

Abstract
A member of the Paramyxoviridae family of RNA viruses, respiratory syncytial virus (RSV), is a leading cause of epidemic respiratory tract infection in children. In children, RSV primarily replicates in the airway mucosa, a process that alters epithelial cell chemokine expression, thereby inducing airway inflammation. We investigated the role of the mitogen-activated protein kinase kinase kinase 14/NF-κB-inducing kinase (NIK) in the activation of NF-κB-dependent genes in alveolus-like A549 cells. RSV infection induces a time dependent increase of NIK mRNA and protein expression that peaks 12 to 24 h after viral exposure. Immunoprecipitation kinase assays indicate that NIK kinase activity is activated even more rapidly (within 6 h of RSV adsorption) associated with an endogenous ∼50-kDa NF-κB2 substrate. Because NIK associates with IKKα to mediate processing of the 100-kDa NF-κB2 precursor into its 52-kDa DNA binding isoform (“p52”), the effects of RSV on NIK complex formation with IKKα and NF-κB2 were determined by coimmunoprecipitation assay. We find that NIK, IKKα, and both 100 kDa- and 52-kDa NF-κB2 isoforms strongly complex 15 h after exposure to RSV at times subsequent to NIK kinase activation. Western immunoblot and microaffinity DNA pull-down assays showed a parallel increase in nuclear translocation and DNA binding of the NF-κB2-Rel B complex. Interestingly, we make the novel observations that NIK also transiently translocates into the nucleus complexed with 52-kDa NF-κB2. Small interfering RNA-mediated NIK “knock-down” blocked RSV-inducible 52-kDa NF-κB2 processing and interfered with the early activation of a subset of NF-κB-dependent genes, indicating the importance of this activation pathway in the genomic NF-κB response to RSV. Together, these data indicate that RSV infection rapidly activates the noncanonical NF-κB activation pathway prior to the more potent canonical pathway activation. This appears to be through a novel mechanism involving induction of NIK kinase activity, expression, and nuclear translocation of a ternary complex with IKKα and processed NF-κB2.
Respiratory syncytial virus (RSV) is an enveloped, negative-sense single-stranded RNA virus of the Paramyxoviridae family that is recognized as a leading cause of respiratory illness in children in the United States and worldwide. RSV infection produces a wide spectrum of diseases in infants and children, including otitis media, mild upper respiratory tract infections, acute laryngotracheobronchitis, and severe lower respiratory tract infections (21). It is estimated that 40 to 60% of children admitted to the hospital with bronchitis and 15 to 35% admitted for pneumonia are infected with RSV; in fact, RSV respiratory infections result in ∼100,000 hospitalizations and 2,000 deaths annually in the United States alone (22, 38). Due to unavailability of any efficacious vaccine for RSV infection and because of its ability to produce lower respiratory tract infections in predisposed infants that results in long-term airway hyper-reactivity, RSV remains a significant health problem worldwide (22, 31, 39, 45, 46). Although the mechanism underlying RSV-induced airway disease is largely unknown, experimental evidence suggests that early inflammatory and immune events of the host in response to RSV may play an important role (16).
The airway epithelium is the main target of RSV infection. After infection, RSV replicates in the respiratory mucosa, leading to epithelial damage (2) and perivascular mononuclear infiltration (12). Infected epithelial cells respond to RSV replication by producing a number of potent immunomodulatory and inflammatory mediators including cytokines (13, 15, 34) and chemokines (5, 35, 52). Recent microarray studies have shown that RSV induces a time dependent increase in the expression of 17 cytokines, including the CC (RANTES, MCP-1, MIP1α, and MIP1β), CXC (growth-regulated oncogene [Gro]-α,-β, and -γ; interleukin-8 [IL-8]; and I-TAC), and CX3C (fractalkine) subclasses of chemokines in lower-airway epithelial cells (43, 52). Although the signaling pathways activated by RSV resulting in an inflammatory response have not been completely characterized, our previous work and that of others have demonstrated that RSV replication activates various transcription factors, including NF-κB (15, 20, 25, 52), a master regulator of inflammation (4, 43). Using a tightly regulated dominant-negative inhibitor of the NF-κB pathway, we recently reported the identification of 144 NF-κB-dependent genes (out of 1,215) whose expression was altered by RSV in epithelial cells that encoded a wide range of functional proteins, including chemokines, NF-κB isoforms, structural proteins, transcription factors involved in interferon signaling and metabolism, and those controlling protein synthesis and turnover (43). Furthermore, in a mouse model of RSV infection, where RSV activates mucosal NF-κB binding, we found that treatment with a specific cell-permeable IκB kinase inhibitor markedly reduced NF-κB DNA-binding activity, chemokine gene expression, and airway inflammation after RSV infection (19). Together, these findings indicate that NF-κB activation is a central mediator of RSV-induced airway inflammation.
NF-κB is a family of highly inducible cytoplasmic DNA-binding proteins that includes the transactivating subunits Rel A, Rel B, and c-Rel and the posttranslationally processed DNA-binding subunits NF-κB1 (p50) and NF-κB2 (p52) (41). The NF-κB dimers remain sequestered in the cytoplasm by interacting with a group of inhibitory ankyrin repeat-containing proteins, collectively referred to as IκBs (IκB-α, -β, -ɛ, p100, and p105) (3). Recently, it has been shown that NF-κB activation can be controlled by at least two separable and independent pathways: (i) the “canonical” pathway mediated by the IκB kinase (IKK), a complex of the IKK-α and -β catalytic subunits with the IKKγ regulatory subunit (36), and (ii) the “noncanonical” pathway mediated by a mitogen-activated protein kinase kinase kinase (MAP3K14)/NF-κB-inducing kinase (NIK)-IKKα complex. Stimuli activating NF-κB via the canonical pathway activate IKK, producing IκBα phosphorylation at specific N-terminal serine residues, thereby targeting it for proteosomal degradation (17). As a result, sequestered Rel A-NF-κB1 complexes are released to enter the nucleus. On the other hand, stimuli activating the noncanonical NF-κB activation pathway include lymphotoxin β (LTβ) (11, 49), CD40 ligand (10), DNA virus infection (47), and B-cell-activating factor (9, 27, 37, 49). Interestingly, neither IKKβ or IKKγ, key regulators of the canonical pathway, is required for activation of the noncanonical pathway (37, 47). Rather, a separate NIK-IKKα kinase complex activates posttranslational processing of the 100-kDa NF-κB2 precursor into the 52-kDa active DNA-binding isoform. Although the mechanisms are less well understood, activators of the noncanonical pathway apparently stimulate IKKα kinase activity, which in turn phosphorylates specific serine residues in p100 NF-κB2, targeting its COOH terminus for ubiquitination and proteasomal processing to generate active p52. Newly formed 52-kDa NF-κB2 then dimerizes with cytoplasmic Rel B, and the complex then translocates into the nucleus. In this pathway, NIK serves to activate IKKα, as well as providing a docking site to recruit both p100 NF-κB2 and IKKα into a complex (48). NIK therefore is an essential component of the noncanonical NF-κB activation pathway.
We have applied discovery-based tools to identify the epithelial response to RSV replication (43, 51, 52). In our microarray analysis of RSV-induced gene network we observed a significant increased expression of NIK. Because the role that NIK plays in the epithelial response to RSV is unknown, we investigated this further in the present study. Our findings indicate that RSV activates NIK expression, as well as its kinase activity, thereby increasing NF-κB2 processing and its nuclear translocation. Short interfering RNA (siRNA)-mediated knock-down of NIK and NF-κB2 further suggest that these proteins are essential for expression of NF-κB-dependent genes in response to RSV infection in lower-airway epithelial cells. These findings are the first to indicate a novel role for inducible NIK expression and activity in RSV infection influencing control of NF-κB-dependent genes.
MATERIALS AND METHODS
Cell culture, reagents, and plasmids.
Human A549 pulmonary type II epithelial cells were obtained from the American Type Culture Collection (Manassas, VA) and grown in DMEM-F12K (Gibco, Grand Island, NY) supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml) in a humidified atmosphere of 5% CO2 (43, 52). The human RSV A2 strain was grown in Hep-2 cells and purified by centrifugation on discontinuous sucrose gradients, and titers were determined by methylcellulose plaque assay (44). No contaminating cytokines, including tumor necrosis factor alpha, IL-1, IL-6, IL-8, granulocyte-macrophage colony-stimulating factor, or interferon were found in these sucrose purified RSV (pRSV) preparations. The virus pools were divided into aliquots, quick-frozen in alcohol-dry ice, and stored at −70°C until use. Before exposure of the cells to RSV, they were placed in F12K medium containing 2% (vol/vol) fetal bovine serum and then adsorbed with pRSV at a multiplicity of infection of 1 for a given time period prior to being harvested and assayed.
