

Abstract
CD163, a macrophage-specific receptor, plays a critical role in scavenging hemoglobin released during hemolysis, protecting against oxidative effects of heme iron. In the bloodstream, hemoglobin is bound by haptoglobin, leading to its immediate endocytosis by CD163. While haptoglobin’s structure and function are well understood, CD163’s structure and its interaction with the haptoglobin-hemoglobin complex have remained elusive. Here, we present the cryo-electron microscopy structure of the entire extracellular domain of human CD163 in complex with haptoglobin-hemoglobin. The structure reveals that CD163 assembles into trimers (and to some extent dimers), binding haptoglobin-hemoglobin in their center. Key acidic residues in CD163 interact with lysine residues from both haptoglobin and hemoglobin. Calcium-binding sites located near the haptoglobin-hemoglobin interface in CD163 provide explanation for the calcium dependence of the interaction. Furthermore, we show that the interaction facilitating CD163 oligomerization mimics ligand binding and is also calcium dependent. This structural insight into CD163 advances our understanding of its role in hemoglobin scavenging as well as its broader relevance to structurally related scavenger receptors.
Subject terms: Cryoelectron microscopy, Blood proteins, Acute inflammation, Protein translocation
CD163, a macrophage receptor, is essential for clearing hemoglobin during hemolysis to prevent oxidative damage. Here, the authors reveal the cryo-electron microscopy structure of CD163 bound to haptoglobin-hemoglobin, uncovering calcium-dependent interactions critical for its function and oligomerization.
Introduction
Hemoglobin (Hb) is important for gas transport (essentially O2, but also CO2, CO and NO), but it can be hazardous if leaking extensively from the red blood cells. This is due to the reactive heme iron, which has the potential to generate reactive oxygen species1,2. Normally, the Hb scavenging system, comprising the acute-phase protein haptoglobin (Hp) and the macrophage-specific receptor CD163, efficiently removes Hb from circulation, thus preventing oxidative damage3,4. However, in various hemolytic conditions (such as in malaria or sickle cell disease) this system can become overloaded, allowing Hb-induced radicals to cause oxidative damage to cells and tissues, in particular the endothelium and kidneys5,6.
Hp serves as the first line of defense against the toxic Hb. It strongly interacts with Hb dimers (αβHb), providing structural stabilization to Hb and shielding against its oxidative reactions7–9. The haptoglobin-hemoglobin complexes (HpHb) formed are immediately recognized by the macrophage-specific receptor CD163 and endocytosed10. Apparently, the binding of αβHb and the endocytosis of the HpHb are only minimally affected by the human Hp phenotypes, which constitute a diverse set of Hp oligomers10–12. These include dimers (Hp1-1 and Hp1-2 phenotypes), circular forms (Hp1-2 and Hp2-2 phenotypes), and linear forms (Hp1-2 phenotypes)3,13. The concentration of Hp is lower in individuals with the Hp2-2 phenotype compared to those with the Hp1-1 and Hp1-2 phenotypes, making them more susceptible to Hb-related oxidative stress14. Within the macrophages, Hp and Hb are degraded by the lysosomal proteases. The released heme is converted into bilirubin, which is secreted and transported to the liver for conjugation and excretion. Hb can also be taken up by CD163 in the absence of Hp, albeit less effectively due to a much lower affinity for CD16315. This pathway might serve as a secondary mechanism during severe hemolysis, where Hp is depleted. Additionally, CD163 plays a role in inflammation regulation concordant with its expression in the anti-inflammatory macrophage subtypes16. Several studies indicate an overall anti-inflammatory effect of CD163. In addition, proinflammatory activation of macrophages downregulates the receptor expression by various means. In humans, this regulation includes shedding and formation of a soluble form of CD163 present in the blood plasma17.
CD163 is composed of nine consecutive Scavenger Repeat Cysteine-Rich (SRCR) domains, followed by a transmembrane segment and a short cytoplasmic tail, which includes a functional internalization motif18,19. Each SRCR domain is a compact and spherical domain of around 110 residues, characterized by a highly conserved structure comprising two β-sheets encasing a single α-helix20. In humans, the SRCR protein family consists of over 20 members, which can be either membrane-bound or secreted proteins. Many function as membrane receptors, where the SRCR domains play a key role in binding to ligands21. In addition to CD163, other notable members of this family include CD163L1, CD6, Mac-2 binding protein, MARCO, Scavenger receptor A1, and SPα. Calcium ions are crucial in the SRCR protein family, facilitating both structural stability and ligand binding, as evidenced in proteins like CD163 and DMBT122,23. Three distinct Ca2+-sites have been identified in structures of SRCR domains23,24. Two sites (denoted site 1 and site 2) are located close to each other with an aspartate residue positioned for coordination of Ca2+-ions at both sites. A third site (denoted site 3) distal to the central α-helix is comprised by two aspartate residues. In addition, mainchain carbonyls participate in the coordination of the Ca2+-ion at site 3.
Ca²⁺-ions are well-established as essential mediators of ligand binding and release in endocytic receptors, such as the Low-Density Lipoprotein Receptor (LDLR)25 and cubilin26. Despite being structurally unrelated, these receptors have evolved to bind ligands through similar mechanisms27. In this process, acidic residues from the receptors are coordinated by Ca2+-ions, positioning them for interaction with positively charged residues from the ligand. Another common feature of these receptors is that multiple low-affinity interactions within the ligand combine to create a high-affinity binding. Ca²⁺ is also critical for HpHb binding to CD16319, and we previously hypothesized that CD163 might utilize a similar ligand-binding mechanism22.
In this study, we present the structure of the human CD163 in complex with HpHb, elucidating both the structural organization of CD163 and the underlying principles of its Ca2+-dependent ligand binding. Our study revealed that CD163 oligomerizes and interacts with both Hp and Hb of the barbell-shaped HpHb complex. The binding of Ca2+-ions to CD163 is crucial not only for ligand binding but also for receptor oligomerization.
Results
Analysis of CD163 oligomerization using multi-angle light scattering
To investigate the oligomerization of CD163, we analyzed two CD163 fragments using size-exclusion chromatography in-line with multi-angle light scattering (SEC-MALS): one encompassing CD163 SRCR1-9 and a shorter fragment comprising CD163 SRCR1-5. The chromatogram for CD163 SRCR1-9 displays a broad peak with molecular weights ranging from 130 kDa to nearly 300 kDa (Fig. 1a). This suggests that CD163 SRCR1-9 (expected molecular weight: 130 kDa) exists in a dynamic equilibrium of monomers, dimers and even trimers, in solution. An identical analysis conducted in the presence of 10 mM EDTA resulted in a single monodisperse peak with a molecular weight of around 130 kDa (Fig. 1a), indicating that the observed oligomerization is dependent on Ca2+ ions. In contrast, CD163 SRCR1-5 (expected molecular weight: 65 kDa) predominantly exists in a monomeric form, though a small fraction seems to form a dimer (Fig. 1b). The peak corresponding to the dimeric CD163 SRCR1-5 is partly reduced in the presence of 10 mM EDTA, suggesting a potential Ca2+-dependence for this dimerization as well.
Fig. 1. CD163 oligomerization and HpHb binding.
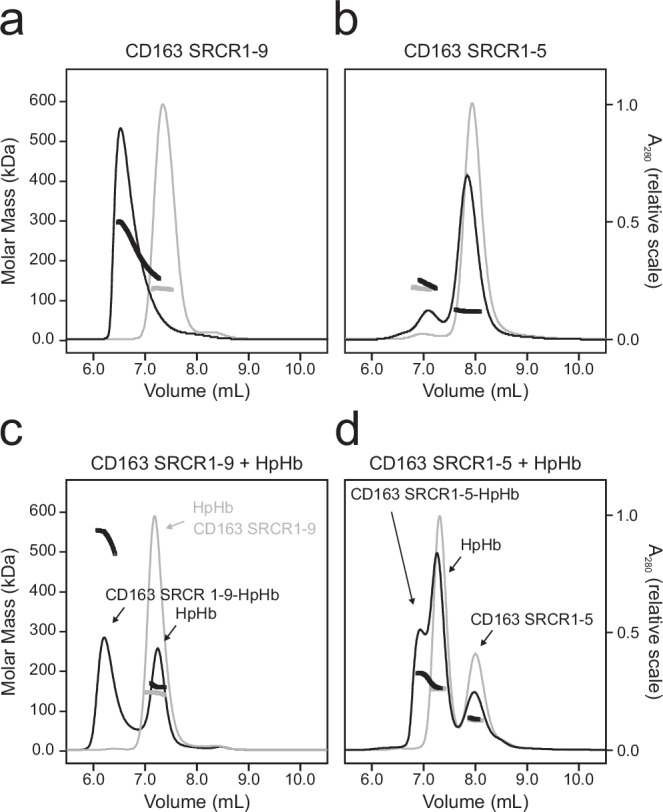
a + b SEC-MALS analysis of CD163 SRCR 1-9 (a) and CD163 SRCR 1-5 (b) in the presence of 5 mM CaCl2 (black lines) or 10 mM EDTA (grey lines). c + d SEC-MALS analysis of CD163 SRCR 1-9 + HpHb (c) and CD163 SRCR 1-5 + HpHb (d) in the presence of 5 mM CaCl2 (black lines) or 10 mM EDTA (grey lines). The measured molecular weights of the chromatogram peaks are shown as thick lines. Theoretical molecular weights (excluding carbohydrates and prosthetic groups): CD163 SRCR 1-9: 105 kDa, CD163 SRCR 1-5: 58 kDa, HpHb: 144 kDa.
