

ABSTRACT
Ever since antibiotics were introduced into human and veterinary medicine to treat and prevent bacterial infections there has been a steady selection and increase in the frequency of antibiotic resistant bacteria. To be able to reduce the rate of resistance evolution, we need to understand how various biotic and abiotic factors interact to drive the complex processes of resistance emergence and transmission. We describe several of the fundamental factors that underlay resistance evolution, including rates and niches of emergence and persistence of resistant bacteria, time- and space-gradients of various selective agents, and rates and routes of transmission of resistant bacteria between humans, animals and other environments. Furthermore, we discuss the options available to reduce the rate of resistance evolution and/ or transmission and their advantages and disadvantages.
Key Words: selection, antibiotic resistance, horizontal gene transfer, transmission, mutation rates, successful clones, mobile genetic elements, SOS, conjugation
INTRODUCTION AND SCOPE
Antibiotics are compounds that inhibit (bacteriostatic drugs) or kill (bactericidal drugs) bacteria by a specific interaction with a specific target in the bacterial cell, and they are arguably the most important medical intervention introduced by humans. Ever since antibiotics were introduced on large scale in the late 1940s to treat human bacterial infectious diseases, there has been a steady selection and increase in the frequency of antibiotic-resistant bacteria, generating a very problematic situation (1–3). Resistance evolution is a complex process that is driven by the interaction between a number of biotic and abiotic factors. Fundamental factors underlying this dynamic are the rates of emergence and persistence of resistant bacterial clones; time and space gradients of antibiotics and other xenobiotics; and transmission rates within human populations and between humans and various other sources, including animals, the environment, food, etc. Furthermore, with the realization that antibiotic resistance has become a serious medical problem, human attempts to reduce transmission of infectious bacteria in general, and resistant ones in particular, by hygienic measures, vaccination, reduced antibiotic pressures, etc., has also influenced this dynamic.
Here we will describe some of the factors that influence the selection and transmission of resistant bacteria and discuss the options available to prevent these processes and reduce the rate of resistance evolution and/or transmission. In this description, we will follow the outline shown in Fig. 1, where we track the initial emergence of resistance in environmental bacteria, its transfer into pathogenic bacteria, and the subsequent transmission of these bacteria between different compartments and environments.
FIGURE 1.

Schematic view of the evolution of antibiotic resistance. Key questions in understanding the emergence and transmission include: (A) What are the origins of resistance genes? (B) Where do resistant pathogens emerge? (C) Which are the most significant selective pressures driving resistance evolution? (D) Which are the biological factors that influence rates of resistance development? (E) What are the routes, directions, and magnitudes of flow of pathogens between humans, animals, and the environment?
WHERE DID RESISTANCE GENES ORIGINATE AND WHY?
The majority of medically relevant antibiotics originate in nature and are synthesized by a variety of species, in particular soil-dwelling bacteria in the genus Streptomyces (4, 5). Both antibiotics and resistance genes are thought to be ancient and far predate the existence of humans. This has been inferred from phylogenetic studies suggesting that class A β-lactamases evolved billions of years ago and were transferred into the Gram-positive bacteria about 800 million years ago and that the progenitors of β-lactamases, like CTX-M’s, diverged 200 million to 300 million years ago (6). Further evidence that resistance genes are old is their presence in environments that have been untouched by humans (7–9), including in isolated caves (>4 million years), Beringian permafrost (30,000 years), and Siberian permafrost (15,000 to 40,000 years old).
The benefits of antibiotics for microbial producers is not entirely clear, but the standard explanation is that the producers use them as ecological weapons to inhibit neighboring competitors in the environment (10, 11). However, they might also have a more benevolent function as signals for cell-to-cell communication in microbial communities. A recent study that distinguished between these hypotheses provided strong evidence that antibiotics are weapons but that their expression is influenced by social interactions between competing strains and species (12). It is expected that the biosynthesis and release of antibiotics in various natural environments will expose many bacteria (both the producers and bystanders) to antibiotics and, as a consequence, select for the evolution of resistance mechanisms to protect against self-destruction (in antibiotic producers), to defend against antibiotics produced by other species, and/or to modulate intermicrobe communication.