Expression vectors encoding full-length FLAG epitope-tagged human NIK were produced by PCR by using the upstream primer 5′-GGTTATGGACTACAAAGACGATGACGATAAGGCAGTGATGGAAATGGCCTGCCCA-3′ and the downstream primer 5′-GGGCCTGTTCTCCAGCTGGCCATG-3′ with either wild-type or dominant-negative NIK cDNA as a template. The PCR products were agarose gel purified, cloned into the pEF6/V5 plasmid (InVitrogen Corp), and sequenced to verify authenticity. Transient transfections in exponentially growing A549 cells were performed by using Lipofectamine reagent (Gibco-BRL, Rockville, MD). All transfections were carried out (in triplicate plates) in three independent experiments for luciferase activity.
Biotinylated DNA pull-down and Western immunoblot assay.
A microaffinity DNA pull-down assay for NF-κB binding proteins was carried out as described earlier (40). In brief, the NF-κB site of IL-8 promoter was chemically synthesized containing 3′ biotin on a flexible linker (Genosys, Inc., The Woodlands, TX). Then, 50 pmol of biotin duplex was incubated with 1 mg of sucrose cushion purified nuclear protein in 1 ml of binding buffer (8% [vol/vol] glycerol, 5 mM MgCl2, 1 mM dithiothreitol [DTT], 150 mM KCl, 1 mM EDTA, and 12 mM HEPES [pH 7.9]) in the presence of 10 μg of dAdT for 1 h at 4°C. After incubation, bound proteins were captured by adding 50 μl of 50% slurry of streptavidin-agarose beads (Pierce, Rockford, IL) for 20 min at 4°C with mixing. The beads were collected by centrifugation at 4,000 × g for 2 min at 4°C and washed twice with binding buffer. NF-κB binding proteins were eluted with 50 μl of 1× sodium-dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer for Western immunoblot analysis.
For Western immunoblotting, the eluted proteins were fractionated by electrophoresis on 10% polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA). Membranes were blocked with a solution of 5% nonfat dried milk dissolved in Tris-buffered saline (TBS) containing 0.1% Tween 20 for 1 h and incubated with indicated affinity-purified antibody for 1 h at room temperature. Antibodies used were either anti-p52 monoclonal (Upstate, Charlottesville, VA) or anti-Rel B polyclonal (Santa Cruz Biotechnology, Santa Cruz, CA). Membranes were washed three times with TBS-0.1% Tween 20 and incubated with either Fluo-700 labeled anti-mouse immunoglobulin G (IgG) or IRDye 800-labeled anti-rabbit IgG antibodies (Rockland Immunochemicals, Gilbertsville, PA) for 1 h. After a wash in TBS-0.1% Tween 20, immune complexes were identified and quantified by using Odyssey Infrared Imaging system (LICOR Biosciences, Lincoln, NE).
Northern blots.
Total RNA was extracted by acid guanidium phenol extraction (Tri Reagent; Sigma). Total RNA (10 μg) was denatured and fractionated by electrophoresis on a 1.2% agarose-formaldehyde gel, capillary transferred to a nylon membrane (Zeta Probe GT; Bio-Rad), and then prehybridized as described previously (7). An NIK cDNA probe was produced by PCR with antisense primer 5′-GGGCCTGTTCTCCAGCTGGCCATG-3′ and PEF-NIKWT as a template with [α-32P]ATP. The membrane was hybridized with 2 × 106 cpm of radiolabeled probe/ml at 60°C overnight in 5% SDS hybridization buffer. The membrane was washed with buffer containing 5% SDS and 1× SSC (0.15 M NaCl plus 0.015 M sodium citrate) for 20 min at room temperature, followed by 30 min at 60°C. Internal control hybridization was carried out with 18S RNA. The image was developed and quantified by exposing the membrane to Molecular Dynamics phosphorimager cassette.
Subcellular fractionation.
A549 cells were resuspended in buffer A (50 mM HEPES [pH 7.4], 10 mM KCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.1 μg of phenylmethylsulfonyl fluoride [PMSF]/ml, 1 μg of pepstatin A/ml, 1 μg of leupeptin/ml, 10 μg of soybean trypsin inhibitor/ml, 10 μg of aprotinin/ml, 20 mM NaF, 1 mM NaP2O7, 1 mM Na3VO3, and 0.5% [vol/vol] IGEPAL CA-630). After 10 min on ice, the lysates were centrifuged at 4,000 × g for 4 min at 4°C. The supernatant was saved as the cytoplasmic fraction. The nuclear pellet was then resuspended in buffer B (buffer A with 1.7 M sucrose) and centrifuged at 15,000 × g for 30 min at 4°C (23). The purified nuclear pellet was then incubated in buffer C (10% glycerol, 50 mM HEPES [pH 7.4], 400 mM KCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.1 μg of PMSF/ml, 1 μg of pepstatin A/ml, 1 μg of leupeptin/ml, 10 μg of soybean trypsin inhibitor/ml, 20 mM NaF, 1 mM NaP2O7, 1 mM Na3VO3, 10 μg of aprotinin/ml), with frequent vortexing for 30 min at 4°C. After centrifugation at 15,000 × g for 5 min at 4°C, the supernatant was saved as nuclear extract (NE). Nuclear proteins prepared in this manner are free of cytoplasmic contamination (8). Both the cytoplasmic and the nuclear fractions were normalized for protein amounts determined by protein assay (Protein Reagent; Bio-Rad, Hercules, CA).
Microscopic imaging.
Acetone-methanol fixed cells were rinsed with phosphate-buffered saline (PBS) containing 50 mM NH4Cl for 10 min and permeabilized in PBS containing 0.1% Triton X-100. For protein-protein interaction studies, cells were subjected to limited protease digestion (100 mg of pepsin/ml; Sigma Biochemicals) in 0.1 N HCl for 15 to 30 min at 37°C. The cells were washed, blocked with preimmune heterologous serum (diluted 1:10 in PBS) for 30 min, and incubated with primary and either fluorescein isothiocyanate (FITC)- or rhodamine-conjugated secondary antibody as indicated. After staining, cells were mounted in antifade mounting solution (Dako, Inc., Carpinteria, CA). Confocal microscopy was performed on a Zeiss LSM510 META System with the 488-nm line of the argon laser for excitation of FITC and helium-neon 543-nm line excitation of rhodamine, combined with appropriate dichroic mirrors, and emission band filters to discriminate between green and red fluorescence. Images were captured at a magnification of ×60. Colocalization was visualized by superimposition of green and red images using MetaMorph software version 4.6r9 (Universal Imaging Corp.).
Electrophoretic mobility shift assay.
A total of 10 μg of NEs from uninfected and 24-h RSV-infected cells were incubated in DNA-binding buffer containing 5% glycerol, 12 mM HEPES, 80 mM NaCl, 5 mM DTT, 5 mM MgCl2, 0.5 mM EDTA, 1 μg of poly(dI-dC), and 40,000 cpm of 32P-labeled double-stranded oligonucleotide in a total volume of 20 μl. For gel mobility supershift assay, the indicated antibodies were added to the binding reactions, followed by incubation on ice for 1 h and fractionation on 6% nondenaturating PAGE. Gels were dried and exposed to BioMax film (Kodak) for autoradiography.
Coimmunoprecipitation.
A549 whole-cell extracts (WCEs) were prepared by using modified radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 0.25% sodium deoxycholate, 1% Nonidet P-40, 1 mM PMSF, 1 mM NaF, 1 mM Na3VO4, and 1 μg each of aprotinin, leupeptin, and pepstatin/ml). When the immunoprecipitation was to be followed by kinase assay, cells were lyzed in RIPA buffer without sodium deoxycholate. For immunoprecipitation, either WCEs or NEs were precleared with protein A-Sepharose 4B (Sigma) for 10 min at 4°C and then immunoprecipitation was carried out for overnight at 4°C with primary antibody. Sources of primary antibody included, anti-NIK (A-12; Santa Cruz Biotech), and anti-p52 (Upstate, Charlottesville, VA). Immune complexes were then captured by adding 50 μl of protein A-Sepharose beads (50% slurry) and rotated for 1 h at 4°C. Beads were washed three times with cold TB buffer (150 mM NaCl, 5 mM EDTA, 50 mM Tris-HCl [pH 7.4], 0.05% IGEPAL CA-630), and immune complexes were separated by SDS-PAGE. Proteins were analyzed by Western immunoblot or kinase assay (below).
NIK immunoprecipitation-kinase assay.