Further experiments were conducted to determine the molar masses of complexes formed when HpHb (expected molecular weight: 170 kDa) was added to both CD163 SRCR1-9 and CD163 SRCR1-5 (Fig. 1c, d). The CD163 SRCR1-9-HpHb complex peaks at a maximal molar mass of approximately 530 kDa, which may correspond to a trimer of CD163 SRCR1-9 in complex with HpHb (Fig. 1c). The peak containing CD163 SRCR1-9-HpHb is not monodisperse and the MALS measurements indicate the presence of smaller complexes. When 10 mM EDTA is added, the resulting peak comprises both CD163 SRCR1-9 and HpHb with a molecular weight of approximately 150 kDa corresponding to the average of the two individual components. This implies that although they elute together, they do not form a complex in the presence of EDTA. CD163 SRCR1-5 and HpHb form a complex with an estimated molecular weight of approximately 200 kDa, which may correspond to HpHb bound to one or two CD163 SRCR 1-5. This interaction is also abolished in the presence of EDTA (Fig. 1d). These findings corroborate previous observations that the recognition of HpHb by CD163 is mediated by the SRCR1-5 domains and is Ca2+-dependent19. Specifically, these studies have shown that 2 mM Ca2+ is sufficient for the complete binding of HpHb to CD163, with a decrease in binding observed at concentrations of 1 mM Ca2+ or lower. Our results further demonstrate that CD163 SRCR1-9 oligomerization is also Ca2+-dependent. Additionally, we observe a similar Ca2+ dependence for CD163 oligomerization as seen with ligand binding (Supplementary Fig. 1), indicating that CD163 oligomerization occurs at physiological Ca2+ concentrations of around 1-2 mM.
Cryo-EM structure determination of CD163 SRCR1-9-HpHb
To gain insights into the structure of CD163 and its interaction with HpHb, we determined the structure of the CD163 ectodomain (SRCR1-9) in complex with HpHb (αβHb bound to dimeric Hp1-1) using single particle cryo-electron microscopy (Supplementary Fig. 2, Supplementary Table 1). Two-dimensional class averages and three-dimensional classification revealed that the purified CD163 SRCR1-9-HpHb complex is a heterogeneous mixture, which includes, dimeric CD163 SRCR1-9 bound to HpHb (denoted as CD1632-HpHb) and trimeric CD163 SRCR1-9 bound to HpHb (denoted as CD1633-HpHb). In our three-dimensional reconstruction, CD1632-HpHb refines to a resolution at around 4.5 Å, while the CD1633-HpHb reached around 5.2 Å (Fig. 2, Supplementary Figs. 2, 3). Applying focused refinement on CD1632-HpHb with a mask covering the serine protease domain of Hp (HpSP), Hb and SRCR2-4 of one CD163 subunit, the resolution improved to 3.8 Å (Fig. 2, Supplementary Figs. 2, 3). Determining the structure of the CD163-HpHb complex proved challenging due to several factors, including the presence of heterogeneous complexes, inherent flexibility, and preferred orientations, all of which contributed to lower-than-expected resolutions for a sample of this size. Surprisingly, the CD1632-HpHb complex yielded slightly higher resolution than the CD1633-HpHb complex. It is possible that the flexible nature of CD163 complicates 3D reconstruction, and that reducing the number of CD163 domains included in the refinement process enhances the resolution obtained.
Fig. 2. Cryo-EM densities of CD163-HpHb.
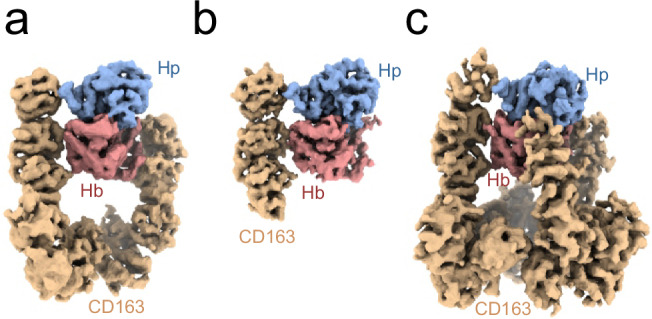
a Cryo-EM density map of dimeric CD163 SRCR 1-9 bound to HpHb (4.5 Å resolution). b Cryo-EM density map from focused refinement of the map shown in a using a mask covering CD163-A SRCR 2-4, Hp and Hb (3.8 Å resolution). c Cryo-EM density map of trimeric CD163 SRCR 1-9 bound to HpHb (5.2 Å resolution). Density for CD163 is shown in orange, Hp in blue and Hb in red.
We docked the structure of human HpHb (RCSB ID: 4WJG) and the AlphaFold228 prediction of human CD163 SRCR1-9 into the densities, followed by rigid-body refinement of individual domains. Generally, the models fitted well with the electron densities, and the linkers between adjacent SRCR domains were unambiguously traceable. A notable exception was the extended linker between SRCR6 and SRCR7, where the density was not well-defined. We observe densities for three N-linked glycosylations at consensus sites in CD163 SRCR3, SRCR7, SRCR9 (Supplementary Fig. 4a–c), confirming the positioning and docking of these domains. The absence of a well-defined density for the CD163 SRCR1 domain in any of our reconstructions led to its exclusion from our models. Additionally, density for the CD163 SRCR2 domain was only observed in one of the three CD163 subunits. These domains are likely not involved in ligand binding and are therefore free to move relative to other regions of CD163, resulting in weaker density.
The electron density map, derived from focused refinement, exhibited sufficient quality to allow for model building and real-space refinement. Within this map, clear and distinct electron densities corresponding to two calcium ions (Ca2+) were observed, and these ions were subsequently incorporated into the structural model (Supplementary Fig. 4d, e).
Structure of CD163
The structure of CD163 SRCR2-9 displays an elongated shape with an approximate length of 160 Å (Fig. 3a). It comprises two distinct segments: a rod-shaped segment formed by SRCR2-4 and a globular base constituted by SRCR5-9. Although the individual SRCR domains of CD163 share structural similarities, there is no clear pattern in the orientation of adjacent SRCR domains. As a result, these domains display a variety of packing arrangements relative to each other. Generally, the interfaces between adjacent SRCR domains are of moderate size and we observed that the electron densities for SRCR domains not directly involved in ligand-binding are less well-defined. This suggests an inherent flexibility within CD163 that could allow for adaptable interactions to accommodate ligand binding. A notable exception is observed at the interface between the ligand-binding SRCR3 and SRCR4, which is significantly larger than the others. In this interface, the β-sheets of SRCR3 and SRCR4 merge, forming an extended β-sheet that spans across the domain interface (Fig. 3b). This fusion likely results in a rigid two-domain architecture potentially important for placing the SRCR domains in optimal position relative to each other for ligand binding.
Fig. 3. Structure of CD163 SRCR 1-9 and Ca2+-binding sites.

a Cartoon representation of CD163 SRCR 1-9 (subunit A). No well-defined density was obtained for SRCR1 and the linker connecting SRCR6 and SRCR7 (represented by a dashed line). N-linked glycans and disulfides are shown as sticks. Ca2+-ions are shown as grey spheres. b Interface between SRCR3 and SRCR4 formed by fusion of two β-sheets. Dashed lines represents hydrogen bonds. c Comparison of the cryo-EM structure of CD163 (orange) with the crystal structure of porcine CD163 SRCR 5-9 (grey, RCSB ID: 8H7J). The structure of the linker region connecting SRCR6 and SRCR7 is shown in the box. d Sequence alignment of Ca2+-binding regions of individual SRCR domains from CD163. Consensus Ca2+-coordinating residues are shown above the alignment, with site 1 residues in magenta, site 2 residues in blue and site 3 residues green. The aspartate residues bridging site 1 and site 2 are shown in black. The boxed residues represent occupied Ca2+-binding sites identified in CD163 SRCR2 and SRCR3. e + f Cartoon representation of CD163 SRCR 2 (e) and SRCR 3 (f). Residues located at the canonical Ca2+-binding sites are shown in sticks. Ca2+-ions are shown as grey spheres.
CD163 SRCR5-9 are organized into a hairpin-like formation (Fig. 3c). Specifically, SRCR5 interacts with SRCR9, and SRCR6 with SRCR8, positioning SRCR7 at the tip of this arrangement. The extended linker connecting SRCR6 and SRCR7 contains two cysteine residues that form a disulfide bond, likely stabilizing this otherwise flexible segment. A recent crystal structure of porcine CD163 SRCR5-9 (RCSB ID 8H7J), corroborates the configuration of CD163 SRCR5-9 observed in the cryo-EM structure and in the Alphafold prediction of CD163 (Fig. 3c). The crystal structure also discloses the structure of the extended linker-region connecting SRCR6 and SRCR7, that is not traceable in the cryo-EM analysis and confirms formation of the disulfide bond.
