms that include decreased proliferation, increased senescence, and increased chemosensitivity34–40.
These studies provide a strong rationale for investigating whether forskolin could be used for HGSOC treatment. Forskolin has been used in the clinic for treatment of glaucoma and is being investigated for treatment of asthma, heart failure, and obesity41–46.However, forskolin is sparingly soluble in aqueous solution and is non-ideal for translation into the clinic. A water-soluble derivative, colforsin daropate (NKH 477, abbreviated here to CF), has been previously synthesized and shown to have similar biological effects47. CF has also been used in the clinical setting for treatment of heart failure, cerebral vasospasm, and cardiac inflammation48–50. The combination of favorable pharmacokinetic properties and low reported toxicity make CF an ideal candidate for drug repurposing. Here, we investigated whether CF can be used for the treatment of HGSOC. We found that it has the potential to be HGSOC cell-selective, mechanistically targeting those with upregulated MYC signaling. It was effective in HGSOC cells that were resistant to a standard ovarian cancer chemotherapeutic and synergized in those that were sensitive. Overall, this study shows that CF should be further explored for the treatment of MYC-abundant HGSOC.
Results
Colforsin daropate induces ovarian cancer cell cycle arrest and cell death
To begin investigating whether CF had potential as a cancer therapeutic, we treated a panel of established HGSOC cell lines (Fig. 1A) with CF in culture and assessed for cytotoxic effects. Cell viability was reduced in the HGSOC cell lines with increasing dose of CF, but with a range of sensitivities among the cell lines, with the half-maximal inhibitory concentration (IC50) values ranging from approximately 0.5 to 40 μM (Fig. 1A). Given the translational potential for CF, we also treated two sets of isogenic cisplatin-sensitive and cisplatin-resistant HGSOC patient-derived xenograft (PDX) cell lines: OV81.2 and OV231 (sensitive) and OV81.2 CP40 and OV231 CP30 (resistant), respectively 51. We confirmed the cisplatin (Cis) resistance of the latter cell lines (fig. S1A) and observed that all 4 PDX cell lines were sensitive to CF, with IC50 values between 10 and 15 μM; the IC50 values of the Cis-resistant cells were not significantly different from those of the Cis-sensitive cells (Fig. 1B). These data suggested that CF was effective at killing HGSOC cells in both Cis resistant and sensitive contexts. To determine whether the cytotoxic effects were selective to cancer cells, we also included a panel of immortalized, non-tumorigenic cell lines [human fibroblasts, fallopian tube secretory epithelial cells (FTSECs), and ovarian surface epithelial cells] (Fig. 1C). We observed that the HGSOC cell lines were more sensitive to CF than the non-tumorigenic cells (IC50 approx. 1-40 μM), which included FTSECs and IMR90 fibroblasts (IC50 approx. 110 to >500 μM) (Fig. 1C). Based on the cell viability response of the HGSOC panel to CF, we selected HEYA8, OVCAR8, and OVCAR4 cells to model sensitive, intermediate, and resistant CF responses, respectively, in subsequent experiments. Next, we investigated the effects of CF on low density cell survival and colony formation using a clonogenic assay (Fig. 1D). We observed a similar pattern of survival inhibition and reduced growth in the results of the clonogenic assays, with HEYA8 being the most sensitive and the two OVCAR cell lines being more resistant (Fig. 1D). It was also observed that the decreases in survival and clonogenic growth were again selective for HGSOC cells over FTSEC cells (Fig. 1, D and E).
Figure 1: Colforsin daropate induces cell cycle arrest and cell death in ovarian cancer cell lines.
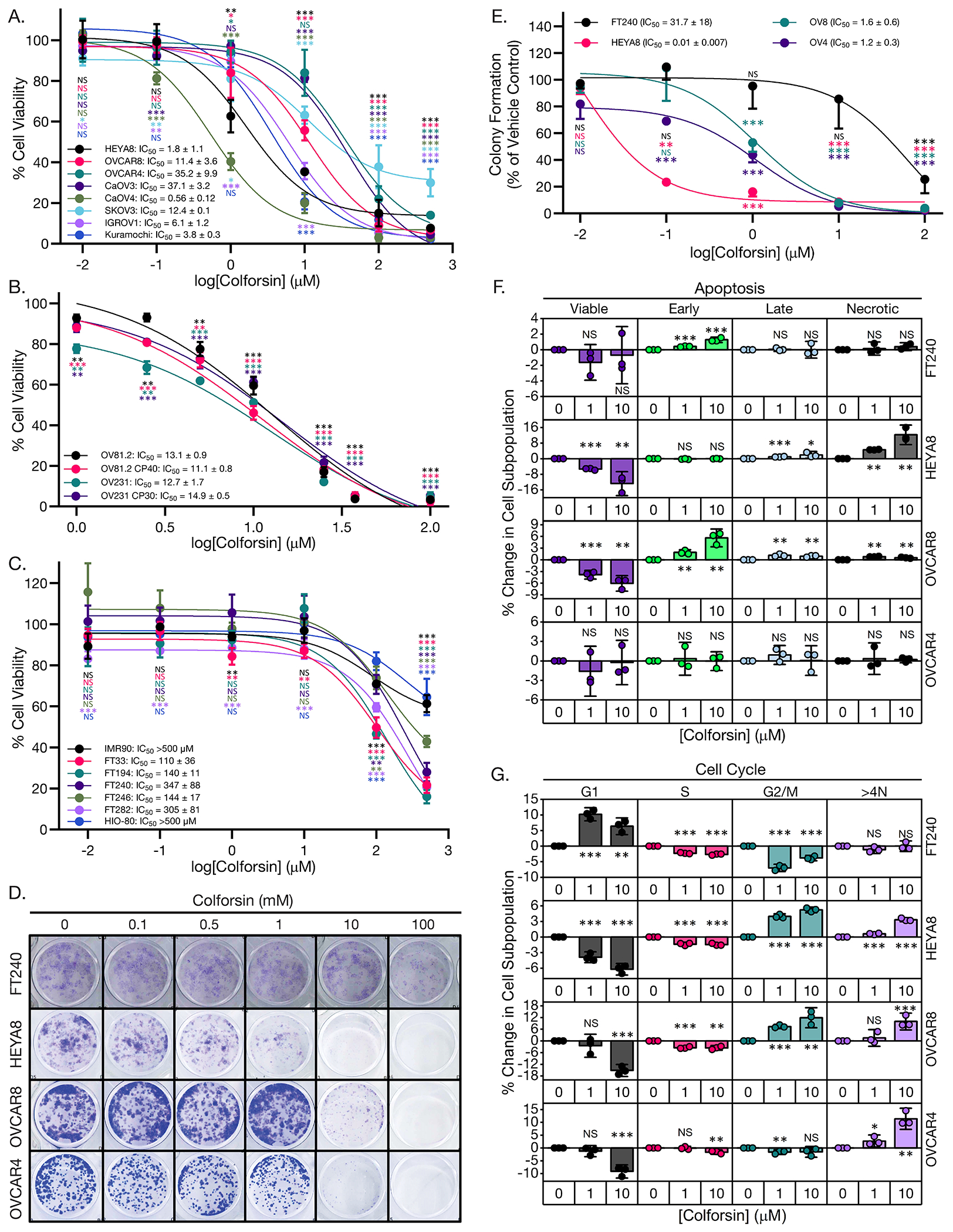
(A) Graph of dose response curves for MTT cell viability assays of HGSOC cell lines treated with colforsin daropate (CF) for 48 hours. All statistical comparisons are between a given CF dose vs vehicle control within each cell line. Mean IC50 values ± SD are shown for each cell line in the legend. (B) Graph of dose response curves for MTT cell viability assays of HGSOC PDX cell lines treated with CF for 48 hours. All statistical comparisons are between a given CF dose vs vehicle control within each cell line. Mean IC50 values ± SD are shown for each cell line in the legend. (C) Graph of dose-response curves for MTT cell viability assays of non-tumorigenic FTSEC, HIO-80, and fibroblast IMR90 cell lines treated with CF for 48 hours. All statistical comparisons are between a given CF dose vs vehicle control within each cell line. Mean IC50 values ± SD are shown for each cell line in the legend. (D) Representative images of day 7 clonogenic survival assays for FTSEC and ovarian cancer cell lines treated with increasing doses of CF. (E) Graph depicting dose-response curves for clonogenic assays of FTSEC and ovarian cancer cell lines. All statistical comparisons are between a given CF dose vs vehicle control within each cell line. Mean IC50 values for each cell line ± SD are displayed in the legend. (F) Graphs showing % change in apoptotic cell subpopulations for FT240, HEYA8, OVCAR8, and OVCAR4 cell lines treated with either 0, 1, or 10 μM CF for 48 hours. (G) Graphs showing % change in cell cycle subpopulations for FT240, HEYA8, OVCAR8, and OVCAR4 cell lines treated with either 0, 1, or 10 μM CF for 48 hours. All statistical comparisons are between CF vs vehicle treatment within a subpopulation for a given cell line. All data (A to G) are representative of N = 3 biological replicates. Statistical comparisons of two groups were performed using student’s t-test and comparisons of three or more groups were performed using one way ANOVA with Tukey multiple testing correction unless otherwise stated.
Following the observation that CF reduced proliferation and colony formation of HGSOC cells, we next wanted to determine whether the specific mechanism was increased cell death, cell cycle arrest, or a combination of both. To answer these questions, we treated our panel of HGSOC and FTSEC cell lines with increasing doses of CF and performed annexin-V/PI staining of live cells to assess changes in apoptosis, as well as PI staining of fixed cells to assess changes in cell cycle populations. In general, we found that CF decreased cell proliferation through a combination of increased cell cycle arrest and increased apoptotic cell death (Fig. 1F, fig. S1, B and C). For the FT240 cells substantial changes in cell death and cell cycle arrest only occurred at the highest dose tested (fig. S1, B and C). HEYA8 cells had significant increases in apoptosis and G2/M arrest starting at doses of 1 μM. OVCAR8 cells displayed a different pattern with most of the cells accumulating in G2/M but with less apoptosis. OVCAR4 cells showed significant accumulation in the >4N or aneuploid subpopulation and did not show increased apoptosis (Fig. 1F). In addition to the experiments performed with CF, we also assessed the anti-tumorigenic properties of the parent compound forskolin. We observed comparable decreases in cell viability and increased cell death and cell cycle arrest for HGSOC cells treated with forskolin (fig. S2, A to D). These data indicated that both compounds produced the same phenotypic response in HGSOC cells, and that the cytotoxic effects were not selective to CF. Taken together these data show that HGSOC cells in general are more sensitive to the cytotoxic effects of CF vs non-transformed FTSEC cell lines, Cis resistant HGSOC cells remain sensitive to CF, and that CF cytotoxicity occurs through a combination of cell cycle arrest and cell death.
Growth of ovarian cancer spheroids is inhibited by colforsin daropate
Given that ovarian cancer cells tend to metastasize as drug resistant spheroids, we also wanted to determine whether CF could cause cell cycle arrest and/or cell death in HGSOC spheroids52,53. To investigate the effects of CF on ovarian cancer spheroids we cultured our panel of HGSOC cell lines under ultra-low attachment conditions in media containing increasing doses of CF. We observed that CF was able to inhibit HGSOC spheroid growth in a dose dependent manner (Fig. 2, A and B). We observed a similar pattern of sensitivity with HEYA8, OVCAR8, and OVCAR4 cells, going from most to least sensitive respectively (Fig. 2, A and B). Analysis of apoptosis and cell cycle for the spheroids following CF treatment showed that HEYA8 cells responded predominantly with G1 cell cycle accumulation along with apoptosis at higher doses (Fig. 2, C and D, and fig. S3, A and B). OVCAR8 cells responded with increases in apoptotic populations and 10-20% G2/M accumulation at high doses (Fig. 2, C and D, and fig. S3, A and B). OVCAR4 cells had predominant change in cell cycle with increased aneuploid accumulation and increases in early apoptosis (Fig. 2, C and D, and fig. S3, A and B). The ability of HGSOC cells to adhere to each other is a key factor necessary for the cells to survive and metastasize as spheroids53. We observed that CF inhibited the ability of HGSOC cells to form tight spheroids (Fig. 2E). Treatment of HGSOC cells in 2D culture with 5 μM CF prior to seeding under ultra-low attachment (ULA) conditions produced a 2 to 3-fold increase in sphere size versus vehicle indicating inhibition of tight sphere formation (Fig. 2F). Pathway analysis data also indicated inhibition of integrin signaling, adherens junction signaling, and adhesion molecules (such as NECTIN1 and TNS1) in response to increasing doses of CF (fig. S9A) Following the observation of CF induced spheroid disruption, we wanted to determine whether this impacted the functional capacity of the spheroids to adhere to and invade an epithelial monolayer. We used an established epithelial clearance assay using vehicle or CF treated HGSOC RFP spheroids seeded on top of a GFP+ HIO-80 ovarian surface epithelial (OSE) cell monolayer54. We observed that CF inhibited the ability of the HGSOC-RFP spheroids to clear and invade the OSE monolayer in a dose-dependent manner (Fig. 2, G and H). Together, these data show that HGSOC spheroids, the metastatic vehicle for ovarian cancer, remain sensitive to CF and that CF inhibits the ability of HGSOC spheroids to invade an epithelial cell monolayer.