The phosphoacceptor site for NIK on IKKβ (amino acids 164 to 206) was PCR amplified with the IKKβ cDNA as a template with the upstream primer 5′-CAAGAATTCTTGACCTAGGATATTGCCAAG-3′ and the downstream primer 5′-GCAAGCTTACCAGTAGTCGACGGTCAC-3′. The 130-nucleotide PCR product was digested with EcoRI and HindIII restriction enzymes (underlined), gel purified, and cloned into the pGEX-KG expression vector digested with the same enzymes. The glutathione S-transferase (GST)-IKKβ(164-206) fusion protein was expressed in Escherichia coli XL-1 Blue by IPTG (isopropyl-β-d-thiogalactopyranoside) induction and purified to homogeneity by glutathione-agarose (Sigma Aldrich, St. Louis, MO) chromatography as described previously (18). Then, 1 mg of WCE from either uninfected or RSV-infected cells was incubated 1 to 2 h at 4°C with 5.0 μg of anti-NIK (Proscie, Poway, CA) in TB buffer. After immunoprecipitation, the immunoprecipitates were washed with TB buffer, followed by a final wash in kinase buffer (20 mM HEPES [pH 7.5], 10 mM MgCl2, 50 mM NaCl, 20 mM β-glycerophosphate, 100 μM Na3VO4, 20 μM ATP, 10 μg of aprotinin/ml, 2 mM DTT). The immunoprecipitates were then incubated for 30 min at 30°C with 1 μCi of [γ-32P] ATP and 2 μg of GST-IKKβ(164-206) substrate in 1× kinase buffer. Reactions were stopped by adding 2× SDS-PAGE sample buffer, followed by boiling for 5 min. Products were separated on 4 to 20% gradient SDS-PAGE (Bio-Rad Laboratories, Hercules, CA), electrophoretically transferred to polyvinylidene difluoride membranes, and exposed for autoradiography.
siRNA-mediated gene silencing.
Short interfering RNA (siRNAs) against human NF-κB2, NIK, and nontargeting (siCONTROL) pools (SMART pools) were commercially obtained (Dharmacon Research, Inc., Lafayette, CO) and transfected at 10 to 100 nM into A549 cells by using a TransIT-TKO transfection kit (Mirus Bio Corp., Madison, WI) according to the manufacturer's instructions. At 72 h after the transfection, cells were either exposed to RSV for the indicated times or lysed in modified RIPA buffer. The samples were then subjected to Western blot analysis with NIK (Santa Cruz Biotechnology, Inc.) or p52 (Upstate, Charlottesville, VA) antibodies.
Quantitative real-time PCR (Q-RT-PCR) primers and probes.
We used Applied Biosystems Assays-on-Demand 20× assay mix of primers and TaqMan MGB probes (FAM dye labeled) for our target genes and predeveloped 18S rRNA (VIC-dye labeled probe) TaqMan assay reagent (P/N 4319413E) for endogenous controls. Separate tubes (singleplex) one-step RT-PCR was performed with 80 ng of RNA. The cycling parameters for one-step RT-PCR were as follows: reverse transcription at 48°C for 30 min, AmpliTaq activation at 95°C for 10 min, denaturation 95°C for 15 s, and annealing-extension at 60°C for 1 min (repeat 40 times) on an ABI 7000 thermocycler. Duplicate CT values were analyzed by using the comparative CT (ΔΔCT) method (Applied Biosystems). The amount of target (2−ΔΔCT) was obtained by normalizing to endogenous 18S RNA reference and relative to a calibrator (untransfected sample).
RESULTS
MAP3K14/NIK expression is induced by RSV infection.
Previously, we used high-density oligonucleotide microarrays to examine the epithelial response to RSV infection. These experiments revealed a set of genes that are induced by RSV in a time-dependent fashion, some of which included critical regulators of the NF-κB pathway itself. Here, we observed that the expression of NIK was significantly induced by replicating RSV (signal intensity increasing from 75 to 3,063 arbitrary units [AU], n = 3), whereas UV-inactivated RSV did not induce NIK expression (signal intensity of 83 AU). We further validated these findings by specifically measuring NIK mRNA and protein expression. A549 cells were exposed to sucrose cushioned purified RSV (multiplicity of infection of 1) for different time intervals, and total RNA was extracted and used for Northern blot hybridization. As shown in Fig. 1A, RSV induces a time-dependent increase in the level of NIK mRNA, which peaks at 24 h, followed by a decline at 36 h. After normalization to an 18S internal control and relative to uninfected cells, NIK mRNA abundance increased by 10-fold at 24 h. This increased level of NIK mRNA was reflected by a parallel time-dependent increase in the protein levels in response to the RSV infection (Fig. 1B). Figure 1B shows a Western immunoblot of WCEs stained with anti-NIK antibody. Relative to β-actin, NIK protein increases by twofold by 24 h after RSV infection. The reduced magnitude of protein induction suggests that RSV may have significant effects on NIK translation or turnover (in other systems NIK is known to be rapidly proteolyzed [28]). Together, these data indicate that the expression of NIK is induced by RSV.
FIG. 1.

Time course of RSV-induced NIK expression in A549 cells. (A) Induction of NIK mRNA by RSV infection. Northern blots of NIK mRNA (top panel) and 18S RNA (bottom panel) levels in RSV-infected cells for the indicated times between 0 and 36 h are shown. Compared to its expression level at 0 h, NIK mRNA increases gradually peaking at 10-fold at 24 h of RSV infection. Similar results were observed by repeating the Northern and microarray studies twice. (B) RSV-induced NIK protein expression. Western immunoblot of WCEs (100 μg) prepared from the A549 cells infected by RSV for indicated times. The top panel shows a blot probed with anti-NIK antibody; for the bottom panel, β-actin was probed as a loading control.
NIK activity is rapidly induced by RSV infection.
The activity, as well as steady-state levels, of NIK is controlled by association with TRAF and other signaling adapters (28). In order to address the question whether the induction of NIK is also associated with the alteration in its kinase activity, we examined the profile of NIK activity after RSV infection. At various time intervals after the RSV adsorption, the cells were lysed in modified RIPA buffer, NIK was immunoprecipitated, and the kinase activity was measured in vitro by determining the [32γ-P] ATP incorporation into the specific GST-IKKβ (164-206) substrate (29). In unstimulated cells, NIK activity was undetectable; however, a transient increase in NIK kinase activity was observed after 6 h of RSV exposure that subsequently declined to control values (Fig. 2A). In addition to radiolabel incorporation into GST-IKKβ, we also observed significant incorporation into coimmunoprecipitating proteins. In particular, we noted that a protein of ∼50 kDa was heavily phosphorylated in the NIK assay. We interpret this to mean that an NIK-associated endogenous substrate was coimmunoprecipitating with NIK in the nondenaturing immunoprecipitation. To identify this protein, we used a sequential immunoprecipitation approach, where a second denaturing immunoprecipitation was performed on the radiolabeled products of the NIK assay. Reasoning that NIK was probably associated with members of the NF-κB pathway, we used antibodies to 48-kDa IKKγ, 50-kDa NF-κB1, and 52-kDa NF-κB2 (i.e., p52). We could not pull down the ∼50-kDa protein by using either anti-p50 or IKKγ antibodies (data not shown); however, anti-p52 specifically immunoprecipitated this phospho-protein (Fig. 2B). The absence of autoradiographic signal in the control IgG lane indicates the specificity of the immunoprecipitation. Together, our data indicate that RSV rapidly and transiently induces NIK activity. Also, p52 associates with NIK and appears to be an endogenous substrate of the activated kinase. Finally, our data also suggest that RSV may be activating the noncanonical pathway.
FIG. 2.
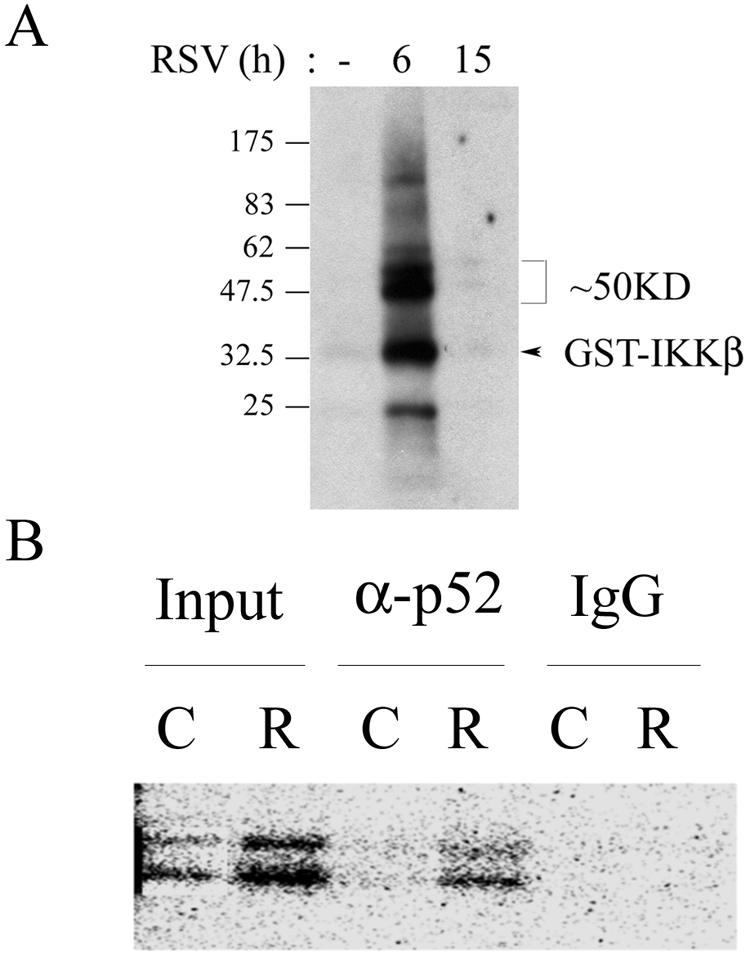
Effect of RSV on NIK activity and phosphorylation of NIK-associated endogenous substrate. (A) RSV induces a transient increase in NIK activity. NIK immunoprecipitation-kinase assay using 1 mg of WCE from either uninfected or RSV-infected cells was performed. An autoradiogram of the radiolabeled products after fractionation by 4 to 20% gradient SDS-PAGE and transfer to polyvinylidene difluoride membrane is shown. On the right, migration of recombinant GST-IKKβ substrate and endogenous ∼50-kDa substrate are shown. This figure represents one of three independent experiments. (B) Identification of p52 as endogenous substrate of NIK complex. Sequential immunoprecipitation of the NIK immunoprecipitation-kinase reaction was performed. Control (C) or RSV-infected (R) WCEs were subjected to NIK immunoprecipitation-kinase reaction. After the immunoprecipitation-NIK assay was performed, the kinase assay was terminated and extracted in glycine extraction buffer (pH 2.5) for 10 min at 4°C. The extracted complex was neutralized by 50 mM Tris-HCl (pH 9.4) and subjected to a second immunoprecipitation with either anti-p52 antibody or IgG (indicated at the top). Phosphoproteins were resolved on SDS-PAGE and detected by autoradiography. Input, fraction of starting immunoprecipitation-kinase assay. Note that the 52-kDa doublet increased by RSV infection was immunoprecipitated by anti-p52 antibody and not by control IgG. The results shown here are representative of two independent experiments.
