

Abstract
Marine cyclopianes are a family of diterpenoid with novel carbon skeleton and diverse biological activities. Herein, we report our synthetic and chemical proteomics studies of cyclopiane diterpenes which culminate in the asymmetric total synthesis of conidiogenones C, K and 12β-hydroxy conidiogenone C, and identification of Immunity-related GTPase family M protein 1 (IRGM1) as a cellular target. Our asymmetric synthesis commences from Wieland-Miescher ketone and features a sequential intramolecular Pauson-Khand reaction and gold-catalyzed Nazarov cyclization to rapidly construct the 6-5-5-5 tetracyclic skeleton. The stereocontrolled cyclopentenone construction is further investigated on complex settings to demonstrate its synthetic utility. Furthermore, using an alkyne-tagged conidiogenone C-derived probe, IRGM1, a master regulator of type I interferon responses, is identified as a key cellular target of conidiogenone C responsible for its anti-inflammatory activity. Preliminary mechanism of action studies shows that conidiogenone C activates IRGM1-mediate dysfunctional mitochondria autophagy to maintain mitochondria quality control of inflammatory macrophages.
Subject terms: Natural product synthesis, Natural product synthesis
The cyclopiane diterpenoids are a class of complex natural products with unique carbon skeleton and diverse biological activities. Herein, the authors report the total synthesis and chemical proteomics studies of cyclopiane diterpenes which culminate in the asymmetric total synthesis of conidiogenones C, K and 12β-hydroxy conidiogenone C, and identification of Immunity-related GTPase family M protein 1 as a cellular target of conidiogenone C.
Introduction
Terpenoid natural products with novel carbon skeletons and diverse biological activities continue to attract attentions from synthetic community1–3. One recent example is the cyclopiane diterpenes4–13. These natural products share a 6-5-5-5 tetracyclic carbon skeleton with 6-9 stereocenters, including 3-4 all-carbon quaternary centers, two of which are vicinal (Fig. 1)14–17. Furthermore, the different oxidation state of A and D ring makes these natural products as challenging synthetic target. Since the first two members conidiogenone (1) and conidiogenol (2) were isolated from the genus Penicillium4, more than 20 congeners have been discovered, some of which displayed important biological activities. For example, conidiogenone (1) and conidiogenol (2) were reported as potent and selective inducers of conidiogenesis in P. cyclopium while conidiogenone C (4) showed potent cytotoxicity against HL60 cell (IC50 = 38 nM) as well as esophageal cancer cell lines5,10. These natural products also exhibited significant activity against methicillin-resistant Staphylococcus aureus (MRSA), with conidiogenone B (3) displaying MIC = 8 µg/mL6. More recently Zhang and co-workers reported several congeners exhibited activity against MRSA, and discussed the structure-antibacterial activity relationship (SAR) of these natural products13. Furthermore, the anti-inflammatory activity of 12β-hydroxy conidiogenone C (9) was also disclosed, which showed comparable potency to indomethacin12.
Fig. 1. Cyclopiane diterpenes.

A Natural products with oxidazed A ring. B Natural Products with oxidazed A and D rings.
Due to the unique skeleton and intriguing biological activities, these natural products quickly became attractive synthetic targets (Fig. 2A)18–22. Tu and co-workers reported the first total syntheses of conidiogenone (1), conidiogenol (2) and conidiogenone B (3) in 24-27 steps through an efficient and well-designed semi-pinacol rearrangement and corrected the absolute configuration of conidiogenone B23. In 2019, Snyder and co-workers coined the “quaternary-centre-guided synthesis” concept and reported the asymmetric total synthesis of conidiogenone B (3) in 13 steps and its conversion into conidiogenone (1) and conidiogenol (2) following Tu’s method24,25. Furthermore, through their excellent synthetic approach, conidiogenones C (4) and D (10) were also obtained in 16 steps by branching off an early-stage intermediate. In 2020, Zhai and co-workers reported26 their successful approach to conidiogenone (1), conidiogenol (2) and conidiogenone B (3) in 14-17 steps featured a late stage Danheiser annulation for constructing tetraquinane skeleton with vicinal all-carbon quaternary centers, followed by ozonolysis and intramolecular aldol condensation to forge the cyclohexanone ring. During the submission process of this manuscript, Han, Lee and coworkers reported their approach to this family. Through trimethylenemethane diyl-mediated cycloaddition, the tetracyclic conidiogenone core was obtained which enabled a divergent synthesis of five conidiogenones and formal synthesis of conidiogenone B in 19-26 steps27. More recently, Carreira and co-workers reported the first total synthesis of the related tetracyclic diterpenoid aberrarone (not shown) through an elegant Nazarov-cyclopropanation-aldol cascade, Jia and co-workers also achieved total synthesis of aberrarone through an impressive Mn-mediated radical cascade cyclization28,29. Beside these total synthesis investigation, a synthetic proposal through computational planning was disclosed by the Grzybowski group30. In addition, several enzymes involved in the biosynthetic pathway of these natural products were recently discovered31–33. Herein, we report a collaborative effect in the asymmetric total synthesis and target identification of conidiogenone C (4), its oxidized congeners conidiogenone K (8) and 12β-hydroxy conidiogenone C (9) and reveal IRGM1 as a key cellular target of conidiogenone C (4) responsible for its anti-inflammatory activity.
Fig. 2. Reported total syntheses and our synthetic strategy of cyclopiane diterpenoids.

A Previous total synthesis of cyclopiane diterpenoids. B Total synthesis of cyclopiane diterpenoids by gold-catalyzed Nazarov cyclization.
Results
Total Synthesis
As conidiogenones C (4), K (8) and 12β-hydroxy conidiogenone C (9) feature an oxidized D ring with an addition all-carbon quaternary center compared to the previously synthesized conidiogenone B, conidiogenone and conidiogenol, we planned to construct this highly substituted five-membered ring (D ring) at late stage by utilizing the cyclopentenone 16 as corresponding precursor (Fig. 2B). This key intermediate 16 is proposed to be accessed from precursor 13 through gold-catalyzed 1,3-acyloxy migration followed by Nazarov cyclization34–40. As shown in Fig. 2B, 1,6-enyne intermediate 11 obtained from Wieland-Miescher ketone through an established synthetic route41, could be advanced to propargylic benzoate 13 through intramolecular Pauson-Khand reaction (to give 12) and Sonogashira cross-coupling. In the key step, 13 would first undergo gold-catalyzed 3,3-rearrangement of enynyl benzoate and after gold catalyst coordinated the formed allenoic benzoate, the resulting “bent-allene” structure 14 would react in a sequence of Nazarov cyclization, protonation and hydrolysis of intermediate 15, to ultimately form the tetracyclic product 16. The challenging all-carbon quaternary centers at C11 and C14 could be constructed through alkylation and conjugate addition by Nagata reagent (Et2AlCN). From 17, oxidation state adjustments in the A and D rings would furnish conidiogenone C (4), conidiogenone K (8) and 12β-hydroxy-conidiogenone C (9). As 21 entities of the cyclopiane diterpenes (25 members in total) exhibit a highly oxidized D ring, the previously inaccessible natural products could be synthesized through this strategy.
The synthetic route commenced from Wieland-Miescher ketone which was prepared in one step by Luo’s procedure (Fig. 3A)42. Following the reported sequence41 including selective ketone protection, Weitz-Scheffer epoxidation, Eschenmoser-Tanabe fragmentation and Wittig olefination, the 1,6-enyne 11 was synthesized. (For details, see page S15-S17 of the Supplementary Information). Through Co2(CO)8-mediated intramolecular Pauson-Khand reaction43–47, the tricyclic skeleton was prepared in only 6 steps from commercially available 2-methyl-1,3-cyclohexanedione. The success in setting the all-carbon quaternary center at C5 may be explained by a Thorpe-Ingold effect imparted by C9 stereocenter. The stereochemistry of C5 was further confirmed by X-ray analysis of crystalline 18 which was synthesized through stereoselective hydrogenation of the Pauson-Khand product 12.
Fig. 3. Total Synthesis of 12β-hydroxy conidiogenone C, conidiogenone C and conidiogenone K.

A Total synthesis of 12β-hydroxy conidiogenone C. B Total synthesis of conidiogenone C and conidiogenone K.