Potential Ca2+-sites are located in SRCR2, SRCR3, SRCR6, SRCR7, and SRCR9 (Fig. 3d). The resolution of the reconstruction covering SRCR5-9 does not allow unambiguous determination of absence or presence of Ca2+-ions. However, our data shows clear density for Ca2+-ions bound at CD163 SRCR2-3 – one at site 2 of SRCR2 and one at site 3 of SRCR3 (Fig. 3d–f and Supplementary Fig. 4d, e). At SRCR2 site 2, the Ca2+-ion is coordinated by Asp186, which can participate in the coordination of Ca2+-ions at both site 1 and site 2 (Fig. 3e). The Ca2+-ion at site 2 is additionally coordinated by Asp224, Asp225, and Asn247. At site 3 of SRCR3, which is located at the end of the central α-helix, the Ca2+-ion is coordinated by Asp296 and Asp299 (Fig. 3f). In addition, the mainchain carbonyls of Gly294 and Val333 participate in coordinating the Ca2+-ion. Although the consensus sequence of site 1 is found in both SRCR2 and SRCR3 of CD163, we do not see any indications of this site being occupied in either of the two domains (Supplementary Fig. 4d, e).
CD163 SRCR1-9 harbors five predicted N-linked glycosylation sites29. Two of these are located at Asn105 and Asn140 in SRCR1 and consequently their presence cannot be verified since well-defined density is not obtained for this domain. At each of the three other sites, Asn320 in SRCR3, Asn767 in SRCR7, and Asn1027 in SRCR9, a single N-acetylgalactosamine is modelled (Supplementary Fig. 4). The N-linked glycosylation in SRCR3, is located opposite the ligand-binding site, protruding into the extracellular space. The two N-linked glycosylation sites in SRCR7 and SRCR9 are both facing the transmembrane region and could serve a function in protecting the receptor from unspecific cleavage by membrane-associated proteases.
CD163 1-9 oligomerizes via SRCR7 and SRCR9 interaction
The trimeric structure of CD163 SRCR1-9 resembles an inverted tripod, with the ligand-binding regions (SRCR2-4) forming elongated, rod-like extensions that project into the extracellular space, enclosing a cavity opposite the triangular base formed by CD163 SRCR5-9 (Fig. 4b). The base is connected to the transmembrane helix of each of the three CD163 subunits through a linker region comprising 21 residues. Notably, in humans this region includes a TACE/ADAM17 cleavage site, crucial for the shedding of CD163’s entire ectodomain30. Although CD163 can form a homotrimer, its ligand-bound state does not exhibit perfect three-fold symmetry. This asymmetry may be an adaptive feature of the flexible, multi-ligand receptor CD163, allowing it to accommodate various structurally diverse ligands within its central binding region. Consequently, the binding of the asymmetric HpHb complex may induce conformational asymmetry in CD163.
Fig. 4. Trimerization of CD163 SRCR 1-9.

a Cartoon representation of trimeric CD163 SRCR 1-9 bound to HpHb. The three CD163 subunits are shown in shades of orange, Hp SP domain in blue and αβHb dimer in red. The entire HpHb complex is superimposed on Hp SP and shown in grey. b Cartoon representation of the base of the CD163 trimer formed by SRCR 7-9. The interfaces between adjacent CD163 subunits are formed by SRCR 7 from one CD163 subunit and SRCR 9 from another subunit. c Close up on the SRCR 7/SRCR 9 interface (boxed area in b). Residues proposed to be responsible for the interaction are shown as sticks. d SEC-MALS analysis of the CD163 SRCR 1-9 K811A/K2012A double mutant in the presence of 5 mM CaCl2 (black lines) or 10 mM EDTA (grey lines). A chromatogram of wild-type CD163 SRCR 1-9 is shown with dashes for comparison.
The trimeric assembly of the CD163 subunits is facilitated through specific intermolecular interactions. Each CD163 subunit utilizes its SRCR7 domain to engage with the SRCR9 domain of an adjacent CD163 molecule (Fig. 4b). This pattern of interaction is found between all three subunits of CD163, resulting in the formation of the triangular base. The interaction between SRCR7 and SRCR9 occurs in a two-fold symmetric arrangement, where their potential Ca2+-binding regions are oriented towards each other (Fig. 4c). In this configuration, Lys811 from SRCR7 is positioned to engage in electrostatic interactions with Asp956 and Glu1022 from SRCR9. Similarly, Lys1021 from SRCR9 is positioned for potential electrostatic interactions with Asp746 and Glu812 of SRCR7, mirroring the interaction pattern observed in the former pair. This indicates that Lys811 and Lys1021 are important for the trimerization of CD163. Hence, we mutated both Lys811 and Lys1021 to alanine and analyzed the oligomerization of CD163 using SEC-MALS (Fig. 4d). Here, we observe a clear effect of the mutagenesis with complete disruption of CD163 oligomerization. Furthermore, mutation of Asp746 and Asp956 to alanine shows a similar result (Supplementary Fig. 5), supporting that the electrostatic interactions between Lys811/Lys1021 and Asp956/A746 is important for CD163 oligomerization.
CD163-HpHb interaction
All three subunits of the CD163 trimer form contacts with HpHb. However, it is only one of the CD163 subunits (CD163A) that simultaneously interacts with both Hp and Hb. Specifically, CD163A SRCR2 is involved in binding with the loop 3 region of Hp, CD163A SRCR3 engages with helix A of αHb (Fig. 5a, b), whereas CD163A SRCR4 forms a limited interface with the AB loop of αHb. In details at SRCR2, Hp Lys321 forms electrostatic interactions with CD163A Asp186 and Glu252 (Fig. 5a). Additionally, there is an electrostatic interaction between Hp Arg311 and CD163A Glu216, and a hydrogen bond links Hp Thr317 with CD163A Asp249. At CD163A SRCR3, αHb Lys11 forms electrostatic interactions with CD163A Asp293 and Glu359 (Fig. 5b), analogous to the interaction observed at SRCR2. Furthermore, CD163A Tyr354 engages in van der Waals interaction with αHb Lys7 and forms a hydrogen bond with αHb Asp74. At CD163A SRCR4, a single electrostatic interaction is formed between CD163A Asp463 and αHb His20 (Fig. 5c).
Fig. 5. CD163 interactions with Hp and Hb.

a CD163A SRCR2 interaction with Hp loop 3 (L3). b CD163A/B/C SRCR3 interaction with αβHb. c. CD163A/B/C SRCR4 interaction with αβHb. The three CD163 subunits are shown in shades of orange, Hp SP domain in blue, αHb in dark red, and βHb in light red. Selected residues involved in interactions are shown as sticks. Dashed black lines represents electrostatic interaction or hydrogen bonds. The resolution of the shown cryo-EM density maps is indicated in the individual close-ups. The highlighted squares show interactions observed in the assembly of dimeric CD163 (CD163A and CD163B) with HpHb, referred to as CD1632-HpHb.
At the two other CD163 subunits (CD163B and CD163C), no well-defined density is observed for SRCR2, suggesting that these domains are not or only loosely associated with HpHb. More or less well-defined density is observed for SRCR3 from both CD163B and CD163C. However, due to the low resolution of the cryo-EM density maps for these domains, their precise interactions with Hb should be interpreted with caution. Despite this limitation, we do observe some shared characteristics in their ligand binding, even though the SRCR3 domains of CD163 recognize different epitopes on αβHb. In both CD163B SRCR3 and CD163C SRCR3, a lysine residue from αβHb is aligned to enable electrostatic interactions with Asp293 and Glu359 of SRCR3 (Fig. 5b). Specifically, Lys90 from the αHb FG region in CD163B and Lys82 from βHb helix F in CD163C are each positioned to interact electrostatically with these SRCR3 residues. Additionally, SRCR3 Tyr354 appears to engage in van der Waals interactions with αHb in CD163B or βHb in CD163C. In both cases, at SRCR4, Asp463 is positioned to interact with αHb His45 (CD163B) or βHb His2 (CD163C), a pattern also observed in CD163A SRCR4 (Fig. 5c). In the CD1632-HpHb reconstruction, electron density is observed only for CD163A and CD163B, while CD163C appears to have dissociated, suggesting a lower affinity for HpHb compared to the other subunits. At CD163B, only SRCR3 engages with HpHb, whereas both SRCR2 and SRCR3 interact at CD163A. These findings indicate that the individual CD163 subunits exhibit varying affinities for HpHb. This differential binding suggests that the subunits contribute unevenly to the overall functional affinity for the ligand, with CD163A providing a stronger interaction, while CD163B and CD163C play more secondary roles.