RSV-induced nuclear translocation and DNA binding of p52 and Rel B.
NIK has been shown to be an indispensable protein in the activation of NF-κB through a noncanonical pathway. In this pathway, NIK is required for the cytoplasmic processing of the 100-kDa NF-κB2 precursor, generating 52-kDa NF-κB2, a DNA-binding subunit that complexes with the transactivating Rel B subunit, translocating into the nucleus. Since we observed a significant increase in the NIK expression, as well as its kinase activity, at 6 h in response to the RSV infection, it was imperative to determine whether RSV also increased 100-kDa NF-κB2 processing and nuclear translocation of the p52-Rel B complex. Nuclear accumulation of p52 and Rel B was assayed by Western immunoblotting in NEs prepared from RSV-infected A549 cells at different times. Sucrose cushion-purified NEs are free of cytoplasmic contamination, as determined by assay of β-tubulin, a highly selective cytoplasmic marker (Fig. 3A) (8). As seen in Fig. 3B, in uninfected cells, the amount of p52 and its precursor were barely detectable. However, at 6 h after RSV infection, a slight increase in p100 and stronger two- to threefold accumulation of p52 was seen, which continued for up to 15 h. In control experiments, p52 processing and nuclear translocation was not induced by mock infection (1.2-fold [data not shown]). Similarly, RSV infection led to a time-dependent increase in the nuclear accumulation of Rel B (Fig. 3C). The nuclear accumulation of Rel B was accompanied by gradual depletion of cytoplasmic Rel B, suggesting that RSV induced Rel B translocation. Together, these data indicate that RSV enhanced NF-κB2 processing and Rel B translocation in a time course subsequent to NIK activation.
FIG. 3.

Effect of RSV infection on the noncanonical NF-κB activation pathway in A549 cells. (A) Western immunoblot for nuclear and cytoplasmic markers. Duplicate samples of cytoplasmic (Cyto) and nuclear (Nuc) extracts were analyzed for lamin B and β-tubulin by Western immunoblotting. Molecular mass markers are indicated on the left (in kilodaltons). β-Tubulin is primarily localized to the cytoplasmic lysates. (B) RSV increases p100 processing and nuclear translocation of p52. The top panel shows a Western immunoblot of 50 μg of NEs prepared from RSV-infected A549 cells for various time periods and probed with anti-p52 monoclonal antibody. The location of unprocessed 100-kDa (p100) and 52-kDa (p52) NF-κB2 are indicated on the right. The first lane shows the molecular mass standards. The apparent sizes (in kilodaltons) are given on the left. In the bottom panel, the blot was probed with β-actin as a loading control. The experiment was repeated more than three times with similar results. (C) Time course of RSV-induced increase in Rel B nuclear translocation. Western immunoblots of 50 μg of cytoplasmic extract or sucrose cushion-purified NE from untreated or RSV-treated A549 cells were probed with anti-Rel B antibody (top panel). The blot was probed with β-actin (bottom panel) as a loading control.
We next assessed whether RSV-induced nuclear translocation of p52 and Rel B participated in the NF-κB binding complex to target DNA. For this purpose, “super-shift” DNA-binding assays were performed with NEs from RSV-infected cells. We found that, although Rel A and 50-kDa NF-κB1 were the major proteins in the NF-κB complex (indicating activation of the canonical NF-κB pathway), RSV exposure also increased p52 and Rel B binding (Fig. 4A). We further confirmed these findings by performing a two-step biotinylated (Bt)-DNA pull-down/Western blot assay on NEs of RSV-infected A549 cells. In this assay, a Bt-NF-κB DNA binding site is used to capture NF-κB isoforms on streptavidin beads. The washed complexes are then fractionated by SDS-PAGE, and the presence of NF-κB isoforms was detected by Western immunoblotting. We have previously shown that this assay accurately records changes in individual NF-κB isoforms in a sequence-specific fashion (7, 26). As observed in Fig. 4B, a time-dependent increase in p52 binding was observed in response to RSV, reaching a maximum by 24 h. A parallel change was observed for Rel B DNA binding (Fig. 4B, bottom). DNA-binding specificity was confirmed by using wild-type or mutant competitors. In this experiment, NEs from RSV-infected cells (24 h) were bound to Bt-DNA in presence of non-Bt competitors; the complexes were then captured on streptavidin beads and used for Western blot assay. We observed enhanced binding of both 100- and 52-kDa NF-κB2 isoforms (Fig. 4C, compare lanes 1 and 2). The binding of both was significantly decreased in the presence of non-Bt wild-type DNA competitor, whereas non-Bt mutant DNA did not affect the binding of either NF-κB2 isoform (Fig. 4C). This finding indicates that the RSV-inducible NF-κB2 binding was sequence specific. Together, these data indicate that the RSV induces NF-κB2 processing and nuclear translocation of the NF-κB2-Rel B DNA-binding complex after activation of NIK activity.
FIG. 4.

Effect of RSV infection on DNA-binding activity of NF-κB isoforms. (A) Gel mobility supershift DNA-binding assays. A total of 10 μg of NEs from uninfected or RSV-infected (24 h) was incubated in DNA-binding buffer and 32P-labeled NF-κB duplex oligonucleotide. Subunit-specific antibodies for the indicated NF-κB family members were added to the binding reactions. After incubation, the complexes were fractionated by 6% nondenaturing PAGE. The top panel shows the results of long autoradiographic exposure to demonstrate the supershifted band. The bottom panel shows the results of short exposure to demonstrate depletion of NF-κB binding complex. *, Location of supershifted band. The experiment was repeated twice showing similar band patterns. (B) Time course of p52 and Rel B DNA binding in response to RSV infection. One milligram of sucrose cushion-purified nuclear protein was assayed for p52 binding by microaffinity DNA pulldown assay. The top panel shows a Western blot of affinity-purified proteins associated with the Bt-NF-κB binding site probed with anti-p52 antibody. The bottom panel shows a Western blot with anti-Rel B antibody. A parallel increase in p52 and Rel B DNA binding was seen. The results here are from one of two independent experiments. (C) DNA binding specificity. Prior to microaffinity pulldown, RSV-infected NEs were first incubated with either wild-type (wt) or mutant (mt) non-Bt DNA duplexes as indicated. Samples were assayed for p52 binding by Western immunoblotting.
RSV-induced interaction of NIK, IKKα, and p52.
The hallmark of the noncanonical NF-κB activation pathway involves complex formation between NIK, IKKα, and 100-kDa NF-κB2 to releasing the proteolytically processed active p52. To determine whether RSV induced interactions between these mediators of the noncanonical pathway, nondenaturing coimmunoprecipitation of WCEs from uninfected or RSV-infected cells was performed. The immune complexes were then assayed by Western blotting with anti-IKKα or anti-p52 antibodies. We observed an exponential increase in IKKα interaction with NIK that peaks at 15 h (eightfold compared to uninfected cells) and further declines at 24 h (fivefold compared to uninfected cells). Recruitment of IKKα in the complex strongly coincides with an increase in NIK bound p52, which also increases tenfold at 24 h compared to uninfected cells (Fig. 5). These findings of NIK-IKKα-NF-κB2 complex formation strongly suggested activation of noncanonical pathway in RSV-infected cells.
FIG. 5.