Ketone 18 was quickly elaborated to key enynyl acetates 21 and 22 by vinyl triflate formation, Sonogashira cross-coupling and acylation. With the two diastereomers in hand, we started investigating the key step (Table 1). When either substrate 21 or 22 were submitted to Zhang’s conditions (entries 1 and 3)34, the cyclopentanone ring was formed as expected, but unsatisfactory diastereomeric ratio was observed. Echavarren’s catalyst (Au(MeCN)(JohnPhos)SbF6)35 yielded only minor improvement (Table 1, entry 2). Apparently, the C6 stereocenter was not able to impart sufficient substrate control as both products 16 and 24 are formed in all cases. Chirality transfer from propargylic alcohol moiety was not efficient either. During this investigation, Zhang and Liu reported48 significant improvement by using enynyl benzoate substrates and, more importantly, identified t-BuBrettphosAuCl as superior catalyst for chirality transfer. Inspired by their work, the enynyl benzoates 13 and 23 were prepared. When 13 was submitted to the reported conditions, to our delight, the desired product 16 was obtained as major product (Table 1, entry 7). The undesired 24 could be synthesized when the “unmatched” diastereomer 23 was used as substrate. (Table 1, entry 6). Further optimization showed that a combination of t-BuBrettphosAuCl and AgSbF6, yielded exclusively the tetracyclic product 25 (Table 1, entry 8). In all cases, the cyclopentanone product was directly formed in the reaction mixture and no benzoate product was observed which also fascinated the overall transformation.
Table 1.
Gold-Catalyzed Nazarov Cyclization[a,b]
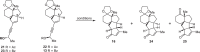 | ||||
|---|---|---|---|---|
| Entry | Substrate | [Au] (5 mol%) | Solvent | Yield (%)[b] |
| 1[c] | 21 | Ph3PAuCl, AgSbF6 | CH2Cl2 | 51% (16:24 = 3:2) |
| 2[d] | 21 | Au(MeCN)(JohnPhos)SbF6 | THF/H2O (0.2% vol%) | 35% (16:24 = 2:1) |
| 3[c] | 22 | Ph3PAuCl, AgSbF6 | CH2Cl2 | 31% (16:24 = 5:1) |
| 4 | 22 | t-BuBrettPhosAuCl, NaBARF | toluene | 43% (16:24 = 3:2) |
| 5[d] | 23 | Au(MeCN)(JohnPhos)SbF6 | THF/H2O (0.2% vol%) | 61% (16:24 = 2:1) |
| 6 | 23 | t-BuBrettPhosAuCl, NaBARF | toluene | 41% (16:24 = 1:10) |
| 7 | 13 | t-BuBrettPhosAuCl, NaBARF | toluene | 42% (16:24 = 9:1) |
| 8[e] | 13 | t-BuBrettPhosAuCl, AgSbF6 | CH2Cl2 | 75% 25 |
aUnless otherwise noted, the reaction was carried out with gold catalyst (5 mol%) at r.t.
bIsolated yield.
cPh3PAuCl (3 mol%), AgSbF6 (3 mol%).
dThe reaction stirred at −10 °C.
eAgSbF6 (10 mol%).
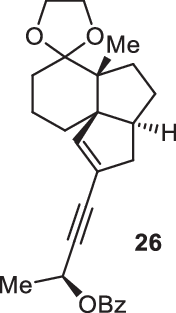
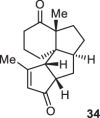
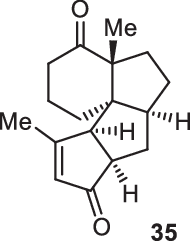
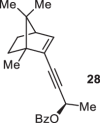
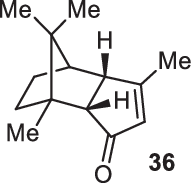
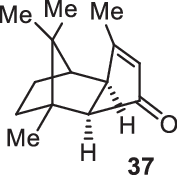
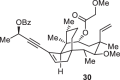
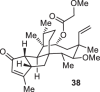
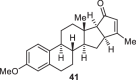
Previously, during the total synthesis of kadcoccinic acid A trimethyl ester37, Trost and co-workers observed that when utilizing the two diastereomers of enynyl acetate as substrates, the Nazarov cyclization led to different cyclopentenone product. However, based on the above result, the cyclopentenones 24 and 25 could be constructed via efficient chirality transfer from the corresponding enynyl benzoate, so we further explored this transformation with other substrates containing multiple stereocenters and functional groups. As shown in Table 2, the stereochemistry of cyclopentenones 34-41 was controlled by the propargylic stereocenter of precursors 26-33, which were prepared from intermediate 18, camphor, pleuromutilin and estrone. We believe this approach could become a valuable option for skeleton construction in complex settings.
Table 2.
Stereocontrolled Cyclopentenone Construction[a,b]
| Substrate | Product | Yield (%) | Substrate | Product | Yield (%) |
|---|---|---|---|---|---|
 |
 |
74% | 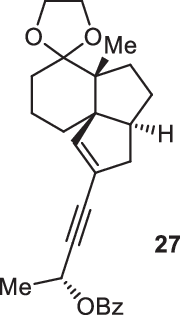 |
 |
75% |
 |
 |
73% | 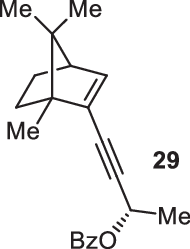 |
 |
70% |
 |
 |
59% | 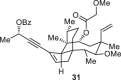 |
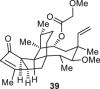 |
62% |
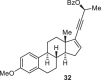 |
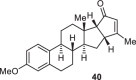 |
68% | 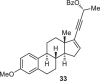 |
 |
71% |
aThe reaction was carried out with t-BuBrettphosAuCl (5 mol%), AgSbF6 (10 mol%) in wet CH2Cl2 at r.t.
bIsolated yield.
After constructing the tetracyclic skeleton in 10 steps, modification of D ring was required (Fig. 3A) for our first target 12β-hydroxy conidiogenone C (8) which has an addition secondary alcohol group comparing conidiogenone C. While constructing the poly-substituted cyclopentane with two all-carbon quaternary centers are challenging, cyclopentanone intermediate 25 severed as a practical precursor. The C11 and C14 quaternary centers were synthesized through alkylation and an efficient 1,4-addition of Nagata reagent49–54, both from the convex face of the cyclopentenone. Notably, freshly prepared Et2AlCN in combination with TMSCl was crucial for high yield and proved uniquely suitable to set this all-carbon quaternary center. In this scenario, the nitrile group serves as masked primary alcohol for our targets. Commenced from key intermediate 43, the initially surveyed metallic sodium or SmI2 mediated reductions for stereoselective construction of the C12 alcohol resulted in no product formation. We then switched to NaBH4 reduction and after screening reaction solvents and temperatures, a separable mixture of 44 and 45 was obtained. The tetracyclic skeleton as well as the stereochemistry from alkylation and 1,4-addition was further confirmed through X-ray of compound 44 and this undesired diastereomer was recycled to 43 through Ley oxidation. Compound 45 features the tetracyclic skeleton and all four all-carbon quaternary stereocenters. From this intermediate, the late-stage modification of A ring inspired by Tu’s synthetic route23 was investigated. Compound 46 was synthesized after removing the ketal protecting group and protected the hydroxy group as propionate. Then through methyl enol ether formation and allylic oxidation as reported by Corey and Yu55, unsaturated ketone was synthesized. From the enone intermediate, after deprotonation by LDA, the α face of in-situ formed enolate was blocked by the adjacent B ring56,57, thereby the C4 methyl group was correctly installed through this stereoselective alkylation. Notably, propionate protection of the C12 hydroxy group proved necessary as the related acetate underwent competitive alkylation during the methylation at C4. With 47 in hand, the planned triple reduction58 of ketone (Stork-Danheiser reductive squence59), ester and nitrile groups were successfully achieved, and the cyclohexanone ring with χ-methyl group was constructed efficiently. The final chemoselective reduction of neopentyl aldehyde 48 in the presence of an unsaturated ketone group was achieved under our previously reported combination60,61 of NaBH4 and acetylacetone and total synthesis of 12β-hydroxy conidiogenone C was achieved.