Figure 2: Colforsin daropate inhibits growth of ovarian cancer spheroids.
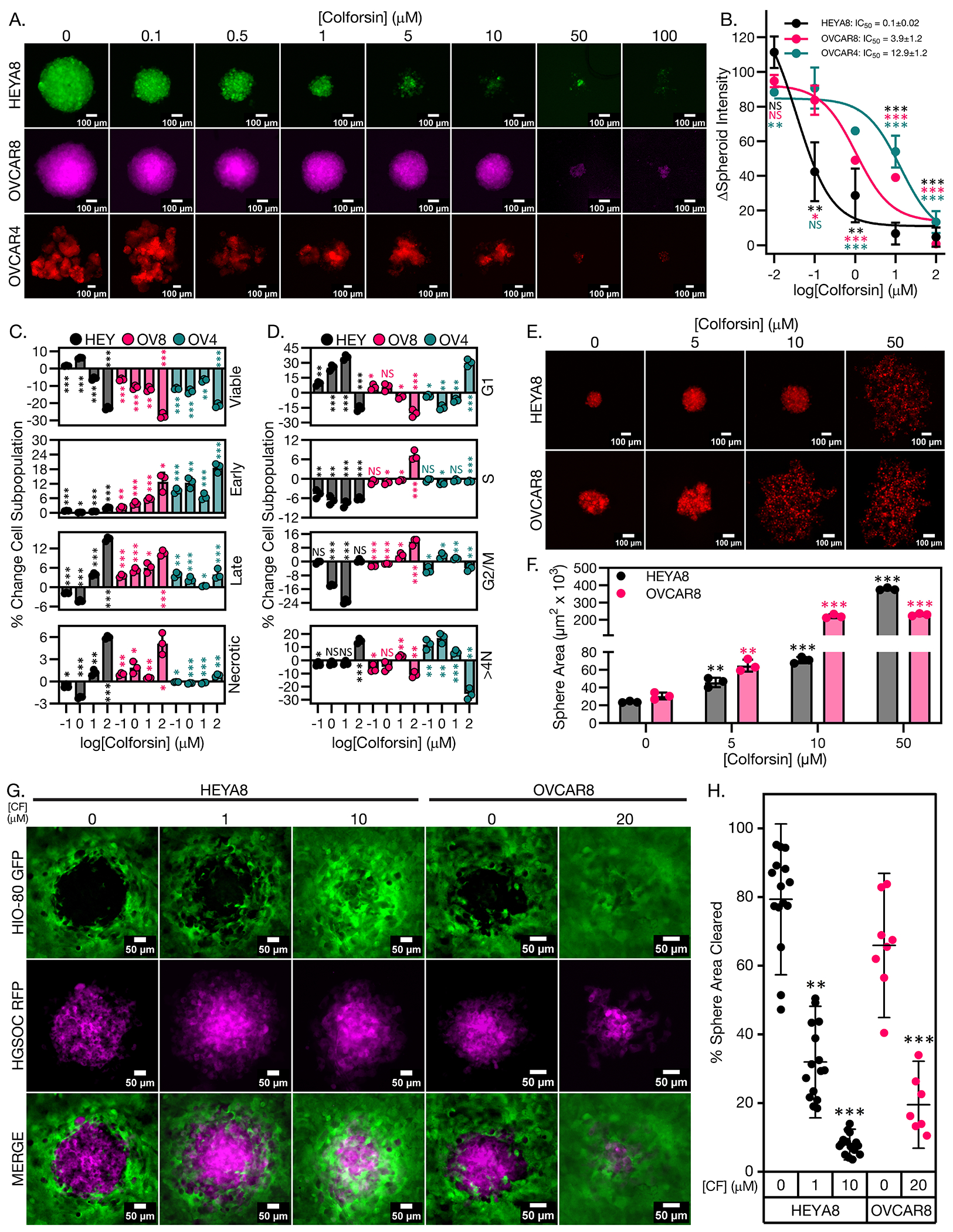
(A) Representative fluorescence micrographs of ovarian cancer spheroids after 7 days of culture under ultra-low attachment (ULA) conditions in media containing either vehicle or increasing doses of CF. Spheroids are shown in pseudocolor to highlight different cell lines. Scale bars for images are 100 μm. A minimum of 3 spheroid formation wells (technical replicates) were analyzed in each of 3 biological replicates. (B) Graph showing dose response curves for ovarian cancer spheroids cultured in either vehicle or increasing doses of CF. Mean IC50 values ± SD are displayed in the upper right corner for each cell line. All statistical comparisons are between a given CF dose vs vehicle control within each cell line. (C) Graphs showing % change in apoptotic cell subpopulations for FTSEC and ovarian cancer spheroids in response to treatment with either vehicle or increasing doses of CF. All statistical comparisons are between a given CF dose vs vehicle control within each cell line. (D) Graphs showing % change in cell cycle subpopulations for FTSEC and ovarian cancer spheroids in response to treatment with either vehicle or increasing doses of CF. All statistical comparisons are between a given CF dose vs vehicle control within each cell line. (E) Representative micrographs of vehicle and CF treated HGSOC spheroid formation at 18 hours under ULA culture conditions. Scale bars for images are 100 μm. A minimum of 3 spheroid formation wells (technical replicates) were analyzed in each of 3 biological replicates. (F) Graph with quantification of sphere area for vehicle and CF treated spheres shown in (E). (G) Representative fluorescence micrographs of vehicle or CF treated HEYA8 or OVCAR8 RFP ovarian cancer spheroid clearance of HIO-80 GFP OSE cell monolayers. Scale bars for images are 50 μm. HGSOC spheroids are shown in pseudocolor. Five spheroid adhesion wells (technical replicates) were analyzed in each of 3 biological replicates. (H) Graph showing quantification of % sphere area cleared for the images shown in (G). All data (A to H) are representative of N = 3 biological replicates. Statistical comparisons of two groups were performed using student’s t-test and comparisons of three or more groups were performed using one way ANOVA with Tukey multiple testing correction unless otherwise stated.
Cisplatin and colforsin daropate synergize to induce ovarian cancer cell death
Based on previous reports that forskolin could have additive or synergistic effects when combined with traditional chemotherapies30, we next investigated the combinatorial treatment of CF with Cis in the OVCAR8 and OVCAR4 cell lines which were less sensitive to CF as a monotherapy. CF displayed synergy with Cis treatment as measured by combinatorial dose-response matrix cell viability assays in OVCAR8 and OVCAR4 cells (Fig. 3, A to C). Synergy scores calculated with SynergyFinderPlus software showed synergistic responses for most drug combinations in both cell lines (Fig. 3B)55. We also calculated synergy for the dose-response matrices using Coefficient of Drug Interaction (CDI)56 and found that most dose combinations resulted in a CDI of <1, confirming synergy between CF and Cis in OVCAR8 and OVCAR4 cells (Fig. 3C). We also examined their synergy in our panel of PDX cell lines, wherein again synergistic responses were observed for most CF + Cis combination doses as calculated with SynergyFinder or CDI (Fig. 3, D and E, and fig. S4A). Notably, synergy scores were higher in the Cis-resistant than in the Cis-sensitive cell lines (Fig. 3E). In addition, synergy between CF and Cis was not observed in normal IMR90 fibroblasts (fig. S4, B and C).
Figure 3: Colforsin daropate synergizes with cisplatin to cause HGSOC cell death in culture.
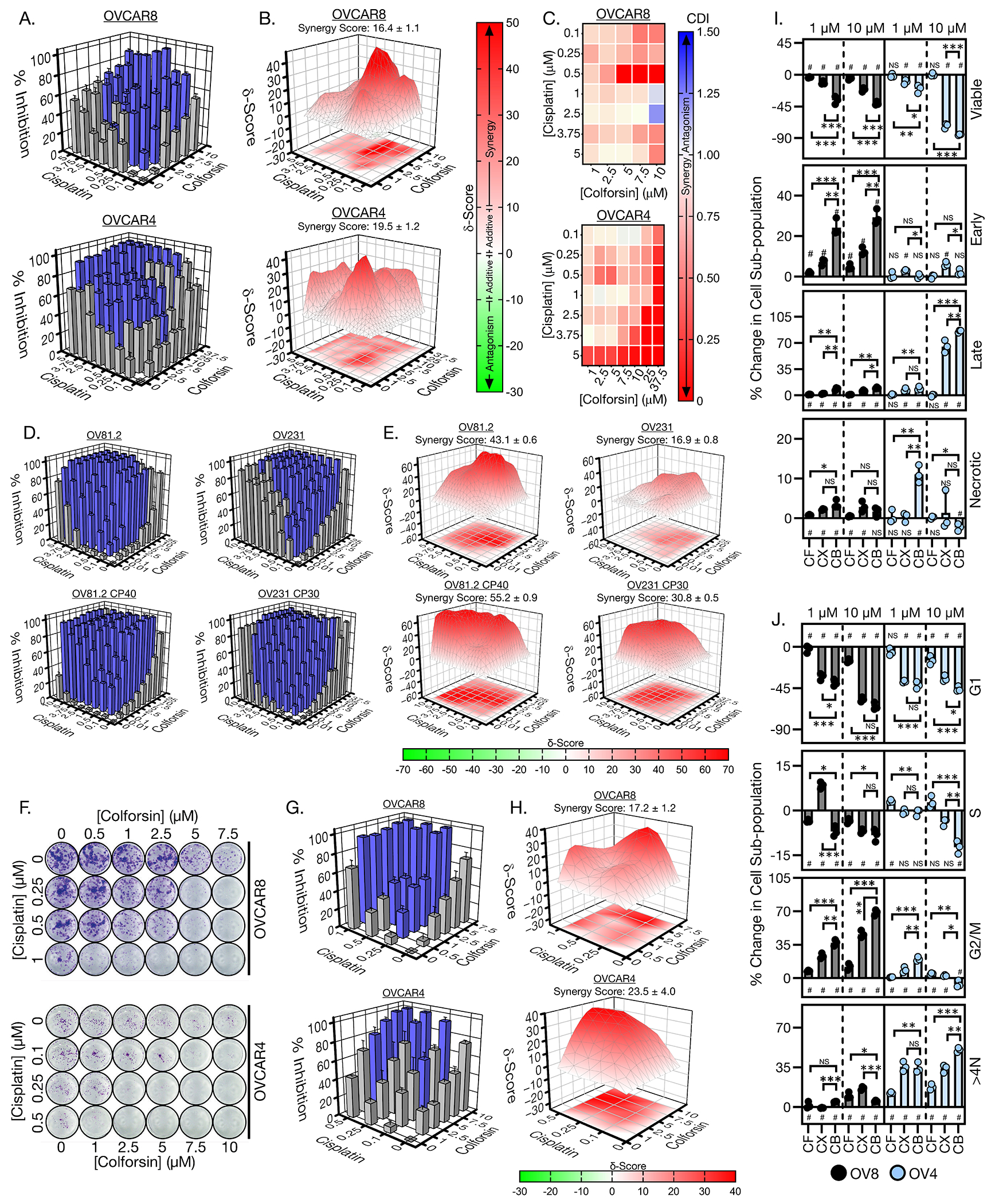
(A) 3D bar graphs showing % cell viability inhibition for CF-Cis combination dose response matrices in OVCAR8 cells (top) and OVCAR4 cells (bottom). Blue bars indicate dose combinations that were significantly different from the corresponding single agent dose. (B) 3D surface colormap for Bliss/Loewe consensus δ synergy scores of CF-Cis combination dose response matrices in OVCAR8 cells (top) and OVCAR4 cells (bottom). Overall synergy scores ± SD are displayed above each graph. Synergy scores less than −10 indicate antagonism, synergy scores from −10 to 0 and 0 to 10 indicate additive interaction in either the negative or positive direction respectively, and synergy scores greater than 10 indicate synergy for a given dose combination. (C) Heatmaps of coefficient of drug interaction (CDI) for OVCAR8 cells (top) and OVCAR4 cells (bottom). CDI values less than 1 indicate synergy, CDI values equal to 1 indicate additive interaction, and CDI values greater than 1 indicate antagonism for a given dose combination. (D) 3D bar graphs showing % cell viability inhibition for CF-Cis combination dose response matrices in OV81.2, OV81.2 CP40, OV231, and OV231 CP30 cells. Blue bars indicate dose combinations that were significantly different from the corresponding single agent dose. (E) 3D surface colormap for Bliss/Loewe consensus δ synergy scores of CF-Cis combination dose response matrices in OV81.2, OV81.2 CP40, OV231, and OV231 CP30 cells. Overall synergy scores ± SD are displayed above each graph. (F) Representative images of wells from OVCAR8 (top) and OVCAR4 (bottom) clonogenic CF-Cis dose-response matrices. (G) 3D bar graphs showing % colony area inhibition for CF-Cis combination dose response matrices in OVCAR8 cells (top) and OVCAR4 cells (bottom). Blue bars indicate dose combinations that were significantly different from the corresponding single agent dose. (H) 3D surface colormap for Bliss/Loewe consensus δ synergy scores of clonogenic CF-Cis combination dose response matrices in OVCAR8 cells (left) and OVCAR4 cells (right). Overall synergy scores ± SD are displayed above each graph. (I) Graphs showing % change in apoptotic subpopulations for OVCAR8 and OVCAR4 cells when treated with either CF, Cis, or combination of CF and Cis at a 1:1 ratio. # indicates statistical significance (p-value ≤ 0.05) for comparison of a particular treatment subpopulation to the vehicle control. (J) Graphs showing % change in cell cycle subpopulations for OVCAR8 and OVCAR4 cells when treated with either CF, Cis, or combination of CF and Cis at a 1:1 ratio. # indicates statistical significance (p-value ≤ 0.05) for comparison of a particular treatment subpopulation to the vehicle control. Abbreviations; colforsin daropate (CF), Cis (CX), combination (CB). All data (A to J) are representative of N = 3 biological replicates. Statistical comparisons of two groups were performed using student’s t-test and comparisons of three or more groups were performed using one way ANOVA with Tukey multiple testing correction unless otherwise stated.