RSV-induced interaction of NIK, IKKα, and p52. Western immunoblot of nondenaturing coimmunoprecipitation. A total of 1 mg of whole-cell lysate from control or RSV-infected cells (for the indicated times) was immunoprecipitated with anti-NIK antibody. Immune complexes were assayed by Western blotting with anti-IKKα (top) or anti-p52 (bottom) antibodies. Immunoprecipitation with IgG demonstrates specificity.
RSV-induced nuclear translocation of NIK.
NIK has been shown to exhibit dynamic nucleocytoplasmic shuttling under the control of functional nuclear import and export signals (6). To investigate whether RSV infection affected the subcellular distribution of NIK, nuclear and cytoplasmic extracts of A549 cells infected with RSV for various time intervals were assayed by Western blotting. In uninfected A549 cells, the amount of nuclear relative to cytoplasmic NIK was low (Fig. 6A). However, after 6 to 12 h of RSV infection, NIK transiently accumulated in the nucleus and then declined after 24 h. Concomitant with the transient nuclear accumulation, a decrease in NIK concentration was observed in the cytosolic fraction 6 to 12 h after RSV adsorption. To validate these dynamic changes in subcellular localization, a time course study of RSV-infected cells subjected to immunofluorescence microscopy with anti-NIK antibodies was carried out. In uninfected A549 cells, NIK was found predominantly in the cytoplasm, enriched in a perinuclear distribution (Fig. 6B). Between 3 and 6 h after RSV infection, NIK is strongly nuclear in localization. Together, these data indicate that RSV infection induces a transient NIK nuclear translocation in A549 cells at various times after activation of its kinase activity.
FIG. 6.

RSV-induced nuclear translocation of NIK. (A) Western immunoblot of 100 μg of NE or CE prepared from uninfected or RSV-infected A549 cells probed for endogenous NIK (top panel). The times of infection are shown at top (h). For the bottom panel, β-actin was probed as a loading control. Cytoplasmic NIK was depleted at 6 and 12 h with a concomitant increase in nuclear NIK. After 24 h, NIK redistributes back into the cytoplasm. (B) RSV-induced NIK nuclear translocation. Immunofluorescence microscopy was performed with anti-NIK antibody in a time course of RSV-infected A549 cells. The time (in hours) for each point is indicated at the top. In parallel, cells were stained with DAPI (4′,6′-diamidino-2-phenylindole) to localize nuclei. NIK is strongly nuclear 6 h after RSV infection. Western blotting and confocal microscopy were repeated twice with similar results.
Interaction of NIK and p52 in response to RSV.
Since RSV induces nuclear translocation of NIK, p52, and Rel B with similar kinetics, all major components of the noncanonical NF-κB activation pathway, we sought to determine whether these proteins interacted within the nuclear compartment. To address these questions, nondenaturing NF-κB2 coimmunoprecipitations from RSV-infected cells were probed with anti-NIK antibody. We observed an exponential increase in the interaction of NIK with p52 in the nucleus; this increase reached a maximum 24 h after RSV exposure (Fig. 7A). To independently validate these findings and exclude artifactual association produced by cell extraction, we performed colocalization experiments in fixed cells by using confocal microscopy. A549 cells were infected with RSV for 6 h, allowing NIK to maximally accumulate in the nucleus (Fig. 6A); the cells were then stained with FITC-labeled anti-NIK- and rhodamine-labeled anti-p52 antibodies (Fig. 7B). This showed that nuclear NIK and p52 are present in large discrete nuclear granules. When the two images were merged, the two proteins were seen to strongly colocalize (Fig. 7B), validating the coimmunoprecipitation experiment (Fig. 7A). Taken together, these findings suggest that NIK complexes with p52 in the nuclei of RSV-infected cells. These data further suggest that NIK plays an important role in regulating the nuclear function of p52 and Rel B in RSV infection.
FIG. 7.

Time course of RSV-induced association of nuclear NIK and p52. (A) Nondenaturing coimmunoprecipitation of NIK-p52 complex. p52 was immunoprecipitated from 1.0 mg of NEs from uninfected or RSV-infected A549 cells. In the top panel, immune complexes were assayed for the presence of NIK by Western immunoblotting. In the lower panel, β-actin staining in the input shows equal amounts of starting protein. (B) Microscopic colocalization of nuclear NIK and p52. A549 cells infected with RSV for 15 h were fixed and treated by limited protease digestion (see Materials and Methods) to isolate nuclei. Cells were stained with primary anti-NIK or p52 antibody and either FITC- or rhodamine-conjugated secondary antibody as indicated. Colocalization was visualized by superimposition of green and red images (right panel). Magnification, ×60.
siRNA-mediated knock-down of NIK blocks RSV-induced p100 processing.
In order to understand the role of the noncanonical pathway in RSV-induced NF-κB-dependent gene expression, we used NIK- and p52-specific siRNAs to knock down the two major components of this pathway. In control experiments, anti-NIK siRNA used at 50 nM could specifically knock down >90% of the steady-state levels of protein compared to control siRNA-transfected cells (Fig. 8A). To determine the role of NIK in RSV-induced p52 nuclear translocation, cells were treated with anti-NIK siRNA and infected with RSV for 6 h, and p52 formation was measured by Western blotting. Figure 8B clearly shows that the NIK siRNA (using either 50 or 100 nM) significantly lowers the ability of RSV to induce nuclear p52 accumulation. Thus, these data suggest the essential role of NIK in RSV-induced activation of NF-κB2 processing (the noncanonical NF-κB activation pathway) in A549 cells.
FIG. 8.

NIK and p52 knock-down by using siRNA. (A) A549 (106) cells were treated with either NIK-specific (NIK) or control nonspecific (C) siRNAs at the indicated concentrations for 72 h. In the top panel is shwon a Western immunoblot probed with anti-NIK antibody. For the bottom panel, the blot was probed with β-actin internal control. The experiment was repeated more than three times with similar results. (B) Effect of NIK knock-down on RSV-induced p52 nuclear translocation. A549 cells were treated with control or NIK-specific siRNA for 72 h, followed by RSV infection for 15 h. Afterward, NEs were prepared, and the level of nuclear p52 protein was determined by Western blotting.
NIK activity influences early RSV-induced NF-κB-dependent gene expression.
To ascertain the role of NIK activity in RSV-induced gene expression at transcription level, A549 cells were transiently transfected with a construct containing human RANTES promoter linked to the luciferase reporter gene in the presence of empty eukaryotic expression vector (pEF) or expression vector encoding a kinase-inactive, dominant-negative NIK (pEF-NIKDN). Transfection of empty vector alone did not affect basal RANTES transcriptional activity; however, basal RANTES activity was significantly reduced by pEF-NIKDN (Fig. 9). When transfectants were exposed to RSV for 24 h, RANTES-dependent luciferase activity was induced by fourfold compared to the vector alone. Cotransfection of pEF-NIKDN completely suppressed this RSV-induced transcriptional activity. These data suggest the importance of NIK activity for basal and RSV-induced transcriptional activation of the human RANTES promoter (Fig. 9).
FIG. 9.

Effect of dominant-negative NIK (DN-NIK) on RSV-induced RANTES promoter activity. A549 cells were transiently transfected with the indicated plasmids, along with the RANTES-220 promoter luciferase construct. At 36 h after transfection, cells were infected with RSV (24 h), and the reporter activity was measured. Each transfection was performed in triplicate, and the experiment was repeated twice. Values are plotted as mean ± the standard deviation. *, P < 0.05; **, P < 0.01 (significantly different from vector transfected cells [two-tailed Student t test]).
We further investigated the role of the noncanonical NF-κB activation pathway on the endogenous expression of RSV-induced NF-κB-dependent genes. These genes are strongly regulated by RSV by the canonical NF-κB activation pathway because their induction is blocked by expression of the nondegradable IκBα mutant (43). For this experiment, siRNA-mediated knock-down for p52 was validated in A549 cells (Fig. 10A). siRNA for p52 potently inhibited nuclear abundance of p52 relative to control siRNA-treated cells. We next transfected A549 with control siRNA or siRNA specific to either NIK or p52 and analyzed the RSV-induced expression of nine different NF-κB-dependent genes (42) determined by Q-RT-PCR. The expression of NF-κB2, IL-8, and CXCL-1 was significantly reduced by either NIK or p52 knock-down at 6 and 12 h after RSV exposure (Fig. 10B). In contrast, the expression of IRF-1, Rel B, and RANTES was significantly lowered by knocking down NIK for 6 and 12 h after RSV infection. In these genes, the effect of p52 knock-down was less, being observed only after 6 h, but was lost at 12 h of RSV infection (Fig. 10C). For another set of NF-κB-dependent genes—IκBα, STAT1, and complement factor B (BF)—the effect of NIK knock-down was also observed at 6 and 12 h of RSV infection, but the effect of p52 was only observed at 6 h after RSV infection (Fig. 10D). These findings suggest that the requirement of NIK for NF-κB-dependent gene expression may not be strictly mediated through the p52 pathway. NIK might be regulating other pathways directly or indirectly, which might be required for the activation of these genes in response to RSV. To determine whether inhibition of p52 or NIK expression affected RSV replication, we measured RSV transcription in control or siRNA transfected cells by Northern blot (Fig. 10E). A strong induction of RSV N mRNA was seen 6 h after RSV infection, whose level of expression was not affected by p52 or NIK siRNA. Taken together, these findings suggest that RSV induces NF-κB-dependent genes in a heterogeneous manner, which may be differentially regulated by NIK, p52 via the noncanonical pathway, or Rel A via the canonical pathway. Studies are under way to further elucidate the contribution of NIK and p52 in the expression of these genes.