Quantification of the CD163 affinity for HpHb, Hp, and Hb
Our structural data shows that HpHb binding can potentially involve interfaces with all three subunits of the trimeric CD163. To measure the affinity of HpHb for stable CD163 trimers, we engineered the ectodomain of CD163 with an established coiled-coil motif (designated CD163-CC) functioning as a trimerization module31. SEC-MALS analysis of CD163-CC revealed a monodisperse peak with molecular weight close to the expected molecular weight of around 420 kDa (Supplementary Fig. 6). Surface Plasmon Resonance (SPR) analysis of immobilized CD163-CC demonstrated that the binding kinetics of HpHb yields an estimated dissociation constant (KD) of 8.5 nM (Fig. 6a). Applying the same method, we also assessed the binding of Hb to trimeric CD163, which similarly adhered to the Langmuir 1:1 model with a KD of 598 nM. In line with previous studies on purified native CD16310,22, Hp alone showed no binding affinity for trimeric CD163-CC (Fig. 6a). To quantify the monomeric interaction of HpHb and Hb to CD163 we immobilized CD163 SRCR 1-5 on the sensor chip. The KD of HpHb binding to CD163 SRCR 1-5 was estimated to 766 nM, while the KD of Hb binding to CD163 SRCR 1-5 was estimated to 43.8 µM (Fig. 6b). These results indicate that the trimerization of CD163 leads to a 70 to 90-fold increase in ligand affinity. This enhanced affinity is likely due to the avidity effect, where multiple interaction sites work together to strengthen binding, as also observed for divalent IgG antibodies32. Quantification of the multimeric Hp(2-2)Hb binding to CD163-CC (Fig. 6c) revealed a significantly higher affinity compared to the dimeric Hp(1-1)Hb. This suggests that the multimeric structure of Hp(2-2), with multiple HpSP domains bound to Hb, can engage with multiple CD163 trimers simultaneously. On the surface of cells expressing CD163, this multivalent interaction would likely enhance the overall avidity of the binding, potentially increasing the efficiency of HpHb clearance. However, in the case where Hp(2-2) is not completely saturated with Hb this increased efficiency is likely lost33. Dimeric Hp(1-1)Hb can in principle also interact simultaneously with two CD163 trimers on the surface of cells. Although we do not observe this type of complex in our cryo-EM data its existence cannot be excluded. Modelling suggest that its formation is possible without steric hindrance (Supplementary Fig. 7).
Fig. 6. Quantification of CD163 interaction with Hp and Hb.

a SPR binding curves of Hp(1-1)Hb (left), Hb (middle) and Hp(1-1) (right) to immobilized CD163-CC. b SPR binding curves of HpHb (top), and Hb (bottom) to immobilized CD163 SRCR 1-5. c SPR binding curves of Hp(2-2)Hb to immobilized CD163-CC. Dashed black lines show the fitting to the binding curves. The dissociation constant (KD) of Hp(1-1) binding to CD163-CC could not be determined (ND). Source data are provided as a Source Data file.
Cellular uptake of HpHb and Hb
To evaluate the impact of CD163 oligomerization on the cellular uptake of HpHb and Hb, we expressed full-length CD163 in HEK293 cells in both its wild-type form (WT) and the monomeric mutant form (K811A/K1021A). Both the WT and mutant CD163 were expressed on the cell surface at nearly identical levels (Fig. 7a). We then quantified the uptake of fluorescently labeled HpHb, Hb, and an anti-CD163 nanobody at a concentration of 10 µg/ml. Our studies show that the cellular uptake the antiCD163 nanobody was similar in cells expressing either WT CD163 or the K811A/K1021A expressing cells (Fig. 7b). However, when measuring HpHb uptake, we observed an approximately 50% reduction in the K811A/K1021A mutant compared to the WT (Fig. 7c), indicating a significant functional role for CD163 oligomerization in HpHb uptake. Despite this reduction, the fact that HpHb uptake is not completely abolished in the mutant suggests that a monomeric CD163 subunit may still support low-level uptake. In contrast, uptake of Hb was reduced to baseline levels in K811A/K1021A-expressing cells, as for non-transfected HEK293 cells. This complete loss of Hb uptake in the mutant implies that Hb uptake relies more on CD163 oligomerization than HpHb uptake.
Fig. 7. CD163 trimerization facilitates uptake of HpHb and Hb.
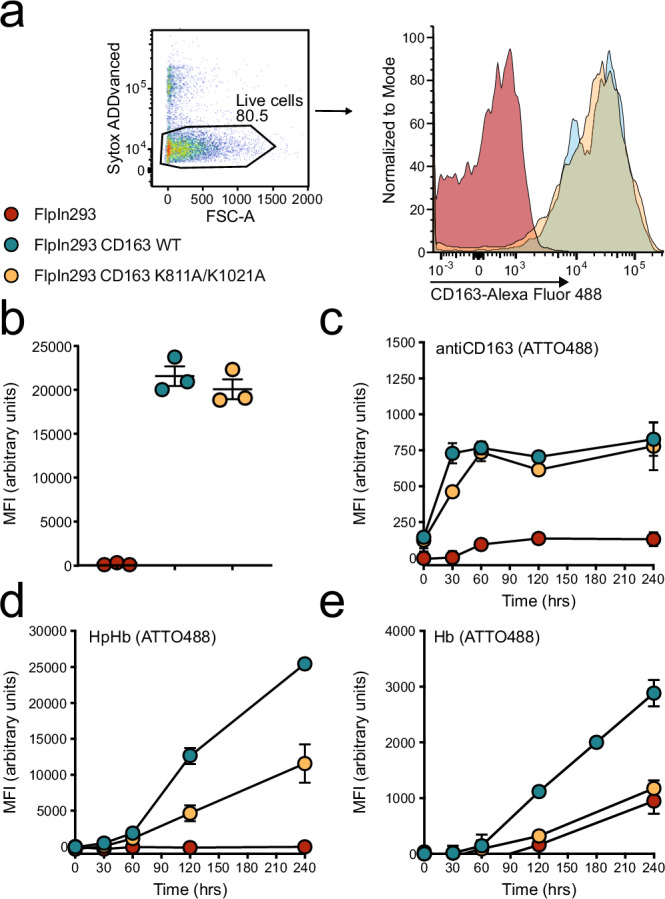
a + b Flow cytometric analysis of CD163 surface expression in FlpIn293 cells either mock transfected (red) or stably expressing wildtype CD163 (blue) or CD163 K811A/K1021A (orange). Live cells are gated as Sytox ADDvanced negative and replotted as histogram (a) followed by calculation of mean fluorescent intensity (MFI) (b). c–e Flow cytometric analysis of ATTO 488 - fluorescently labeled ligand uptake in FlpIn293 cells either mock transfected (red) or stably expressing wildtype CD163 (blue) or CD163 K811A/K1021A (orange) using either anti-CD163 nanobody (c), haptoglobin-hemoglobin complex (d) or hemoglobin alone (e). Data points represent the mean of three technical replicates, and error bars show the standard deviation (n = 3). Source data are provided as a Source Data file.
Discussion
The data presented here reveals the structural basis for CD163-mediated macrophage clearance of toxic Hb released from red blood cells. Our data also reveals that CD163 forms Ca2+-dependent oligomers with a central ligand-binding site that engages in multiple interactions, primarily with basic residues of HpHb, facilitating its binding and efficient clearance.
SEC-MALS analysis showed that the recombinantly expressed extracellular part of CD163 in solution exists in a dynamic equilibrium of monomers, dimers, and trimers. However, full-length CD163, when expressed on the macrophage surface in its native environment, may behave differently. The membrane environment may favor CD163 oligomerization due to spatial constraints and a higher receptor density, increasing the likelihood of trimer formation compared to CD163 in solution. However, it remains possible that dimeric and monomeric forms coexist on the macrophage membrane.
Our uptake studies suggest that CD163 oligomerization on the cell membrane plays an important role in the uptake of both HpHb and Hb. However, oligomerization does not seem to be strictly necessary for HpHb uptake, implying that a monomeric form of CD163 may suffice at the tested concentration. In contrast, the disruption of CD163 oligomerization has a more significant impact on Hb uptake. This is consistent with our observation that the lower affinity of Hb for CD163 makes it more dependent on CD163 oligomerization for efficient uptake.
The oligomerization of CD163 is mediated by interactions between SRCR7 and SRCR9 from adjacent CD163 subunits. This oligomerization relies on pairwise interactions, where residues Lys811 (in SRCR7) and Lys1021 (in SRCR9) appear to play crucial roles by interacting with the negatively charged residues at site 1 of the opposing SRCR domain. We speculate that other SRCR domains with a lysine in this position could be involved in similar SRCR-SRCR interactions, either as a homodimer or heterodimer as in CD163. Analysis of the human SRCR sequences revealed lysine residues at this position in CD6 (SRCR2), SCART1 (SRCR6), Neurotrypsin (SRCR2) and CD163L1 (SRCR10 and SRCR12) (Fig. 8a). These domains also show the canonical residues at site 1, which is likely a prerequisite for dimerization. The transmembrane receptor CD6 is expressed on T-cells and plays an important role in T-cell activation, proliferation, and differentiation by mediating cell-cell interactions through its binding to CD16634. CD6 harbors three SRCR domains and its structure has been determined using x-ray crystallography (RCSB ID 5A2E)35. By analyzing the crystal packing of CD6, we found an interaction between two CD6 molecules that display an interface remarkably like the interface between CD163 SRCR7 and SRCR6, which likely reveals the molecular basis for CD6 dimerization (Fig. 8b, c). This observation also suggests that SCART1, neurotrypsin, and CD163L1 may dimerize through a similar interface.