Next, we investigated the effects of CF-Cis combination treatment on clonogenic survival and colony formation for OVCAR8 and OVCAR4 cells (Fig. 3, F to H). We again observed synergistic response to combinations of CF and Cis (Fig. 3H). Analysis of apoptosis for the combination treatment of OVCAR8 and OVCAR4 cells demonstrated increases in apoptotic subpopulations for both cell lines compared to either single agent, particularly at lower doses (Fig. 3I). In the OVCAR8 cells, early apoptotic cells increased approximately 2- to 3-fold for the combination vs CF or Cis alone at a dose of 1 μM. For OVCAR4 cells, there was a notable increase in necrotic cells at 1 μM combination (10% subpopulation change) versus either single agent (0.3% and 0.5% for CF and Cis, respectively). With respect to cell cycle, increases in G2/M accumulation were increased in the combination versus single-agent treated OVCAR8 cells (Fig. 3J). The OVCAR4 cells also showed increased aneuploid accumulation when treated with combination versus either single agent (Fig. 3J). Combined, these data indicate that CF can synergize with Cis to induce cell death and cell cycle arrest in HGSOC cell lines.
Colforsin daropate inhibits ovarian cancer growth in vivo
After observing the capacity of CF to induce HGSOC cell death in culture, we next investigated whether CF could inhibit tumor growth in vivo. For these experiments we utilized a subcutaneous tumor formation model using the more sensitive HEYA8 cells to determine efficacy of CF in a single, localized tumor and determine whether CF and Cis displayed any synergistic activity in vivo. We also used an orthotopic intraperitoneal (IP) tumor formation model with CF resistant OVCAR4 cells to determine efficacy of systemic CF administration in the context of metastatic dissemination. For the subcutaneous model, HEYA8 tumors were allowed to grow to 100 mm3, randomized, and then treated with either PBS vehicle, CF, Cis, or CF-Cis combination via intratumor injection (Fig. 4A). We found that CF treatment was able to inhibit tumor growth (Fig. 4, B and C). Initially, CF treatment was able to cause partial regression of tumor size for the first 2 weeks before tumors started to grow beyond baseline (Fig. 4C). Cis monotherapy performed comparably to CF in terms of tumor kinetics, particularly at later timepoints (Fig. 4C). Combination treatment of CF and Cis together showed the best response with a more pronounced inhibition of tumor growth (Fig. 4C). The combination treatment also had the best profile with respect to tumor progression with regression in tumor size that was sustained for approximately 3 weeks (Fig. 4C). We analyzed the subcutaneous tumor kinetics data with combPDX software to determine whether CF and Cis were synergistic in vivo58. Combination indices (CI) for the combined CF + Cis treatment arm were greater than 1 (indicating synergy) from days 14 to 48 when generated using either the Bliss Independence or Highest Single Agent method (Fig. 4D). In terms of histology, all tumors had morphology features consistent with HGSOC and stained positive for the mullerian marker PAX8 (Fig. 4E). However, it was observed that vehicle-treated tumors had a higher percentage of proliferative (Ki-67-positive) cells than CF-treated tumors (Fig. 4, E and F). In addition, the combination-treated tumors had a lower percentage of Ki-67–positive cells than tumors treated with either the CF or Cis alone (Fig. 4, E and F). With respect to final tumor burden, we found that CF+Cis combination treatment followed by CF monotherapy, then Cis monotherapy produced the largest to smallest decrease in final tumor mass, respectively (Fig. 4G and fig.S5A).
Figure 4: Colforsin daropate inhibits HGSOC tumor growth in vivo.
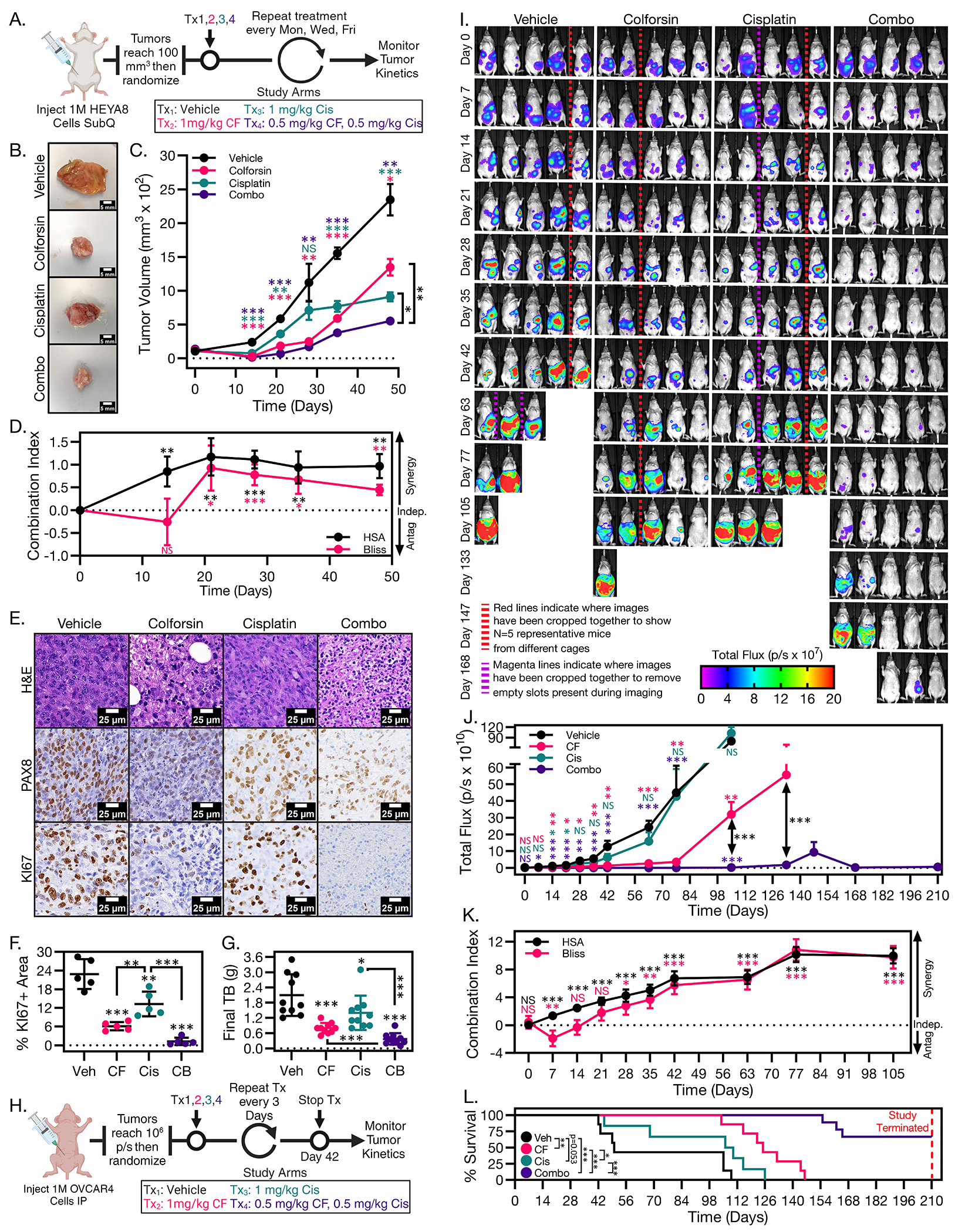
(A) Diagram depicting dosing schedule for HEYA8 subcutaneous tumor formation experiment. (B) Representative images of subcutaneous HEYA8 tumors harvested from mice treated with either vehicle, CF, Cis, or 1:1 CF-Cis combination. (C) Graph of tumor kinetics for mice treated with either vehicle, CF, Cis, or 1:1 CF-Cis combination. Error bars represent ± SEM. (D) Graph of Bliss and HSA in vivo synergy combination indices (CI) for CF-Cis treated HEYA8 tumors. A CI greater than 0 represents synergy between the two treatment groups, a CI equal to 0 represents independent effects for the two treatment groups, and a CI less than 0 represents antagonism between the two treatment groups. (E) Representative micrographs of H&E, PAX8, and KI67 staining of vehicle, CF, Cis, and 1:1 CF-Cis combination treated tumors. Scale bars for images are 25 μm. (F) Graph showing % positive area for KI67 in vehicle, CF, Cis, or 1:1 CF-Cis combination treated tumors. (G) Graph showing final tumor burden for vehicle, CF, Cis, or 1:1 CF-Cis combination treated tumors. (H) Diagram depicting dosing schedule for OVCAR4-Luc IP tumor formation experiment. (I) Representative bioluminescence images of IP tumor kinetics at Day 0-168 for mice treated with either vehicle, CF, Cis, or 1:1 CF-Cis combination. Red lines indicate where images have been cropped together to show N=5 representative mice from different cages. Magenta lines indicate where images have been cropped together to remove empty slots present during imaging. (J) Graph of IP tumor kinetics for mice treated with either vehicle, CF, Cis, or 1:1 CF-Cis combination. Error bars represent ± SEM. (K) Graph of Bliss and HSA in vivo synergy combination indices for CF-Cis treated OVCAR4-Luc tumors. (L) Kaplan-Meier survival curves for OVCAR4-Luc mice treated with either vehicle, CF, Cis, or 1:1 CF-Cis combination. Statistical comparisons between treatment groups are shown next to the legend and were performed using the log-rank test. Dashed red line indicates the timepoint where the study was terminated and all surviving mice were euthanized. All data (A to L) are representative of at least N = 3 biological replicates. For all in vivo experiments N ≥ 5 mice for each treatment group. Statistical comparisons of two groups were performed using student’s t-test and comparisons of three or more groups were performed using one way ANOVA with Tukey multiple testing correction unless otherwise stated.
We also observed synergy between CF and Cis in the OVCAR4 intraperitoneal (i.p.) model of metastatic dissemination. After i.p. injection and randomization, we observed that CF treatment produced a robust growth inhibition response, whereas tumor growth inhibition was less pronounced in Cis treated mice (Fig 4, H to J, and fig. S5B). Combination treatment produced the greatest response outperforming either monotherapy (Fig 4, I and J, and fig. S5B). Combination treatment of tumors produced sustained tumor growth regression that persisted even after treatment was stopped (Fig 4, I and J, and fig. S5B). Analysis of tumor kinetic data with combPDX showed combination indices greater than 1 from day 21 onward using both Bliss and Highest Single Agent methods (Fig. 4K). In addition, survival analysis demonstrated that CF monotherapy significantly extended survival compared to vehicle or Cis (Fig. 4L). Combination treatment resulted in the largest survival increase outperforming either monotherapy (Fig. 4L). Notably, we did not observe any overt drug toxicity in CF- or combination-treated mice, and there were no statistically significant differences in body weight between the drug-treated vs vehicle-treated mice when treatment was stopped (fig. S5C) Together, these data show that CF can inhibit HGSOC tumor growth, and that combination treatment with CF and Cis can generate more durable tumor growth inhibition than either monotherapy.