FIG. 10.

Effect of siRNA-mediated NIK or p52 knock-down on RSV-induced expression of NF-κB-dependent genes. (A) Efficiency of p52 siRNA knock-down. A549 cells were treated with either NF-κB2 (p52) or control nonspecific (C) siRNAs at the indicated concentrations for 72 h.The top panel shows a Western immunoblot probed with anti-NF-κB2 antibody. For the bottom panel, the blot was probed with anti-β-actin as an internal control. (B) Effect of NIK or p52 knock-down on RSV-induced expression on NF-κB2, IL-8, and CXCL-1. A549 cells were treated with 50 nM concentrations of control or NIK- or p52-specific siRNA for 72 h. After RSV infection for the indicated times, total RNA was isolated and subjected to quantitative real-time PCR. Values were normalized to 18S internal control and plotted as means ± the standard deviations relative to control samples. *, P < 0.05; **, P < 0.01 (significantly different from control siRNA transfected cells [two-tailed Student t test]). Experiments were repeated twice with similar results. (C) Effect of NIK or p52 knock-down on IRF-1, Rel B, and RANTES. The experiment was as described in Fig. 10B. (D) Effect of NIK or p52 knock-down on IκBα, STAT1, and complement B (Comp B). The experiment was as described in Fig. 10B. (E) Effect of NIK or p52 knock-down on RSV transcription. Cells were transfected with control (C), NIK (N), or p52 (P) siRNA and infected with RSV. A Northern blot of RSV N transcript expression at the indicated times (in hours) after RSV adsorption is shown. For the bottom panel, thymosin β hybridization was used as a loading control. siRNA treatment had no effect on RSV N expression.
DISCUSSION
RSV is a major cause of epidemic respiratory disease in the United States and worldwide (22, 46). In natural infections, RSV only replicates in the airway mucosa, eliciting inflammatory responses by producing cytokines (13, 15, 34) and chemokines (5, 35, 52). Although RSV replication activates diverse intracellular signaling pathways, we have found that NF-κB activation is the central mediator of RSV-induced airway inflammation in vivo by its ability to activate expression of gene networks in airway epithelium (15, 43). In the present study, we examine the serendipitous finding that RSV induces expression of NIK in alveolus-like A549 cells, a critical upstream mediator of the noncanonical NF-κB activation pathway. We have observed that RSV-induces NIK activity rapidly at times that precede maximal complex formation with IKKα and NF-κB2. Moreover, we find that NIK is required for both processing the 100-kDa NF-κB2 precursor into the 52-kDa NF-κB2 DNA-binding protein and subsequent translocation of the p52-Rel B complex into the nucleus. We make the novel observations that p52 is an endogenous substrate of the activated NIK complex and that the NIK-p52 complex transiently translocates into the nucleus of RSV-infected cells. That NIK is essential for activation of NF-κB2 processing and influences expression of NF-κB-dependent genes in a heterogeneous manner is indicated by specific siRNA-mediated knock-down of NIK in RSV-infected cells. These studies extend our previous understanding for the signaling pathways controlling NF-κB activation after RSV infection.
Earlier we showed that RSV infection induces activation of the “classical” or “canonical” NF-κB activation pathway in A549 cells. The hallmark of the canonical NF-κB activation pathway is the activation of IKKβ, the major kinase responsible for coupled phosphorylation-degradation of the IκBα inhibitor. This process releases Rel A to translocate into the nucleus, bind DNA, and activate the expression of genetic targets (14, 24, 32, 33). Evidence for the activation of the canonical pathway has been observed both in vitro and in vivo. In vitro, RSV induces a time-dependent induction of IκBα proteolysis (24) and Rel A translocation (15). Moreover, transient expression of the nondegradable IκBα mutant that potently inhibits the canonical NF-κB activation pathway significantly blocks expression of 144 NF-κB-dependent genes, including chemokines (IL-8 and RANTES), transcription factors controlling innate immunity (STAT-1/IRF-1), and other diverse functions (43). In vivo, RSV infection activates mucosal IKKβ kinase and Rel A translocation in a manner that was inhibited by administration of a cell-permeable inhibitor of the IKKγ-β association (20). Here, selective inhibition of the canonical pathway reduced expression of RSV-induced chemokine production and peribronchial airway inflammation (19). Together, these findings indicate that the canonical NF-κB activation pathway is strongly activated by RSV infection and that its genetic targets play an important role in RSV-mediated lung inflammation. However, our kinetic studies have consistently indicated that IκBα proteolysis occurs quite slowly, being consistently detectable 24 h after RSV adsorption, whereas activation of NF-κB-dependent targets precedes IκBα proteolysis (24, 43).
Our findings here that activation of NIK activity and NF-κB2 processing precedes activation of the canonical pathway can help to reconcile our previous disparate observation between the kinetics of classic NF-κB activation and activation of NF-κB-dependent genes. The “alternative” or “noncanonical” NF-κB pathway activation is independent of the IKKβ and -γ kinases (37, 47), relying instead on NIK and IKKα (37, 49). Here, NIK has a central role in inducible 100-kDa NF-κB2 processing, where it functions as a docking or scaffold protein to recruit IKKα to p100 (48), and its kinase activity phosphorylates and activates IKKα on its activation loop (29). Activated IKKα, in turn, phosphorylates specific COOH-terminal serine residues of p100, targeting it for ubiquitination and subsequent 26S proteasomal processing mediated by the β-TrCP ubiquitin ligase and 26S proteasome (48). The requirement for NIK activity in this process is further shown by the fact that the p100 processing is impaired in mice expressing a mutant NIK, known as the alymphoplasia mutant (37, 49). Our kinetic experiments indicate that NIK activity precedes significant 52-kDa NF-κB2 processing and nuclear translocation of the p52-Rel B complex. Our findings that siRNA-mediated knock-down of NIK expression blocks RSV-induced p52 formation and nuclear translocation clearly suggests the central role of NIK in the RSV-induced noncanonical pathway. Although the canonical and noncanonical pathways are mediated by separate intracellular kinases, a requirement for the activity of both can be identified on the NF-κB-dependent genes. We show here that inhibition of the noncanonical pathway interferes with early NF-κB-dependent gene activation over the first 6 to 12 h of RSV infection, with the effect of NIK knock-down being more significant than that of p52. Later, at 24 h, when IκBα proteolysis is observed, the more potent canonical pathway predominates, producing robust induction of gene expression (24, 43). The two pathways thus appear to interact at temporally distinct phases on the same genes during the evolution of the RSV response. These interrelationships are shown schematically in Fig. 11. Although the contribution of the NIK/p52 pathway on the magnitude of NF-κB-dependent gene expression appears to be minor, our data show that the kinetic response of the NF-κB-dependent chemokine genes is markedly affected, with the expression profile being significantly slower in the absence of NIK. The rate of chemokine expression may have significant implications for host response to viral infection, a topic that needs to be explored further.
FIG. 11.

Temporal interrelationships between the noncanonical and canonical NF-κB activation pathways in RSV-infected cells. Schematic diagram of early (6-h) and late (24-h) responses to RSV replication. (Left panel) Early in the course of RSV infection, the noncanonical NF-κB pathway is activated, resulting in initial activation of NF-κB-dependent genes. This pathway involves NIK activation, NF-κB2 processing, and nuclear translocation of the NIK-p52 complex. (Right panel) Later in the course of RSV infection, the canonical pathway is activated mediated by IKKβ activation, IκBα proteolysis, and Rel A translocation. At this point, NIK is redistributed back into the cytoplasm.