Fig. 8. Dimerization of CD6 is structurally similar to CD163 trimerization.
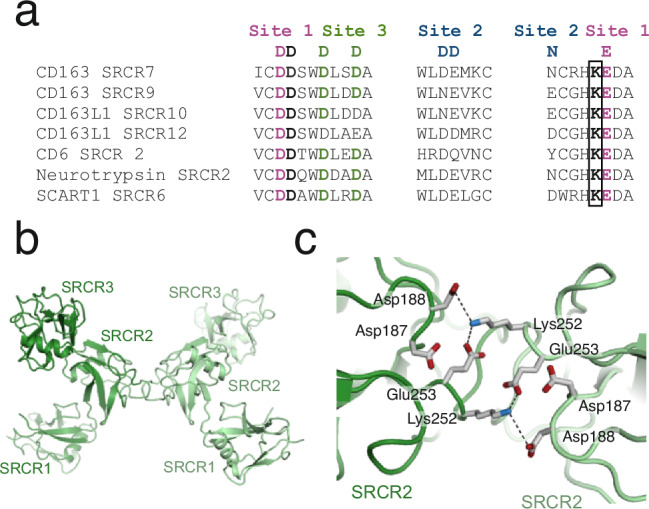
a Alignment of human SRCR domains with a lysine residue on the N-terminal side of the glutamate residues in site 1 (marked by a black box). b Cartoon representation of a potential CD6 dimer. c Close-up on the interaction between the two CD6 subunits in the potential CD6 dimer shown in b. Residues from site 1 and the lysine residues are shown as sticks. Dashed black lines represent electrostatic interactions.
The human genome harbors 27 genes encoding proteins with SRCR domains, which include a diverse set of membrane-associated or secreted proteins. In several of these proteins, SRCR domains have been shown to mediate Ca2+-dependent interaction with ligands such as apolipoproteins and ferritin36,37. Furthermore, these studies demonstrate that mutations in site 1 or site 2 reduce ligand affinity, highlighting the importance of these regions in ligand binding and suggesting a potential shared mechanism among SRCR proteins.
The structure of CD163 in complex with HpHb provides the structural evidence of SRCR domains bound to a ligand, unveiling key ligand recognition mechanisms. Across all four observed ligand interaction sites and in the oligomerization sites, a lysine residue from the interacting partner aligns for electrostatic interactions with the aspartate residue linking Ca2+-binding sites 1 and 2 and with the glutamate residue at Ca2+-binding site 1. This observation confirms the hypothesis that basic residues from the ligand interact with acidic residues associated with the putative Ca²⁺-binding sites of the receptor. Moreover, the structure of CD163 uncovers a mechanism of receptor oligomerization, which is mediated through similar types of interactions.
Our SEC-MALS studies show that both ligand binding and oligomerization is dependent on the presence of Ca2+-ions. Based on the structural data, we can now pinpoint the Ca2+-binding sites located in the ligand-binding region of CD163. CD163 SRCR2 and SRCR3 each bind a single Ca2+ ion, which may mediate both ligand binding and release in conjunction. At SRCR2, a Ca2+ ion is bound at site 2, directly coordinating Asp185 that interacts with Hp Lys321. At SRCR3, a Ca2+ ion is bound at site 3, further away from the ligand-binding site. Although not directly involved in ligand binding, this Ca2+ ion may serve an indirect role by stabilizing the structural integrity of SRCR3. Both SRCR2 and SRCR3 display Ca2+ binding consensus residues at site 1, located adjacent to site 2, yet no density for a Ca2+ ion is observed at these sites. Structural analysis of SRCR domains available in the Protein Data Bank shows that all canonical Ca2+-binding sites are occupied when Ca2+ is present during crystallization (Supplementary Table 2). However, this does not appear to be the case for CD163 SRCR2 and SRCR3. There are several potential reasons for this discrepancy. Firstly, the structural conformations of SRCR2 and SRCR3 may not be favorable to Ca2+ binding at site 1. Secondly, the relatively low resolution of the cryo-EM maps might obscure the detection of a partially occupied site. Finally, it is possible that HpHb binding displaces the Ca2+ ions from site 1.
Observing variations in Ca2+-binding sites across these SRCR domains suggests that the precise nature of Ca2+-binding might be less critical for direct interactions. Instead, the bound Ca2+-ions likely play a supporting role in domain stabilization, regardless of the specific sites involved.
Due to the low resolution of the density maps covering SRCR7 and SRCR9, we cannot identify the Ca2+-binding site mediating oligomerization. However, sequence analysis predicts that potential Ca2+-binding sites are present in SRCR6 (site 3), SRCR7 (site 1/site 3), and SRCR9 (site 1/site 3). Any of these sites could, in principle, be responsible for the Ca2+-dependent oligomerization of CD163. To further understand the mechanism behind Ca²⁺-dependent CD163 oligomerization, high-resolution structures of CD163 are required.
The present structural data on CD163 also provides a potential explanation for the release of HpHb from CD163 in endosomes, driven by the gradual removal of Ca2+. This may promote the release of HpHb from CD163 by a combined process of two distinct mechanisms. Firstly, Ca2+ efflux leads to reduced affinities for HpHb at the individual binding sites. Secondly, the dissociation of the trimer causes the disassembly of the three subunits forming the composite binding site for HpHb. This abolishes the avidity effect caused by multiple interactions, thereby further reducing the affinity of HpHb for CD163. In addition to these mechanisms, a reduction in endosomal pH could also contribute to HpHb release, likely through the protonation of functionally important histidine residues i.e., αHb His20, αHb His45, and βHb His2.
In the absence of Hp, Hb is recognized and taken up by CD163, albeit at a much lower level compared to the HpHb complex. This reduced uptake by CD163 is likely due to Hb’s lower affinity for the receptor. It is currently unknown whether CD163 takes up tetrameric or dimeric Hb, as these two forms exist in a dynamic equilibrium in the serum. In principle, dimeric αβHb could bind at the same site as the HpHb complex. The absence of the Hp binding site interaction, mediated by CD163 SRCR2, could explain the lower affinity compared to that of HpHb.
Hp is present in serum at high concentrations, poised for the rapid clearance of Hb in instances of increased hemolysis. However, in the absence of hemolysis, the uptake of Hp by the CD163 receptor on macrophages is not a favorable process. Accordingly, Hp alone does not appear to bind to CD163, even though it has a significant interface with SRCR2. The structure of human Hp without Hb shows that no conformational changes occur upon Hb binding (Supplementary Table 3 and Supplementary Fig. 8). This observation suggests that the increased affinity of the HpHb complex for CD163, compared to Hp alone, is not due to structural alterations in Hp that expose binding sites on the protein. Rather, it appears that it is the interaction with additional binding sites located on αβHb that leads to an enhanced affinity for CD163 and drives the cellular uptake.
A key aspect of CD163’s role in regulating inflammation involves its proteolytic cleavage, which releases soluble CD163 (sCD163) into the plasma. sCD163 is present at concentrations around 2 µg/ml, and studies have shown that it does not interfere with the uptake of HpHb by membrane-bound CD16317. This may be due to sCD163 dissociating into monomers at such low concentrations, resulting in a significantly lower affinity for HpHb, and therefore being unable to compete with the high-affinity oligomeric CD163 on the surface of macrophages.
In conclusion we have now described how the HpHb complexes engage with CD163 at the molecular level. Importantly, the study describes the crucial role of calcium in this process and how Hp binding to Hb, without conformational changes to individual components of the complex, increases the affinity towards the receptor by contributing with more CD163-binding sites. The functional CD163 is likely an oligomer forming a central ligand-binding site. The oligomerization of CD163 also depends on calcium that regulates the assembly via the SRCR7 and 9 domains. The calcium sensitivity suggests that the receptor constitutively changes between the oligomeric form when exposed outside to plasma and the monomeric form when present intracellularly in endosome and recycling vesicles (Fig. 9). To our knowledge, this distinctive dynamic represents an unprecedented mechanism by which Ca²⁺-ions contributes to receptor-ligand coupling and uncoupling.
Fig. 9. Proposed mechanism of CD163-mediated endocytosis of Hb-bound Hp oligomers.
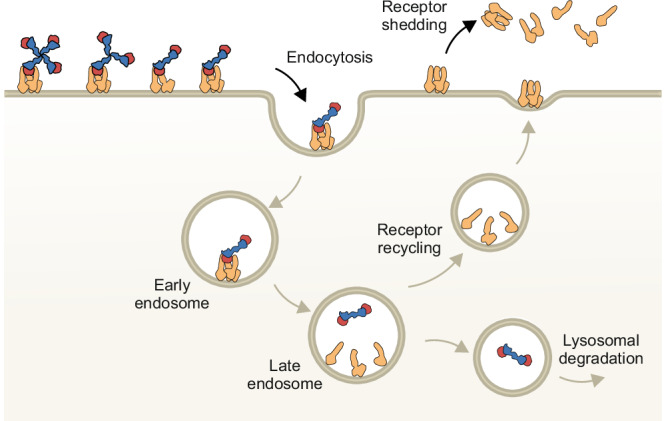
On the surface of the cell, CD163 forms Ca2+-dependent oligomers that binds Hp(1-1)Hb, Hp(2-1)Hb, or Hp(2-2)Hb. After endocytosis, lowering of the Ca2+ concentration and pH in the late endosome causes CD163 dissociation and HpHb release. Monomeric CD163 is recycled back to the plasma membrane where the CD163 trimer is reestablished while HpHb is degraded in the lysosome. CD163 shed from the membrane may dissociate into CD163 monomers with low affinity for HpHb.