With a synthetic route to 12β-hydroxy conidiogenone C established, we directed our attention to conidiogenones C and K (Fig. 3B). To reach these two natural products, the C12 carbonyl group needed to be removed. Attempts at direct conversion to methylene through Huang Minlon modification of Wolf-Kishner reduction or Caglioti reduction resulted in recovered starting material 43. The failure is likely due to the sterically hindered environment of the C12 carbonyl group, which preclude direct nucleophilic attack by hydrazine hydrate or NH2NHTs. To overcome this problem, the carbonyl group was deprotonated at C13 by KHMDS and transformed into the vinyl triflate with Comins’ reagent. The planned two-step procedure consisting of palladium-catalyzed reductive cross-coupling and hydrogenation were attempted, but neither formic acid nor Et3SiH provided conversion, indicating the steric hindrance preclude oxidative addition of palladium catalyst. Ultimately, we found direct hydrogenolysis62–65 of the vinyl triflate 49 enabled the transformation into 50 after treatment with acid. A plausible pathway involves hydrogenation of the double bond followed by substitution of the labile triflate intermediate. From 50, through a similar sequence as above including modification of A ring and chemoselective aldehyde reduction, conidiogenone C (4) was synthesized. As the NaBH4-mediated transformation of conidiogenone C (4) to conidiogenol D (5) was reported10, our work also constituted a formal synthesis of conidiogenol D (5). After Weitz-Scheffer epoxidation of conidiogenone C, the first total synthesis of conidiogenone K was completed (Fig. 3B). The spectra of those three natural products were identical in all respects to those previously reported7,12,24.
Biological evaluation and probe synthesis
After completing our synthesis, we firstly verified the cytotoxicity of these three cyclopiane diterpenes against HL60 cells5. Disappointedly, none of them probed to be toxic even at 40 µM concentration (Fig. S1). Later on, we evaluated the anti-inflammatory activity of these three cyclopiane diterpenes in type I interferon production in LPS-stimulated RAW264.7 cells12. Much to our delight, conidiogenone C (4), rather than conidiogenone K (8) and 12β-hydroxy conidiogenone C (9), downregulated the mRNA expression levels of IFN-stimulated genes (ISGs) Ifnb, Mx1 (Figs. 4A and S2). Of note, a series of both human and mouse cancer (mostly leukemia) cell lines (Raji, HL-60, Jurkat, K562, A20, etc.) have also been evaluated, and no inhibition of IFN-stimulated genes (ISGs) (Ifnb, Mx1) expression by conidiogenone C (4) was observed (Fig. S3). Taken together, since the anti-inflammatory activity was very specific and selective in inflammatory macrophages, thereby it would be worthwhile to further decipher the cellular mechanism of action (MOA) of immunosuppressant conidiogenone C (4), given that IFN-I plays a central role in the pathogenesis of autoimmune diseases66.
Fig. 4. Identification of IRGM1 as a cellular target.

A Mx1 and Ifnb1 expression in Raw264.7 treated with conidiogenone C by quantitative RT-PCR (n = 3). B The synthesis of conidiogenone C probe (51). C Mx1 and Ifnb1 expression in Raw264.7 treated with conidiogenone C or conidiogenone C probe (51) by quantitative RT-PCR (n = 3). D Concentration-dependent, Time-dependent and Competitive labeling of conidiogenone C probe (51) in living Raw264.7 cells. E Workflow for the quantitative chemoproteomic profiling of conidiogenone C probe (51) in inflammatory macrophages. F Competitive labeling of probe (51) by conidiogenone C (4). Rho: Rhodamine. CBB: Coomassie Brilliant Blue. G Overlap of the proteins identified by conidiogenone C probe (51) H The shortlist of 5 target proteins with enrichment ratio L/H > 10 and competition ratio L/M > 1.0 in three replicates. L/H: the enrichment ratio of the “light” group using 20 µM of conidiogenone C probe (51) versus the “heavy” group using 20 µM of conidiogenone C (4); L/M: the competition ratio of the “light” group using 20 µM of conidiogenone C probe (51) versus the “medium” group using 20 µM of conidiogenone C probe (51) together with 20 times excess of conidiogenone C (4) for competition. I In vitro competitive pull-down assay using conidiogenone C probe, followed by western blot to confirm that conidiogenone C directly binds to IRGM1 protein. Con C = conidiogenone C. J The representative MS/MS spectrum for the identification of conidiogenone C-labeled IRGM1 peptide containing one of the binding residue C373, MS/MS spectrum of conidiogenone C-labeled IRGM1 peptide containing binding residues C374 and C375 had been showed in Supplementary Information. The yellow symbol refers to the protein IRGM1. K, L In vitro pull-down assay of conidiogenone C probe (51) with WT IRGM1 and Cys-to-Ala mutants (n = 3). All measurements are presented as mean ± SD for three biological replicates (a, c, l), n = 3 independent experiments with similar results (d, f, i). Statistical differences were determined by one-way ANOVA followed by Dunnett’s multiple comparison tests (l). Source data are provided as a Source Data file.
Therefore, we introduced an alkyne handle and synthesized an activity-based conidiogenone C probe (51) (Fig. 4B), which could be subsequently linked with azide-fluorescent dye or azide-biotin to label the direct cellular protein targets. Again, the mRNA expression levels of ISGs after treatment with conidiogenone C (4) or probe (51) were tested, and conidiogenone C probe (51) downregulated the mRNA expression of ISGs in LPS-stimulated RAW264.7 cells, similar to conidiogenone C (4) (Fig. 4C).
Chemical proteomics and target identification
Next, we conducted conidiogenone C probe (51) labeling experiments in LPS-stimulated RAW264.7 living cells or cell lysates (Figs. 4D and S4A). Initial Raw264.7 living cell labeling by treating with conidiogenone C probe (51) indicated a decrease in signal intensity with increasing probe concentrations, while time-dependent labeling proved to be almost empty (Fig. S4A). The unsatisfactory live cell labeling experiments might be largely due to the facile hydrolysis of the probe ester bond in the Raw264.7 cells. Later on, upon 3 h treatment of LPS-stimulated RAW264.7 cell lysates with 20 µM of conidiogenone C probe (51), we observed strong probe labeling based on in-gel fluorescence scanning. Subsequent competitive probe labeling upon coincubation with increasing concentrations of conidiogenone C (4) indicated that conidiogenone C probe (51) binds to similar cellular targets with conidiogenone C (4), as labeling fluorescence intensity decreased accordingly.
As shown in Figs. 4E, F, we then proceeded to competitive chemoproteomic profiling with conidiogenone C probe (51) in LPS-stimulated RAW264.7 cell lysates. The lysis was then reacted with azide-biotin, and after enrichment and digestion, the peptides were labeled as “light, “medium” or “heavy” by dimethyl labeling. The labeled proteins were identified and quantified by SDS-PAGE or LC-MS/MS analysis. In all three replicates, the enrichment ratio L/H was assigned as the “light” group by using 20 µM of conidiogenone C probe (51) versus the “heavy” group by using 20 µM of conidiogenone C (4), while the competition ratio L/M was calculated as the “light” group by using 20 µM of conidiogenone C probe (51) versus the “medium” group by using 20 µM of conidiogenone C probe (51) together with 20 times excess of conidiogenone C (4) for competition. Overall, after performing three replicates, 262, 145, and 145 proteins with both L/H enrichment ratio and L/M competition ratio over 1.0 have been found respectively (for more detail, see additional Excel file “proteomics tables”), among them 59 proteins have been found in all three biological replicates and thereby assigned as potential targets (Fig. 4G and Table S7). Of note, further Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis and Gene ontology (GO enrichment analysis of these 59 proteins using DAVID (Fig. S4C, D) provided limited information for our target protein assessment66.
Finally, as seen in Fig. 4H, a shortlist including XPO1, AT1A1, IRGM1, GNAI2, and TNPO1 was selected out of 59 proteins with enrichment ratio L/H > 10 and competition ratio L/M > 1.0 in all three replicates. Among them, Immunity-related GTPase family M protein 1 (IRGM1), an important protein which regulates innate immunity and inflammatory response67–70, was identified as the potential promising cellular target (Fig. 4H). Competitive pull-down assay by coincubation with conidiogenone C (4) either in lysates or living cells (Fig. 4I and S4B) suggested that conidiogenone C (4) directly binds to IRGM1 protein.
