

ABSTRACT
Fungi have emerged as premier opportunistic microbes of the 21st century, having a considerable impact on human morbidity and mortality. The huge increase in incidence of these diseases is largely due to the HIV pandemic and use of immunosuppressive therapies, underscoring the importance of the immune system in defense against fungi. This article will address how the mammalian immune system recognizes and mounts a defense against medically relevant fungal species.
CONCEPTUAL FRAMEWORK FOR IMMUNOLOGY
The immune system has over millennia evolved strategies to discriminate between what is “self” and what is foreign (nonself) as well as “normal self” and “injured or altered self,” with the ultimate purpose being to defend and repair self. First, to fully appreciate the framework that governs most of our current understanding of how the immune system operates, it is imperative to be cognizant of some of the immunological models that have laid the foundation on which we stand.
Self-Nonself Discrimination Models
The 1960 Nobel Prize in Physiology or Medicine was awarded to F. MacFarlane Burnet and Peter Medawar for the discovery of immunological tolerance. Burnet suggested that each B lymphocyte, an immune cell responsible for the production of antibodies (or immunoglobulins), expresses numerous clones of cell surface immunoglobulin receptors specific for a foreign body (antiforeign body) and that when antibody receptor recognizes its cognate antigen, an immune response is initiated (1). This idea was solidified by Billingham, Brent, and Medawar’s findings, which demonstrated that adult mice “tolerate” skin grafts if they are transplanted at birth; that is, the immune system can learn to tolerate the foreign antigens until defensive intolerance is acquired (2).
The self-nonself discrimination model became the modus operandi of the immune system until it was modified in 1969 by Bretscher and Cohn, who postulated that a quiescent B lymphocyte requires interaction with a “helper” cell (now known as helper T lymphocyte) specific for the same antigen to induce an activation signal, whereas inactivation ensues when antigen is recognized in the absence of the helper cell (3). In 1975, Lafferty and Cunningham introduced a requirement for a costimulation signal and proposed that recognition of antigen by the helper T cell (which provides signal 1) together with a “stimulator cell” (signal 2) (known now as antigen-presenting cell [APC]) induced activation of the immune system (4, 5). Over a decade later, Jenkins and Schwartz provided experimental evidence for the requirement of costimulation for activation of resting lymphocytes (6). However, the prerequisite of costimulation introduced a conundrum because this meant that immune activation hinges on APCs that capture and present both self and nonself antigens; therefore, immunity could not be solely targeted against the invading foreigners.
Noninfectious Self and Infectious Nonself Model and the Inception of Pattern Recognition Receptors
Charles Janeway provided an elegant solution to the costimulation conundrum mediated by APCs. In his 1989 article Janeway suggested that the immune system has evolved specifically to recognize and respond to infectious organisms and that this involves recognition not only of antigenic determinants but also of certain characteristics or patterns common in infectious organisms but absent from the host, and he coined the term “pattern recognition receptor” (PRR) (7). He also pointed out that these germline-encoded receptors would act early during an immune response and imagined that they also play a role in shaping the adaptive T and B cell responses. Another piece of the puzzle was added by Polly Matzinger, who in 1994 proposed that resting APCs are activated by stimuli associated with host-cell damage, which she termed “danger signals” (8). She posited that by definition, pathogens damage the host and that recognition of damage could be instrumental in validating initiation of an immune response. A summary of the history of the immune models discussed above is depicted in Fig. 1.
FIGURE 1.
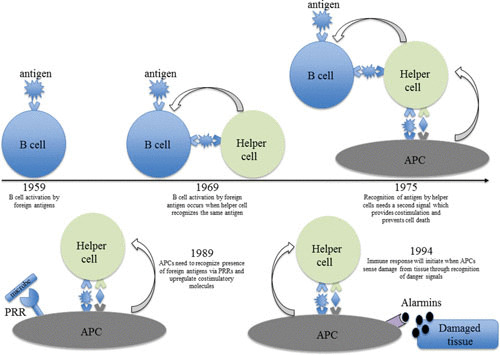
More than half a century of immunogical theories.
PRRs INVOLVED IN SENSING FUNGI
Several classes of PRRs have been discovered since their initial identification, providing experimental support for Janeway’s theory. It is noteworthy that pioneering work emanating from the use of fungi and their antigenic determinants has contributed significantly to our understanding of the initiation of an immune response (for a detailed timeline, see reference 9). Here, we will discuss PRRs that have been demonstrated to sense the presence of fungi and activate antifungal host immune responses as well as the implications of defective receptor/signaling networks on susceptibility to fungal infections.
Due to the ubiquitous presence of fungi in the environment, humans are constantly exposed to them; in fact, it has been proposed that we have coevolved with commensals such as Candida spp. and Malassezia (10). Despite this, fungi are associated with various diseases ranging from asthma to mucocutaneous infections to severe life-threatening systemic infections in patients with a compromised immune system (e.g., organ tissue transplant patients and HIV-infected individuals).
Fungi are eukaryotes and thus are similar to mammalian cells; however, they possess a cell wall, which is absent in mammalian cells. The cell wall is thought to provide several advantages to fungi including protection against environmental stresses and structural rigidity for invading ecological niches (11). For many fungal species, the molecular details of the structural composition and organization of cell wall components have not been fully elucidated. However, for most of the medically relevant fungi, polysaccharides account for more than 90% of the wall content, with the central core comprising branched β-1,3 and β-1,6 glucan that associate with chitin via a β-1,4 linkage (11). The core structure is decorated by unique extensions depending on the pathogen; for instance, O- or N-linked mannans and mannoproteins are found in Candida yeasts, while glucuronoxylomannan and galactoxylomannan coat Cryptococcus (12). Because of their exclusivity, the components of the fungal cell wall contribute a large reservoir of Janeway’s “patterns,” which are now commonly termed “pathogen-associated molecular patterns” (PAMPs). Chitin, mannans, and β-glucan are the three major PAMPs present in all fungi that cause human disease, with the possible exception of Pneumocystis jirovecii which lacks chitin (13, 14). Thus, the framework for induction of an immune response to most fungi follows similar sets of rules and involves recognition of PAMPs by several classes of germ-line-encoded PRRs expressed by innate immune cells, including C-type lectin receptors (CLRs), Toll-like receptors (TLRs), nucleotide-oligomerization domain (NOD)-like receptors (NLRs), and retinoic acid-inducible gene 1 (RIG-I)-like receptors (RLRs), discussed below.
TLRs and Fungal Sensing
By the mid-1980s, the nuclear factor-κB (NF-κB) was known to mediate upregulation of many genes involved in immunity and stress in mammalian cells (15). In Drosophila melanogaster, the NF-κB homologue, Dorsal, was shown by Nüsslein-Volhard and colleagues to drive expression of genes involved in development (16). Furthermore, activation of Toll by its ligand Spätzle was found to be the critical receptor involved in triggering the signaling cascade resulting in activation and nuclear translocation of Dorsal (17). Notably, work emanating from the Hoffman laboratory also demonstrated the expression and nuclear translocation of Dorsal upon microbial challenge (18). Importantly, this group demonstrated that the Spätzle-Toll-Dorsal signaling axis mediated expression of the antifungal peptide drosomycin, with Toll mutant flies succumbing to infection with Aspergillus fumigatus (19). Of interest to note is that Drosophila possesses only an innate immune system, while mammals also have an adaptive immune system. Medzhitov and Janeway subsequently demonstrated that a human cDNA clone of Toll homologue (now known as TLR4) also activated NF-κB and induced expression of costimulatory molecule B7.1 required for activating the adaptive immune responses (20). Seminal findings from Buetler’s laboratory using mice with genetic mutations that abolished recognition of lipopolysaccharide, a major PAMP conserved in Gram-negative bacteria, established that TLR4 is the innate immune sensor of infection critical for host immunity (21).
Since the discovery of TLR4, over 12 mammalian TLRs have been described and their ligands and functions in immunity extensively studied (22). Similar to Drosophila Toll, the extracellular domain of TLRs contains leucine-rich repeat motifs and the intracellular domain structure that is homologous to the interleukin-1 (IL-1) receptor and pathogen resistance proteins found in plants—hence the name TIR (Toll/IL-1 receptor/resistance domain) (23, 24). TLRs transmit signals by interacting with several TIR-containing proteins such as myeloid differentiation primary response (MyD88), TIR domain-containing adaptor protein inducing interferon-β (IFN-β) (TRIF), and TRIF-related adaptor molecule (Fig. 2). With the exception of TLR3 and to some degree TLR4, all TLRs couple the MyD88 adaptor, which interacts with the IL-1 and IL-4 receptor-associated kinase (IRAK1/4) via its N-terminus death domain, resulting in downstream activation of several transcription factors including NF-κB, activation protein 1, interferon regulatory factor (IRF) 3, IRF5, IRF7, and mitogen-activated protein kinases that induce expression of inflammatory mediators (25).
FIGURE 2.
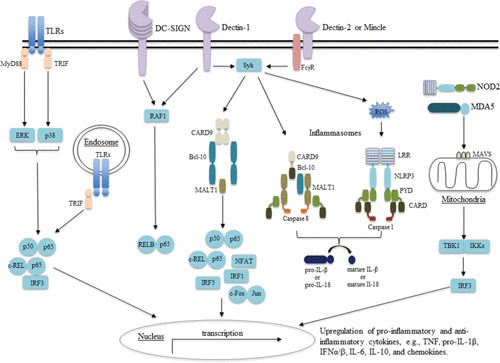
Signaling pathways involved in antifungal immunity.