EIF2 and MYC signaling are inhibited in colforsin daropate treated ovarian cancer cells
Following the observations of therapeutic efficacy for CF in cell culture and in vivo, we next wanted to elucidate which signaling pathways were altered in CF-treated HGSOC cells that could drive the observed anti-tumorigenic phenotypes. To identify key signaling pathways in CF vs vehicle-treated HGSOC cells, we treated HEYA8 cells with either vehicle or increasing doses of CF and then performed bulk RNA-sequencing to profile transcriptional changes in the HEYA8 cells at the various CF doses (Fig. 5A). We then applied a criterion filter to the sequencing data so that only genes whose expression was significantly increased or decreased by CF treatment vs the vehicle by a fold change of 1.5 (≤ −1.5 or ≥ 1.5, with a multiple testing corrected p-value of p<0.01) were included in further analysis. The total number of differentially expressed genes (DEGs) increased as a function of CF dose, with 1 μM CF having approximately 600 DEGs and 10 μM having approximately 2300 DEGs (Fig. 5B), indicating a dynamic response in the transcriptional profile of the cells to CF. Varying degrees of DEG overlap were observed between the different CF doses, with a core network of 460 DEGs common to all 3 doses (Fig. 5C).
Figure 5: EIF2 and MYC signaling are downregulated in colforsin daropate treated HGSOC cells.
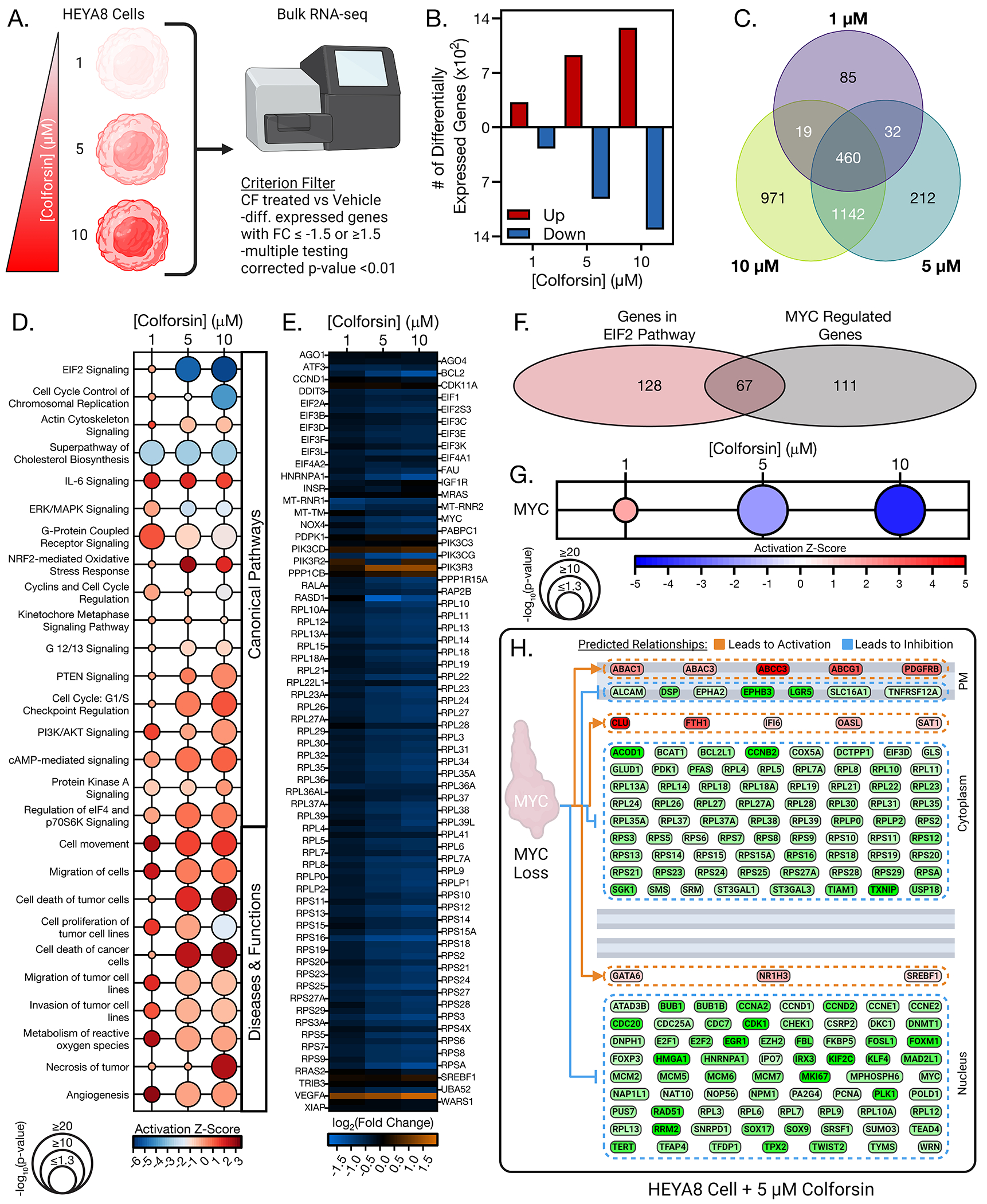
(A) Diagram of experimental setup for bulk RNA sequencing of HEYA8 cells treated with either vehicle, 1, 5, or 10 μM CF. (B) Graph showing number of up and downregulated differentially expressed genes for each CF dose displayed below. (C) Venn diagram of differentially expressed gene overlap for each CF dose. (D) Matrix bubble graph showing top differentially regulated canonical and functional pathways for CF treated HEYA8 cells versus vehicle treated HEYA8 cells. Significance values and activation z-scores for each pathway were computed using Ingenuity Pathway Analysis software. (E) Heatmap showing gene expression changes of CF vs vehicle treated HEYA8 cells for genes in the EIF2 pathway. (F) Venn diagram showing overlap of differentially expressed genes common to both the EIF2 signaling pathway and MYC upstream regulator pathway that are present in HEYA8 cells treated with 10 μM CF. (G) Matrix bubble graph showing statistical significance and predicted activation z-score for the MYC upstream regulator pathway in CF treated HEYA8 cells. Significance values and activation z-scores for each pathway were computed using IPA software. (H) Diagram showing RNAseq expression levels of MYC targets in HEYA8 cells treated with 5 μM CF. All data (A to H) are representative of N = 3 biological replicates. Statistical comparisons of two groups were performed using student’s t-test and comparisons of three or more groups were performed using one way ANOVA with Tukey multiple testing correction unless otherwise stated.
Next, we examined the DEGs of each CF dose using Ingenuity Pathway Analysis (IPA) to identify which signaling pathways were differentially altered in CF- vs vehicle-treated HEYA8 cells. Comparison analysis of canonical signaling pathways in the different CF-treated samples identified several hits with relevance to our observed phenotypes (Fig. 5D). The top two canonical pathways that had significant overlap with the DEGs of the CF-treated samples were “EIF2 Signaling” and “Cell Cycle Control of Chromosomal Replication”, both of which were predicted to be downregulated in response to CF treatment (Fig. 5D). EIF2 signaling is a key pathway involved in ribosome assembly and protein translation that is frequently dysregulated in cancer58. The “Cell Cycle Control of Chromosomal Replication pathway” contains key signaling molecules that facilitate DNA replication and G1/S transition. Notably, inhibition of both pathways can lead to the cell death and cell cycle arrest phenotypes we observed when HGSOC cells were treated with CF. We also observed upregulation of cell cycle checkpoint pathways such as G1/S Checkpoint Regulation (Fig. 5D). Curiously, we did not see strong activation of cAMP or PKA signaling pathways (contrary to expectations, given the canonical function of CF as an AC activator), although there was significant overlap between these pathways and the DEGs of CF-treated HEYA8 cells (Fig. 5D, fig. S6A). More detailed functional analysis confirmed that CF-treated HEYA8 cells did not produce increased intracellular cAMP as measured by cAMP Glo assay (fig. S6B). Levels of phosphorylated CREB also did not increase in CF treated HGSOC cells (fig. S6, C and D). These data suggested that CF was increasing cell cycle arrest and cell death in the HEYA8 cells through a non-canonical mechanism. Alternatively, there may be strong negative feedback by phosphodiesterase (PDE)-mediated hydrolysis, as there was a 3- to 10-fold upregulation of multiple PDEs, including cAMP-selective PDE4 and PDE7, in CF-treated cells (fig. S6A).
Further analysis of the functional signaling pathways that were altered in the CF-treated cells again produced several hits relevant to our observed phenotype. This included predicted activation, and significant overlap, of several cell death pathways such as “Cell death of cancer cells” and “Necrosis of tumor” (Fig. 5D, and fig. S7, A to C). It also included predicted inhibition of cell proliferation and migration/invasion pathways (Fig. 5D, and fig. S7, A to C). Following the analysis of CF induced pathway alterations at a global level, we focused on EIF2 signaling in more detail to further elucidate which components of the pathway were downregulated. We found that several eIF family members were downregulated including eIF1, eIF2A, and several subunits of eIF3 (Fig. 5E). In addition, many ribosomal proteins were downregulated in response to CF treatment including several members of both the small and large ribosomal subunit complexes (Fig. 5E). Based on these data, we used IPA to identify predicted upstream regulators that could modulate expression of both eIFs and ribosomal proteins as well as the CF induced pathway changes we observed. The top predicted upstream regulator for these criteria was the well-known oncogenic transcription factor c-MYC. Sixty-seven of the DEGs present in the CF-treated HEYA8 cells (~34 to 37%) were common to both the EIF2 signaling pathway and MYC upstream regulator pathway (Fig. 5F). MYC RNA levels were decreased in response to CF treatment (Fig. 5E), and MYC signaling was predicted to be inhibited in response to CF treatment (Fig. 5G). Together, the results of our pathway analysis indicated that CF-induced cell cycle arrest and cell death were likely mediated through inhibition of MYC and EIF2 signaling.
Colforsin daropate inhibits MYC to induce ovarian cancer cell death
The results of our pathway analysis indicated that CF may be inhibiting MYC signaling to drive HGSOC cell cycle arrest/cell death. Therefore, we next investigated the functional relationship between CF and MYC. MYC is an established driver of HGSOC and is amplified at the copy number and protein level in up to 50% of HGSOC patients59. We observed that several MYC targets were decreased at the mRNA level in HEYA8 cells treated with CF (Fig. 5H). We next examined protein levels of downstream MYC targets involved in translation and cell cycle progression whose downregulation could contribute to the cell cycle arrest/cell death phenotypes we observed in CF treated HGSOC cells. We observed decreased levels of cell cycle regulator proteins, such as BUB1 and Cdc20, and of several MYC-regulated translation factors, such as E2F1, eIF2α, eIF4E, RPS12 and RPS16, as well as of phosphorylated eukaryotic translation initiation factor 4E binding protein-1 (p-4EBP1), a master regulator of protein synthesis (Fig. 6A). Phosphorylation of 4EBP1 prevents it from binding to eukaryotic translation initiation factor 4E (eIF4E) and inhibits formation of the translation initiation complex60. Notably, increased levels of non-phosphorylated 4EBP1, observed in the CF-treated HEYA8 cells (Fig. 6A), have been linked to promoting cell cycle arrest and apoptosis in other cancer contexts61–64. In OVCAR8 and OVCAR4 cells, MYC and its downstream targets were less responsive to CF treatment (Fig. 6B).
Figure 6: Colforsin daropate decreases MYC levels to facilitate HGSOC cell death.