Earlier work has shown that the noncanonical pathway can be activated in response to specific stimuli, including LTβ (11, 49), CD40 ligand (10), DNA virus infection with Epstein-Barr virus (30), and B-cell activating factor (9, 27). Presently, it is thought that NIK activity is negatively controlled by signal adapters, including tumor necrosis factor receptor-associated factors (TRAFs) (28). Ligands binding LTβ or CD40 ligand rapidly induce TRAF ubiquitination and proteasomal degradation. Depletion of TRAF then allows activation of NIK through a poorly understood process involving autophosphorylation and oligomerization. In the case of Epstein-Barr virus, a specific oncoprotein termed the latent membrane protein 1 (LMP-1) binds TRAFs and, in so doing, activates the noncanonical NF-κB activation pathway in infected cells. The mechanism by which RSV induces NIK need to be examined further. In our preliminary Western blot studies, we have found that RSV does not induce TRAF degradation in a time frame that would be consistent with NIK activation (data not shown). Moreover, RSV, a member of the Paramyxoviridae family of negative-sense RNA viruses, has no LMP-1 homolog, and intracellular sequestration of TRAF does not appear to be a likely mechanism. Additional research is required to determine exactly how RSV-mediated NIK activation occurs. Finally, our observation that NIK gene expression is induced by RSV is novel. We note that the induction of NIK expression occurs temporally after the major activation of its kinase activity, and therefore this phenomenon of enhanced gene expression is apparently unrelated to early NF-κB2 processing (Fig. 3), complex formation with p52 and IKKα, and nuclear translocation (Fig. 6). Because the magnitude of NIK protein expression is less than that of its mRNA induction, RSV may be enhancing NIK protein turnover. More studies are needed to identify the signal transduction mechanisms controlling its expression or to determine the consequences of enhanced NIK production for the RSV-infected cell.
We report here our observations that RSV induces transient nuclear accumulation of an NIK · 52-kDa NF-κB2 complex in A549 cells. Recently, it has been shown that NIK, like other NF-κB family members, has a classical nuclear localization signal and dynamically shuttles between the cytoplasm and nucleus (6). The effects of various stimuli on changing the equilibrium of NIK nuclear transport have not been systemically studied to our knowledge. Our findings here that siRNA-mediated knock-down of NIK and NF-κB2 resulted in decreased RSV-induced expression of nine different NF-κB-dependent genes suggests a potential functional nuclear role of this NIK-52-kDa NF-κB2 complex. Recently, the role of IKKα in controlling NF-κB-dependent gene expression was suggested from chromatin immunoprecipitation assays (1, 50). IKKα was implicated as having a secondary activity by phosphorylating regulatory histones on target gene promoters. These studies indicated that the regulatory kinases of the IKK pathway may have dual functions in the cytoplasm and nucleus. A similar, as-yet-undiscovered, role may also be possible for NIK.
The NF-κB family of transcription factors are extensively phosphorylated in ways that modify their nuclear localization, transcriptional potency, or protein processing. In immunoprecipitation-kinase assays, we tentatively identified p52 as an endogenous substrate for the RSV-activated NIK complex and showed that its phosphorylation occurs in parallel with IKKβ-directed NIK activity. The kinase responsible for p52 phosphorylation cannot be determined from our data and may represent NH2-terminal p52 phosphorylation by IKKα. However, we note that NIK activation and p52 phosphorylation occurs prior to strong complex association with IKKα (compare Fig. 2 and 5), so our data are not entirely consistent with IKKα mediating this role. Irrespective of the kinase responsible, the functional consequences of p52 phosphorylation have not been systematically studied, but this may affect its DNA-binding properties or its ability to interact with coactivators. To determine this will require further study.
In conclusion, RSV infection activates NF-κB by both canonical and noncanonical NF-κB activation pathways in a temporally defined cascade. Our findings here indicate that NIK may play a central role in this process, being essential for RSV-induced 100-kDa NF-κB2 processing into p52, forming nuclear complexes with p52, and influencing early NF-κB-dependent gene expression.
Acknowledgments
We thank Ping Liu for discussions and David Goeddel for the kind gift of NIK cDNA.
This project was supported by NIAID grant R01AI40218 (A.R.B.). Core laboratory support was by NIEHS grant P30 ES06676 (J. Halpert, UTMB).
REFERENCES
- 1.Anest, V., J. L. Hanson, P. C. Cogswell, K. A. Steinbrecher, B. D. Strahl, and A. S. Baldwin. 2003. A nucleosomal function for IκB kinase-alpha in NF-κB-dependent gene expression. Nature 423:659-663. [DOI] [PubMed] [Google Scholar]
- 2.Atreya, P. L., and S. Kulkarni. 1999. Respiratory syncytial virus strain A2 is resistant to the antiviral effects of type I interferons and human MxA. Virology 261:227-241. [DOI] [PubMed] [Google Scholar]
- 3.Baldwin, A. S. J. 1996. The NF-κB and I κB proteins: new discoveries and insights. Annu. Rev. Immunol. 14:649-683. [DOI] [PubMed] [Google Scholar]
- 4.Barnes, P. J., and M. Karin. 1997. Nuclear factor-κB: a pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 336:1066-1071. [DOI] [PubMed] [Google Scholar]
- 5.Becker, S., W. Reed, F. W. Henderson, and T. L. Noah. 1997. RSV infection of human airway epithelial cells causes production of the β-chemokine RANTES. Am. J. Physiol. 272:L512-L520. [DOI] [PubMed] [Google Scholar]
- 6.Birbach, A., P. Gold, B. R. Binder, E. Hofer, R. de Martin, and J. A. Schmid. 2002. Signaling molecules of the NF-κB pathway shuttle constitutively between cytoplasm and nucleus. J. Biol. Chem. 277:10842-10851. [DOI] [PubMed] [Google Scholar]
- 7.Brasier, A. R., M. Jamaluddin, A. Casola, W. Duan, Q. Shen, and R. Garofalo. 1998. A promoter recruitment mechanism for TNFα-induced IL-8 transcription in type II pulmonary epithelial cells: dependence on nuclear abundance of RelA, NF-κB1, and c-Rel transcription factors. J. Biol. Chem. 273:3551-3561. [DOI] [PubMed] [Google Scholar]
- 8.Brasier, A. R., H. Spratt, Z. Wu, I. Boldogh, Y. Zhang, R. P. Garofalo, A. Casola, J. Pashmi, A. Haag, B. Luxon, and A. Kurosky. 2004. Nuclear heat shock response and novel nuclear domain 10 reorganization in respiratory syncytial virus-infected A549 cells identified by high-resolution two-dimensional gel electrophoresis. J. Virol. 78:11461-11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Claudio, E., K. Brown, S. Park, H. Wang, and U. Siebenlist. 2002. BAFF-induced NEMO-independent processing of NF-κB2 in maturing B cells. Nat. Immunol. 3:958-965. [DOI] [PubMed] [Google Scholar]
- 10.Coope, H. J., P. G. Atkinson, B. Huhse, M. Belich, J. Janzen, M. J. Holman, and S. C. Ley. 2002. CD40 regulates the processing of NF-κB2 p100 to p52. EMBO J. 21:5375-5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dejardin, E., N. M. Droin, M. Delhase, E. Haas, Y. Cao, C. Makris, Z. W. Li, M. Karin, C. F. Ware, and D. R. Green. 2002. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-κB pathways. Immunity 17:525-535. [DOI] [PubMed] [Google Scholar]
- 12.Ferris, J. A., W. A. Aherne, and W. S. Locke. 1973. Sudden and unexpected deaths to infants: histology and virology. BMJ 2:439-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fiedler, M. A., K. Wernke-Dollries, and J. M. Stark. 1995. Respiratory syncytial virus increases IL-8 gene expression and protein release in A549 cells. Am. J. Physiol. 269:L865-L872. [DOI] [PubMed] [Google Scholar]
- 14.Fiedler, M. A., K. Wernke-Dollries, and J. M. Stark. 1996. Inhibition of viral replication reverses respiratory syncytial virus-induced NF-kB activation and interleukin-8 gene expression in A549 cells. J. Virol. 70:9079-9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garofalo, R., M. Sabry, M. Jamaluddin, R. K. Yu, A. Casola, P. L. Ogra, and A. R. Brasier. 1996. Transcriptional activation of the interleukin-8 gene by RSV infection in alveolar epithelial cells: nuclear translocation of the Rel A transcription factor as a mechanism producing airway mucosal inflammation. J. Virol. 70:8773-8781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garofalo, R. P., and H. Haeberle. 2000. Epithelial regulation of innate immunity to respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 23:581-585. [DOI] [PubMed] [Google Scholar]
- 17.Ghosh, S., and D. Baltimore. 1990. Activation in vitro of NF-κB by phosphorylation of its inhibitor I κB. Nature 344:678-682. [DOI] [PubMed] [Google Scholar]
- 18.Guan, K. L., and J. E. Dixon. 1991. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal. Biochem. 192:262-267. [DOI] [PubMed] [Google Scholar]
- 19.Haeberle, H., A. Casola, Z. Gatalica, S. Petronella, H.-J. Dieterich, P. B. Ernst, A. R. Brasier, and R. P. Garofalo. 2004. IκB kinase is a critical regulator of chemokine expression and lung inflammation in respiratory syncytial virus infection. J. Virol. 78:2232-2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haeberle, H., R. Takizawa, A. Casola, A. R. Brasier, H.-J. Dieterich, N. van Rooijen, Z. Gatalica, and R. P. Garofalo. 2002. Respiratory syncytial virus-induced activation of NF-κB in the lung involves alveolar macrophages and Toll-like receptor 4-dependent pathways. J. Infect. Dis. 186:1199-1206. [DOI] [PubMed] [Google Scholar]
- 21.Hall, C. B. 2001. Respiratory syncytial virus and parainfluenza virus. N. Engl. J. Med. 344:1917-1928. [DOI] [PubMed] [Google Scholar]
- 22.Hall, C. B., and C. A. McCarthy. 1995. Respiratory syncytial virus, p. 1501-1519. In G. L. Mandel, J. E. Bennett, and R. Dolin (ed.), Principles and practice of infectious diseases. Churchill Livingston, New York, N.Y.