Mice lacking MyD88 have been shown to be sensitive to infections with various pathogens, presumably, in addition to TLR unresponsiveness, defective immune activation due to a lack of IL-1R, IL-18R, and IL-33R signaling, which also utilize the TIR-MyD88 module (26–28). In experimental settings, MyD88-deficient mice displayed broad susceptibility to bacterial, viral, parasitic, and fungal pathogens, although only selected bacterial infections were observed in humans with deficiencies in MyD88 or IRAK4 (see reference 29 for a detailed review). Nonetheless, polymorphisms in TLR4 were found to be associated with pulmonary infections with Aspergillus and systemic candidiasis (30–32). Additionally, mutations in TLR3 resulting in reduced activation of NF-κB and decreased production of IFN-γ were associated with cutaneous candidiasis (33). Furthermore, defective TLR9 expression was implicated in allergic bronchopulmonary aspergillosis in humans, suggesting a possible accessory role of TLRs in antifungal mammalian immunity (32). Thus far, fungal PAMPs including α-1,4-glucan, glucuronoxylo/phospholipido-mannans, cytosolic nucleic acids, and O-linked/rhamnomannans have been reported to be recognized by TLRs (12) (Fig. 3).
FIGURE 3.
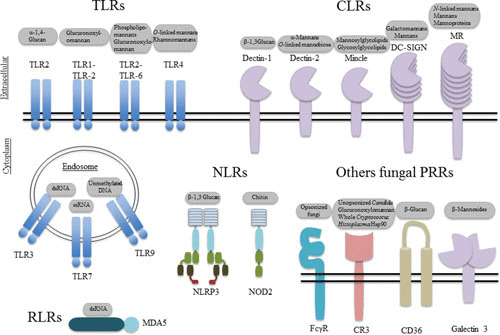
PRRs and the fungal components they recognize. (Adapted with modifications from reference 12).
Contribution of NLRs and RLRs
NLRs comprise a large family of cytoplasmic receptors made up of three characteristic domains (NH2-terminal protein-protein interaction domain, a central NOD, and a C-terminal leucine-rich repeat) and can be grouped into subfamilies based on the nature of the NH2-terminal domain structure (34). Of particular interest to antifungal immunity is NALP3—or NACHT (NAIP [neuronal apoptosis inhibitor], CIITA [MHC class II transcription activator], HET-E [heterokaryon incompatibility protein], and TP1 [telomerase-associated protein])–LRR (leucine-rich repeats)–PYD (pyrin domains)-containing protein 3—which activates a multiprotein complex known as the inflammasome by recruiting the adaptor molecule apoptosis-associated speck-like protein containing a caspase-associated recruitment domain (CARD) to the protein-protein interaction pyrin domain that subsequently activates caspase-1-mediated cleavage of the inflammatory cytokines, pro-IL-1β and pro-IL-18, to their mature forms (35–38) (Fig. 2). To this end, several studies have linked defective NALP3-mediated IL-1β release with susceptibility to several fungi including Candida albicans and A. fumigatus (39–42). Interestingly, the main component of Cryptococcus neoformans capsule, glucoronoxylomannan, was suggested to facilitate intracellular survival of this pathogen via inhibition of Syk-mediated NALP3 inflammasome activation (43).
RLRs are another important family of cytosolic sensors responsible for detecting double-stranded RNA and are distinct from TLR detection of endosome-associated RNA (22). Members of this family include RIG-I, melanoma differentiation-associated gene 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2). All these proteins have a central ATPase-containing DExD/H box helicase domain as a common feature (22). RIG-I and MDA5 also contain N-terminal CARD domains that facilitate downstream signaling via an adaptor molecule mitochondrial antiviral signal, resulting in activation of mitogen-activated protein kinase, NF-κB, type I IFNs (IFNα/β), and IFN-stimulated genes (44–46) (Fig. 2).
To date, MDA5 is the only member of the RLR family suggested to play a role in antifungal defense. In both mice and humans, altered expression or function of MDA5 correlated with chronic mucocutaneous candidiasis and was associated with susceptibility to systemic Candida infections, presumably due to reduced expression of type I IFNs (47). Although the C. albicans ligand(s) that mediates activation of MDA5 is currently unknown, there is a possibility that fungal components gain access to the cytosol and induce immune responses, but this warrants further investigation. The role of type I IFNs, IRFs, and IFN-stimulated genes in antifungal immunity also needs more clarity. For instance, a protective role of IFNs was reported in mice and humans infected with C. albicans (48, 49), whereas in a different study using Candida glabrata, these cytokines were associated with susceptibility to infection (50).
CLRs Are Central for Recognition of Fungi and Antifungal Host Defense
CLRs constitute a superfamily of more than 1,000 soluble and membrane-bound proteins classically characterized by the presence of at least one C-type lectin domain (CTLD) and can be clustered into at least 17 subgroups based on architecture of the CTLD (51). The EPN (Glu-Pro-Asn) motif within the CTLD specifies binding to mannose, N-acetylglucosamine, l-fucose, and glucose, whereas QPD (Gln-Pro-Asp) confers specificity to galactose and N-acetylgalactosamine (52). While calcium-dependent binding of carbohydrates is commonly associated with the function of the CTLD, many CTLDs are known to bind a vast array of noncarbohydrate ligands including proteins, lipids, and lipoproteins (51). Of note are membrane CLRs expressed on myeloid cells that function as PRRs and prime the immune system such as the dendritic cell-associated C-type lectin 1 (Dectin-1, also known as CLEC7A), discovered in 2001 by Brown and Gordon as the receptor for β-1,3-glucan (a PAMP expressed in many, if not all, medically important fungi) (53).
Dectin-1 binding of β-glucan results in phosphorylation of the integral immunoreceptor tyrosine-based-like motif embedded in the cytoplasmic tail providing docking sites for the Src homology 2 domain of spleen tyrosine kinase (Syk) (54). Syk subsequently activates the signaling scaffold CARD9-Bcl-10-Malt-1 through PKCδ and a recently described complex of CARD9-H-Ras-Ras-GRF-1, which promotes nuclear translocation of several transcription factors including NF-κB, nuclear factor and activator of transcription, IRF1, IRF5, and the activation of extracellular-regulated kinases pathway (55). In addition, unlike the Syk pathway, which activates both classical NF-κB (p65 and c-Rel) and noncanonical NF-κB (RelB), Dectin-1 also activates the Raf-1 kinase pathway that sequesters RelB by forming inactive p65/RelB dimers, thereby promoting Syk-mediated activation of p65 (56) (Fig. 2). Signaling via Dectin-1 regulates several processes including phagocytosis, autophagy, NETosis, respiratory burst, and gene expression of proinflammatory mediators including cytokines such as tumor necrosis factor (TNF), IL-23, IL-6, and chemokines (C-C motif) CCL2, CCL3, and (C-X-C motif) CXCL1 (57). Activation of the Dectin-1-Syk-CARD9 signaling pathway by fungi upregulates IL-1β and IL-18 transcripts and is implicated in “priming” of the NALP3/caspase 1 inflammasome, while reactive oxygen species, potassium efflux, and cathepsin B activate inflammasome activity (58, 59). More recently, the Dectin-1-Syk pathway was reported to activate a noncanonical inflammasome comprising MALT1-caspase 8 and apoptosis-associated speck-like protein containing a CARD domain that cleaved pro-IL-1β (60).
Several studies in mice have demonstrated a critical role of Dectin-1 in host defense against fungal infections. In mice lacking Dectin-1, a failure to sense the presence of fungi results in tissue overgrowth of C. albicans or A. fumigatus and is associated with an overall failure to induce appropriate inflammatory responses (such as IL-6 and granulocyte colony-stimulating factor [G-CSF]) that would normally result in the recruitment of monocytes and neutrophils to the site of infection (61–64). Importantly, individuals with polymorphisms in Dectin-1 that affect the ability of leukocytes to bind β-glucan display increased susceptibility to chronic mucocutaneous candidiasis, recurrent episodes of vulvovaginal candidiasis, onychomycosis, and intra-abdominal Candida infections and a reduced Th17 cell response (discussed below) (65).
CLRs such as Dectin-2, macrophage-inducible C-type lectin (Mincle), dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN), and the mannose receptor have also been shown to elicit an immune response against fungi (66–68). Dectin-2 recognizes α-mannans from several fungi including C. albicans, Histoplasma capsulatum, and C. neoformans, as well as glycoproteins containing O-linked mannobiose-rich residues from Malassezia (69, 70). Signals transmitted via Dectin-2 activate the Syk-CARD9 pathway through association with an integral immunoreceptor tyrosine-based-bearing Fc receptor γ chain (FcRγ) adaptor molecule, which results in improved reactive oxygen species production by neutrophils, expression of IL-23p19, and IL-1β release by APCs (71, 72). Dectin-2-deficient mice display increased susceptibility to infection with C. albicans (Fig. 2) (73, 74). Similar to Dectin-2, Mincle recognition of glucosyl- and mannosylglycolipids from Malassezia and ill-defined ligands from C. albicans and Fonsecaea pedrosoi also activates the Syk-CARD9 pathway via the FcRγ adaptor (75). However, the role of Mincle in antifungal immunity may be context dependent. In a recent report, interaction of Mincle with the causative agent of chromoblastomycosis, Fonsecaea monophora, was shown to suppress IL-12p35 (an important cytokine required for T helper cell polarization) via Syk-CARD9 activation of E3 ubiquitin ligase Mdm2-dependent degradation of the transcription factor IRF-1 (76).