Methods
Expression and purification of human CD163 SRCR1-9
Expression and purification of human CD163 SRCR 1-9 was performed as described by Madsen et. al.19. A construct encoding CD163 SRCR 1-9 (residues 1-1029) was subcloned into the pcDNA5/FRT vector (cat. no. V601020, Thermo Fisher). Flp-In chinese hamster ovary (CHO) cells (cat. no. R75807, ThermoFisher) were transfected using the FuGENE 6 transfection reagent (cat. no. E2691, Promega) and clones were grown in serum-free HyClone medium for CHO cells (cat. no SH30549, Cytiva). CD163 SRCR 1-9 was purified from the cultured medium using HpHb affinity chromatography. The affinity column was generated by coupling 30 mg HpHb to CNBr-activated sepharose 4B (cat. no. 17043001, Cytiva). After binding to the immobilized HpHb, CD163 SRCR 1-9 was eluted with a pH 4.0 buffer containing 10 mM EDTA. The collected fractions were immediately neutralized by Tris-HCl pH 8.0 and CaCl2 added to 10 mM. The sample was further purified using a Superdex 200 INCREASE 10/300 GL column (cat. no. GE17-5175-01, Cytiva) equilibrated in a buffer containing 20 mM Hepes pH 7.6, 75 mM KCl and 5 mM CaCl2. Fractions containing essentially pure CD163 SRCR1-9 were pooled and concentrated using a 10 kDa molecular weight cut-off (MWCO) Amicon Ultra centrifugal filter (cat. no. UFC8010, Millipore).
Purification of HpHb and Hp from human blood
The purification of HpHb from human blood was performed as described by Stødkilde et al. 38. Anti-coagulants were added to human blood (Hp1-1 genotype) to a final concentration of 15 mM trisodium citrate and 0.15 mM EDTA. Plasma and blood cells were separated by centrifugation at 4000 g for 20 min. The blood cell fraction was lysed by addition of water (1:1 ratio) and cell debris was removed by centrifugation at 8,000 g for 15 min. Clotting factors were removed from the plasma by addition of 80 mM BaCl2, followed by incubation on ice for 1 h and centrifugation at 27,000 g for 15 min. For purification of HpHb, serum and blood cell fractions were mixed in a ratio of 25:1 and incubated at 4 °C overnight. The sample was diluted 1:2 in 20 mM Tris–HCl pH 7.6 and loaded on a HiTrap Blue HP column (cat. no. 17041301, Cytiva) equilibrated in 50 mM KCl, 20 mM Tris–HCl pH 7.6. The flow-through was collected and loaded on a Q Sepharose Fast Flow column (Cytiva) equilibrated with buffer Q-A (50 mM KCl, 20 mM Tris–HCl pH 7.6). A gradient from 10 to 70% buffer Q-B (500 mM KCl, 20 mM Tris–HCl pH 7.6) was applied. Fractions containing HpHb or Hp were pooled, and ammonium sulphate added to 55% saturation. The sample was centrifuged at 27,000 g for 15 min and the supernatant loaded on a Source 15 Iso column (Cytiva) equilibrated with buffer Iso-A (55% ammonium sulphate, 20 mM Tris–HCl, pH 7.6). A gradient from 0 to 100% buffer Iso-B (20 mM Tris pH 7.6) was applied. Eluted fractions were further purified using a Superdex 200 INCREASE 10/300 GL column equilibrated in 75 mM KCl, 20 mM Tris–HCl pH 7.6 and 5 mM CaCl2. Fractions containing HpHb or Hp were pooled and concentrated using an Amicon Ultra centrifugal filter (10 kDa MWCO).
Cloning, expression and purification of CD163 SRCR1-9 K811A/K1021A
Constructs expressing CD163 SRCR1-9 K811A/K1021A and CD163 SRCR1-9 D745A/746 A/D955A/D956A were generated as follows. A His-tag followed by an enterokinase cleavage site was inserted after the signal peptide of CD163 by site-directed mutagenesis of the CD163 SRCR1-9 pcDNA5/FRT19 vector using the Q5 Site-directed Mutagenesis kit (cat. no. E0554S, New England Biolabs). The sequences of the applied primers are provided in the Source Data. Following the instructions of the manufacturer, the full-length gene was transferred into the pcDNA3.4 vector (cat. no. A14697, ThermoFisher) by TOPO cloning and mutations were introduced using two rounds of site-directed mutagenesis. The construct was transfected into ExpiCHO-S cells (cat. no. A29133, ThermoFisher) using ExpiFectamine CHO reagent (cat. no. A29131, ThermoFisher) and cells grown according to the manufacturer’s instruction. The cultured medium was applied to a 5 ml cOmpleteTM His His-tag purification column (cat. no. 06781535001, Roche diagnostics) equilibrated in 20 mM Hepes pH 7.6, 500 mM KCl, 5 mM CaCl2 and eluted using a buffer containing 400 mM Imidazole, 20 mM Hepes pH 7.6, 500 mM KCl and 5 mM CaCl2. Fractions containing CD163 SRCR1-9 K811A/K1021A or CD163 SRCR1-9 D745A/746 A/D955A/D956A were pooled, dialyzed against a buffer containing 20 mM Hepes pH 7.6, 75 mM KCl and 5 mM CaCl2 and concentrated using an Amicon Ultra centrifugal filter (10 kDa MWCO).
Cloning, expression and purification of CD163 SRCR1-5
A construct encoding CD163 SRCR 1-5 (residues 42-578) with an N-terminal mouse Ig kappa signal peptide and a His-tag followed by a thrombin cleavage site was cloned into the BamHI site of the pcDNA5/FRT. The construct was transfected into Flp-In CHO cells using the FuGENE 6 transfection reagent and clones were grown in serum-free HyClone medium for CHO cells. The cultured medium was applied to a 5 ml cOmpleteTM His-tag purification column equilibrated in 20 mM Hepes pH 7.6, 500 mM KCl, 5 mM CaCl2 and eluted using a buffer containing 400 mM Imidazole, 20 mM Hepes pH 7.6, 500 mM KCl and 5 mM CaCl2. Fractions containing CD163 SRCR1-5 were pooled and dialyzed against a buffer containing 20 mM Hepes pH 7.6, 75 mM KCl and 5 mM CaCl2. Thrombin (cat. no. 112374, Sigma-Aldrich) was added in a 1:200 w/w ratio and the sample was incubated over-night at 25 °C. As a final step of purification, the sample was applied on a Superdex 200 INCREASE 10/300 GL column equilibrated in a buffer containing 20 mM Hepes pH 7.6, 75 mM KCl and 5 mM CaCl2. Fractions containing CD163 SRCR1-5 were pooled and concentrated using an Amicon Ultra centrifugal filter (10 kDa MWCO).
Expression and purification of CD163-CC
A construct encoding CD163 SRCR 1-9 with a C-terminal coiled-coil motif (sequence: EPPTQKPKKIVNAKKDVVNTKMFEELKSRLDTLAQEVALLKEQQALQTV) and a V5-tag was assembled using the In-fusion HD cloning kit (cat. no. 102518, Clontech). Sequences of applied primers are listed in Source Data. The construct was transfected into Flp-In CHO cells using the FuGENE 6 transfection reagent and clones were grown in serum-free HyClone medium for CHO cells. The cultured medium was applied to a 5 ml cOmpleteTM His-tag purification column equilibrated in 20 mM Hepes pH 7.6, 500 mM KCl, 5 mM CaCl2 and eluted using a buffer containing 400 mM Imidazole, 20 mM Hepes pH 7.6, 500 mM KCl and 5 mM CaCl2. Fractions containing CD163-CC were applied on a Superdex 200 INCREASE 10/300 GL column equilibrated in a buffer containing 20 mM Hepes pH 7.6, 75 mM KCl and 5 mM CaCl2.