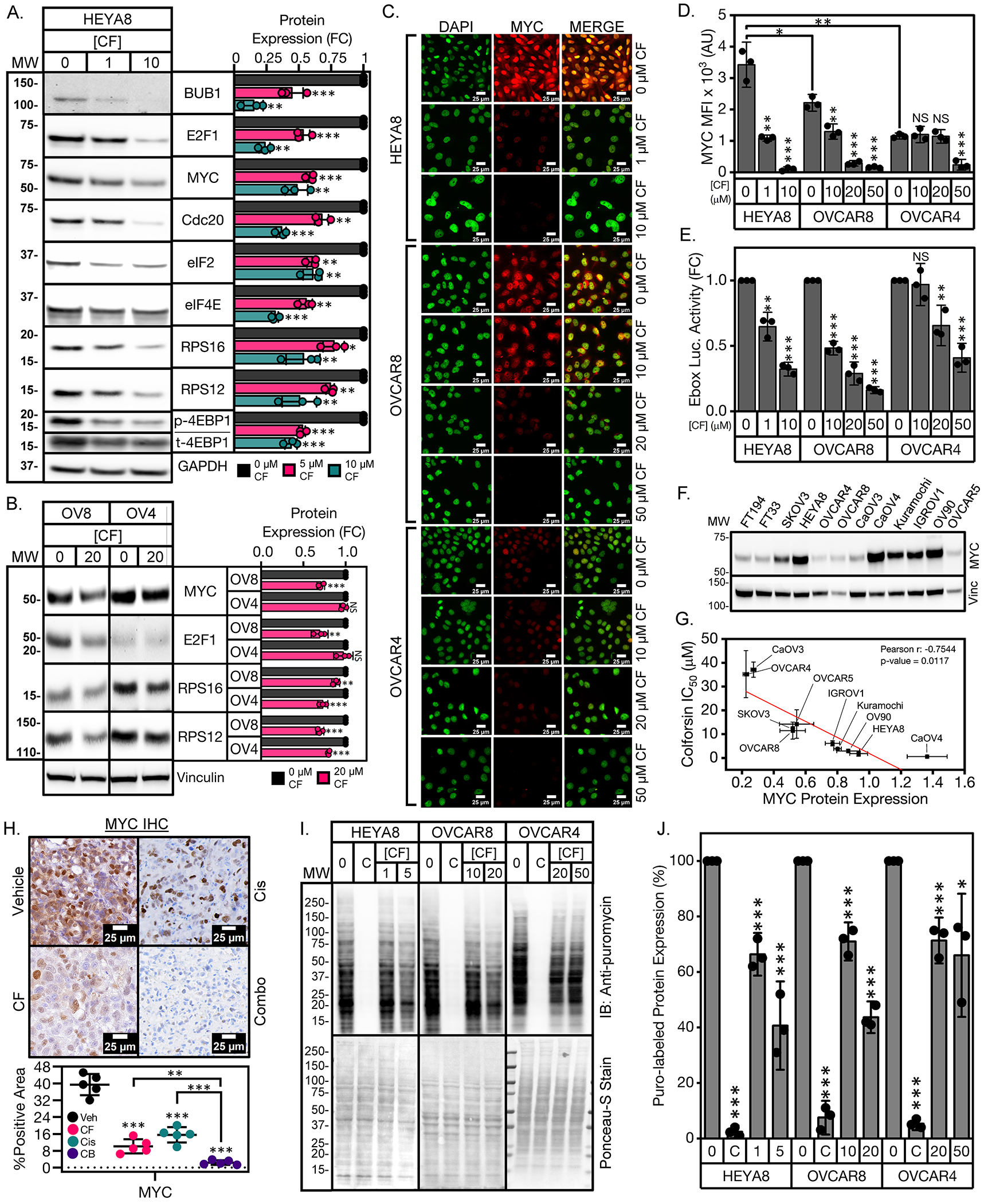
(A) Representative western blots of HEYA8 cells showing dose-dependent decreased levels of MYC protein and downstream MYC regulated targets in response to CF treatment with quantification (right). Immunoblot of phosphorylated 4EBP1 (p-4EBP1) is shown above immunoblot of total 4EBP1 (t-4EBP1) in a split panel. For quantification of p4EBP1, p4EBP1 levels were normalized to total 4EBP1 levels in each sample prior to normalization of CF treated samples to vehicle. (B) Representative western blots of OVCAR8 and OVCAR4 cells showing levels of MYC protein and downstream MYC regulated targets in response to CF treatment with quantification (right). (C) Representative immunofluorescence images of MYC staining for HGSOC cells treated with CF for 48 hours. Scale bars for images are 25 μm. Five fields of at least 20 cells were analyzed in each of 3 biological replicates. (D) Quantification of nuclear MYC fluorescence for HGSOC cells treated with CF for 48 hours. (E) Quantification of E-box luciferase activity for HGSOC cells treated with CF for 48 hours. (F) Western blot of MYC expression across a panel of FTSEC and HGSOC cell lines. (G) Correlation plot of CF IC50 values vs MYC protein expression for the panel of HGSOC cell lines shown in (F). Pearson r correlation coefficient along with corresponding p-value are shown in the upper right corner of the graph. (H) Representative micrographs of MYC staining of HEYA8 vehicle, CF, Cis, and 1:1 CF-Cis combination treated tumors with quantification below. Scale bars for images are 25 μm. (I) Detection of nascent polypeptides via puromycin labeling of HGSOC cells treated with either vehicle, CF, or 50 μg/mL cycloheximide (abbrev. “C”). Western blot of lysates using anti-puromycin antibody (top), Ponceau-S staining of lysates measuring total protein levels (bottom). (J) Graph showing quantification of % puromycin labeling of drug treated lysates relative to vehicle. All data (A to J) are representative of N = 3 biological replicates. Statistical comparisons of two groups were performed using student’s t-test and comparisons of three or more groups were performed using one way ANOVA with Tukey multiple testing correction unless otherwise stated.
MYC nuclear localization was also decreased in response to CF treatment as measured by immunofluorescence (Fig 6, C and D). We then validated that the decreases in MYC protein levels caused by CF corresponded to decreased MYC transcriptional activity using an established E-box luciferase reporter. We found that E-box reporter activity decreased in response to increasing CF dose in HGSOC cells (Fig. 6E). MYC protein levels correlated with CF sensitivity in a panel of HGSOC cell lines (Fig. 6, F and G). Notably, two of the most CF-sensitive cell lines, CaOV4 and Kuramochi, had MYC amplification (in copy number, protein expression, and signaling activity; table S1). In addition, MYC levels were reduced in the CF- and Cis (single agent) treated HEYA8 tumors by approx. 50% and in combined-treated tumors by >90% compared to vehicle-treated tumors (Fig. 6H). These data indicated that CF and Cis acted synergistically to decrease MYC in the HEYA8 tumors. To validate that CF was inhibiting translation, we performed a puromycin labeling assay to identify nascent polypeptides in vehicle- and CF-treated HGSOC cells. We observed that CF treatment reduced global protein synthesis in HGSOC cells in a dose-dependent manner (Fig. 6, I and J). In addition, the levels of translation inhibition achieved with CF treatment corresponded to the relative CF sensitivity of the HGSOC cell lines tested (Fig. 6, G, I, and J).
To investigate the functional relationship between CF and MYC, we profiled the response of isogenic FTSECs that were transformed into HGSOC with overexpression of either MYC or a different oncogene. We hypothesized that because the antitumorigenic effects of CF are mediated at least in part by decreasing MYC expression, then FTSECs transformed with MYC should be more sensitive to CF than those transformed with a different oncogene. Specifically, the FTSEC lines used included FT33 +EV (immortalized with SV40 Large T antigen, not transformed, EV indicates empty vector), FT33 +MYC (immortalized with SV40 Large T antigen, transformed with degradable MYC overexpression), FT33 +Ras (immortalized with SV40 Large T antigen, transformed with Ras overexpression), FT194 +EV (immortalized with SV40 Large T antigen, not transformed), FT194 +MYC (immortalized with SV40 Large T antigen, transformed with degradable MYC overexpression), FT194 +YAP (immortalized with SV40 Large T antigen, transformed with YAP overexpression). We observed that the MYC transformed FT cell lines were more sensitive to CF treatment than the Ras- or YAP-transformed cell lines (Fig. 7A). As expected, the non-transformed FT cell lines were the least responsive to CF and had the highest IC50 values (Fig. 7A). The CF-treated, MYC-transformed FTSEC cell lines also exhibited increased levels of apoptosis compared to the Ras- or YAP-transformed cell lines (Fig. 7, B and C). With respect to MYC signaling, we found that the MYC transformed FT33 cells had larger decreases in MYC and MYC target levels than either the FT33 +Ras or FT33 +EV cells when treated with CF (Fig. 7, D and E). We also investigated the effects of transient MYC depletion on HGSOC sensitivity to CF treatment. HEYA8 and CaOV4 cells were transfected with MYC siRNA then cultured in either vehicle- or CF-containing media. We observed a baseline reduction in proliferation of vehicle-treated HGSOC cell lines when MYC was knocked down (fig. S8, A to C). Notably, transient MYC knockdown diminished the ability of CF to inhibit proliferation of the HGSOC cells, as indicated by the increase in IC50 values in the siMYC-transfected cells (fig. S8D). Together, these data show that CF facilitates cell cycle arrest and cell death in HGSOC at least in part by decreasing MYC expression.
Figure 7: Overexpression of MYC in FTSECs sensitizes them to colforsin daropate treatment.
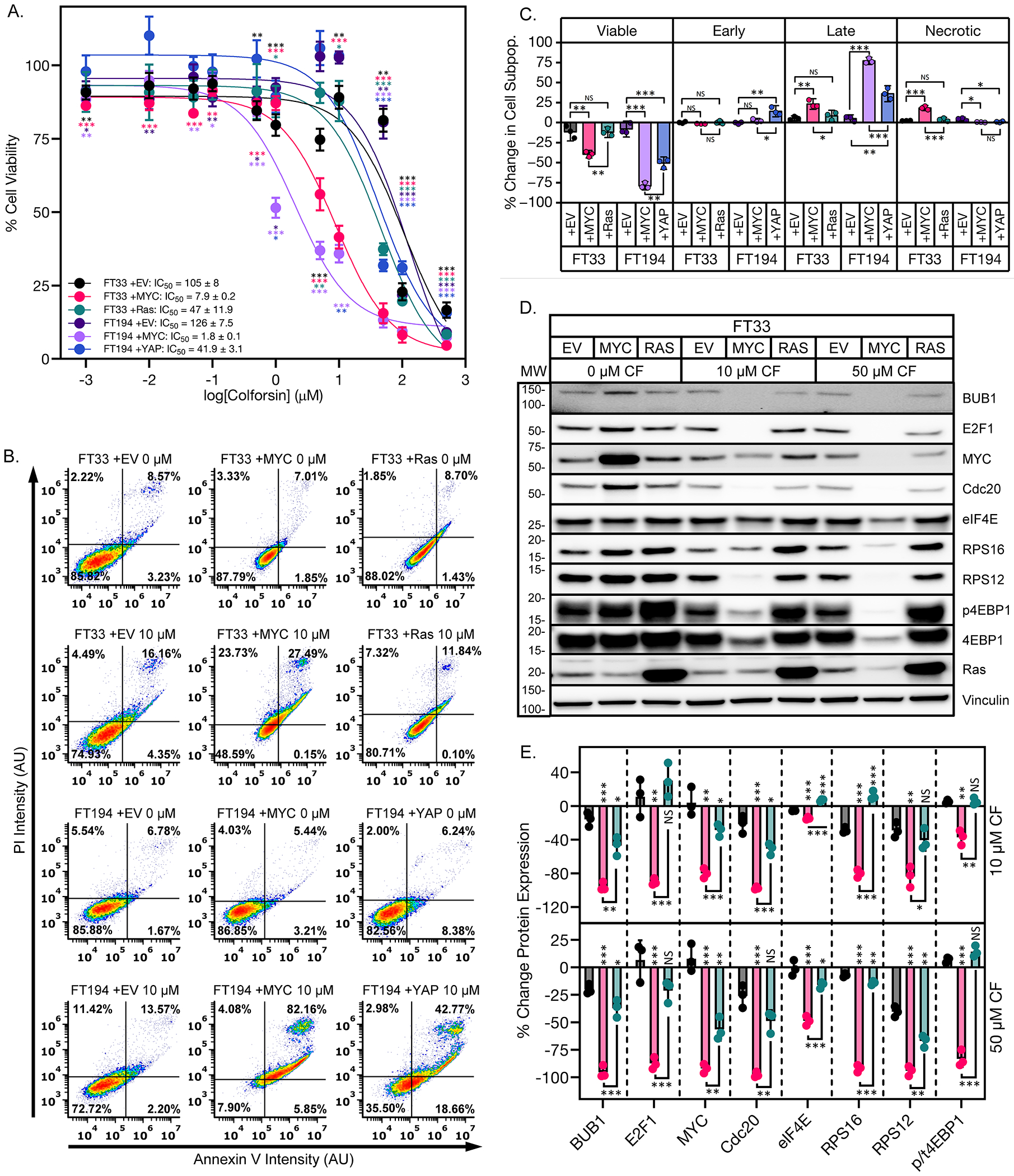
(A) Dose response curves of % cell viability for FT33 +TAg, FT33 +MYC, FT33 +Ras, FT194 +TAg, FT194 +MYC, and FT194 +YAP cells treated with increasing doses of CF. IC50 values ± SD are shown in the legend. (B) Representative graphs showing Annexin V-PI staining for the FTSEC panel in response to treatment with 10 μM CF. (C) Graph showing % change in apoptotic cell subpopulations for the FTSEC panel in response to treatment with 10 μM CF. (D) Representative western blots of FTSEC panel showing dose-dependent decreased levels of MYC protein and downstream MYC regulated targets in response to CF treatment. (E) Graph showing quantification of MYC and MYC-regulated targets in FT33 cells treated with CF. p/t4EBP1 corresponds to the ratio of phosphorylated to total 4EBP1. All data (A to E) are representative of N = 3 biological replicates. Statistical comparisons of two groups were performed using student’s t-test and comparisons of three or more groups were performed using one way ANOVA with Tukey multiple testing correction unless otherwise stated.