DC-SIGN and the mannose receptor recognize branched N-linked mannans and have been implicated in trafficking of mannosylated fungal antigens into the APC endocytic pathway, facilitating antigen processing and presentation to T helper cells (77, 78). In mice, a lack of mannose receptor was shown to result in susceptibility to infection with C. neoformans but not C. albicans (79, 80).
Although mouse models and in vitro studies with human leukocytes provide experimental support for a role in immunity of CLRs, mentioned above, to date, only polymorphisms in Dectin-1 and mutations in the signaling adaptor molecule CARD9 (which acts downstream of Syk-coupled CLRs) have been associated with susceptibility to fungal infections in humans. While polymorphisms in Dectin-1 were predominantly linked with superficial infections, CARD9 deficiency in both mice and humans was found to be critical for protection against systemic Candida and Exophiala infection, particularly of the central nervous system (49, 65, 81–83). CARD9-mediated protection against Candida spinal osteomyelitis and meningoencephalitis was shown to require neutrophil chemotaxis into the central nervous system (49, 65, 81, 84, 85).
EFFECTOR MECHANISMS DRIVING HOST PROTECTION FROM FUNGI
Epithelium
Apart from the obvious physical barrier function, the epithelium, through secretion of factors such as mucins and antimicrobial peptides, plays an important role in protection against microbial tissue invasion (86). Salivary mucins in host defense and disease prevention were shown to repress formation of C. albicans hyphae (a morphological form thought to be associated with host invasion) and displayed direct candidacidal activities in the oral cavity, respectively (87).
Although research into “self versus nonself” interactions has for the most part focused on pathogens, commensal microorganisms, including the mycobiota (fungal communities), colonize mucosal surfaces such as the gut, and their role in health and disease is slowly gaining attention (88). For instance, increased Candida gut colonization and differential fungal profiles were observed in patients with inflammatory bowel disease and in ulcerative colitis patients, respectively (89–91). Notably, in these studies, an absence of Dectin-1 signaling correlated with severe forms of ulcerative colitis in both mice and humans. Intriguingly, Dectin-1 and CARD9 have also been implicated in regulation of intestinal homeostasis by modulating gut microbiota. Dectin-1-mediated induction of antimicrobial peptides S100A8 and S100A9 paralleled decreases in Lactobacilli numbers; these are commensal gut bacteria best known for metabolizing tryptophan into aryl hydrocarbon receptor ligands and generation of regulatory helper CD4 T cells (discussed below) (92, 93). By contrast, gut microbiota from CARD9-deficient mice failed to metabolize tryptophan and consequently displayed decreased aryl hydrocarbon receptor ligands and IL-22 and increased sensitivity to colitis (94).
Phagocytes
Derived from the Greek term phagein, meaning “to eat,” phagocytes are critical for clearing foreign bodies and debris from dead or dying cells. Tissue-resident macrophages and neutrophils are key phagocytic cells that mediate host protection against fungi (95). Here, a few examples of the mechanisms deployed by phagocytic cells in antifungal immunity are described. Upon sensing fungi, macrophages recruit other immune cells to sites of infection through secretion of proinflammatory cytokines and chemokines, discussed above (95). In vivo depletion of macrophages or deficiency of CX3C-chemokine receptor 1 (CX3CR1), which is associated with poor survival of macrophages, was observed to result in unrestricted fungal growth in tissues and heightened mortality rates in mice systemically challenged with C. albicans (96). Importantly, patients with decreased expression of CX3CR1 are highly susceptible to disseminated candidiasis (96). In the lung, alveolar macrophages kill inhaled A. fumigatus conidia and Pneumocystis carinii cysts through mechanisms involving reactive oxidant intermediates (97, 98). As immunologists increasingly define functional “subsets” of cells, specific populations of monocytes and macrophages that protect against fungi are being described. For instance, several studies have reported a protective role of CCR2+Ly6hi inflammatory monocytes in systemic candidiasis (99, 100). In mice, depletion of this subset resulted in lethal invasive aspergillosis (101).
The importance of neutrophils is underscored by the fact that neutropenia is one of the major risk factors driving disseminated fungal infections in mouse models and in humans (95, 102, 103). Neutrophils accomplish their potent fungicidal activities through generation of reactive oxygen radicals and nonoxidative effector mechanisms including secretion of lysozyme, lactoferrin, elastase, β-defensin, gelatinases, and cathepsin G (104). Neutrophil extracellular traps, released during the last stages of neutrophil active cell death, known as NETosis, have been implicated as another strategy of antifungal defense (105–107). Neutrophil extracellular trap formation coincides with neutrophil degranulation and release of microbicidal factors such as cathelicidin (108). Proteinase 3-mediated cleavage of cathelicidin into LL-37 was shown to interfere with fungal cell membranes, suppress formation of biofilms and fungal attachment, induce reactive oxygen species production, enhance chemotaxis, and suppress neutrophil apoptosis (109–114).
Reinforcing Effectors: Cytokines
The basics of the immune process as we currently understand it were discussed in the preceding sections; that is, PAMPs are recognized by PRRs expressed by innate immunocytes, resulting in activation of the innate immune system. As described above, the bridge between innate and adaptive host immunity is mediated by professional APCs, dendritic cells, discovered in 1973 by Steinman and Cohn (115). Dendritic cells provide signal 1 (antigen-presentation) and signal 2 (costimulation) to naive T helper CD4 cells (bearing αβ T cell receptors) initiating “effector” antigen-specific helper CD4 T cell responses (116, 117). Other aspects including generation of cytotoxic CD8 T cells and B cell development into immunoglobulin-producing plasma cells are also activated but will be not be discussed here. In the last section, we discuss the nature of helper cells induced upon fungal recognition and the type of help they provide (Fig. 4).
FIGURE 4.
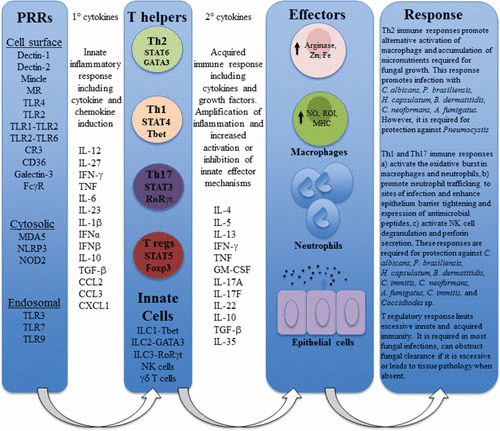
Schematic representation of the sequential inflammatory immune reaction involved in antifungal immune responses.
Cytokines produced by APCs in response to PAMPs provide signal 3i, which instructs polarity of T cells (Th) into functionally distinct helper subsets. Members of the IL-12 family of cytokines are known critical mediators of Th polarization, and these include IL-12 (IL-12p35:IL-12p40), IL-23 (IL-12p19:IL-12p40), IL-27 (IL-12p28:EBi3), and IL-35 (IL-12p35:EBi3) (118). Binding of these cytokines to their cell surface receptors activates the Janus kinase and signal transducer and activator of transcription (STAT) signaling pathways, trans-activating gene expression, and in the case of naive T cells, differentiation (119).
IL-12 and IL-27 secretion by innate immune cells as well as innate lymphocyte-derived IFN-γ skew naive cells toward a Th1 differentiation program driven by STAT1, STAT4, and T box transcription factor T-bet (120, 121). Classically, Th1 cells are defined by production of mainly IFN-γ but also produce other important proinflammatory cytokines such as TNF and GM-CSF. The role of Th1 cells in protective immunity against most fungal pathogens in both experimental mouse models and humans is well established. Several laboratories have shown that IFN-γ and TNF stimulate macrophages to release nitric oxide and reactive oxygen species, causing intracellular growth arrest of several fungal species such as H. capsulatum (122–124). An absence of Th1 immunity or expression of the effector cytokines as demonstrated in IL-12, IFN-γ, or TNF deficiency in both mice and humans results in increased susceptibility to a myriad of fungal infections, including C. neoformans. Patients on IFN-γ immunotherapy show augmented protection against aspergillosis, cryptococcosis, and coccidioidomycosis (125). GM-CSF, on the other hand, was shown to mediate antifungal responses in macrophages by a mechanism involving sequestering intracellular zinc (a micronutrient required by yeast cells), stimulating reactive oxygen species, as well as neutrophil recruitment (126, 127). In contrast, several laboratories reported that STAT1 (signal transducer of IL-27, type I IFNs, and IFN-γ) gain of function mutations in humans resulted in autosomal dominant chronic mucocutaneous candidiasis, potentially due to excess induction of type 1 IFNs which promote IL-27 and IL-10, resulting in inhibition IL-17-producing T cells (128, 129) (Table 1).
TABLE 1.