Multi-angle light scattering in-line with size-exclusion chromatography
Purified samples (CD163 SRCR 1-9 (40 µg), CD163 SRCR 1-5 (40 µg) with/without HpHb (40 µg), CD163 SRCR 1-9 K811A/K1021A (100 µg), CD163 SRCR 1-9 D745A/D746A/D955A/D956A (100 µg), CD163-CC (100 µg)) were diluted in a buffer containing 20 mM Hepes pH 7.6, 75 mM KCl and 5 mM CaCl2/10 mM EDTA to a final volume of 100 µl and incubated for 2 h at 25 °C. Samples were subsequently analyzed by size-exclusion chromatography (using buffer containing 20 mM Hepes pH 7.6, 75 mM KCl and 5 mM CaCl2/10 mM EDTA) using a Dionex UltiMate 3000 HPLC (100 µl loop) equipped with a Wyatt SEC Analytical Column (WTC-030S5, Wyatt Technology Europe GmbH) or a Bio-Rad NGC Chromatography System (1 ml loop) equipped with a Superdex 200 Increase 10/300 GL column. Multi-angle light scattering was measured in-line with an Optilab T-rEX Refractive Index Detector (model no. WTREX-02, Wyatt Technology Europe GmbH) and DAWN 8+ Multi-Angle Light Scattering Detector (model no. WH2-06, Wyatt Technology Europe GmbH). Bovine serum albumin (cat. no. A1900, Sigma-Aldrich) was used to calibrate the system. The experimental data were recorded and processed by ASTRA software (Wyatt Technology).
Cryo-EM data collection
CD163 SRCR 1-9 and HpHb were mixed in a 1:2 w/w ratio to saturate CD163 with HpHb. The sample was incubated for 2 h at 25 °C prior to size-exclusion chromatography using a Superdex 200 INCREASE column equilibrated in 20 mM Hepes pH 7.6, 75 mM KCl and 5 mM CaCl2. Cryo-EM grids were prepared by applying purified CD163-HpHb to glow-discharged C-FLAT carbon grids (Protochips), blotting with a Vitrobot (FEI) and plunging into liquid ethane. Data were collected on a Titan Krios (FEI) equipped with a Quantum GIF energy filter (Gatan) operating in zero-loss mode with a 20-eV slit width. Movies of 50 frames were acquired on a K2 Summit Direct Detect camera in super-resolution mode with a pixel size of 0.525 Å and a total dose of 90 e-/Å-2. Movies were collected with a defocus range of -0.5 to -1.5 µm. Leginon software was used for automated data collection39.
Cryo-EM image processing and reconstructions
Movies were imported into CryoSPARC240 and subjected to Patch Motion correction and Patch CFT estimation. Initial particle picking was performed using Blob picker and a subset of the resulting 2D classes were used for template-based picking. After several rounds of 2D classification, the remaining particles were used to generate ab initio models in three classes with 0.3 similarity. Particles from each class were further refined using heterogeneous refinement and non-uniform refinement. To further improve the resolution, we applied local refinement using a mask covering αβHb, Hp SP and CD163A SRCR2-4. Resolutions were estimated using the Fourier shell correlation cut-off of 0.143 (Supplementary Fig. 3b). Figures of cryo-EM maps were prepared in ChimeraX41.
Model building and refinement
Cryo-EM maps were sharpened using Phenix.autosharpen42 and LocalDeblur43. Individual SRCR domains of the AlphaFold2 prediction of human CD163 and the structure of human HpHb (RCSB ID 4WJG) were manually docked into the density and subjected to automated molecular dynamics flexible fitting followed by real-space refinement using Namdinator pipe-line tool44. The locally refined map covering CD163A SRCR2-4 and HpHb, was further subjected to manual rebuilding in Coot and subsequent real-space refinement in Phenix45.
Surface plasmon resonance
Surface plasmon resonance (SPR) experiments were carried out on a Biacore 3000 (Cytiva). Binding analysis was performed at 25 °C, and data were collected at a rate of 1 Hz. For the development of a double capture experiment, flow cells 1 and 2 (FC1 and 2) of a CM5 chip (cat. no. BR100399, Cytiva) were coated with protein G (cat. no. 08062, Sigma-Aldrich) using standard amine chemistry (EDC/NHS, cat. no. BR100050, Cytiva), according to the instructions of the manufacturer. Residual reactive groups were blocked by a 7 min injection of 1 M ethanolamine pH 8.5. Monoclonal anti-V5 tag antibody (cat. no. MCA1360, Bio-Rad) or MAC2-158 anti-CD163 antibody (Trillium Diagnostics) was captured in both flow cells to approximately 3000 RU. In FC2 only, recombinant human CD163-CC or CD163 SRCR 1-5 was captured to a level of ~300 or 1200 RU, for the binding analyses of HpHb and Hb, respectively. Next, using a flow rate of 30 μl/min, purified analytes were injected over both surfaces for 180 s followed by a 300 s dissociation phase. At the end of each binding cycle, non-covalently attached molecules were removed from both surfaces by a 30 s injection of 10 mM glycine, pH 1.5. All proteins were diluted in running buffer (10 mm HEPES, pH 7.5, 150 mm NaCl, 2 mM CaCl2 and 0.05% Tween 20). Using the BIAevaluation 4.1.1 software (Cytiva), recorded signals from FC2 were double referenced; the response from the in-line reference flow cell (FC1) was subtracted as was the response from a blank run (0 nM analyte). Affinities were estimated using fitting to the Langmuir 1:1 model.
Structure determination of human Hp
Crystals of Hp were obtained by mixing 2 µl protein with 2 µl reservoir solution (6% 2-propanol, 0.1 M sodium acetate trihydrate pH 4.5 and 26% PEG MME 550) in sitting-drop vapor diffusion experiments at 4°C. Prior to flash-freezing in liquid nitrogen, crystals were transferred to cryo-protection buffer (6% 2-propanol, 0.1 M sodium acetate trihydrate pH 4.5 and 40% PEG MME 550). X-ray diffraction data were collected at the I911-2 beam line at MAX-Lab using a wavelength of 1.03841 Å and at a temperature of 100 K. Data were indexed, integrated and scaled with the XDS package46. The structure was solved by molecular replacement in PHASER47 with human Hp as search model. Model building was done in program Coot48 followed by iterative refinement cycles in PHENIX45. Data collection, phasing and refinement statistics are shown in Supplementary Table 1.
Establishment of stable cell line expressing wildtype and K811A/K1021A full-length human CD163
FlpIn293 cell line either mock transfected or stably expressing full length human CD163 or full-length human CD163 K811A/K1021A was established essentially as described in Etzerodt et al.49. In brief, FlpIn293 cells were transfected with pcDNA5/FRT either empty (mock) or containing cDNA encoding full-length human CD163 or full-length human CD163 K811A/K1021. Stable transfected cell lines were subsequently generated by antibiotic selection using 150 µg/ml Hygromycin B (cat. no. 10687010, Thermo Fisher).
Quantification of cellular uptake and CD163 surface expression levels
To quantify CD163 surface expression in established cell lines, adherent cells were harvested by 5 min incubation with 10 mM EDTA and 4 mg/ml Lidocaine HCl at 37 °C and subsequently collected in DMEM followed by centrifugation at 400 g for 5 min at 4 °C. For CD163 surface staining, cell pellets were resuspended in FACS buffer (1x PBS pH 7.4, 1 mM EDTA, 2 % FCS, 0,09% NaN3) and stained with Alexa Flour 488 conjugated anti-human CD163 mAb (cat. no. 53-1637-42, Invitrogen) for 30 min on ice. Cells were subsequently washed twice with FACS buffer and surface expression of CD163 was estimated by measuring fluorescent intensity on a Novocyte Quanteon Flow cytometer (Agilent). To exclude dead cells, Sytox AAdvanced was added to each sample prior to running the sample on the flow cytometer. Following, fluorescent intensity was estimated in FlowJo by initially gating on live cells (Sytox AAdvanced negative) and subsequently estimating mean fluorescent intensity of Alexa Flour 488. The experiment was conducted with three technical replicates for each condition.
Quantification of cellular uptake of fluorescently labeled ligands was essentially performed as described for estimating CD163 surface levels. Before fluorescent assays, Hb or anti-human CD163 nanobody was conjugated with ATTO488 (cat. no. 41698, Sigma-Aldrich) according to the instructions of the manufacturer. For quantification of ligand uptake, FlpIn293 cell lines were seeded in 24 well plates and incubated O/N. Cells were then incubated with Nb-ATTO488, Hb-ATTO488 or preformed HpHb-ATTO488 at a concentration of 10 µg/ml for varying time points 0, 30, 60, 120, 180 or 240 min. Cells were then washed thrice with DMEM and harvested as described above after which fluorescent ligand uptake was estimated on Novocyte Quanteon Flow cytometer using Sytox AAdvanced to exclude dead cells.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Source data
Acknowledgements
We are grateful to G. Ratz for technical assistance and the staff at MAX-lab beamlines for help with data collection. We thank J. Quispe for assistance with electron microscopy. The research was supported by the Lundbeck Foundation (R291-2018-49, C.B.F.A), Ragna Rask-Nielsens Grundforskningsfond (C.B.F.A.), Direktør Ib Henriksens Fond (C.B.F.A.) and the Danish National Research Foundation to Center for Functional Genomics and Tissue Plasticity (DNRF141, J.H.G.).
Author contributions
A.E.: flow cytometry, study design. J.H.M.: cloning, expression, mutagenesis, purification, binding studies. M.T.J.: purification, crystallization. D.H.: cloning, expression, purification. T.B.: electron microscopy. J.H.G.: cloning, expression, purification. S.K.M.: manuscript preparation and study design. J.M.K.: electron microscopy. C.B.F.A.: light scattering analysis, structure determination and analysis, electron microscopy, manuscript preparation and study design.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Data availability
The atomic structure factors and coordinates for the structure of human Hp are available at the Protein Data Bank under accession number 9FNM. The Cryo-EM maps and coordinates for the structure of CD163-HpHb are available at the EMDB under accession numbers 9FHB, 9FMU, and 9FNO. Other data are available from the corresponding author upon request. Source data are provided with this paper.