Discussion
We wanted to determine whether CF, a water-soluble derivative of forskolin, could be repurposed for HGSOC treatment. CF induced cell death in established HGSOC cell lines, including clinically relevant PDX cell lines. Notably, immortalized, non-transformed FTSECs were far less sensitive to CF than HGSOC cells demonstrating that CF was selective for cancer vs “normal” cells of the reproductive tract. Interestingly, we observed differences in the type of cell cycle arrest/cell death for the different HGSOC cell lines that roughly corresponded to their sensitivity to CF overall. These data highlight the ability of CF to consistently produce a therapeutic response in a heterogeneous panel of HGSOC cell lines suggesting that it will be effective in most ovarian cancer patients.
It has been previously reported that ovarian cancer spheroids are protected from chemotherapy and demonstrate increased drug resistance compared to their 2D counterparts65,66. Intriguingly, the HGSOC spheroids were more sensitive to CF than the corresponding 2D culture. We observed here that the CF-treated spheroids showed a reduction in adhesion signaling and tended to be less compact than vehicle-treated spheroids. It is likely that CF decreases important intrinsic pro-survival signals in the spheroids that depend on cell-cell adhesion. This also allows the drug greater access to more tumor cells than would otherwise be possible. Future studies will need to investigate in detail the mechanisms by which CF disrupts ovarian cancer sphere adhesion.
Combination treatment of our HGSOC cell lines with CF + Cis produced synergism in therapeutic response for CF resistant cells. In addition, CF-Cis synergy was more pronounced in the Cis-resistant versus sensitive PDX isogenic cell lines. These data suggest that CF may also have utility in resensitizing resistant disease to Cis. Forskolin treatment has been shown to produce increased Cis accumulation within ovarian cancer cells30. Given the synergism observed between CF and Cis in our experiments, it is likely this phenomenon occurs with CF as well. It will be important to confirm whether CF can increase Cis accumulation in HGSOC cells, elucidate the mechanism by which this occurs, and determine whether CF-enhanced Cis accumulation selectively occurs in cancer cells vs normal tissue.
We next wanted to determine CF efficacy for treating HGSOC in vivo. In the subcutaneous and IP models CF was effective as a monotherapy in reducing tumor kinetics and tumor burden, but CF-Cis combination therapy produced the largest and most durable response. Combination treatment was able to cause sustained IP tumor regression that persisted even after treatment was stopped. Notably, analysis of tumor kinetics data with combPDX showed strong in vivo synergy for CF and Cis. These data provide proof-of-concept for the use of CF as an adjuvant therapy that can be incorporated into standard HGSOC chemotherapy regimens. Our results indicate that CF can inhibit the capacity of HGSOC spheroids to invade OSE monolayers, thus we predict that CF will also be able to inhibit intraperitoneal metastasis as well.
Several changes in pathway expression were observed with CF treatment of HEYA8 cells that were consistent with increased tumor cell death and cell cycle arrest. The top pathway alteration was a significant downregulation of EIF2 signaling, the primary pathway that controls translation and protein production. Further analysis revealed downregulation of several ribosomal proteins and eIFs that are required to assemble ribosomal subunits. These changes in signaling are consistent with the observed CF-induced cell cycle arrest and cell death. Decreased ribosome production has been previously linked to increases in cell cycle arrest and cell death67–71. In addition, inhibition of ribosome biogenesis as a cancer therapy is an active area of research70.
Pathway analysis indicated the top upstream signaling regulator was the transcription factor MYC, an established oncogene and master regulator of ribosome biogenesis. CF treatment was predicted to inhibit MYC activity, and MYC directly regulates many of the same genes found in the EIF2 pathway. CF treatment decreased MYC protein levels and transcriptional activity in HGSOC. Overexpression of MYC in isogenic FTSEC lines selectively sensitized them to CF versus other oncogenes. These results demonstrate that the anti-cancer effects of CF are mediated at least in part by inhibition of MYC. The CF induced signaling changes we observed are particularly interesting because they indicate the predominant mechanism of HGSOC cell death is non-canonical and independent of adenylyl cyclase activation. Forskolin has been shown to bind to targets other than adenylyl cyclase and this may occur with CF as well18.
This study establishes that CF has anti-cancer activity in HGSOC. Such activity is not, to our knowledge at the time of publication, demonstrated in any other cancer. Our data indicate that CF has robust cytotoxic effects that are selective for cancer cells versus non-transformed cells. The ability of CF to synergize with Cis opens the possibility for its use as an adjuvant to boost the efficacy of Cis in treating patients. CF inhibition of MYC protein expression is also highly relevant to HGSOC, as the MYC gene is amplified in approximately 50% of HGSOCs. HGSOC cells in general are highly dependent on MYC for survival, however, therapeutic targeting of MYC has remained a challenge as the important functional domains of MYC are intrinsically disordered and not suitable for small molecule binding72,73. Most drugs target MYC indirectly and only a few that can induce decreases in MYC protein levels72. CF is therefore a novel and valuable addition to this sub-class of drugs that can decrease MYC protein levels.
Several questions remain that will need to be addressed in future studies. The details of how CF decreases MYC levels in HGSOC will need to be elucidated and whether additional factors besides MYC are important CF targets. Further study will also be needed to determine the direct binding targets of CF in HGSOC cells. It will be important to determine if CF derivatization can uncouple the anti-cancer effects from canonical effects to improve tolerability and minimize off-target effects. These questions will follow from our study’s findings, which establish CF as a potential, novel therapeutic for treatment of MYC-driven HGSOC.
Materials and Methods
Annexin V apoptosis assay
Cells were plated in 6 well plates at a density of 200,000 cells per well. The next day cells were treated with media containing either CF, Cis, or 1:1 CF-Cis combination for 48 hours. After drug treatment, the drug containing media was collected from each well to collect floating cells. Each well was then trypsinized to collect adherent cells and the two cell fractions (floating + adherent) were pooled for each well. Detection of apoptosis was performed using the FITC Annexin V Apoptosis Detection Kit (BD, 556547) according to the manufacturer’s protocol. Annexin V-FITC and propidium iodide emissions were measured for all samples using a BD Accuri flow cytometer with a minimum number of 10,000 cells measured per sample. Analysis and quantification of annexin V staining populations was performed using FCS Express software (DeNovo Software).
cAMP Glo assay
Cells were plated in 6-well plates at 100,000 cells/well and treated the next day with either vehicle or CF for 72 hours. After 72 hours the cells were assayed to quantify intracellular cAMP levels according to the manufacturer’s instructions.
Cell culture and reagents
Cells were cultured in 10mm plates in a humidified atmosphere (5% CO2) at 37°C. At 70-90% confluence, trypsin (0.25%)/EDTA solution was used to split the cells which were used until passage 20. Cell culture medium (Corning) was supplemented with 10% FBS (Gibco) and 1% Penicillin-Streptomycin (PS) (Gibco) [HEYA8 cells (DMEM), FTSE cells (DMEM-F12), OVCAR8, OVCAR4 cells (RPMI)]. The following reagents were used in the study: Cisplatin (Penn Hospital Pharmacy), Colforsin daropate (Tocris, cat. #1099). All cell lines used in the study tested negative for mycoplasma and were validated using IDEXX CellCheck.
Cell cycle assay
Cells were plated in 10 cm dishes at a density of 500,000 cells per well. The next day cells were treated with media containing either CF, Cis, or 1:1 CF-Cis combination for 48 hours. After drug treatment, the drug containing media was collected from each plate to collect floating cells. Each well was then trypsinized to collect adherent cells and the two cell fractions (floating + adherent) were pooled for each well. Cells were washed in 1X PBS and then fixed in 70% EtOH overnight at −20°C. Propidium iodide staining was performed using FxCycle™ PI/RNAse staining solution (Thermo Fisher, F10797) according to the manufacturer’s protocol. Propidium iodide emissions were measured for all samples using a BD Accuri flow cytometer with a minimum number of 10,000 cells measured per sample. Analysis and quantification of PI staining populations was performed using FCS Express software (DeNovo Software).
Cell viability assay
For the single-agent CF treatments, cells were plated in a 12-well plate at 50,000 cells/well and treated the next day with CF. After 48 hours, cells were then incubated with 3-(4,5-dimethylthiazolyl) for 2 hours and absorbance was measured at 600nm. IC50 values were calculated from dose response curves using GraphPad Prism software.
For the CF-Cis combination treatments, cells were plated in a 96-well plate at 2,000 cells/well and treated the next day with either CF, Cis, or CF-Cis combination at the stated ratio. After 48 hours, cells were then incubated with CellTiterGlo Reagent (Promega) according to the manufacturer’s protocol. Synergy scores were calculated using either SynergyFinderPlus software or Coefficient of Drug Interaction (see Statistics section for details).
Clonogenic assay
Cells were trypsinized and counted then resuspended in media containing CF, Cis, or the stated dose combination of CF and Cis at a density of 1000 cells/mL. 1 mL of cells (1000 cells/well) were plated in one well of a 12-well plate (single agent studies) or 24-well plate (drug combination studies) for each drug concentration analyzed. Colonies were allowed to form for 7 days and were then stained with crystal violet staining solution (1% Paraformaldehyde(v/v), 10% methanol (v/v), 0.05% crystal violet (w/v), in 1X PBS). Plates were imaged and staining quantified using the ColonyArea ImageJ plugin74. Synergy scores were calculated using SynergyFinderPlus software (see Statistics section for details).
Immunoblotting
Cell pellets were resuspended in 1X RIPA buffer (Millipore, 20-188) containing protease inhibitor (Roche, 11697498001) and phosphatase inhibitor (Roche, 4906845001) cocktail, incubated on ice for 30 min and then sonicated to lyse cells. Lysates were spun down for 20 min at 12,000 rpm at 4°C and the supernatant was collected. Protein concentration of lysates was estimated using bicinchoninic acid (BCA) assay (Thermo Fisher Scientific, 23227). Thirty micrograms of sample protein were mixed with sample buffer then loaded and separated using Criterion™ XT 4-12% Bis-Tris polyacrylamide gels (Bio-Rad, 3450123) and XT MES Buffer (Bio-Rad, 1610789). Transfer of separated samples from gels to nitrocellulose membranes was performed with the Bio-Rad Trans-Blot Turbo system (Bio-Rad, 1704156). Primary antibodies to BUB1 (Proteintech, 13330-1-AP), E2F1 (CST, 3742), Cdc20 (Proteintech, 10252-1-AP), eIF2α (CST, 9722), eIF4E (CST, 9742), RPS12 (Proteintech, 16490-1-AP), RPS16 (Proteintech, 15603-1-AP), c-Myc (CST, 5605), phospho-4EBP1 (CST, 2855), 4EBP1 (CST, 9644), anti-Puromycin (Sigma, MABE343) and Vinculin (CST, 13901) were diluted 1:1000 in tris buffered saline containing 0.1% Tween 20 (TBST) and 5% (w/v) nonfat milk and incubated with membranes overnight at 4°C. Membranes were then washed 3X in TBST followed by incubation with HRP-conjugated anti-rabbit IgG antibody (CST, 7074) for 1 hour at room temperature. Western blot images were acquired by chemiluminescence using SuperSignal™ West Pico Plus (Thermo Fisher, 34577). Quantification of signal from western blot bands was performed using ImageLab densitometry software.
Immunohistochemistry
HEYA8 subcutaneous mouse tumors were processed as previously reported75. Immunohistochemical staining was performed using a 1:500 dilution of antibodies to PAX8 (Novus, NBP1-32440), KI67 (CST, 12202), and MYC (CST, 5605). Slides were scanned using a 3D Histech Panoramic Midi scanner. IHC staining was quantified using Panoramic SlideViewer software using the densitometry function. For Ki-67 staining,total number of positive (brown) pixels within nuclei were quantified for the scanned area of the tumor and were reported as a percentage of the total area scanned. For MYC staining, total number of cytoplasmic and nuclear positive (brown) pixels were quantified for the scanned area of the tumor and were reported as a percentage of the total area scanned.
Lentiviral transduction
For lentiviral transfection, Lenti Starter Kit (System Biosciences, CA) was used. Briefly, 293T cells (ATCC) were transduced with 2μg plasmid and 10μg of pPACKH1-plasmid mix with Lipofectamine 2000 (Life Technologies, CA). 48hr later, virus particles were harvested and precipitated. Target cells were transduced by plating 100,000 cells/well in a 6 well plate with virus particles 4μg/mL polybrene (Santa Cruz Biotechnologies, CA)
Luciferase reporter assay
Cells were plated in 6-well plates at 70,000 cells/well. The next day, regular media was replaced with CF containing media and the cells were transfected with the 7X E-box luciferase reporter plasmid (Addgene, plasmid #124532). 24 hours after transfection the cells were lysed and luminescence was measured using components from the Promega Dual Luciferase Reporter Assay System according to the manufacturer’s protocol (Promega, E1910).