Selected human genetic associations with fungal susceptibility discussed in this article
| Gene | Immunological phenotype | Associated disease |
|---|---|---|
| Dectin-1 | Dimunition of cytokine responses | Chronic mucocutaneous candidiasis, vulvovaginal candidiasis, onychomycosis, and intra-abdominal Candida |
| CARD9 | Dimunition of cytokine responsesDefective neutrophil chemotaxis | Disseminated candidiasis |
| TLR1 | Decreased IL-1β, IL-6, and IL-18 | Candidemia |
| TLR2 | Decreased IFN-γ and IL-8 | Candidemia |
| TLR3 | Reduced activation of NF-κB and decreased IFN-γ | Cutaneous candidiasis |
| NALP3 | Decreased IL-1β? | Candidiasis-mediated vestibulitis |
| MDA5 | Decreased type I IFNs | Systemic candidiasis |
| STAT1 | Decreased IL-17 production, elevated cell response to IFNs, IFN-γ,s and IL-27Defective IL-12R and IL-23R signaling | Chronic mucocutaneous candidiasis, coccidioidomycosis, histoplasmosis |
| STAT3 | Defective Th17 polarization, decreased IL-17/IL-22-expressing T cells, decreased IL-17-induced antimicrobial peptides | Chronic mucocutaneous candidiasis, coccidioidomycosis, histoplasmosis, cryptococcosis, aspergillosis, nail dermatophytosis |
| Autoimmune regulator | Neutralizing antibodies to IL-17 and IL-22 | Chronic mucocutaneous candidiasis |
| IL-17/IL-17R | Lack of IL-17 cellular responses | Chronic mucocutaneous candidiasis |
| IL-12RA | Impaired IL-12 and IL-23-mediated expression of IFN-γ | Coccidioidomycosis, paracoccidioidomycosis, histoplasmosis, cryptococcosis |
It is now well established that IL-17-producing T cells play a significant role in the clearance of fungi, particularly at the mucosae. This lineage differs from Th1 in that it requires STAT3 activation by IL-6 and IL-21 as well as transforming growth factor β induction of retinoid-related orphan receptor γt, while IL-23 is required for stabilization of the phenotype (130). Effector cytokines, particularly IL-17 and IL-22, were shown to promote the release of various antimicrobial peptides by keratinocytes and epithelial cells as well as strengthening of the mucosal barrier preventing disease dissemination (131–134). The importance of this lineage is made obvious by the heightened susceptibility to a wide range of fungal infections in individuals with loss of function mutations in STAT3 or defects in the IL-17 signaling axis (135–138). In addition, mutations in the autoimmune regulator gene that result in antibodies targeted at Th17 cytokines predispose to chronic and recurrent mucocutaneous candidiasis as noted in individuals with autoimmune polyendocrinopathy with candidiasis and ectodermal dystrophy (APECED) (125). Decreased antimicrobial peptide secretion as well as reduced neutrophil-attracting CXC chemokines due to a lack of Th17-mediated immune responses are thought to be key drivers of susceptibility to mucosal fungal infections (135, 138).
Polarization toward the Th2 subset requires IL-4-mediated STAT6 activation and GATA3 expression, which drive induction of the signature cytokines: IL-4, IL-5, and IL-13. Th2-cytokines are considered important for promoting alternatively activated macrophages reported to permit intracellular growth of fungi due to decreased nitric oxide expression (139). In mouse models, a pathogenic role of an elevated Th2 immune response is best exemplified by the observation of progressive lung infections with C. neoformans and H. capsulatum (69).
Regulatory T cells (T regs) are unique helper cells that play the essential role of restraining immune responses limiting collateral damage to the host. Mechanisms of action mediated by T regs include secretion of inhibitory cytokines such as IL-10, transforming growth factor β, IL-27, IL-35, contact-dependent mechanisms, and sequestration of IL-2 (125). Decreased T reg numbers as observed in TLR2- or CCR5-deficient mice are thought to drive efficient clearance of several fungal infections including C. albicans (140, 141). Interestingly, defective natural T reg development is associated with pathogenesis of chronic mucocutaneous candidiasis in APECED patients, highlighting the importance of the delicate balance between host-driven immune pathology and resolution of infection (142).
AIDS and susceptibility to fungal infections such as Cryptococcus, Candida, Pneumocystis, and Histoplasma have focused attention on the critical contribution of T cells to the host defense arsenal against fungal pathogens (143). Yet cells of the innate immune system such as natural killer cells, γδ T cells, and the recently described innate lymphoid cells also contribute to the effector cytokine pool, and the contribution of these cells in antifungal defense is under intense investigation (133). A case in point is work from LeinbundGut-Landmann’s laboratory demonstrating that antibody depletion of innate lymphoid cells in Rag-deficient mice (which lack mature classical T helper cells) resulted in attenuation of IL-17 and increased susceptibility to oropharyngeal candidiasis infection (144). Moreover, this group also reported that IL-17-mediated immune responses are critical for natural killer cell activation and secretion of GM-SCF, which is essential for antifungal immunity (145). Another study reported natural killer cell direct killing of C. neoformans and Cryptococcus gattii through mechanisms that involved perforin (146). The contribution of these cells to invasive fungal disease and their relevance in human disease are still under investigation.
IMMUNE-BASED THERAPIES AGAINST FUNGAL INFECTIONS
Vaccines
A major hurdle in this area has been largely impacted by the lack of understanding of the requirements for induction of protective immunity and defining surrogate markers for these responses. This is made apparent by the lack of any fungal human vaccine approved for clinical use to date. However, there are exciting developments including studies showing that mice immunized with β1-3-d-glucan-laminarin-diphtheria toxoid conjugate vaccine or antigens encapsulated with glucan particles induce a strong antibody response and a long-term antifungal T cell response that protects mice against several fungal species including Cryptococcus and Candida (147, 148). The use of a live vaccine strategy has also shown promising results in several settings. Examples such as immunization with a C. neoformans-expressing IFN-γ strain offer advantages for developing therapeutic vaccines that offer protection in conditions where CD4 helper T cells are deficient (149).
Adaptive Cell Therapies
Fungal-specific T cell transfer studies have been shown to improve disease outcomes in high-risk individuals such as hematopoietic transplant patients by providing enhanced control of Aspergillus antigenemia and reducing the mortality rate (150). More recently, T cells bearing modified chimeric antigen receptors have been investigated for potential use in fungal diseases (for details about chimeric antigen receptor technology see reference 151). In a proof-of-concept study, T cells bearing Dectin-1 (directing T cell specificity to fungal cell wall glucans) were shown to mediate protection against A. fumigatus (152).
As discussed above, neutropenia is associated with susceptibility to fungal infections, and approaches that reduce the duration of neutropenia in chemotherapy patients have obvious attractive therapeutic implications. Thus, using G-CSF and GM-CSF to increase the yield of granulocytes from donors is gaining a lot of interest in clinical settings where granulocyte transfusions are frequently used as supportive therapy (153). Another exciting approach is immune modulation via exogenous provision of costimulation to effectively activate the immune responses. This was demonstrated by a recent study showing that chronicity of chromoblastomycosis caused by F. pedrosoi was due to lack of costimulation with TLRs; thus, topical application of TLR ligands such as imiquimod (which signals via TLR7) resulted in an adequate inflammatory response that resolved the infection (154).
FUTURE PERSPECTIVES
In the 27 years since the inception of the PRR theory, much progress has been accomplished. We now know that there are other pathogen-sensing mechanisms exemplified by ideas such as the guard theory, which posits that the mammalian immune system detects cellular processes that are targeted by the pathogen’s virulence factors. Furthermore, identification of the cell biology components such as compartmentalization of sensing receptors into membrane-bound, cytosolic, or endosomal modules has emerged as an important framework for the activity of the innate immune system. Understanding how all of these pathways are integrated and controlled from different compartments in the context of fungal infections will be essential for advancing our understanding of antifungal immune responses and developing future immunotherapies. Moreover, additional efforts using a systems biology approach to better define immunological, cellular, and molecular pathways involved in antifungal responses at different anatomical sites will also expand our knowledge. Studies characterizing the complex interplay between host genetics, microbiota, and the virome in health and disease will help decipher an age-old question: what is the basis for mammalian protective immunity?
ACKNOWLEDGMENTS
We thank the Wellcome Trust Strategic Award, Wellcome Trust and Medical Research Council Centre for Medical Mycology, and the University of Aberdeen for funding.