- 23.Han, Y., and A. R. Brasier. 1997. Mechanism for biphasic Rel A:NF-κB1 nuclear translocation in tumor necrosis factor α-stimulated hepatocytes. J. Biol. Chem. 272:9823-9830. [DOI] [PubMed] [Google Scholar]
- 24.Jamaluddin, M., A. Casola, R. P. Garofalo, Y. Han, T. Elliott, P. L. Ogra, and A. R. Brasier. 1998. The major component of IκBα proteolysis occurs independently of the proteasome pathway in respiratory syncytial virus-infected pulmonary epithelial cells. J. Virol. 72:4849-4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jamaluddin, M., R. Garofalo, P. L. Ogra, and A. R. Brasier. 1996. Inducible translational regulation of the NF-IL6 transcription factor by respiratory syncytial virus infection in pulmonary epithelial cells. J. Virol. 70:1554-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jamaluddin, M., T. Meng, J. Sun, I. Boldogh, Y. Han, and A. R. Brasier. 2000. Angiotensin II induces nuclear factor (NF)-κB1 isoforms to bind the angiotensinogen gene acute-phase response element: a stimulus-specific pathway for NF-κB activation. Mol. Endocrinol. 14:99-113. [DOI] [PubMed] [Google Scholar]
- 27.Kayagaki, N., M. Yan, D. Seshasayee, H. Wang, W. Lee, D. M. French, I. S. Grewal, A. G. Cochran, N. C. Gordon, J. Yin, M. A. Starovasnik, and V. M. Dixit. 2002. BAFF/BLyS receptor 3 binds the B-cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-κB2. Immunity 17:515-524. [DOI] [PubMed] [Google Scholar]
- 28.Liao, G., M. Zhang, E. W. Harhaj, and S. C. Sun. 2004. Regulation of the NF-κB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J. Biol. Chem. 279:26243-26250. [DOI] [PubMed] [Google Scholar]
- 29.Ling, L., Z. Cao, and D. V. Goeddel. 1998. NF-κB inducing kinase activates IKKα phosphorylation of Ser-176. Proc. Natl. Acad. Sci. USA 95:3792-3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luftig, M., T. Yasui, V. Soni, M. S. Kang, N. Jacobson, E. Cahir-McFarland, B. Seed, and E. Kieff. 2004. Epstein-Barr virus latent infection membrane protein 1 TRAF-binding site induces NIK/IKKα-dependent noncanonical NF-κB activation. Proc. Natl. Acad. Sci. USA 101:141-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacDonald, N. E., C. B. Hall, and S. C. Suffin. 1982. Respiratory syncytial viral infection in infants with congenital heart disease. N. Engl. J. Med. 307:397-400. [DOI] [PubMed] [Google Scholar]
- 32.Mastronarde, J. G., B. He, M. M. Monick, N. Mukaida, K. Matsushima, and G. W. Hunninghake. 1996. Induction of interleukin (IL)-8 gene expression by respiratory syncytial virus involves activation of nuclear factor (NF)-κB and NF-IL-6. J. Infect. Dis. 174:262-267. [DOI] [PubMed] [Google Scholar]
- 33.Mastronarde, J. G., M. M. Monick, N. Mukaida, K. Matsushima, and G. W. Hunninghake. 1998. Activator protein-1 is the preferred transcription factor for cooperative interaction with nuclear factor-κB in respiratory syncytial virus-induced interleukin-8 gene expression in airway epithelium. J. Infect. Dis. 177:1275-1281. [DOI] [PubMed] [Google Scholar]
- 34.Noah, T. L., and S. Becker. 1993. Respiratory syncytial virus-induced cytokine production by a human bronchial epithelial cell line. Am. J. Physiol. 265:L472-L478. [DOI] [PubMed] [Google Scholar]
- 35.Olzewska, B., A. Casola, T. Saito, R. Alam, S. Crowe, F. Mei, P. L. Ogra, and R. Garofalo. 1998. Cell-specific expression of RANTES, MCP-1, and MIP-1α by lower airway epithelial cells and eosinophils infected with respiratory syncytial virus. J. Virol. 72:4756-4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rothwarf, D. M., E. Zandi, G. Natoli, and M. Karin. 1998. IKK-γ is an essential regulatory subunit of the IκB kinase complex. Nature 395:297-300. [DOI] [PubMed] [Google Scholar]
- 37.Senftleben, U., Y. Cao, G. Xiao, F. R. Greten, G. Krahn, G. Bonizzi, Y. Chen, Y. Hu, A. Fong, S. C. Sun, and M. Karin. 2001. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science 293:1495-1499. [DOI] [PubMed] [Google Scholar]
- 38.Shay, D. K., R. C. Holman, R. D. Newman, L. L. Liu, J. W. Stout, and L. J. Anderson. 1999. Bronchiolitis-associated hospitalizations among US children, 1980-1996. JAMA 282:1440-1446. [DOI] [PubMed] [Google Scholar]
- 39.Shay, D. K., R. C. Holman, G. E. Roosevelt, M. J. Clarke, and L. J. Anderson. 2001. Bronchiolitis-associated mortality and estimates of respiratory syncytial virus-associated deaths among US children, 1979-1997. J. Infect. Dis. 183:16-22. [DOI] [PubMed] [Google Scholar]
- 40.Sherman, C. T., and A. R. Brasier. 2001. Role of STAT1 and STAT3 in inducible expression of the human angiotensinogen gene by interleukin-6. Mol. Endocrinol. 15:441-457. [DOI] [PubMed] [Google Scholar]
- 41.Siebenlist, U., G. Franzoso, and K. Brown. 1994. Structure, regulation and function of NF-κB. Annu. Rev. Cell Biol. 10:405-455. [DOI] [PubMed] [Google Scholar]
- 42.Tian, B., and A. R. Brasier. 2003. Identification of an NF-κB-dependent gene network. Rec. Prog. Hormone Res. 58:95-130. [DOI] [PubMed] [Google Scholar]
- 43.Tian, B., Y. Zhang, B. A. Luxon, R. P. Garofalo, A. Casola, M. Sinha, and A. R. Brasier. 2002. Identification of NF-κB-dependent gene networks in respiratory syncytial virus-infected cells. J. Virol. 76:6800-6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ueba, O. 1978. Respiratory syncytial virus: I. concentration and purification of the infectious virus. Acta Med. Okayama 32:265-272. [PubMed] [Google Scholar]
- 45.Webb, M. S. C., R. L. Henry, and A. D. Milner. 1985. Continuing respiratory problems three and a half years after acute viral bronchiolitis. Arch. Dis. Child 60:1064-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wyde, P. R. 1998. Respiratory syncytial virus (RSV) disease and prospects for its control. Antiviral Res. 39:63-79. [DOI] [PubMed] [Google Scholar]
- 47.Xiao, G., M. E. Cvijic, A. Fong, E. W. Harhaj, M. T. Uhlik, M. Waterfield, and S. C. Sun. 2001. Retroviral oncoprotein Tax induces processing of NF-κB2/p100 in T cells: evidence for the involvement of IKKα. EMBO J. 20:6805-6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiao, G., A. Fong, and S. C. Sun. 2004. Induction of p100 processing by NF-κB-inducing kinase involves docking IκB kinase alpha (IKKα) to p100 and IKKα-mediated phosphorylation. J. Biol. Chem. 279:30099-30105. [DOI] [PubMed] [Google Scholar]
- 49.Xiao, G., E. W. Harhaj, and S. C. Sun. 2001. NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol. Cell 7:401-409. [DOI] [PubMed] [Google Scholar]
- 50.Yamamoto, Y., U. N. Verma, S. Prajapati, Y. T. Kwak, and R. B. Gaynor. 2003. Histone H3 phosphorylation by IKKα is critical for cytokine-induced gene expression. Nature 423:655-659. [DOI] [PubMed] [Google Scholar]
- 51.Zhang, Y., M. Jamaluddin, B. Tian, S. Wang, A. Casola, R. P. Garofalo, and A. R. Brasier. 2003. Ribavirin treatment upregulates antiviral gene expression via the IFN-stimulated response element in RSV-infected epithelial cells. J. Virol. 78:5933-5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang, Y., B. A. Luxon, A. Casola, R. P. Garofalo, M. Jamaluddin, and A. R. Brasier. 2001. Expression of RSV-induced chemokine gene networks in lower airway epithelial cells revealed by cDNA microarrays. J. Virol. 75:9044-9058. [DOI] [PMC free article] [PubMed] [Google Scholar]