MYC siRNA transfection with and without colforsin daropate treatment
For MYC depletion experiments, HEYA8 or CaOV4 cells were seeded in 2D culture at a density of 40,000 cells per well in 12 well tissue culture plates. After 24 hours the cells were transfected with either siscram (cat. # 4390843, Thermo Fisher), siMYC #1 (Assay ID s9129, Thermo Fisher), or siMYC #2 (Assay ID s9131, Thermo Fisher) according to the manufacturer’s instructions. 24 hours after transfection, the transfection media was removed and media containing either vehicle or CF was added to the appropriate wells. Cells were then incubated for 72 hours in vehicle- or CF-containing media. After incubation with or without CF, cell viability was assessed via MTT assay. IC50 values were calculated from dose response curves using GraphPad Prism software. To assess MYC knockdown efficiency, vehicle-treated cells transfected with scrambled siRNA (siscram) or one of two MYC-target siRNAs (siMYC #1 or siMYC #2) were collected 48 hours post-transfection and MYC levels were quantified via immunoblotting. Quantification of signal from western blot bands was performed using ImageLab densitometry software.
Protein synthesis assay
Assay was performed using a modification of the SUnSET assay described in 76. Cells were plated in 6-well plates at 100,000 cells/well. The next day cells were treated with either vehicle or increasing doses of CF for 48 hours, after which, media from all wells was removed and replaced with drug-free media containing 10 μg/mL puromycin (Sigma-Aldrich, P8833). Cells were incubated in puromycin media for 10 min to label nascent polypeptide chains (pulse). Puromycin media was removed and all wells were washed 1X with ice cold PBS. Puromycin free drug containing media was then replaced on cells and cells were incubated for 50 min at 37°C (chase). Cells were then harvested and puromycin labeling was measured via western blotting techniques. As a positive control, cells were also treated with 50 μg/mL cycloheximide protein synthesis inhibitor for 24 hours prior to puromycin pulse/chase in all experiments. Total protein was measured by Ponceau S staining.
Spheroid clearance assay
To measure the ability of HGSOC spheroids to clear a monolayer of ovarian surface epithelial cells, HEYA8-RFP or OVCAR8-RFP cells were plated in 2D adherent conditions (6-well plate, 50,000 cells per well). 24 hours after seeding, the HGSOC cells were treated with either vehicle or CF for 48 hours. Following drug treatment, the cells were trypsinized and plated into 96 well round bottom ULA plates in drug-free media at a density of 500 cells per well. The cells were allowed to form spheroids for 18 to 24 hours. On the same day as sphere formation HIO-80 GFP cells were seeded into a 96 well plate at a density of 40,000 cells per well to form a confluent monolayer. The HGSOC RFP spheroids were then transferred into the 96-well plate containing HIO-80 GFP cells (one spheroid per well) and were allowed to adhere to the monolayer for 24 hours. After 24 hours the wells containing the monolayer with spheroids were washed with 1X PBS 3 times to remove any non-attached spheroids. Attached spheroids and monolayer were then imaged using a Nikon Ti2e inverted fluorescence microscope. OSE clearance was calculated by measuring the GFP negative area within the circumference of the attached spheroid using NIS Elements image analysis software (Nikon).
Spheroid formation assay
To measure spheroid formation, HEYA8-RFP cells were plated in 2D adherent conditions (6-well plate, 50,000 cells per well). 24 hours after seeding, the cells were treated with either vehicle or CF for 48 hours. Following drug treatment, the cells were trypsinized and plated into 96-well round bottom ULA plates in drug-free media at a density of 500 cells per well. The cells were allowed to form spheroids for 18 to 24 hours and then the spheroids were imaged using a Nikon Ti2e inverted fluorescence microscope. The area of each spheroid was measured using NIS Elements image analysis software (Nikon).
Spheroid growth assay
To measure spheroid growth, fluorescently labeled cells were counted then resuspended in media containing CF, Cis, or a 1:1 combination of CF and Cis at the appropriate dose at a density of 100 cells per 0.2 mL of media. 0.2 milliliters of drug + cells media were then added in technical triplicate to the wells of a 96-well ultra-low attachment round bottom plate (Corning, 4515). Spheroids were grown for 7 days and then imaged using a Nikon Ti2e inverted fluorescence microscope. Fluorescence intensity was measured for each spheroid using NIS Elements image analysis software (Nikon). Spheroid fluorescence intensity values for biological replicates (average of technical triplicates) were used to generate dose response curves and IC50 values using GraphPad Prism software.
RNA sequencing and IPA
For RNA sequencing, HEYA8 ovarian cancer cells were plated in 10 cm dishes under adherent conditions at a density of 500,000 cells per well. The next day cells were treated with media containing CF at concentrations of 0, 1, 5, and 10 μM for 48 hours. After drug treatment, the cells were collected, and total RNA was extracted from each sample using the Norgen Total RNA Purification Kit (17200). Samples were prepared in collaboration with Children’s Hospital of Philadelphia Center for Applied Genomics, and only samples with OD260/280 = 1.9 to 2.1 and RNA integrity number (RIN) scores >7 were used for sequencing. Samples were prepared using the TrueSeq Total RNA library (RS-122-2001) with ribosomal depletion. Sequencing was performed using an S2 Flow Cell with 3.3 – 4.1 billion read cluster capacity. Bioinformatics analysis was executed using the R package DESeq2 (version 3.16) and was performed in collaboration with the Translational Molecular Medicine group at Saint John’s Cancer Institute. Significant changes in gene expression were classified as fold change less than/equal to −1.5 or greater than/equal to 1.5 with multiple testing corrected p-value ≤ 0.01. Ingenuity Pathway Analysis (Qiagen) was used to compare samples and identify changes in canonical pathways, diseases & functions, and predicted upstream regulators that occurred in response to treatment with increasing CF dose.
Statistical analysis
Unless otherwise noted, data are presented as mean ± SD from three-independent experiments. All statistical significance tests were carried out on a single factor (e.g. protein expression) compared between two biological groups (e.g. vehicle vs CF treated). Statistical comparisons of one-factor data sets with only two biological groups were performed using student’s t-test. Statistical comparisons of one factor datasets with three or more biological groups were performed using one way ANOVA with Tukey multiple testing correction unless otherwise stated. A p-value < 0.05 was considered statistically significant for both t-test and one way ANOVA. To calculate synergy scores for combination dose-response matrices analyzing cell viability, luminescence values for each drug combination were converted to % inhibition using the formula (combination luminescence – vehicle luminescence/vehicle luminescence) x −100. Combination dose response matrices of % inhibition for N = 3 biological replicates were then uploaded into the SynergyFinder software. Synergy scores for each dose combination were calculated using Bliss/Loewe consensus scoring which combines multiple synergy reference models (Bliss, Loewe, and HSA) with outlier correction turned on56. Synergy scores for clonogenic combination dose-response matrices were generated using % area values calculated by the ColonyArea ImageJ plugin. % Area values for each drug combination were converted to % inhibition using the formula (combination % Area – vehicle % Area/vehicle %Area) x −100. Synergy scores were then calculated with the SynergyFinder program using the same steps as the cell viability analysis described above. Coefficient of Drug Interaction was calculated for each dose combination using the following formula; CDI = AB/(A × B) where AB is the ratio of the two-drug combination luminescence to the vehicle control luminescence, A is the ratio of the single-agent CF luminescence to the vehicle control luminescence, and B is the ratio of the single-agent Cis luminescence to the vehicle control luminescence. CDI <1 indicates synergism, CDI <0.7 significant synergism, CDI = 1 additivity, and CD>1 antagonism.
Tumor formation assays
All in vivo tumor formation experiments were performed under a protocol reviewed and approved by the Penn Institutional Animal Care and Use Committee (Protocol # 806138). To investigate the effects of CF ± Cis treatment on in vivo subcutaneous tumor growth, 40 NOD scid gamma (NSG) mice (Jackson Laboratory, strain no. 005557) were each injected subcutaneously with 1,000,000 HEYA8 cells suspended in a 1:1 mixture of DMEM to Matrigel (Corning, 354234) on the lower right flank. Tumors were allowed to grow to 100 mm3 in size then mice were randomized into four treatment groups: (i) vehicle (PBS)-treated, (ii) CF treated (1 mg/kg), (iii) Cis treated (1 mg/kg), and (iv) combination treated (0.5 mg/kg CF, 0.5 mg/kg Cis). Each treatment group was dosed 3X per week (Mon/Wed/Fri) via intratumoral injection for the duration of the study. Tumor volume was measured using calipers every 7 days. All mice were euthanized on day 50 post-randomization when the majority (6 out of 10) of vehicle-treated mice either had tumors >2000 mm3 or had been euthanized for animal welfare reasons.
For intraperitoneal tumor formation, athymic nude mice were used (Jackson Labs, strain # 007850). All mice were each injected intraperitoneally with 1,000,000 luciferized OVCAR4 cells. Intraperitoneal tumors were allowed to grow to ≥1 × 106 photons per second (p/s) in size, and then mice were randomized into four treatment groups as above for subcutaneous tumor assays. Each treatment group was dosed every 3 days via IP injection for the duration of the study. Tumor volume was measured using bioluminescence imaging every 7 days. All mice were euthanized for animal welfare reasons as they became moribund with tumor burden.
Supplementary Material
Acknowledgements:
We thank Joe Salvino and Christine Eischen for critical review of the manuscript. We thank the Saint John’s Cancer Institute sequencing center for providing RNA sequencing services. We thank Mei Zheng for assistance with histology.
Funding:
This study was supported by the National Institutes of Health P50 SPORE CA228991 (RD), the Department of Defense W81XWH-21-1-0643 (MK), W81XWH-22-1-0852 (RD), the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (RD, DH), the Honorable Tina Brozman Foundation for Ovarian Cancer Research (RD), the Claneil Foundation (RD), the Gray Foundation (RD), the Carl H. Goldsmith Ovarian Cancer Translational Research Fund (RD), the Marjorie S. Stanek and Lowell H. Dubrow Ovarian Cancer Research Center Endowed Fund (RD), the Mike and Patti Hennessy Foundation (RD), the Monica K. Young Foundation (RD), Susan and David Wolk (RD), and the Basser Center for BRCA (MK, RD).
Footnotes
Competing interests: RD serves on the scientific advisory board of VOC Health and Repare Therapeutics. The other authors declare that they have no competing interests.
Data and materials availability:
The RNA-seq dataset (GSE) have been deposited in GEO, accession ID GSE280516. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Cancer Tomorrow. https://gco.iarc.fr/tomorrow/en/dataviz/trends?types=0_1&sexes=2&mode=population&group_populations=0&multiple_populations=1&multiple_cancers=0&cancers=25&populations=900.