REFERENCES
- 1.Burnet FM. 1959. The Clonal Selection Theory of Acquired Immunity. Vanderbilt University Press, Nashville, TN. 10.5962/bhl.title.8281 [PubMed] [DOI] [Google Scholar]
- 2.Billingham RE, Brent L, Medawar PB. 1953. Actively acquired tolerance of foreign cells. Nature 172:603–606 10.1038/172603a0. [DOI] [PubMed] [Google Scholar]
- 3.Bretscher PA, Cohn M. 1970. A theory of self-nonself discrimination. Science 169:1042–1049. [DOI] [PubMed] [Google Scholar]
- 4.Lafferty KJ, Cunningham AJ. 1975. A new analysis of allogeneic interactions. Aust J Exp Biol Med Sci 53:27–42 10.1038/icb.1975.3. [PubMed] [DOI] [PubMed] [Google Scholar]
- 5.Lafferty KJ, Warren HS, Woolnough JA, Talmage DW. 1978. Immunological induction of T lymphocytes: role of antigen and the lymphocyte costimulator. Blood Cells 4:395–406. [PubMed] [PubMed] [Google Scholar]
- 6.Jenkins MK, Schwartz RH. 1987. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med 165:302–319 10.1084/jem.165.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Janeway CA Jr. 1989. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 54:1–13 10.1101/SQB.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Matzinger P. 1994. Tolerance, danger, and the extended family. Annu Rev Immunol 12:991–1045 10.1146/annurev.iy.12.040194.005015. [PubMed] [DOI] [PubMed] [Google Scholar]
- 9.Brown GD. 2010. How fungi have shaped our understanding of mammalian immunology. Cell Host Microbe 7:9–11 10.1016/j.chom.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 10.Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, White TC. 2012. Hidden killers: human fungal infections. Sci Transl Med 4:165rv13 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 11.Latgé JP. 2007. The cell wall: a carbohydrate armour for the fungal cell. Mol Microbiol 66:279–290 10.1111/j.1365-2958.2007.05872.x. [DOI] [PubMed] [Google Scholar]
- 12.Erwig LP, Gow NA. 2016. Interactions of fungal pathogens with phagocytes. Nat Rev Microbiol 14:163–176 10.1038/nrmicro.2015.21. [PubMed] [DOI] [PubMed] [Google Scholar]
- 13.Latgé JP. 2010. Tasting the fungal cell wall. Cell Microbiol 12:863–872 10.1111/j.1462-5822.2010.01474.x. [DOI] [PubMed] [Google Scholar]
- 14.Levitz SM. 2010. Innate recognition of fungal cell walls. PLoS Pathog 6:e1000758 10.1371/journal.ppat.1000758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sen R, Baltimore D. 1986. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 46:705–716 10.1016/0092-8674(86)90346-6. [DOI] [PubMed] [Google Scholar]
- 16.Nüsslein-Volhard C, Lohs-Schardin M, Sander K, Cremer C. 1980. A dorso-ventral shift of embryonic primordia in a new maternal-effect mutant of Drosophila. Nature 283:474–476 10.1038/283474a0. [DOI] [PubMed] [Google Scholar]
- 17.Anderson KV, Bokla L, Nüsslein-Volhard C. 1985. Establishment of dorsal-ventral polarity in the Drosophila embryo: the induction of polarity by the Toll gene product. Cell 42:791–798 10.1016/0092-8674(85)90275-2. [DOI] [PubMed] [Google Scholar]
- 18.Reichhart JM, Georgel P, Meister M, Lemaitre B, Kappler C, Hoffmann JA. 1993. Expression and nuclear translocation of the rel/NF-kappa B-related morphogen dorsal during the immune response of Drosophila. C R Acad Sci III 316:1218–1224. [PubMed] [PubMed] [Google Scholar]
- 19.Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. 1996. The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86:973–983 10.1016/S0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 20.Medzhitov R, Janeway CA Jr. 1997. Innate immunity: impact on the adaptive immune response. Curr Opin Immunol 9:4–9 10.1016/S0952-7915(97)80152-5. [DOI] [PubMed] [Google Scholar]
- 21.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085–2088 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 22.Kawai T, Akira S. 2009. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol 21:317–337 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gay NJ, Packman LC, Weldon MA, Barna JC. 1991. A leucine-rich repeat peptide derived from the Drosophila Toll receptor forms extended filaments with a beta-sheet structure. FEBS Lett 291:87–91 10.1016/0014-5793(91)81110-T. [DOI] [PubMed] [Google Scholar]
- 24.O’Neill LA, Bowie AG. 2007. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 7:353–364 10.1038/nri2079. [PubMed] [DOI] [PubMed] [Google Scholar]
- 25.Takeda K, Akira S. 2004. TLR signaling pathways. Semin Immunol 16:3–9 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9:143–150 10.1016/S1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 27.Li X, Qin J. 2005. Modulation of Toll-interleukin 1 receptor mediated signaling. J Mol Med (Berl) 83:258–266 10.1007/s00109-004-0622-4. [PubMed] [DOI] [PubMed] [Google Scholar]
- 28.Ali S, Huber M, Kollewe C, Bischoff SC, Falk W, Martin MU. 2007. IL-1 receptor accessory protein is essential for IL-33-induced activation of T lymphocytes and mast cells. Proc Natl Acad Sci USA 104:18660–18665 10.1073/pnas.0705939104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Bernuth H, Picard C, Puel A, Casanova J-L. 2012. Experimental and natural infections in MyD88- and IRAK-4-deficient mice and humans. Eur J Immunol 42:3126–3135 10.1002/eji.201242683. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van der Graaf CA, Netea MG, Morré SA, Den Heijer M, Verweij PE, Van der Meer JW, Kullberg BJ. 2006. Toll-like receptor 4 Asp299Gly/Thr399Ile polymorphisms are a risk factor for Candida bloodstream infection. Eur Cytokine Netw 17:29–34. [PubMed] [PubMed] [Google Scholar]
- 31.Bochud P-Y, Chien JW, Marr KA, Leisenring WM, Upton A, Janer M, Rodrigues SD, Li S, Hansen JA, Zhao LP, Aderem A, Boeckh M. 2008. Toll-like receptor 4 polymorphisms and aspergillosis in stem-cell transplantation. N Engl J Med 359:1766–1777 10.1056/NEJMoa0802629. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carvalho A, Pasqualotto AC, Pitzurra L, Romani L, Denning DW, Rodrigues F. 2008. Polymorphisms in toll-like receptor genes and susceptibility to pulmonary aspergillosis. J Infect Dis 197:618–621 10.1086/526500. [PubMed] [DOI] [PubMed] [Google Scholar]
- 33.Nahum A, Dadi H, Bates A, Roifman CM. 2012. The biological significance of TLR3 variant, L412F, in conferring susceptibility to cutaneous candidiasis, CMV and autoimmunity. Autoimmun Rev 11:341–347 10.1016/j.autrev.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 34.Motta V, Soares F, Sun T, Philpott DJ. 2015. NOD-like receptors: versatile cytosolic sentinels. Physiol Rev 95:149–178 10.1152/physrev.00009.2014. [DOI] [PubMed] [Google Scholar]
- 35.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. 2004. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20:319–325 10.1016/S1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 36.van de Veerdonk FL, Kullberg BJ, van der Meer JWM, Gow NAR, Netea MG. 2008. Host-microbe interactions: innate pattern recognition of fungal pathogens. Curr Opin Microbiol 11:305–312 10.1016/j.mib.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 37.Gross O, Poeck H, Bscheider M, Dostert C, Hannesschläger N, Endres S, Hartmann G, Tardivel A, Schweighoffer E, Tybulewicz V, Mocsai A, Tschopp J, Ruland J. 2009. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 459:433–436 10.1038/nature07965. [PubMed] [DOI] [PubMed] [Google Scholar]
- 38.Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD, Fitzgerald KA. 2009. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 5:487–497 10.1016/j.chom.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lev-Sagie A, Prus D, Linhares IM, Lavy Y, Ledger WJ, Witkin SS. 2009. Polymorphism in a gene coding for the inflammasome component NALP3 and recurrent vulvovaginal candidiasis in women with vulvar vestibulitis syndrome. Am J Obstet Gynecol 200:303.e1-6. 10.1016/j.ajog.2008.10.039. [DOI] [PubMed] [Google Scholar]
- 40.Saïd-Sadier N, Padilla E, Langsley G, Ojcius DM. 2010. Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PLoS One 5:e10008 10.1371/journal.pone.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tavares AH, Magalhães KG, Almeida RD, Correa R, Burgel PH, Bocca AL. 2013. NLRP3 inflammasome activation by Paracoccidioides brasiliensis. PLoS Negl Trop Dis 7:e2595 10.1371/journal.pntd.0002595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kistowska M, Fenini G, Jankovic D, Feldmeyer L, Kerl K, Bosshard P, Contassot E, French LE. 2014. Malassezia yeasts activate the NLRP3 inflammasome in antigen-presenting cells via Syk-kinase signalling. Exp Dermatol 23:884–889 10.1111/exd.12552. [DOI] [PubMed] [Google Scholar]
- 43.Guo C, Chen M, Fa Z, Lu A, Fang W, Sun B, Chen C, Liao W, Meng G. 2014. Acapsular Cryptococcus neoformans activates the NLRP3 inflammasome. Microbes Infect 16:845–854 10.1016/j.micinf.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 44.Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M Jr, Akira S, Yonehara S, Kato A, Fujita T. 2005. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol 175:2851–2858 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- 45.Cui S, Eisenächer K, Kirchhofer A, Brzózka K, Lammens A, Lammens K, Fujita T, Conzelmann KK, Krug A, Hopfner KP. 2008. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol Cell 29:169–179 10.1016/j.molcel.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 46.Takahasi K, Yoneyama M, Nishihori T, Hirai R, Kumeta H, Narita R, Gale M Jr, Inagaki F, Fujita T. 2008. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol Cell 29:428–440 10.1016/j.molcel.2007.11.028. [PubMed] [DOI] [PubMed] [Google Scholar]