Another hypothesis is that resistance genes originally performed metabolic functions unrelated to antibiotics and that they had weak secondary promiscuous activities that conferred a low-level resistance that was exapted and further evolved to become bona fide antibiotic resistance functions. For example, aminoglycoside acetylate-modifying enzymes could originally have been involved in sugar metabolism and modification of complex sugars. Similarly, the plasmid-borne, dual-activity fluoroquinolone acetylate-modifying enzyme AAC(6′)-Ib-cr (13–15) belongs to the GNAT (GCN5-related N-acetyltransferase) superfamily, with 10,000 known enzymes that perform a variety of coenzyme A-dependent acetylation reactions (16). Considering the low activity of this enzyme on fluoroquinolones (only a fewfold increase in MIC of fluoroquinolones), it is likely that this represents a weak promiscuous activity. Another example is the class A, C, and D (Ser-OH) β-lactamases that have been suggested to originate from penicillin-binding proteins (PBPs) by acquiring the capability to hydrolyze the acyl-enzyme bond between the β-lactam ring and the hydroxyl group of the PBP’s active-site serine (17, 18). This notion is supported by experimental data showing that PBPs can evolve weak β-lactamase activity after mutagenesis (19–21). A final example of where a function might have been coopted is the TetX enzyme, a flavin-dependent monooxygenase that inactivates all known tetracyclines, including tigecycline. This enzyme belongs to the flavoprotein monooxygenase group, whose native metabolic function is in the hydroxylation and degradation of phenolic compounds (22).
Our possibilities to interfere with and slow down the rate by which novel resistance mechanisms evolve in nonpathogenic bacteria (e.g., Streptomyces) are at present nonexistent, but this type of knowledge is still useful since it provides the tools that allow us to explore the intrinsic potential of a bacterium to acquire a high-level resistance mechanism to a novel antibiotic by evolving an existing weak promiscuous activity into a more efficient enzyme. For example, by directed in vitro evolution of a suspected candidate enzyme or an adaptive evolution experiment with whole cells, one can explore the likelihood and efficiency of such a process (23).
TRANSFER FROM NONPATHOGENS TO PATHOGENS
Antibiotic resistance can propagate and transmit through either vertical transfer, whereby mutations or resistance genes are transfered to the offspring, or horizontal gene transfer (HGT), whereby antibiotic resistance genes are acquired from close relatives or other species and subsequently propagated to the offspring. The comprehensive genomic characterization of human pathogens during the last decades shows that horizontal transfer of preexisting genes contributes to the majority of current resistance problems generated by our use of antibiotics (24). The vast pool of existing resistance determinants present in the biosphere, known as the resistome, provides an evolutionary toolbox of ready-made genes that has the potential to be transferred within and between species and to confer resistance to any antibiotic that might be used against human and animal pathogens. Transfer mechanisms include conjugative transfer of plasmids and conjugative transposons, transduction via bacteriophages, and transformation of naked DNA taken up from the environment (discussed further in “Transmission of Resistance Genes,” below). Apart from HGT, resistance can also arise by mutations (including point mutations, gene rearrangements, and amplification) of native resident genes.
Many horizontally transferred resistance genes are thought to have their origin in various environmental bacteria (25, 26). For example, the widespread and clinically relevant CTX-M class of genes was probably imported to pathogens from the chromosome of different species of the environmental genus Kluyvera (27–29). This particular case is especially interesting since it illustrates the importance of a transposable genetic element (insertion sequence ISEcp1). This element facilitated selection and transfer by allowing both expression (by providing a promoter) and transposition of the progenitor resistance gene from Kluyvera into other species (30). Furthermore, the plasmid-encoded qnrA gene was most likely transferred from marine and freshwater Shewanella algae into various Enterobacteriaceae species, emphasizing the role of aquatic environments as resistance reservoirs (31). Similarly, Shewanella oneidensis is the natural reservoir of OXA-48, a plasmid-encoded, carbapenem-hydrolyzing β-lactamase gene found in Klebsiella pneumoniae, further indicating gene exchange between Shewanella spp. and Enterobacteriaceae (32). Another more recent example is provided by the tetX gene, which has been found in obligate anaerobes such as Bacteroides (33–37), in the soil bacterium Sphingobacterium sp. (38), and in manure-treated soil (39) and activated sludge from sewage treatment plants (40). TetX has been suggested to have its immediate origin in Bacteroides fragilis, where it was found to be located in two different transposons (34, 35). Up until very recently the tetX gene had not been found in human pathogens, but a study in a Sierra Leone hospital from 2013 showed that 21% of Gram-negative bacteria (e.g., Escherichia coli, K. pneumoniae, and Enterobacter sp.) isolated from urine carried the tetX gene, demonstrating that this particular resistance gene had rapidly entered into significant human pathogens from other bacteria (41). That this recent acquisition of resistance genes in human pathogens is driven by anthropogenic use of antibiotics is substantiated by, for example, the demonstration that in agricultural soil the frequency of resistance genes (in particular genes conferring resistance to β-lactams and tetracycline) increased significantly from the 1940s (pre-antibiotics) to 2008 (42). Similarly, plasmids isolated before antibiotics were used by humans lacked resistance genes, indicating that mobilization of resistance genes to plasmids is a recent event (43).