Covalent IRGM1 modification
In possession of an electrophilic enone moiety which very often serves as reactive warhead that can covalently react with nucleophilic amino acid residues (i.e. cysteine) on target proteins71–75, the potential covalent IRGM1 modifications of conidiogenone C (4) has been further investigated. To decipher the specific covalent binding sites of IRGM1, conidiogenone C (4) was incubated with purified recombinant IRGM1 protein for 1 h at 29 °C. Then the conidiogenone C-IRGM1 complex was digested with trypsin and the conidiogenone C-labeled IRGM1 fragment was further detected by LC-MS/MS analysis. Much to our delight, one specific peptide 368FLPCVCCCLR377 was identified which contains three conidiogenone C-modified residues C373, C374, C375 (Fig. 4J and S6). Later on, the three identified binding sites (C373, C374, C375) of IRGM1 were mutated and then subjected to the pull-down experiments. Whereas site mutagenesis of single cysteine residue (C373A, C374A, C375A) to Ala partially weakened the binding ability of conidiogenone C with IRGM1, the whole mutagenesis of all 3 cysteine residues to Ala (3CA) drastically interrupted the interaction (Fig. 4K–L). Therefore, cysteine residues (C373A, C374A, C375 A) were identified as nucleophilic amino acid hotspots responsible for the covalent modification of conidiogenone C (4) on IRGM1.
IRGM1 modulation
IRGM1, one of Immunity-related GTPase family protein, is a master suppressor of type I interferonopathy67–70. To confirm if IRGM1 is directly responsible for the type I IFN suppression of conidiogenone C (4) observed in inflammatory macrophages, cellular IRGM1 was knocked down by transfecting corresponding small interfering RNA (siRNA) into RAW264.7 cells (Fig. S7). Indeed, upon silencing of IRGM1, no more inhibition of IFN-stimulated genes (ISGs) (Ifnb, Mx1) expression by conidiogenone C (4) was observed (Fig. 5A). Of note, upon treatment with conidiogenone C (4), both mRNA and protein expression levels of IRGM1 showed no dramatic change, nor did the cellular thermal shift of IRGM1 (Fig. S8A, B).
Fig. 5. Evaluation of IRGM1 activation by conidiogenone C (4).

A Ifnb1 and Mx1 expression in Raw264.7 transfected using IRGM1 siRNA or negative siRNA, followed by LPS and conidiogenone C (4) treatment (n = 3). B Ifnb1 expression in Raw264.7 with or without mtDNA depletion by ethidium bromide (EtBr), followed by LPS and conidiogenone C treatment (n = 3). C Cytosolic mtDNA expression in Raw264.7 pretreated with LPS and conidiogenone C (n = 3). D, E Mitochondrial membrane potential in Raw264.7 stained with the potentiometric dye JC-1 measured by flow cytometry. Cells were treated with DMSO or conidiogenone C after transfected using Irgm1 siRNA or negative siRNA. Representative dot plot in (D), quantification in (E) (n = 3). F, G Mitochondrial membrane potential in Raw264.7 stained with the potentiometric dye JC-1 measured by flow cytometry. Cells were treated with Bafilomycin A1(Bar A1) or conidiogenone C. Representative dot plot in (F), quantification in (G) (n = 3). H A schematic depiction of MOA that conidiogenone C modulates IRGM1-mediated type I interferon production. mtDNA: Mitochondrial DNA. All measurements are presented as mean ± SD for three biological replicates (a–c, e, g). Statistical differences were determined by a one-way ANOVA followed by Dunnett’s multiple comparison tests(a) or a two-sided Student’s t-test (b, c, e, g). Source data are provided as a Source Data file.
Previous studies have identified that IRGM impacts mitochondrial DNA (mtDNA)-dependent induction of type I interferon (IFN-I) responses mainly via regulating mitophagy flux67,68. Released mtDNA in cytosol, would act as danger-associated molecular patterns (DAMPs), thereby induce activation of the type I IFN response67,68,76. To see whether the suppression of IFN-I by conidiogenone C (4) depends on released mtDNA or not, we depleted mtDNA by cell culture in ethidium bromide (EtBr). Not surprisingly, conidiogenone C (4) no longer inhibited the expression of Ifnb1 after depleting mtDNA (Fig. 5B). Meanwhile, we also observed the significant decrease of cytosolic mtDNA in the LPS-stimulated RAW264.7 cells upon treatment with conidiogenone C (4) (Fig. 5C). Collectively, conidiogenone C (4) suppressed the type I IFN response by reducing the release of mtDNA into cytosol.
Since LPS treatment normally induces mitochondrial dysfunction77, we argued that conidiogenone C (4) might activate IRGM1-mediate dysfunctional mitochondria autophagy, to rescue LPS-induced mitochondrial dysfunction. Indeed, after measuring mitochondrial membrane potential in LPS-stimulated RAW264.7 cells, we noticed that conidiogenone C (4) significantly rescued the low mitochondrial membrane potential induced by LPS treatment (Fig. 5F, G). In addition, either inhibition of lysosomal function with autophagy inhibitor bafilomycin A1 or silencing of IRGM1 would drastically weakened the rescue effect caused by conidiogenone C (4) (Fig. 5D–G). Thus, in inflammatory macrophages, conidiogenone C (4) could activate IRGM1-mediated mitochondria autophagy, thereby maintain the mitochondria quality control.
Overall, as shown in Fig. 5H, in LPS-stimulated RAW264.7 cells, owing to mitochondrial dysfunction, released mtDNA in cytosol constantly serves as DAMPs to further enhance type I interferon production. Upon treatment with conidiogenone C (4), it directly binds to IRGM1 to promote IRGM1-mediate dysfunctional mitochondria autophagy, thus eliminates cytosolic mtDNA and suppresses the type I interferon response in inflammatory macrophages. Our results also demonstrate that conidiogenone C (4) is the first small molecular activator of IRGM1.
Discussion
In summary, we completed the asymmetric total syntheses of conidiogenone C, conidiogenone K and 12β-hydroxy conidiogenone C. Our synthesis features a gold-catalyzed 1,3-acyloxy migration and Nazarov cyclization with efficient chirality transfer. Meanwhile, the hydrogenation of vinyl triflate and chemoselective reduction of aldehyde through combination of NaBH4 and acetylacetone can also be envisaged to find further application in total synthesis. Furthermore, biological evaluation disclosed conidiogenone C (4) as anti-inflammatory agent. By performing chemical proteomic profiling with conidiogenone C probe (51) in LPS-stimulated RAW264.7 cell, we identified IRGM1 as a key cellular target of conidiogenone C (4). Preliminary MOA studies reveal that conidiogenone C (4) suppresses the type I interferon production by activating IRGM1-mediate dysfunctional mitochondria autophagy, thereby maintain mitochondria quality control of inflammatory macrophages. Our present total synthesis has opened the gate for SAR studies and further preparation of conidiogenone C (4) analogues. Meanwhile, since IRGM1 is a guardian of mitochondrial DAMPs–mediated autoinflammation67, the identification of conidiogenone C as the very first small-molecule activator of IRGM1 could potentially shed light on the further investigation of IRGM1 biology and potential therapeutical application for the treatment of a series of autoimmune diseases (IBD, Psoriasis, SLE, etc.)66.
Method
General information for organic synthesis
All reactions involving air or moisture sensitive reagents were carried out under an argon atmosphere with dry solvents or anhydrous conditions, unless otherwise noted. A heat-gun at 650 °C under vacuum was used to further dry glassware (over 70 °C). Subsequently, stuffed with the rubber stopper, back-filling with inert gas was carried out three times. Solids were dissolved with the appropriate solvent and added to reaction under inert gas counter flow. Liquids reagents were transferred to reaction vessels by oven-dried stainless-steel cannulas or nitrogen flushed syringes. Low temperature reactions were carried out in a Dewar vessel filled with ethanol (−78 °C) or distilled water/ice (0 °C). High temperature reactions equipped with a reflux condenser were heated with silicon oil bath.
NMR spectra were recorded on a Brucker Avance III HD 500 MHz and 400 MHz spectrometer equipped with a CroProbe TM. Chemical shifts were reported in parts per million (ppm) respectively to the residual solvent signal (1H NMR: CDCl3 (7.26 ppm); CD3OD (3.31 ppm). 13C NMR: CDCl3 (77.2 ppm); CD3OD (49.0 ppm)). The reported data is represented as follows: chemical shift in parts per million (ppm, δ scale), multiplicity, coupling constants J in Hz, integration intensity and proton assignment. Abbreviations used for analysis of multiplets are as follows: s (singlet), br (broad singlet), d (doublet), t (triplet), q (quartet), quin (quintet), h (hextet), and m (multiplet). Variable temperature NMR spectroscopy was performed at the Northwest A&F University NMR facility.