- 47.Jaeger M, van der Lee R, Cheng SC, Johnson MD, Kumar V, Ng A, Plantinga TS, Smeekens SP, Oosting M, Wang X, Barchet W, Fitzgerald K, Joosten LA, Perfect JR, Wijmenga C, van de Veerdonk FL, Huynen MA, Xavier RJ, Kullberg BJ, Netea MG. 2015. The RIG-I-like helicase receptor MDA5 (IFIH1) is involved in the host defense against Candida infections. Eur J Clin Microbiol Infect Dis 34:963–974 10.1007/s10096-014-2309-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.del Fresno C, Soulat D, Roth S, Blazek K, Udalova I, Sancho D, Ruland J, Ardavín C. 2013. Interferon-β production via Dectin-1-Syk-IRF5 signaling in dendritic cells is crucial for immunity to C. albicans. Immunity 38:1176–1186 10.1016/j.immuni.2013.05.010. [PubMed] [DOI] [PubMed] [Google Scholar]
- 49.Smeekens SP, van de Veerdonk FL, Kullberg BJ, Netea MG. 2013. Genetic susceptibility to Candida infections. EMBO Mol Med 5:805–813 10.1002/emmm.201201678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bourgeois C, Majer O, Frohner IE, Lesiak-Markowicz I, Hildering KS, Glaser W, Stockinger S, Decker T, Akira S, Müller M, Kuchler K. 2011. Conventional dendritic cells mount a type I IFN response against Candida spp. requiring novel phagosomal TLR7-mediated IFN-β signaling. J Immunol 186:3104–3112 10.4049/jimmunol.1002599. [DOI] [PubMed] [Google Scholar]
- 51.Zelensky AN, Gready JE. 2005. The C-type lectin-like domain superfamily. FEBS J 272:6179–6217 10.1111/j.1742-4658.2005.05031.x. [DOI] [PubMed] [Google Scholar]
- 52.Drickamer K, Fadden AJ. 2002. Genomic analysis of C-type lectins. Biochem Soc Symp 69:59–72 10.1042/bss0690059. [PubMed] [DOI] [PubMed] [Google Scholar]
- 53.Brown GD, Gordon S. 2001. Immune recognition. A new receptor for beta-glucans. Nature 413:36–37 10.1038/35092620. [PubMed] [DOI] [PubMed] [Google Scholar]
- 54.Sancho D, Reis e Sousa C. 2012. Signaling by myeloid C-type lectin receptors in immunity and homeostasis. Annu Rev Immunol 30:491–529 10.1146/annurev-immunol-031210-101352. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dambuza IM, Brown GD. 2015. C-type lectins in immunity: recent developments. Curr Opin Immunol 32:21–27 10.1016/j.coi.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gringhuis SI, den Dunnen J, Litjens M, van der Vlist M, Wevers B, Bruijns SC, Geijtenbeek TB. 2009. Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat Immunol 10:203–213 10.1038/ni.1692. [DOI] [PubMed] [Google Scholar]
- 57.Hardison SE, Brown GD. 2012. C-type lectin receptors orchestrate antifungal immunity. Nat Immunol 13:817–822 10.1038/ni.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. 2007. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 14:1583–1589 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 59.Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. 2009. Mitochondria and reactive oxygen species. Free Radic Biol Med 47:333–343 10.1016/j.freeradbiomed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 60.Gringhuis SI, Kaptein TM, Wevers BA, Theelen B, van der Vlist M, Boekhout T, Geijtenbeek TB. 2012. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat Immunol 13:246–254 10.1038/ni.2222. [DOI] [PubMed] [Google Scholar]
- 61.LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, Schweighoffer E, Tybulewicz V, Brown GD, Ruland J, Reis e Sousa C. 2007. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol 8:630–638 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 62.Saijo S, Fujikado N, Furuta T, Chung SH, Kotaki H, Seki K, Sudo K, Akira S, Adachi Y, Ohno N, Kinjo T, Nakamura K, Kawakami K, Iwakura Y. 2007. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat Immunol 8:39–46 10.1038/ni1425. [DOI] [PubMed] [Google Scholar]
- 63.Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H, Haynes K, Steele C, Botto M, Gordon S, Brown GD. 2007. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat Immunol 8:31–38 10.1038/ni1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Werner JL, Metz AE, Horn D, Schoeb TR, Hewitt MM, Schwiebert LM, Faro-Trindade I, Brown GD, Steele C. 2009. Requisite role for the dectin-1 beta-glucan receptor in pulmonary defense against Aspergillus fumigatus. J Immunol 182:4938–4946 10.4049/jimmunol.0804250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ferwerda B, Ferwerda G, Plantinga TS, Willment JA, van Spriel AB, Venselaar H, Elbers CC, Johnson MD, Cambi A, Huysamen C, Jacobs L, Jansen T, Verheijen K, Masthoff L, Morré SA, Vriend G, Williams DL, Perfect JR, Joosten LA, Wijmenga C, van der Meer JW, Adema GJ, Kullberg BJ, Brown GD, Netea MG. 2009. Human dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med 361:1760–1767 10.1056/NEJMoa0901053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kerscher B, Willment JA, Brown GD. 2013. The Dectin-2 family of C-type lectin-like receptors: an update. Int Immunol 25:271–277 10.1093/intimm/dxt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Plato A, Hardison SE, Brown GD. 2015. Pattern recognition receptors in antifungal immunity. Semin Immunopathol 37:97–106 10.1007/s00281-014-0462-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Barreto-Bergter E, Figueiredo RT. 2014. Fungal glycans and the innate immune recognition. Front Cell Infect Microbiol 4:145 10.3389/fcimb.2014.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wüthrich M, Deepe GS Jr, Klein B. 2012. Adaptive immunity to fungi. Annu Rev Immunol 30:115–148 10.1146/annurev-immunol-020711-074958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ishikawa T, Itoh F, Yoshida S, Saijo S, Matsuzawa T, Gonoi T, Saito T, Okawa Y, Shibata N, Miyamoto T, Yamasaki S. 2013. Identification of distinct ligands for the C-type lectin receptors Mincle and Dectin-2 in the pathogenic fungus Malassezia. Cell Host Microbe 13:477–488 10.1016/j.chom.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 71.Goodridge HS, Wolf AJ, Underhill DM. 2009. Beta-glucan recognition by the innate immune system. Immunol Rev 230:38–50 10.1111/j.1600-065X.2009.00793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Taylor PR, Roy S, Leal SM Jr, Sun Y, Howell SJ, Cobb BA, Li X, Pearlman E. 2014. Activation of neutrophils by autocrine IL-17A-IL-17RC interactions during fungal infection is regulated by IL-6, IL-23, RORγt and dectin-2. Nat Immunol 15:143–151 10.1038/ni.2797. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Robinson MJ, Osorio F, Rosas M, Freitas RP, Schweighoffer E, Gross O, Verbeek JS, Ruland J, Tybulewicz V, Brown GD, Moita LF, Taylor PR, Reis e Sousa C. 2009. Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J Exp Med 206:2037–2051 10.1084/jem.20082818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Saijo S, Ikeda S, Yamabe K, Kakuta S, Ishigame H, Akitsu A, Fujikado N, Kusaka T, Kubo S, Chung SH, Komatsu R, Miura N, Adachi Y, Ohno N, Shibuya K, Yamamoto N, Kawakami K, Yamasaki S, Saito T, Akira S, Iwakura Y. 2010. Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity 32:681–691 10.1016/j.immuni.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 75.Ishikawa E, Ishikawa T, Morita YS, Toyonaga K, Yamada H, Takeuchi O, Kinoshita T, Akira S, Yoshikai Y, Yamasaki S. 2009. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J Exp Med 206:2879–2888 10.1084/jem.20091750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wevers BA, Kaptein TM, Zijlstra-Willems EM, Theelen B, Boekhout T, Geijtenbeek TB, Gringhuis SI. 2014. Fungal engagement of the C-type lectin mincle suppresses dectin-1-induced antifungal immunity. Cell Host Microbe 15:494–505 10.1016/j.chom.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 77.Cambi A, Netea MG, Mora-Montes HM, Gow NA, Hato SV, Lowman DW, Kullberg BJ, Torensma R, Williams DL, Figdor CG. 2008. Dendritic cell interaction with Candida albicans critically depends on N-linked mannan. J Biol Chem 283:20590–20599 10.1074/jbc.M709334200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tacken PJ, Ginter W, Berod L, Cruz LJ, Joosten B, Sparwasser T, Figdor CG, Cambi A. 2011. Targeting DC-SIGN via its neck region leads to prolonged antigen residence in early endosomes, delayed lysosomal degradation, and cross-presentation. Blood 118:4111–4119 10.1182/blood-2011-04-346957. [DOI] [PubMed] [Google Scholar]
- 79.Dan JM, Kelly RM, Lee CK, Levitz SM. 2008. Role of the mannose receptor in a murine model of Cryptococcus neoformans infection. Infect Immun 76:2362–2367 10.1128/IAI.00095-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee SJ, Zheng NY, Clavijo M, Nussenzweig MC. 2003. Normal host defense during systemic candidiasis in mannose receptor-deficient mice. Infect Immun 71:437–445 10.1128/IAI.71.1.437-445.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Glocker EO, Hennigs A, Nabavi M, Schäffer AA, Woellner C, Salzer U, Pfeifer D, Veelken H, Warnatz K, Tahami F, Jamal S, Manguiat A, Rezaei N, Amirzargar AA, Plebani A, Hannesschläger N, Gross O, Ruland J, Grimbacher B. 2009. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med 361:1727–1735 10.1056/NEJMoa0810719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Alves de Medeiros AK, Lodewick E, Bogaert DJ, Haerynck F, Van Daele S, Lambrecht B, Bosma S, Vanderdonckt L, Lortholary O, Migaud M, Casanova JL, Puel A, Lanternier F, Lambert J, Brochez L, Dullaers M. 2016. Chronic and invasive fungal infections in a family with CARD9 deficiency. J Clin Immunol 36:204–209 10.1007/s10875-016-0255-8. (Erratum, 36:528. doi:10.1007/s10875-016-0283-4.) [DOI] [PubMed] [Google Scholar]