- 2.Karst AM & Drapkin R Ovarian cancer pathogenesis: a model in evolution. J Oncol 2010, 932371 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lheureux S, Gourley C, Vergote I & Oza AM Epithelial ovarian cancer. The Lancet 393, 1240–1253 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Torre LA et al. Ovarian cancer statistics, 2018. CA Cancer J Clin 68, 284–296 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aghajanian C et al. Final overall survival and safety analysis of OCEANS, a phase 3 trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent ovarian cancer. Gynecol Oncol 139, 10–16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perren TJ et al. A Phase 3 Trial of Bevacizumab in Ovarian Cancer. New England Journal of Medicine 365, 2484–2496 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Burger RA et al. Incorporation of Bevacizumab in the Primary Treatment of Ovarian Cancer. New England Journal of Medicine 365, 2473–2483 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Maxwell KN & Domchek SM Cancer treatment according to BRCA1 and BRCA2 mutations. Nature Reviews Clinical Oncology vol. 9 520–528 Preprint at 10.1038/nrclinonc.2012.123 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Mirza MR et al. The forefront of ovarian cancer therapy: update on PARP inhibitors. Annals of oncology : official journal of the European Society for Medical Oncology Preprint at 10.1016/j.annonc.2020.06.004 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Kurnit KC, Avila M, Hinchcliff EM, Coleman RL & Westin SN PARP inhibition in the ovarian cancer patient: Current approvals and future directions. Pharmacology and Therapeutics vol. 213 107588 Preprint at 10.1016/j.pharmthera.2020.107588 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rossi L et al. Bevacizumab in ovarian cancer: A critical review of phase III studies. Oncotarget vol. 8 12389–12405 Preprint at 10.18632/oncotarget.13310 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tewari KS et al. Final overall survival of a randomized trial of bevacizumab for primary treatment of ovarian cancer. Journal of Clinical Oncology 37, 2317–2328 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sapio L et al. The Natural cAMP Elevating Compound Forskolin in Cancer Therapy: Is It Time? J Cell Physiol 232, 922–927 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Bhat SV, Bajqwa BS, Dornauer H, do Scusa NJ & Fehlhaber HW Structures and stereochemistry of new labdane diterpiniods from coleus forskohlii briq. Tetrahedron Lett 18, 1669–1672 (1977). [Google Scholar]
- 15.Kavitha C, Rajamani K & Vadivel E Coleus forskohlii - a comprehensive review on morphology, phytochemistry and pharmacological aspects. Journal of Medicinal Plants Research (2010). [Google Scholar]
- 16.Seamon KB, Padgett W & Daly JW Forskolin: unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proceedings of the National Academy of Sciences 78, 3363–3367 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pinto C et al. Activation and Inhibition of Adenylyl Cyclase Isoforms by Forskolin Analogs. Journal of Pharmacology and Experimental Therapeutics 325, 27–36 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Laurenza A, Sutkowski EMH & Seamon KB Forskolin: a specific stimulator of adenylyl cyclase or a diterpene with multiple sites of action? Trends Pharmacol Sci 10, 442–447 (1989). [DOI] [PubMed] [Google Scholar]
- 19.Sassone-Corsi P The Cyclic AMP Pathway. Cold Spring Harb Perspect Biol 4, a011148 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan K, Gao LN, Cui YL, Zhang Y & Zhou X The cyclic AMP signaling pathway: Exploring targets for successful drug discovery (review). Mol Med Rep 13, 3715–3723 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kilanowska A, Ziółkowska A, Stasiak P & Gibas-Dorna M cAMP-Dependent Signaling and Ovarian Cancer. Cells 2022, Vol. 11, Page 3835 11, 3835 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piontek M, Hengels KJ, Porschen R & Strohmeyer G Protein kinase C and adenylate cyclase as targets for growth inhibition of human gastric cancer cells. J Cancer Res Clin Oncol 119, 697–699 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gützkow KB, Naderi S & Blomhoff HK Forskolin-mediated G1 arrest in acute lymphoblastic leukaemia cells: phosphorylated pRB sequesters E2Fs. J Cell Sci 115, 1073–1082 (2002). [DOI] [PubMed] [Google Scholar]
- 24.McEwan DG et al. Chemoresistant KM12C Colon Cancer Cells Are Addicted to Low Cyclic AMP Levels in a Phosphodiesterase 4–Regulated Compartment via Effects on Phosphoinositide 3-Kinase. Cancer Res 67, 5248–5257 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Agarwal KC & Parks RE Forskolin: A potential antimetastatic agent. Int J Cancer 32, 801–804 (1983). [DOI] [PubMed] [Google Scholar]
- 26.Burdyga A et al. cAMP inhibits migration, ruffling and paxillin accumulation in focal adhesions of pancreatic ductal adenocarcinoma cells: Effects of PKA and EPAC. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1833, 2664–2672 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ou Y et al. Activation of cyclic AMP/PKA pathway inhibits bladder cancer cell invasion by targeting MAP4-dependent microtubule dynamics. Urologic Oncology: Seminars and Original Investigations 32, 47.e21-47.e28 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Dong H, Claffey KP, Brocke S & Epstein PM Inhibition of breast cancer cell migration by activation of cAMP signaling. Breast Cancer Res Treat 152, 17–28 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Pattabiraman DR et al. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science (1979) 351, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mann SC, Andrews PA & Howell SB Modulation of cis-diamminedichloroplatinum(II) accumulation and sensitivity by forskolin and 3-isobutyl-1-methylxanthine in sensitive and resistant human ovarian carcinoma cells. Int J Cancer 48, 866–872 (1991). [DOI] [PubMed] [Google Scholar]
- 31.Warrenfeltz SW, Lott SA, Palmer TM, Gray JC & Puett D Luteinizing Hormone–Induced Up-Regulation of ErbB-2 Is Insufficient Stimulant of Growth and Invasion in Ovarian Cancer Cells. Molecular Cancer Research 6, 1775–1785 (2008). [DOI] [PubMed] [Google Scholar]
- 32.D’Souza T, Agarwal R & Morin PJ Phosphorylation of Claudin-3 at Threonine 192 by cAMP-dependent Protein Kinase Regulates Tight Junction Barrier Function in Ovarian Cancer Cells. Journal of Biological Chemistry 280, 26233–26240 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Kamrava M et al. Lysophosphatidic acid and endothelin-induced proliferation of ovarian cancer cell lines is mitigated by neutralization of granulin–epithelin precursor (GEP), a prosurvival factor for ovarian cancer. Oncogene 2005 24:47 24, 7084–7093 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Takahashi A et al. Induction of senescence by progesterone receptor-B activation in response to cAMP in ovarian cancer cells. Gynecol Oncol 113, 270–276 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Shaw TJ, Keszthelyi EJ, Tonary AM, Cada M & Vanderhyden BC Cyclic AMP in Ovarian Cancer Cells Both Inhibits Proliferation and Increases c-KIT Expression. Exp Cell Res 273, 95–106 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Gong S et al. Roflumilast enhances cisplatin-sensitivity and reverses cisplatin-resistance of ovarian cancer cells via cAMP/PKA/CREB-FtMt signalling axis. Cell Prolif 51, e12474 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mistarz A et al. Induction of cell death in ovarian cancer cells by doxorubicin and oncolytic vaccinia virus is associated with CREB3L1 activation. Mol Ther Oncolytics 23, 38–50 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu P et al. GANT61 elevates chemosensitivity to cisplatin through regulating the Hedgehog, AMPK and cAMP pathways in ovarian cancer. Future Med Chem 14, 479–500 (2022). [DOI] [PubMed] [Google Scholar]
- 39.Dimitrova N et al. InFlo: a novel systems biology framework identifies cAMP-CREB1 axis as a key modulator of platinum resistance in ovarian cancer. Oncogene 2017 36:17 36, 2472–2482 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tong L, Wang Y, Ao Y & Sun X CREB1 induced lncRNA HAS2-AS1 promotes epithelial ovarian cancer proliferation and invasion via the miR-466/RUNX2 axis. Biomedicine & Pharmacotherapy 115, 108891 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Godard MP, Johnson BA & Richmond SR Body Composition and Hormonal Adaptations Associated with Forskolin Consumption in Overweight and Obese Men. Obes Res 13, 1335–1343 (2005). [DOI] [PubMed] [Google Scholar]
- 42.The Effect of Coleus Forskohlii Extract on the Risk Factors of Metabolic Syndrome - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02143349. [Google Scholar]
- 43.González-Sánchez R et al. Forskolin versus Sodium Cromoglycate for Prevention of Asthma Attacks: A Single-blinded ClinicalTrials.gov. 10.1177/147323000603400210 34, 200–207 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Bauer K, Dietersdorfer F, Sertl K, Kaik B & Kaik G Pharmacodynamic effects of inhaled dry powder formulations of fenoterol and colforsin in asthma. Clin Pharmacol Ther 53, 76–83 (1993). [DOI] [PubMed] [Google Scholar]
- 45.Wagh VD, Patil PN, Surana SJ & Wagh KV Forskolin: Upcoming antiglaucoma molecule. J Postgrad Med 58, 199 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Retinal Nerve Fibres Layers Thickness Study in Glaucomatous Patients - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/study/NCT01254006. [Google Scholar]
- 47.Khandelwal Y et al. Articles. J Med Chem 31, 1872–1879 (1988). [DOI] [PubMed] [Google Scholar]
- 48.Suzuki S, Ito O, Sayama T & Goto K Intra-arterial colforsin daropate for the treatment of cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Neuroradiology 52, 837–845 (2010). [DOI] [PubMed] [Google Scholar]
- 49.Hayashida N et al. Antiinflammatory effects of colforsin daropate hydrochloride, a novel water-soluble forskolin derivative. Ann Thorac Surg 71, 1931–1938 (2001). [DOI] [PubMed] [Google Scholar]
- 50.Kikura M, Morita K & Sato S Pharmacokinetics and a simulation model of colforsin daropate, new forskolin derivative inotropic vasodilator, in patients undergoing coronary artery bypass grafting. Pharmacol Res 49, 275–281 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Nagaraj AB et al. Critical role of Wnt/β-catenin signaling in driving epithelial ovarian cancer platinum resistance. Oncotarget 6, 23720–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yee C, Dickson KA, Muntasir MN, Ma Y & Marsh DJ Three-Dimensional Modelling of Ovarian Cancer: From Cell Lines to Organoids for Discovery and Personalized Medicine. Front Bioeng Biotechnol 10, 116 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shield K, Ackland ML, Ahmed N & Rice GE Multicellular spheroids in ovarian cancer metastases: Biology and pathology. Gynecol Oncol 113, 143–148 (2009). [DOI] [PubMed] [Google Scholar]
- 54.Iwanicki MP et al. Mutant p53 regulates ovarian cancer transformed phenotypes through autocrine matrix deposition. JCI Insight 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ianevski A, Giri AK & Aittokallio T SynergyFinder 3.0: an interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res 50, W739–W743 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim H et al. Targeting the ATR/CHK1 axis with PARP inhibition results in tumor regression in BRCA-mutant ovarian cancer models. Clinical Cancer Research 23, 3097–3108 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang L et al. CombPDX: a unified statistical framework for evaluating drug synergism in patient-derived xenografts. Scientific Reports 2022 12:1 12, 1–10 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hao P et al. Eukaryotic translation initiation factors as promising targets in cancer therapy. Cell Communication and Signaling 18, 1–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reyes-González JM & Vivas-Mejía PE c-MYC and Epithelial Ovarian Cancer. Front Oncol 11, 524 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Musa J et al. Eukaryotic initiation factor 4E-binding protein 1 (4E-BP1): a master regulator of mRNA translation involved in tumorigenesis. Oncogene 2016 35:36 35, 4675–4688 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Elia A et al. Implication of 4E-BP1 protein dephosphorylation and accumulation in pancreatic cancer cell death induced by combined gemcitabine and TRAIL. Cell Death & Disease 2017 8:12 8, 1–11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Y et al. 4EBP1 senses extracellular glucose deprivation and initiates cell death signaling in lung cancer. Cell Death & Disease 2022 13:12 13, 1–13 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fingar DC et al. mTOR Controls Cell Cycle Progression through Its Cell Growth Effectors S6K1 and 4E-BP1/Eukaryotic Translation Initiation Factor 4E. 10.1128/MCB.24.1.200-216.2004 24, 200–216 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pourdehnad M et al. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc Natl Acad Sci U S A 110, 11988–11993 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Makhija S, Taylor DD, Gibb RK & Gerçel-Taylor Ç Taxol-induced Bcl-2 phosphorylation in ovarian cancer cell monolayer and spheroids. Int J Oncol 14, 515–521 (1999). [DOI] [PubMed] [Google Scholar]
- 66.L’Espérance S, Bachvarova M, Tetu B, Mes-Masson AM & Bachvarov D Global gene expression analysis of early response to chemotherapy treatment in ovarian cancer spheroids. BMC Genomics 9, 1–21 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Turi Z, Lacey M, Mistrik M & Moudry P Impaired ribosome biogenesis: mechanisms and relevance to cancer and aging. Aging 11, 2512–2540 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stedman A et al. Ribosome biogenesis dysfunction leads to p53-mediated apoptosis and goblet cell differentiation of mouse intestinal stem/progenitor cells. Cell Death & Differentiation 2015 22:11 22, 1865–1876 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen FW & Ioannou YA Ribosomal Proteins in Cell Proliferation and Apoptosis. 10.3109/08830189909088492 18, 429–448 (2009). [DOI] [PubMed] [Google Scholar]
- 70.Lindström MS, Bartek J & Maya-Mendoza A p53 at the crossroad of DNA replication and ribosome biogenesis stress pathways. Cell Death & Differentiation 2022 29:5 29, 972–982 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lessard F et al. Senescence-associated ribosome biogenesis defects contributes to cell cycle arrest through the Rb pathway. Nature Cell Biology 2018 20:7 20, 789–799 (2018). [DOI] [PubMed] [Google Scholar]
- 72.Llombart V & Mansour MR Therapeutic targeting of “undruggable” MYC. EBioMedicine 75, 103756 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zeng M et al. Targeting MYC dependency in ovarian cancer through inhibition of CDK7 and CDK12/13. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guzmán C, Bagga M, Kaur A, Westermarck J & Abankwa D ColonyArea: An ImageJ Plugin to Automatically Quantify Colony Formation in Clonogenic Assays. PLoS One 9, e92444 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hooda J et al. Early loss of histone H2B monoubiquitylation alters chromatin accessibility and activates key immune pathways that facilitate progression of ovarian cancer. Cancer Res 79, 760–772 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schmidt EK, Clavarino G, Ceppi M & Pierre P SUnSET, a nonradioactive method to monitor protein synthesis. Nature Methods 2009 6:4 6, 275–277 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq dataset (GSE) have been deposited in GEO, accession ID GSE280516. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.