Mass spectroscopy (MS) experiments were performed in high resolution with an AB SCIEX Triple TOF 5600+ spectrometer (AB SCIEX, Boston, MA, USA). IR spectra were recorded on a Perkin-Elmer Frontier FT-IR spectrometer.
Cell culture
RAW264.7 cells were cultured in DMEM (Gibco) supplemented with 10% FBS (Excell) and Penicillin/Streptomycin (Gibco). Cells were grown at 37 °C, 5% CO2 in a 95% humidified atmosphere. In vitro study, RAW264.7 cells were pre-stimulated with 100 ng/mL LPS (Sigma) for 3 h, and then treated with Conidiogenone C or Conidiogenone C probes.
Cell viability assay
RAW264.7 cells were seeded into 96-well plate overnight at 5000 cells per well The culture medium was replaced with medium containing varying concentrations of Conidiogenone C, 12β-Conidiogenone C or Conidiogenone K for 24 h. Cell numbers were assessed using the CCK-8 reagent at OD 450.
Fluorescence labeling experiments
For the labeling of proteins in cell lysates, frozen RAW264.7 cells were resuspended in ice-cold 0.1% Triton/PBS buffer. The cells were lysed by sonication in ice and cell lysates were collected by centrifugation (20,000 g, 30 min) at 4 °C to remove the debris. The protein concentration was determined by using the PierceTM BCA Protein Assay kit (Thermo Fisher Scientific).100 µL of cell lysates (2 mg/ml) were incubated with Conidiogenone C or Conidiogenone C probes. The resulting lysates were precipitated by 400 µL methanol, 100 µL chloroform and 300 µL Milli-Q water. For visualizing the probe labeling efficiency by in-gel fluorescence, the precipitated proteins were resuspended in 100 µL PBS containing 0.4% SDS, 1 mM CuSO4, 100 µM TBTA ligand, 100 µM N3-TMARA, and 1 mM TCEP for 1 h at room temperature. The reacted samples were resolved on 10% SDS-PAGE gels and imaged by a ChemiDocTM MP imaging system (Bio-Rad). The gels were then stained by Coomassie brilliant blue to demonstrate equal loading.
Quantitative profiling of Conidiogenone C-interacting proteins
For the identification of Conidiogenone C -interacting proteins, the cells were lysed by sonication in ice-cold 0.1% Triton/PBS buffer and cell lysates were collected by centrifugation (20000 g, 30 min) at 4 °C to remove the debris. The protein concentration was determined by using the PierceTM BCA Protein Assay kit (Thermo Fisher Scientific). 1 mL cell lysates (2 mg/mL) were incubated with Conidiogenone C or Conidiogenone C probes. The resulting lysates were precipitated by 4 mL methanol, 1 mL chloroform and 3 mL Milli-Q water. The precipitated proteins were resuspended in 100 µL PBS containing 0.4% SDS. The resulting lysates reacted with 1 mM CuSO4, 100 µM TBTA ligand, 100 µM azide-biotin, and 1 mM TCEP for 1 h at room temperature. The resulting click-labeled lysates were centrifuged at 8000 g for 5 min at 4 °C and washed twice with 1 mL cold methanol. The proteins were resuspended in 1 mL PBS containing 1.2% SDS. 100 µL streptavidin beads (Thermo Fisher Scientific) were washed for three times with 1 mL PBS, and resuspended in 5 mL PBS, which was added to the protein solution. The resulting solution was incubated for 4 h at 29 °C, followed by washing with 5 mL PBS for three times, and 5 mL distilled water for three times. The resulting beads were resuspended in 500 µl PBS containing 6 M urea and 10 mM dithiothreitol and incubated at 37 °C for 30 min, followed by addition of 20 mM iodoacetamide for 30 min at 37 °C in the dark. The beads were then collected by centrifugation and resuspended in 200 µL PBS containing 2 M urea, 1 mM CaCl2 and 10 ng/µL trypsin (Enzyme& Spectrum). Trypsin digestion was performed at 37 °C with rotation 16 h and the beads were washed with 200 µl distilled water for three times. For dimethyl labeling, per 100 µL peptides were reacted with 4 µL of 4% “light” formaldehyde (Sigma) and 4 µL of 0.6 M Sodium cyanotrihydridoborate (Sigma), “Medium” formaldehyde (CD2O) (Sigma) and 4 µL of 0.6 M Sodium cyanotrihydridoborate, “heavy” formaldehyde (13CD2O) (Sigma) and 4 µL of 0.6 M Sodium cyanoborodeuteride (Sigma), respectively.
The resulting solution incubated at room temperature for 2 h. The reaction was quenched by adding 16 µL 1% ammonia and 8 µL 5% formic acid. The “light”, “Medium” and “heavy” samples were 1:1:1 mixed and the mixture was collected and desalted with C18 column (Thermo Fisher Scientific). Eventually, 3 biological replicated samples were identified by Orbitrap Fusion™ Lumos™ (Thermo scientific, USA).
For in vitro pull down-Western blot experiment, the procedures of pull down assay were largely the same as described above. After washing beads, 2 × loading buffer were added into beads, and then denatured for 5 min at 95 °C with the detection being western blot.
The LC-MS/MS analysis of digestion samples was performed on LC-MS/MS using Orbitrap Fusion™ Lumos™ (Thermo scientific, USA). The raw data was analyzed using MSFragger V22. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD047981 (https://proteomecentral.proteomexchange.org/).
In vivo pull down assay
For the labeling of Conidiogenone C targets in living cells, the Raw264.7 cells were grown to 80% confluence. The cells were stimulated with LPS (100 ng/mL) for 24 h. Then, the cells were incubated with Conidiogenone C or Conidiogenone C probes for 10 min. The cells were collected by PBS wash for three times and centrifugation at 188 g for 3 min. The cells were lysed by sonication in ice-cold 0.1% Triton/PBS buffer and cell lysates were collected by centrifugation (20000 g, 30 min) at 4 °C to remove the debris. The protein concentration was determined by using the PierceTM BCA Protein Assay kit (Thermo Fisher Scientific). 1 mL cell lysates (2 mg/mL) reacted with 1 mM CuSO4, 100 µM TBTA ligand, 100 µM azide-biotin, and 1 mM TCEP for 1 h at room temperature. The resulting click-labeled lysates were centrifuged at 8000 g for 5 min at 4 °C and washed twice with 1 mL cold methanol. The proteins were resuspended in 1 mL PBS containing 1.2% SDS. 100 µL streptavidin beads (Thermo Fisher Scientific) were washed for three times with 1 mL PBS, and resuspended in 5 mL PBS, which was added to the protein solution. The resulting solution was incubated for 4 h at 29 °C, followed by washing with 5 mL PBS for three times, and 5 mL distilled water for three times. 2 × loading buffer were added into beads, and then denatured for 5 min at 95 °C with the detection being western blot.
Western blot analysis
Cell lysates were run on a 10% gradient Bis Tris gel and transferred to a PVDF membrane using standard methods. The membrane was probed with GAPDH (Abcam ab181602; 1:1000), IRGM (CST #14979; 1:1000). Samples were incubated with horseradish peroxidase-conjugated secondary antibodies (CST #7074, 1:5000) for 2 h at RT, then washed twice with TBST. The signals were visualized by enzyme-linked chemiluminescence (Yeasen). Protein band was analyzed by using image J software. The uncropped and unprocessed scans of blots are provided as a Source Data file.
Quantitative RT-PCR
Total RNA was extracted from cells using Fast Pure Cell/Tissue Total RNA Isolation Kit (Vazyme), and cDNA was prepared using HiScript III All-in-one RT SuperMix Perfect for qPCR (Vazyme) according to the manufacturer’s guideline. Quantitative RT-PCR was performed using Taq Pro Universal SYBR qPCR Master Mix (Vazyme). Relative gene expression was determined using the standard 2(-△△Ct) calculations by normalizing to GAPDH. The primer pairs of all genes used in this study are shown in Supplementary Data 1.
siRNA transfection
For RNA interference, RAW264.7 cells were transfected in a 12-well plate with 12.5 pmol targeted siRNA and negative control siRNA using GP transfect mate (Genepharma) following the manufacturer’s instructions. The siRNA used in the present study are as follows: negative control siRNA (Genepharma, A06001); Mouse IRGM1 siRNA (sense 5′- GCACAUGUGACUUAUUCAUTT -3′, anti-sense 5′- AUGAAUAAGUCACAUGUGCTT -3′).