- 83.Lanternier F, Barbati E, Meinzer U, Liu L, Pedergnana V, Migaud M, Héritier S, Chomton M, Frémond ML, Gonzales E, Galeotti C, Romana S, Jacquemin E, Angoulvant A, Bidault V, Canioni D, Lachenaud J, Mansouri D, Mahdaviani SA, Adimi P, Mansouri N, Jamshidi M, Bougnoux ME, Abel L, Lortholary O, Blanche S, Casanova JL, Picard C, Puel A. 2015. Inherited CARD9 deficiency in 2 unrelated patients with invasive Exophiala infection. J Infect Dis 211:1241–1250 10.1093/infdis/jiu412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gavino C, Cotter A, Lichtenstein D, Lejtenyi D, Fortin C, Legault C, Alirezaie N, Majewski J, Sheppard DC, Behr MA, Foulkes WD, Vinh DC. 2014. CARD9 deficiency and spontaneous central nervous system candidiasis: complete clinical remission with GM-CSF therapy. Clin Infect Dis 59:81–84 10.1093/cid/ciu215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Drummond RA, Collar AL, Swamydas M, Rodriguez CA, Lim JK, Mendez LM, Fink DL, Hsu AP, Zhai B, Karauzum H, Mikelis CM, Rose SR, Ferre EM, Yockey L, Lemberg K, Kuehn HS, Rosenzweig SD, Lin X, Chittiboina P, Datta SK, Belhorn TH, Weimer ET, Hernandez ML, Hohl TM, Kuhns DB, Lionakis MS. 2015. CARD9-dependent neutrophil recruitment protects against fungal invasion of the central nervous system. PLoS Pathog 11:e1005293 10.1371/journal.ppat.1005293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liévin-Le Moal V, Servin AL. 2006. The front line of enteric host defense against unwelcome intrusion of harmful microorganisms: mucins, antimicrobial peptides, and microbiota. Clin Microbiol Rev 19:315–337 10.1128/CMR.19.2.315-337.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Frenkel ES, Ribbeck K. 2015. Salivary mucins in host defense and disease prevention. J Oral Microbiol 7:29759 10.3402/jom.v7.29759. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Underhill DM, Iliev ID. 2014. The mycobiota: interactions between commensal fungi and the host immune system. Nat Rev Immunol 14:405–416 10.1038/nri3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ott SJ, Kühbacher T, Musfeldt M, Rosenstiel P, Hellmig S, Rehman A, Drews O, Weichert W, Timmis KN, Schreiber S. 2008. Fungi and inflammatory bowel diseases: alterations of composition and diversity. Scand J Gastroenterol 43:831–841 10.1080/00365520801935434. [DOI] [PubMed] [Google Scholar]
- 90.Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, Strom SP, Brown J, Becker CA, Fleshner PR, Dubinsky M, Rotter JI, Wang HL, McGovern DP, Brown GD, Underhill DM. 2012. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 336:1314–1317 10.1126/science.1221789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Moyes DL, Naglik JR. 2012. The mycobiome: influencing IBD severity. Cell Host Microbe 11:551–552 10.1016/j.chom.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 92.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. 2008. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453:65–71 10.1038/nature06880. [PubMed] [DOI] [PubMed] [Google Scholar]
- 93.Tang C, Kamiya T, Liu Y, Kadoki M, Kakuta S, Oshima K, Hattori M, Takeshita K, Kanai T, Saijo S, Ohno N, Iwakura Y. 2015. Inhibition of dectin-1 signaling ameliorates colitis by inducing lactobacillus-mediated regulatory T cell expansion in the intestine. Cell Host Microbe 18:183–197 10.1016/j.chom.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 94.Lamas B, Richard ML, Leducq V, Pham H-P, Michel M-L, Da Costa G, Bridonneau C, Jegou S, Hoffmann TW, Natividad JM, Brot L, Taleb S, Couturier-Maillard A, Nion-Larmurier I, Merabtene F, Seksik P, Bourrier A, Cosnes J, Ryffel B, Beaugerie L, Launay J-M, Langella P, Xavier RJ, Sokol H. 2016. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med 22:598–605 10.1038/nm.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brown GD. 2011. Innate antifungal immunity: the key role of phagocytes. Annu Rev Immunol 29:1–21 10.1146/annurev-immunol-030409-101229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lionakis MS, Swamydas M, Fischer BG, Plantinga TS, Johnson MD, Jaeger M, Green NM, Masedunskas A, Weigert R, Mikelis C, Wan W, Lee CC, Lim JK, Rivollier A, Yang JC, Laird GM, Wheeler RT, Alexander BD, Perfect JR, Gao JL, Kullberg BJ, Netea MG, Murphy PM. 2013. CX3CR1-dependent renal macrophage survival promotes Candida control and host survival. J Clin Invest 123:5035–5051 10.1172/JCI71307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Steele C, Marrero L, Swain S, Harmsen AG, Zheng M, Brown GD, Gordon S, Shellito JE, Kolls JK. 2003. Alveolar macrophage-mediated killing of Pneumocystis carinii f. sp. muris involves molecular recognition by the Dectin-1 beta-glucan receptor. J Exp Med 198:1677–1688 10.1084/jem.20030932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Philippe B, Ibrahim-Granet O, Prévost MC, Gougerot-Pocidalo MA, Sanchez Perez M, Van der Meeren A, Latgé JP. 2003. Killing of Aspergillus fumigatus by alveolar macrophages is mediated by reactive oxidant intermediates. Infect Immun 71:3034–3042 10.1128/IAI.71.6.3034-3042.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Quintin J, Saeed S, Martens JH, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, Jacobs L, Jansen T, Kullberg BJ, Wijmenga C, Joosten LA, Xavier RJ, van der Meer JW, Stunnenberg HG, Netea MG. 2012. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 12:223–232 10.1016/j.chom.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ngo LY, Kasahara S, Kumasaka DK, Knoblaugh SE, Jhingran A, Hohl TM. 2014. Inflammatory monocytes mediate early and organ-specific innate defense during systemic candidiasis. J Infect Dis 209:109–119 10.1093/infdis/jit413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Espinosa V, Jhingran A, Dutta O, Kasahara S, Donnelly R, Du P, Rosenfeld J, Leiner I, Chen CC, Ron Y, Hohl TM, Rivera A. 2014. Inflammatory monocytes orchestrate innate antifungal immunity in the lung. PLoS Pathog 10:e1003940 10.1371/journal.ppat.1003940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kullberg BJ, van ’t Wout JW, van Furth R. 1990. Role of granulocytes in increased host resistance to Candida albicans induced by recombinant interleukin-1. Infect Immun 58:3319–3324. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Horn DL, Ostrosky-Zeichner L, Morris MI, Ullmann AJ, Wu C, Buell DN, Kovanda LL, Cornely OA. 2010. Factors related to survival and treatment success in invasive candidiasis or candidemia: a pooled analysis of two large, prospective, micafungin trials. Eur J Clin Microbiol Infect Dis 29:223–229 10.1007/s10096-009-0843-0. [DOI] [PubMed] [Google Scholar]
- 104.Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. 2012. Neutrophil function: from mechanisms to disease. Annu Rev Immunol 30:459–489 10.1146/annurev-immunol-020711-074942. [DOI] [PubMed] [Google Scholar]
- 105.McCormick A, Heesemann L, Wagener J, Marcos V, Hartl D, Loeffler J, Heesemann J, Ebel F. 2010. NETs formed by human neutrophils inhibit growth of the pathogenic mold Aspergillus fumigatus. Microbes Infect 12:928–936 10.1016/j.micinf.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 106.Menegazzi R, Decleva E, Dri P. 2012. Killing by neutrophil extracellular traps: fact or folklore? Blood 119:1214–1216 10.1182/blood-2011-07-364604. [DOI] [PubMed] [Google Scholar]
- 107.Kobayashi Y. 2015. Neutrophil biology: an update. EXCLI J 14:220–227. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brinkmann V, Zychlinsky A. 2012. Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol 198:773–783 10.1083/jcb.201203170. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sørensen OE, Follin P, Johnsen AH, Calafat J, Tjabringa GS, Hiemstra PS, Borregaard N. 2001. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood 97:3951–3959 10.1182/blood.V97.12.3951. [DOI] [PubMed] [Google Scholar]
- 110.Kahlenberg JM, Kaplan MJ. 2013. Little peptide, big effects: the role of LL-37 in inflammation and autoimmune disease. J Immunol 191:4895–4901 10.4049/jimmunol.1302005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang X, Oglęcka K, Sandgren S, Belting M, Esbjörner EK, Nordén B, Gräslund A. 2010. Dual functions of the human antimicrobial peptide LL-37-target membrane perturbation and host cell cargo delivery. Biochim Biophys Acta 1798:2201–2208 10.1016/j.bbamem.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 112.Tsai PW, Yang CY, Chang HT, Lan CY. 2011. Human antimicrobial peptide LL-37 inhibits adhesion of Candida albicans by interacting with yeast cell-wall carbohydrates. PLoS One 6:e17755 10.1371/journal.pone.0017755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Alalwani SM, Sierigk J, Herr C, Pinkenburg O, Gallo R, Vogelmeier C, Bals R. 2010. The antimicrobial peptide LL-37 modulates the inflammatory and host defense response of human neutrophils. Eur J Immunol 40:1118–1126 10.1002/eji.200939275. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dürr UHN, Sudheendra US, Ramamoorthy A. 2006. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim Biophys Acta 1758:1408–1425 10.1016/j.bbamem.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 115.Steinman RM, Cohn ZA. 1973. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med 137:1142–1162 10.1084/jem.137.5.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pasare C, Medzhitov R. 2004. Toll-dependent control mechanisms of CD4 T cell activation. Immunity 21:733–741 10.1016/j.immuni.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 117.Manicassamy S, Pulendran B. 2009. Modulation of adaptive immunity with Toll-like receptors. Semin Immunol 21:185–193 10.1016/j.smim.2009.05.005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vignali DA, Kuchroo VK. 2012. IL-12 family cytokines: immunological playmakers. Nat Immunol 13:722–728 10.1038/ni.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shuai K, Liu B. 2003. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol 3:900–911 10.1038/nri1226. [PubMed] [DOI] [PubMed] [Google Scholar]