Competing interests
A.E. and J.H.G. and S.K.M reports to be minority shareholders of OncoSpear ApS that develops CD163 antibodies for the treatment of cancer. The remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-55171-4.
References
- 1.Quaye, I. K. Extracellular hemoglobin: the case of a friend turned foe. Front. Physiol.6, 96 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buehler, P. W. & D’Agnillo, F. Toxicological consequences of extracellular hemoglobin: biochemical and physiological perspectives. Antioxid. Redox Signal12, 275–291 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Andersen, C. B. F. et al. Haptoglobin. Antioxid. Redox Signal26, 814–831 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Madsen, M., Graversen, J. H. & Moestrup, S. K. Haptoglobin and CD163: captor and receptor gating hemoglobin to macrophage lysosomes. Redox Rep.6, 386–388 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Gwozdzinski, K., Pieniazek, A. & Gwozdzinski, L. Reactive Oxygen Species and Their Involvement in Red Blood Cell Damage in Chronic Kidney Disease. Oxidat. Med. Cell. Longev.2021, 6639199 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rifkind, J. M., Mohanty, J. G. & Nagababu, E. The pathophysiology of extracellular hemoglobin associated with enhanced oxidative reactions. Front Physiol.5, 500 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pimenova, T. et al. Quantitative mass spectrometry defines an oxidative hotspot in hemoglobin that is specifically protected by haptoglobin. J. Proteome Res.9, 4061–4070 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Kapralov, A. et al. Peroxidase activity of hemoglobin-haptoglobin complexes: covalent aggregation and oxidative stress in plasma and macrophages. J. Biol. Chem.284, 30395–30407 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vallelian, F. et al. Spin trapping combined with quantitative mass spectrometry defines free radical redistribution within the oxidized hemoglobin:haptoglobin complex. Free Radic. Biol. Med.85, 259–268 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Kristiansen, M. et al. Identification of the haemoglobin scavenger receptor. Nature409, 198–201 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Okazaki, T. & Nagai, T. Difference in hemoglobin-binding ability of polymers among haptoglobin phenotypes. Clin. Chem.43, 2012–2013 (1997). [PubMed] [Google Scholar]
- 12.Lim, S. K., Ferraro, B., Moore, K. & Halliwell, B. Role of haptoglobin in free hemoglobin metabolism. Redox Rep.6, 219–227 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Wejman, J. C., Hovsepian, D., Wall, J. S., Hainfeld, J. F. & Greer, J. Structure and assembly of haptoglobin polymers by electron microscopy. J. Mol. Biol.174, 343–368 (1984). [DOI] [PubMed] [Google Scholar]
- 14.Vlierberghe, H. V., Langlois, M. & Delanghe, J. Haptoglobin polymorphisms and iron homeostasis in health and in disease. Clin. Chim. Acta345, 35–42 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Schaer, D. J. et al. CD163 is the macrophage scavenger receptor for native and chemically modified hemoglobins in the absence of haptoglobin. Blood107, 373–380 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Moestrup, S. K. & Møller, H. J. CD163: a regulated hemoglobin scavenger receptor with a role in the anti-inflammatory response. Ann. Med.36, 347–354 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Møller, H. J., Peterslund, N. A., Graversen, J. H. & Moestrup, S. K. Identification of the hemoglobin scavenger receptor/CD163 as a natural soluble protein in plasma. Blood99, 378–380 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Nielsen, M. J., Madsen, M., Møller, H. J. & Moestrup, S. K. The macrophage scavenger receptor CD163: endocytic properties of cytoplasmic tail variants. J. Leukoc. Biol.79, 837–845 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Madsen, M. et al. Molecular characterization of the haptoglobin.hemoglobin receptor CD163. Ligand binding properties of the scavenger receptor cysteine-rich domain region. J. Biol. Chem.279, 51561–51567 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Hohenester, E., Sasaki, T. & Timpl, R. Crystal structure of a scavenger receptor cysteine-rich domain sheds light on an ancient superfamily. Nat. Struct. Biol.6, 228–232 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Bowdish, D. M. E. & Gordon, S. Conserved domains of the class A scavenger receptors: evolution and function. Immunol. Rev.227, 19–31 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Nielsen, M. J., Andersen, C. B. F. & Moestrup, S. K. CD163 Binding to Haptoglobin-Hemoglobin Complexes Involves a Dual-point Electrostatic Receptor-Ligand Pairing. J. Biol. Chem.288, 18834–18841 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reichhardt, M. P., Loimaranta, V., Lea, S. M. & Johnson, S. Structures of SALSA/DMBT1 SRCR domains reveal the conserved ligand-binding mechanism of the ancient SRCR fold. Life Sci. Alliance3, e201900502 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ojala, J. R. M., Pikkarainen, T., Tuuttila, A., Sandalova, T. & Tryggvason, K. Crystal structure of the cysteine-rich domain of scavenger receptor MARCO reveals the presence of a basic and an acidic cluster that both contribute to ligand recognition. J. Biol. Chem.282, 16654–16666 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Zhao, Z. & Michaely, P. The role of calcium in lipoprotein release by the low-density lipoprotein receptor. Biochemistry48, 7313–7324 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andersen, C. B. F., Madsen, M., Storm, T., Moestrup, S. K. & Andersen, G. R. Structural basis for receptor recognition of vitamin-B(12)-intrinsic factor complexes. Nature464, 445–448 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Andersen, C. B. F. & Moestrup, S. K. How calcium makes endocytic receptors attractive. Trends Biochem. Sci.39, 82–90 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta, R. & Brunak, S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac. Symp. Biocomput. Pac. Symp. Biocomput. 310–22 (2002). [PubMed]
- 30.Etzerodt, A., Maniecki, M. B., Møller, K., Møller, H. J. & Moestrup, S. K. Tumor necrosis factor α-converting enzyme (TACE/ADAM17) mediates ectodomain shedding of the scavenger receptor CD163. J. Leukoc. Biol.88, 1201–1205 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Graversen, J. H. et al. Trimerization of Apolipoprotein A-I Retards Plasma Clearance and Preserves Antiatherosclerotic Properties. J. Cardiovasc. Pharmacol.51, 170–177 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Tang, Y. et al. Regulation of Antibody-Dependent Cellular Cytotoxicity by IgG Intrinsic and Apparent Affinity for Target Antigen. J. Immunol.179, 2815–2823 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Skytthe, M. K. et al. Re-evaluation of the measurement of haptoglobin in human plasma samples. Scand. J. Clin. Lab. Investig.82, 467–473 (2022). [DOI] [PubMed] [Google Scholar]
- 34.Santos, R. F., Oliveira, L. & Carmo, A. M. Tuning T Cell Activation: The Function of CD6 At the Immunological Synapse and in T Cell Responses. Curr. Drug Targets17, 630–639 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Chappell, P. E. et al. Structures of CD6 and Its Ligand CD166 Give Insight into Their Interaction. Struct. (Lond., Engl.:1993)23, 1426–1436 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu, B. et al. Interactions of ferritin with scavenger receptor class A members. J. Biol. Chem.295, 15727–15741 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng, C. et al. Recognition of lipoproteins by scavenger receptor class A members. J. Biol. Chem.297, 100948 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stodkilde, K., Torvund-Jensen, M., Moestrup, S. K. & Andersen, C. B. Structural basis for trypanosomal haem acquisition and susceptibility to the host innate immune system. Nat. Commun.5, 5487 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Suloway, C. et al. Automated molecular microscopy: The new Leginon system. J. Struct. Biol.151, 41–60 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci.30, 70–82 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terwilliger, T. C., Sobolev, O. V., Afonine, P. V. & Adams, P. D. Automated map sharpening by maximization of detail and connectivity. Acta Crystallogr. Sect. D.74, 545–559 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramírez-Aportela, E. et al. Automatic local resolution-based sharpening of cryo-EM maps. Bioinformatics36, 765–772 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kidmose, R. T. et al. Namdinator – automatic molecular dynamics flexible fitting of structural models into cryo-EM and crystallography experimental maps. IUCrJ6, 526–531 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. Biol. Crystallogr66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kabsch, W. XDS. Acta Crystallogr D. Biol. Crystallogr66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCoy, A. J. et al. Phaser crystallographic software. J. Appl Crystallogr40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. Biol. Crystallogr60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Etzerodt, A. et al. Plasma clearance of hemoglobin and haptoglobin in mice and effect of CD163 gene targeting disruption. Antioxid. Redox Signal18, 2254–2263 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic structure factors and coordinates for the structure of human Hp are available at the Protein Data Bank under accession number 9FNM. The Cryo-EM maps and coordinates for the structure of CD163-HpHb are available at the EMDB under accession numbers 9FHB, 9FMU, and 9FNO. Other data are available from the corresponding author upon request. Source data are provided with this paper.