Mitochondrial membrane potential
Raw264.7 cells were incubated in 5 μg/mL Jc-1 (Invitrogen) in serum-free DMEM for 20 min at 37 °C. Cells were washed twice, resuspended in PBS, and then were analyzed on a flow cytometer CytoFLEX (Beckmen).
Binding sites identification by LC-MS/MS
Recombinant IRGM1 protein (YEASEN, YSGF-XS-202402-G0042) was solubilized in 100 μL PBS to a final concentration of 1 mg/ml. The proteins was then incubated with 10 μM Conidiogenone C or DMSO at 29 °C for 3 h. After washing with 200 μL PBS for three times, the proteins were resuspended in 100 μL ddH2O of containing 6 M urea and 10 mM DTT and incubated at 37 °C for 30 min, followed by addition of 20 mM IAA for 30 min at 37 °Cin the dark. After washing with 400 μL 50 mM NH4HCO3 for three times, the proteins were resuspended in 200 μL digestion buffer (1 M Urea, 50 mM NH4HCO3), 0.5 μg Lys-C and 2.5 μg trypsin were added. After an 16 h digest at 37 °C, the peptides were eluted by 400 μL ddH2O. The sample was dried in a vacuum centrifuge, desalted with an analytical C18 column (Agela Technologies) using Agilent HPLC system. Mobile phase A: 2% ACN–98% H2O (adjusted to pH 10 with NH3·H2O); B: 98% ACN–2% H2O (adjusted to pH 10 with NH3·H2O). Samples were separated using a 20 min gradient of buffer B at a flow rate of 1.5 mL/min, as follows: 0 min 0% B; 0.1 min 5% B; 1 min 8% B; 7 min 14% B; 12 min 24% B; 16 min 40% B; 18 min 95% B; 20 min 15% B. The columns were operated at 25 °C and the temperature was controlled by a built-in column heater. The 20 fractions were combined into 5 fractions and dried in a SpeedVac. The fractions resuspended in 0.1% formic acid in ddH2O for LC-MS/MS analysis. The LC-MS/MS analysis of digestion samples was performed on LC-MS/MS using Orbitrap Fusion™ Lumos™ (Thermo scientific, USA). The raw data was analyzed using MSFragger V22 with setting a variable modification of +359.2459 Da and 302.2245 Da, selecting Default as the workflow. The sequence fasta file of IRGM1 was used as the sequence searching database.The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD056084 (https://proteomecentral.proteomexchange.org).
Pulldown assay for Binding sites validation
Flag-tagged IRGM1 WT or mutations were transiently transfected in HEK293FT cells. The HEK293FT cell lysates incubated with 5 μM Conidiogenone C or Conidiogenone C probes at 29 °C for 1 h. The resulting lysates were precipitated by 4 mL methanol, 1 mL chloroform and 3 mL Milli-Q water. The precipitated proteins were resuspended in 100 µL PBS containing 0.4% SDS. The resulting lysates reacted with 1 mM CuSO4, 100 µM TBTA ligand, 100 µM azide-biotin, and 1 mM TCEP for 1 h at room temperature. The resulting click-labeled lysates were centrifuged at 8000 g for 5 min at 4 °C and washed twice with 1 mL cold methanol. The proteins were resuspended in 1 mL PBS containing 1.2% SDS. 100 µL streptavidin beads (Thermo Fisher Scientific) were washed for three times with 1 mL PBS, and resuspended in 5 mL PBS, which was added to the protein solution. The resulting solution was incubated for 4 h at 29 °C, followed by washing with 5 mL PBS for three times. 2 × loading buffer were added into beads, and then denatured for 5 min at 95 °C with the detection being western blot. Uncropped and unprocessed scans are provided in source data file.
Data analysis
The isotopic modifications (28.0313, 32.0564 and 36.07566 Da for light, medium and heavy labeling, respectively) are set as static modifications on the N-terminal of a peptide and lysines. The ratios of reductive dimethylation were quantified by the CIMAGE software as described previously.
Statistical analysis
Results are expressed as mean ± s.d. Fold change in relation to control groups of three independent cell culture and subsequent procedures. We analyzed the data in GraphPad Prism (GraphPad Software 9.5). The respective figure legends provide information on the specific statistical test used for each assay.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Source data
Acknowledgements
H. Hao thanks Prof. Dirk Trauner, Prof. Zhen Yang and Prof. Yikang Wu for continuous support. We are grateful for financial support from Natural Science Foundation of China (Grant No. 21901211) to H. Hao, National Key Research and Development Plan (Grant No. 2021YFC2102900) to H. Li, the Natural Science Research Program of Shaanxi Province of China (2019JQ-355), and Open Fund of State Key Laboratory of Chemical Oncogenomics (PKU, Shenzhen) to H. Hao. H. Hao thank Dr. Felix Hartrampf for helpful discussions during the preparation of this manuscript. We sincerely acknowledge all the reviewers for their valuable comments and suggestions.
Author contributions
H. Hao.; H. Li.; W. Zhou. designed and supervised the project. Synthetic experimental work was conducted by T. Li.; Y. Dai.; H. Guo.; Liang. Shi.; X. Sang.; L. Ren.; and Lili. Shi. Chemical probe was synthesized by J. Wang; Chemical proteomics and subsequent biological validation experimental work was conducted by S. Jiang and X. Wu. All the authors were involved the analysis of results and discussions of the project. Tian Li. Shan Jiang, Yuanhao Dai and Xia Wu contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Data availability
The data supporting the findings of this study, including experimental procedures, NMR spectra and target identification are available within the article and its Supplementary Information files. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 2164144 and 2164173. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD047981 and PXD056084 [https://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD047981] (proteomics profiling experiments in RAW264.7 cells). [https://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD056084] (Binding site identification experiments). All primer data generated in this study are provided in the Supplementary Data 1. LC-MS/MS data of chemo-proteomic profiling related to Fig. 4H and Fig. 4G are provided in the Supplementary Data 2. LC-MS/MS data of binding site identification related to Fig. 4J–L are provided in the Supplementary Data 3. The uncropped and unprocessed scans of blots are provided in the Source Data. All data are available from the corresponding author upon request. Source data are provided with this paper.
Competing interests
The authors declare no competing interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Tian Li, Shan Jiang, Yuanhao Dai, Xia Wu.
Contributor Information
Wenming Zhou, Email: zhouwenming2008@nwafu.edu.cn.
Houhua Li, Email: lihouhua@pku.edu.cn.
Hong-Dong Hao, Email: hongdonghao@nwafu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-55189-8.