- 120.Schulz O, Edwards AD, Schito M, Aliberti J, Manickasingham S, Sher A, Reis e Sousa C. 2000. CD40 triggering of heterodimeric IL-12 p70 production by dendritic cells in vivo requires a microbial priming signal. Immunity 13:453–462 10.1016/S1074-7613(00)00045-5. [DOI] [PubMed] [Google Scholar]
- 121.Takeda A, Hamano S, Yamanaka A, Hanada T, Ishibashi T, Mak TW, Yoshimura A, Yoshida H. 2003. Cutting edge: role of IL-27/WSX-1 signaling for induction of T-bet through activation of STAT1 during initial Th1 commitment. J Immunol 170:4886–4890 10.4049/jimmunol.170.10.4886. [PubMed] [DOI] [PubMed] [Google Scholar]
- 122.Beaman L. 1987. Fungicidal activation of murine macrophages by recombinant gamma interferon. Infect Immun 55:2951–2955. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brummer E, Stevens DA. 1995. Antifungal mechanisms of activated murine bronchoalveolar or peritoneal macrophages for Histoplasma capsulatum. Clin Exp Immunol 102:65–70 10.1111/j.1365-2249.1995.tb06637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Novak ML, Koh TJ. 2013. Macrophage phenotypes during tissue repair. J Leukoc Biol 93:875–881 10.1189/jlb.1012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Verma A, Wüthrich M, Deepe G, Klein B. 2014. Adaptive immunity to fungi. Cold Spring Harb Perspect Med 5:a019612 10.1101/cshperspect.a019612. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Subramanian Vignesh K, Landero Figueroa JA, Porollo A, Caruso JA, Deepe GS Jr. 2013. Granulocyte macrophage-colony stimulating factor induced Zn sequestration enhances macrophage superoxide and limits intracellular pathogen survival. Immunity 39:697–710 10.1016/j.immuni.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Laan M, Prause O, Miyamoto M, Sjöstrand M, Hytönen AM, Kaneko T, Lötvall J, Lindén A. 2003. A role of GM-CSF in the accumulation of neutrophils in the airways caused by IL-17 and TNF-alpha. Eur Respir J 21:387–393 10.1183/09031936.03.00303503. [PubMed] [DOI] [PubMed] [Google Scholar]
- 128.Zhang L, Yuan S, Cheng G, Guo B. 2011. Type I IFN promotes IL-10 production from T cells to suppress Th17 cells and Th17-associated autoimmune inflammation. PLoS One 6:e28432 10.1371/journal.pone.0028432. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liu L, et al. 2011. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 208:1635–1648 10.1084/jem.20110958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Stockinger B, Veldhoen M. 2007. Differentiation and function of Th17 T cells. Curr Opin Immunol 19:281–286 10.1016/j.coi.2007.04.005. [PubMed] [DOI] [PubMed] [Google Scholar]
- 131.Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. 2006. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203:2271–2279 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gaffen SL, Hernández-Santos N, Peterson AC. 2011. IL-17 signaling in host defense against Candida albicans. Immunol Res 50:181–187 10.1007/s12026-011-8226-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Conti HR, Gaffen SL. 2010. Host responses to Candida albicans: Th17 cells and mucosal candidiasis. Microbes Infect 12:518–527 10.1016/j.micinf.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.De Luca A, Zelante T, D’Angelo C, Zagarella S, Fallarino F, Spreca A, Iannitti RG, Bonifazi P, Renauld JC, Bistoni F, Puccetti P, Romani L. 2010. IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunol 3:361–373 10.1038/mi.2010.22. [DOI] [PubMed] [Google Scholar]
- 135.Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, Anderson VL, Darnell DN, Welch PA, Kuhns DB, Frucht DM, Malech HL, Gallin JI, Kobayashi SD, Whitney AR, Voyich JM, Musser JM, Woellner C, Schäffer AA, Puck JM, Grimbacher B. 2007. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 357:1608–1619 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 136.Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, Migaud M, Israel L, Chrabieh M, Audry M, Gumbleton M, Toulon A, Bodemer C, El-Baghdadi J, Whitters M, Paradis T, Brooks J, Collins M, Wolfman NM, Al-Muhsen S, Galicchio M, Abel L, Picard C, Casanova JL. 2011. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 332:65–68 10.1126/science.1200439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Puel A, Cypowyj S, Maródi L, Abel L, Picard C, Casanova JL. 2012. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr Opin Allergy Clin Immunol 12:616–622 10.1097/ACI.0b013e328358cc0b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ling Y, Cypowyj S, Aytekin C, Galicchio M, Camcioglu Y, Nepesov S, Ikinciogullari A, Dogu F, Belkadi A, Levy R, Migaud M, Boisson B, Bolze A, Itan Y, Goudin N, Cottineau J, Picard C, Abel L, Bustamante J, Casanova JL, Puel A. 2015. Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J Exp Med 212:619–631 10.1084/jem.20141065. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Davis MJ, Tsang TM, Qiu Y, Dayrit JK, Freij JB, Huffnagle GB, Olszewski MA. 2013. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. MBio 4:e00264-13 10.1128/mBio.00264-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Netea MG, Sutmuller R, Hermann C, Van der Graaf CA, Van der Meer JW, van Krieken JH, Hartung T, Adema G, Kullberg BJ. 2004. Toll-like receptor 2 suppresses immunity against Candida albicans through induction of IL-10 and regulatory T cells. J Immunol 172:3712–3718 10.4049/jimmunol.172.6.3712. [DOI] [PubMed] [Google Scholar]
- 141.Moreira AP, Cavassani KA, Massafera Tristão FS, Campanelli AP, Martinez R, Rossi MA, Silva JS. 2008. CCR5-dependent regulatory T cell migration mediates fungal survival and severe immunosuppression. J Immunol 180:3049–3056 10.4049/jimmunol.180.5.3049. [DOI] [PubMed] [Google Scholar]
- 142.Kekäläinen E, Tuovinen H, Joensuu J, Gylling M, Franssila R, Pöntynen N, Talvensaari K, Perheentupa J, Miettinen A, Arstila TP. 2007. A defect of regulatory T cells in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Immunol 178:1208–1215 10.4049/jimmunol.178.2.1208. [DOI] [PubMed] [Google Scholar]
- 143.Shoham S, Levitz SM. 2005. The immune response to fungal infections. Br J Haematol 129:569–582 10.1111/j.1365-2141.2005.05397.x. [DOI] [PubMed] [Google Scholar]
- 144.Gladiator A, Wangler N, Trautwein-Weidner K, LeibundGut-Landmann S. 2013. Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J Immunol 190:521–525 10.4049/jimmunol.1202924. [DOI] [PubMed] [Google Scholar]
- 145.Bär E, Whitney PG, Moor K, Reis e Sousa C, LeibundGut-Landmann S. 2014. IL-17 regulates systemic fungal immunity by controlling the functional competence of NK cells. Immunity 40:117–127 10.1016/j.immuni.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 146.Islam A, Li SS, Oykhman P, Timm-McCann M, Huston SM, Stack D, Xiang RF, Kelly MM, Mody CH. 2013. An acidic microenvironment increases NK cell killing of Cryptococcus neoformans and Cryptococcus gattii by enhancing perforin degranulation. PLoS Pathog 9:e1003439 10.1371/journal.ppat.1003439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Specht CA, Lee CK, Huang H, Tipper DJ, Shen ZT, Lodge JK, Leszyk J, Ostroff GR, Levitz SM. 2015. Protection against experimental cryptococcosis following vaccination with glucan particles containing Cryptococcus alkaline extracts. MBio 6:e01905-15 10.1128/mBio.01905-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Torosantucci A, Bromuro C, Chiani P, De Bernardis F, Berti F, Galli C, Norelli F, Bellucci C, Polonelli L, Costantino P, Rappuoli R, Cassone A. 2005. A novel glyco-conjugate vaccine against fungal pathogens. J Exp Med 202:597–606 10.1084/jem.20050749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wozniak KL, Young ML, Wormley FL Jr. 2011. Protective immunity against experimental pulmonary cryptococcosis in T cell-depleted mice. Clin Vaccine Immunol 18:717–723 10.1128/CVI.00036-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Perruccio K, Tosti A, Burchielli E, Topini F, Ruggeri L, Carotti A, Capanni M, Urbani E, Mancusi A, Aversa F, Martelli MF, Romani L, Velardi A. 2005. Transferring functional immune responses to pathogens after haploidentical hematopoietic transplantation. Blood 106:4397–4406 10.1182/blood-2005-05-1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sadelain M, Brentjens R, Rivière I. 2013. The basic principles of chimeric antigen receptor design. Cancer Discov 3:388–398 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kumaresan PR, Manuri PR, Albert ND, Maiti S, Singh H, Mi T, Roszik J, Rabinovich B, Olivares S, Krishnamurthy J, Zhang L, Najjar AM, Huls MH, Lee DA, Champlin RE, Kontoyiannis DP, Cooper LJ. 2014. Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc Natl Acad Sci USA 111:10660–10665 10.1073/pnas.1312789111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Averbuch D, Engelhard D, Pegoraro A, Cesaro S. 2016. Review on efficacy and complications of granulocyte transfusions in neutropenic patients. Curr Drug Targets. [Epub ahead of print.] 10.2174/1389450117666160201113612. [PubMed] [DOI] [PubMed] [Google Scholar]
- 154.de Sousa MG, Belda W Jr, Spina R, Lota PR, Valente NS, Brown GD, Criado PR, Benard G. 2014. Topical application of imiquimod as a treatment for chromoblastomycosis. Clin Infect Dis 58:1734–1737 10.1093/cid/ciu168. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]