References
- 1.Brill, Z. G., Condakes, M. L., Ting, C.-P. & Maimone, T. J. Navigating the chiral pool in the total synthesis of complex terpene natural products. Chem. Rev.117, 11753–11795 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Urabe, D., Asaba, T. & Inoue, M. Convergent strategies in total syntheses of complex terpenoids. Chem. Rev.115, 9207–9231 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Maimone, T. J. & Baran, P. S. Modern synthetic efforts toward biologically active terpenes. Nat. Chem. Bio.3, 396–407 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Roncal, T., Cordobes, S., Ugalde, U., He, Y.-H. & Sterner, O. Novel diterpenes with potent conidiation inducing activity. Tetrahedron Lett.43, 6799–6802 (2002). [Google Scholar]
- 5.Du, L. et al. New alkaloids and diterpenes from a deep ocean sediment derived fungus Penicillium sp. Tetrahedron65, 1033–1039 (2009). [Google Scholar]
- 6.Gao, S.-S., Li, X.-M., Zhang, Y., Li, C.-S. & Wang, B.-G. Conidiogenones H and I, Two New Diterpenes of Cyclopiane Class from a Marine-Derived Endophytic Fungus Penicillium chrysogenum QEN-24S. Chem. Biodivers.8, 1748–1753 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Niu, S.-W. et al. Spirograterpene A, a tetracyclic spiro-diterpene with a fused 5/5/5/5 ring system from the deep-sea-derived fungus Penicillium granulatum MCCC 3A00475. J. Nat. Prod.80, 2174–2177 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Niu, S.-W. et al. Cyclopiane-type Diterpenes from The Deep-Sea-Derived Fungus Penicillium commune MCCC 3A00940. Tetrahedron Lett.59, 375–378 (2018). [Google Scholar]
- 9.Cheng, Z.-B. et al. Two new meroterpenoids and two new monoterpenoids from the deep sea-derived fungus Penicillium sp. YPGA11. Fitoterapia133, 120–124 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Cheng, Z.-B. et al. Three new cyclopiane-type diterpenes from a deep-sea derived fungus Penicillium sp. YPGA11 and their effects against human esophageal carcinoma cells. Bioorg. Chem.91, 103129 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Chen, H.-Y., Liu, T.-K., Shi, Q. & Yang, X.-L. Sesquiterpenoids and Diterpenes with Antimicrobial Activity from Leptosphaeria sp. XL026, an Eendophytic Fungus in Panax notoginseng. Fitoterapia137, 104243 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Li, F.-L. et al. New cyclopiane diterpenes with anti-inflammatory activity from the sea sediment-derived fungus Penicillium sp. TJ403-2. Chin. Chem. Lett.31, 197–201 (2020). [Google Scholar]
- 13.Zhang, S.-T. et al. Bioassay-directed isolation of antibacterial metabolites from an arthropod-derived Penicillium chrysogenum. J. Nat. Prod.83, 3397–3403 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Xin, Z., Wang, H., He, H. & Gao, S. Recent advances in the total synthesis of natural products bearing the contiguous all-carbon quaternary stereocenters. Tetrahedron Lett.71, 153029 (2021). [Google Scholar]
- 15.Eggert, A., Etling, C., Lübken, D., Saxarra, M. & Kalesse, M. Contiguous quaternary carbons: a selection of total syntheses. Molecules25, 3841 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Büschleb, M. et al. Synthetic strategies toward natural products containing contiguous stereogenic quaternary carbon atoms. Angew. Chem. Int. Ed.55, 4156–4186 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long, R., Huang, J., Gong, J. & Yang, Z. Direct construction of vicinal all-carbon quaternary stereocenters in natural product synthesis. Nat. Prod. Rep.32, 1584–1601 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Jeon, H. J. & Winkler, J. D. Synthesis of cyclohexane-angularly-fused triquinanes. Synthesis53, 475–488 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh, V., Singh, R. B. & Mobin, S. M. Cycloaddition of Annulated Cyclo-hexa-2,4-Dienones and Novel Reduction of Halogen at Bridge-head: an Expedient Route to Tetracyclo[6.5.2.02,7.09,13]Pentadec-2(7),11-Dien-14-One and Framework of Conidiogenol and Conidiogenone. Tetrahedron65, 7969–7974 (2009). [Google Scholar]
- 20.Behera, T. K., Jarhad, D. B., Mobin, S. M. & Singh, V. Molecular complexity and diversity from aromatics intramolecular cycloaddition of cyclo-hexa-2,4-dienones and sigmatropic shift in excited state: a unified approach towards synthesis of polycyclic frameworks related to crotogoudin, conidiogenol, and crinipellins. Tetrahedron72, 5377–5393 (2016). [Google Scholar]
- 21.Donahue, M. G., Jentsch, N. G. & Realini, E. C. An Intramolecular Para-Phenolic Allylation Ffree Radical Cyclization Strategy For the Synthesis of Alkaloids and Terpenes with Spiro[4.5]Decane Architectures. Tetrahedron Lett.58, 3219–3222 (2017). [Google Scholar]
- 22.Kim, M. J., Lee, S., Kang, T., Baik, M.-H. & Lee, H.-Y. Unexpected Selectivity of Intramolecular [3+2] Cycloaddition of Trimethylenemethane (TMM) Diyl toward Total Synthesis of Conidiogenone B. Eur. J. Org. Chem.2020, 609–617 (2020). [Google Scholar]
- 23.Hou, S.-H. et al. Total Syntheses of the Tetracyclic Cyclopiane Diterpenes Conidiogenone, Conidiogenol, and Conidiogenone B. Angew. Chem. Int. Ed.55, 4456–4460 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Hu, P.-F. et al. Quaternary-Centre-Guided Synthesis of Complex Polycyclic Terpenes. Nature569, 703–707 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chi, H.-M., Cole, C. J., Hu, P.-F., Taylor, C. A. & Snyder, S. A. Total Syntheses of Spiroviolene and Spirograterpene A: a Structural Reas-signment with Biosynthetic Implications. Chem. Sci.11, 10939–10944 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu, B., Xun, W., Su, S.-B. & Zhai, H.-B. Total Syntheses of (−)-Conidiogenone B, (−)-Conidiogenone, and (−)-Conidiogenol. Angew. Chem. Int. Ed.59, 16475–16479 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Kim, J., Lee, S., Han, S. & Lee, H.-Y. Divergent Synthesis of Conidiogenones B-F and 12β-hydroxy Conidiogenone C. Chem9, 1270–1280 (2023). [Google Scholar]
- 28.Amberg, W. M. & Carreira, E. M. Enantioselective Total Synthesis of (+)-Aberrarone. J. Am. Chem. Soc.144, 15475–15479 (2022). [DOI] [PubMed] [Google Scholar]
- 29.Wang, Y., Su, Y. & Jia, Y. Total Synthesis of (+)-Aberrarone. J. Am. Chem. Soc.145, 9459–9463 (2023). [DOI] [PubMed] [Google Scholar]
- 30.Mikulak-Klucznik, B. et al. Computational planning of the synthesis of complex natural products. Nature588, 83–88 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Hewage, R. T. et al. An Enzyme-Mediated Aza-Michael addition is involved in the biosynthesis of an imidazoyl hybrid product of conidiogenone B. Org. Lett.23, 1904–1909 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Shiina, T. et al. Biosynthetic study of conidiation-inducing factor conidiogenone: heterologous production and cyclization mechanism of a key bifunctional diterpene synthase. Biosci., Biotech., Biochem83, 192–201 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Mitsuhashi, T. et al. Crystalline sponge method enabled the investigation of a prenyltransferase-terpene synthase chimeric enzyme, whose product exhibits broadened NMR signals. Org. Lett.20, 5606–5609 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Zhang, L.-M. & Wang, S.-Z. Efficient Synthesis of Cyclopentenones from Enynyl Acetates via Tandem Au(I)-Catalyzed 3,3-Rearrangement and the Nazarov Reaction. J. Am. Chem. Soc.128, 1442–1443 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Brandstätter, M., Freis, M., Huwyler, N. & Carreira, E. M. Total Synthesis of (−)-Merochlorin A. Angew. Chem. Int. Ed.58, 2490–2494 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Brandstätter, M., Huwyler, N. & Carreira, E. M. Gold (I)-Catalyzed Stereoselective Cyclization of 1,3-Enyne Aldehydes by a 1,3-Acyloxy Migration/Nazarov Cyclization/Aldol Addition Cascade. Chem. Sci.10, 8219–8223 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trost, B. M., Zhang, G., Gholami, H. & Zell, D. Total synthesis of kadcoccinic acid a trimethyl ester. J. Am. Chem. Soc.143, 12286–12293 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Shi, F.-Q., Li, X., Xia, Y., Zhang, L. & Yu, Z.-X. DFT Study of the Mechanisms of In Water Au (I)-Catalyzed Tandem [3,3]-Rearrangement/Nazarov Reaction/[1,2]-Hydrogen Shift of Enynyl Acetates: A Proton-Transport Catalysis Strategy in the Water-Catalyzed [1,2]-Hydrogen Shift. J. Am. Chem. Soc.129, 15503–15512 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Gandon, V., Lemiere, G., Hours, A., Fensterbank, L. & Maiacria, M. The role of bent acyclic allene gold complexes in axis-to-center chirality transfers. Angew. Chem. Int. Ed.47, 7534–7538 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Lemiere, G. et al. Generation and Trapping of Cyclopentenylidene Gold Species: Four Pathways to Polycyclic Compounds. J. Am. Chem. Soc.131, 2993–3006 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Jung, M.-E. & Hatfield, G. L Facile Synthesis of (3as)-1,3a-Dimethyl-2,3,3a,5,6,7-Hyxahydroinden-4(5h)-One, an Intermediate for Steroid Synthesis. Tetrahedron Lett.24, 3175–3178 (1983). [Google Scholar]
- 42.Xu, C.-M., Zhang, L., Zhou, P.-X., Luo, S.-Z. & Chen, J.-P. A practical protocol for asymmetric synthesis of wieland-miescher and hajos–parrish ketones catalyzed by a simple chiral primary amine. Synthesis45, 1939–1945 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Yang, Z. Navigating the Pauson–Khand reaction in total syntheses of complex natural products. Acc. Chem. Res.54, 556–568 (2021). [DOI] [PubMed] [Google Scholar]
- 44.Chen, S.-J., Jiang, C.-G., Zheng, N., Yang, Z. & Shi, L.-L. Evolution of Pauson-Khand reaction: strategic applications in total syntheses of architecturally complex natural products (2016–2020). Catalysts10, 1199 (2020). [Google Scholar]
- 45.Ishizaki, M., Iwahara, K., Kyoumura, K. & Hoshino, O. An effective synthesis of angular tricyclic compounds having two contiguous quaternary centers by intramolecular pauson-khand reaction of exocyclic enynes. Synlett1999, 587–589 (1999). [Google Scholar]
- 46.Ishizaki, M., Iwahara, K., Niimi, Y., Satoh, H. & Hoshino, O. Investigation of the synthesis of angular tricyclic compounds by intra-molecular pauson–khand reaction of exo- and endo-cyclic enynes. Tetrahedron57, 2729–2738 (2001). [Google Scholar]
- 47.Wang, X. et al. A Synthetic Route to the Core Structure of (−)-Retigeranic Acid A. Org. Lett.23, 5092–5097 (2021). [DOI] [PubMed] [Google Scholar]
- 48.Zhao, K., Hsu, Y.-C., Yang, Z., Liu, R.-S. & Zhang, L. Gold catalyzed synthesis of chiral cyclopentadienyl esters via chirality transfer. Org. Lett.22, 6500–6504 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nagata, W., Yoshioka, M. & Hirai, S. Hydrocyanation. IV. New Hydrocyanation Methods Using Hydrogen Cyanide and an Alkyl-aluminum, and an Alkylaluminum Cyanide. J. Am. Chem. Soc.94, 4635–4643 (1972). [Google Scholar]
- 50.Qu, P. & Snyder, S. A. Concise and Stereoselective Total Syntheses of Annotinolides C, D, and E. J. Am. Chem. Soc.143, 11951–11956 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murphy, S. K., Zeng, M.-S. & Herzon, S. B. A modular and enantioselective synthesis of the pleuromutilin antibiotics. Science356, 956–959 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Min, S.-J. & Danishefsky, S. J. Total synthesis of paecilomycine A. Angew. Chem. Int. Ed.46, 2199–2202 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Waters, S. P., Tian, Y., Li, Y.-M. & Danishefsky, S. J. Total Synthesis of (−)-Scabronine G, an Inducer of Neurotrophic Factor Production. J. Am. Chem. Soc.127, 13514–13515 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Overman, L. E., Ricca, D. J. & Tran, V. D. Total Synthesis of (±)-Scopadulcic Acid B. J. Am. Chem. Soc.119, 12031–12040 (1997). [Google Scholar]
- 55.Yu, J.-Q., Wu, H.-C. & Corey, E. J. Pd(OH)2/C-mediated selective oxidation of silyl enol ethers by tert-butylhydroperoxide, a useful method for the conversion of ketones to α,β-Enones or β-Silyloxy-α,β-Enones. Org. Lett.7, 1415–1417 (2005). [DOI] [PubMed] [Google Scholar]
- 56.Tanaka, R. et al. Synthetic study of pyrrocidines: first entry to the decahydrofluorene core of pyrrocidines. Org. Lett.14, 4886–4889 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Srikrishna, A. & Mahesh, K. Enantiospecific first total synthesis of ent-Allothapsenol. Synlett23, 1021–1024 (2012). [Google Scholar]
- 58.Parr, B. T., Economou, C. & Herzon, S. B. A Concise Synthesis of (+)-Batzelladine B from simple pyrrole-based starting materials. Nature525, 507–510 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stork, G. & Danheiser, R. L. Regiospecific alkylation of cyclic beta-diketone enol ethers. general synthesis of 4-Alkylcyclohexenones. J. Org. Chem.38, 1775–1776 (1973). [Google Scholar]
- 60.Sui, G.-Q. et al. Chemoselective reduction of aldehydes via a combination of NaBH4 and Acetylacetone. New. J. Chem.43, 15793–15796 (2019). [Google Scholar]
- 61.Han, Y.-J., Zhu, L.-Z., Gao, Y. & Lee, C.-S. A Highly Convergent Cascade Cyclization to cis-Hydrindanes with All-Carbon Quaternary Centers and Its Application in the Synthesis of the Aglycon of Dendronobiloside A. Org. Lett.13, 588–591 (2011). [DOI] [PubMed] [Google Scholar]
- 62.Jigajinni, V. B. & Wightman, R. H. Hydrogenolysis of Enol Triflates; A New Method for the Reduction of Ketones to Methylene Compounds. Tetrahedron Lett.23, 117–120 (1982). [Google Scholar]
- 63.Gouault, N., Roch, M. L., Pinto, G. C. & David, M. Total synthesis of dendrobate alkaloid (+)-241D, isosolenopsin and isosolenopsin a: application of a gold-catalyzed cyclization. Org. Biomol. Chem.10, 5541–5546 (2012). [DOI] [PubMed] [Google Scholar]
- 64.Kurosu, M., Marcin, L. R., Grinsteiner, T. J. & Kishi, Y. Total Synthesis of (±)-Batrachotoxinin A. J. Am. Chem. Soc.120, 6627–6628 (1998). [Google Scholar]
- 65.Polniaszek, R. P. & Dillard, L. W. Stereospecific total syntheses of decahydroquinoline alkaloids (±)-195A and (±)-2-Epi-195A. J. Org. Chem.57, 4103–4110 (1992). [Google Scholar]
- 66.Vashkiv, L. B. & Donlin, L. T. Regulation of type I Interferon Responses. Nat. Rev. Immunol.14, 36–49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rai, P. et al. IRGM1 links mitochondrial quality control to autoimmunity. Nat. Immunol.22, 312–321 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kaufman, B. A. & Mora, A. L. IRGM1, a Guardian of Mitochondrial DAMP–mediated Autoinflammation. Nat. Immunol.22, 272–273 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jena, K. K. et al. Autoimmunity Gene IRGM Suppresses cGAS-STING and RIG-I-MAVS Signaling to Control Interferon Response. EMBO Rep.21, e50051 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mehto, S. et al. The Crohn’s Disease Risk Factor IRGM Limits NLRP3 inflammasome activation by impeding its assembly and by mediating its selective autophagy. Mol. Cell.73, 429–445 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jackson, P. A., Widen, J. C., Harki, D. A. & Brummond, K. M. Covalent Modifiers: A Chemical Perspective on the Reactivity of α,β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions. J. Med. Chem.60, 839–885 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Berdan, C. A. et al. Parthenolide covalently targets and inhibits focal adhesion kinase in breast cancer cells. Cell Chem. Biol.26, 1027–1035 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Davis, D. C. et al. Total synthesis, biological evaluation, and target identification of rare Abies Sesquiterpenoids. J. Am. Chem. Soc.140, 17465–17473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lagoutte, R. et al. Divergent synthesis and identification of the cellular targets of deoxyelephantopins. Nat. Commun.7, 12470 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cui, C. et al. Total synthesis and target identification of the curcusone diterpenes. J. Am. Chem. Soc.143, 4379–4386 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rongvaux, A. et al. Apoptotic caspases prevent the induction of type i interferons by mitochondrial DNA. Cell159, 1563–1577 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lin, A. J. et al. δPKC-Mediated DRP1 phosphorylation impacts macrophage mitochondrial function and inflammatory response to endotoxin. SHOCK57, 435–443 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The data supporting the findings of this study, including experimental procedures, NMR spectra and target identification are available within the article and its Supplementary Information files. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 2164144 and 2164173. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD047981 and PXD056084 [https://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD047981] (proteomics profiling experiments in RAW264.7 cells). [https://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD056084] (Binding site identification experiments). All primer data generated in this study are provided in the Supplementary Data 1. LC-MS/MS data of chemo-proteomic profiling related to Fig. 4H and Fig. 4G are provided in the Supplementary Data 2. LC-MS/MS data of binding site identification related to Fig. 4J–L are provided in the Supplementary Data 3. The uncropped and unprocessed scans of blots are provided in the Source Data. All data are available from the corresponding author upon request. Source data are provided with this paper.