These findings show that many resistance genes have their origins in environmental bacteria and that they have been mobilized by transposons and plasmids for transfer into pathogens. A key question with regard to our ability to influence and reduce these types of HGT events is where these transfer events from environmental bacteria to pathogens occurred. Unfortunately, we do not have any solid data regarding this question, but potential environments include sewage treatment plants, manure tanks, and manure-treated soils. In all these environments, one would expect both environmental bacteria and pathogens (in particular Enterobacteriaceae) to be present at high densities, at favorable temperatures (at least in sewage plants and manure tanks), and with weak antibiotic selective pressures present, all conditions that would be conducive to selection of rare HGT events of, for example, resistance plasmids. Several experimental studies have indeed shown that horizontal transfer of antibiotic resistance genes is a common occurrence in such environments (for reviews, see references 44 to 48). Another potential environment could be in humans and animals where environmental bacteria could transiently (e.g., via ingestion of food contaminated with environmental bacteria or mixed wound infections) be physically near pathogens for transfer. Demonstration that HGT can occur is a first step in demonstrating their role, but even with experimental demonstration of transfer it is still difficult to extrapolate from such data to assess which environments are the most relevant and where to make potential interventions. Apart from generally reducing the selective pressures provided by antibiotics and other potential selectors (biocides, heavy metals) to reduce enrichment for rare HGT events in these types of environments, it would also be possible to break potential transmission pathways by, for example, treating the water exiting from sewage plants with ozone or similar oxidative compounds (49, 50). This would have several beneficial effects in that it would reduce the levels of antibiotics (and other pharmaceuticals) released and also kill off most types of microorganisms present in the outgoing water.
THE ROLE OF SELECTIVE PRESSURES
Antimicrobial Drugs as Selectors
The rate of fixation of a neutral HGT event or a mutation = HGT/mutation rate, and classical theoretical work suggests the fixation time to be 4Ne generations (where Ne is the effective population size) (51). Ne in bacterial populations (e.g., E. coli) is estimated to be in the range of 105 to 109 (52, 53), implying that the fixation of a neutral resistance mutation/gene is unlikely and will take a very long time without selection. Also, most resistance mutations/genes are deleterious in the absence of antibiotics (reviewed in reference 54), which will further reduce the likelihood of their fixation. Thus, a selective pressure(s) must have driven the rise in antibiotic resistance over the last century. Obviously, human use of antibiotics in various settings has been the main driver for the rise in the frequency of resistant bacteria, and it is clear from studies at different geographic levels (e.g., county and country) that the frequency of resistant bacteria is positively correlated with the antibiotic pressure, i.e., the amount of antibiotic used (55–57). Antibiotics are present in humans, animals, and the environments at varying concentrations, and traditionally it has been assumed that selection of resistant bacteria only occurs at antibiotic concentrations between the MIC of the susceptible wild-type population (MICsusc) and that of the resistant population (MICres), as outlined in the mutant selective window hypothesis (58). Recent studies have examined the selective potential of sub-MIC antibiotic concentrations and shown that drug concentrations several hundredfold below the MICsusc can in fact be selective (59–62). Thus, the determined minimal selective concentrations (MSCs) were in the range of nanograms to picograms per milliliter for certain antibiotics (e.g., fluoroquinolones and tetracyclines), showing that the selective window is much wider than previously thought and expands far below the MICsusc (reviewed in reference 63).
Apart from antibiotics, other potential selectors for antibiotic resistance include biocides (including heavy metals) that are widespread and extensively used in numerous types of settings (64, 65). Biocides could select for antibiotic resistance in at least three different ways: (i) by cross-resistance, where a biocide resistance mechanism confers resistance to an antibiotic, e.g., via upregulation of an efflux pump (66) or a specific target modification (67, 68); (ii) by coselection, where genetic colocalization (e.g., on a plasmid or in a clone) of biocide and antibiotic resistance determinants drives indirect enrichment of antibiotic resistance genes by virtue of biocide selection (69); and (iii) by the ability of biocides to reduce the MSC of antibiotics. With regard to the latter, it has been shown that certain heavy metals (e.g., silver) can strongly potentiate the selective effect of certain antibiotics and thereby cause a significant reduction in the MSC (L. M. Albrecht and D. I. Andersson, unpublished data). Such interactive (synergistic or antagonistic) effects on selection of combinations of different agents (antibiotics and biocides) present at sub-MIC levels are largely unexplored, but as suggested by recent experiments (Albrecht and Andersson, unpublished), the synergistic effects can be considerable. Thus, mixes of up to seven different selectors (antibiotics and heavy metals) reduced the MSC of each individual compound at least 4-fold. As natural environments (e.g., wastewater, soils) do generally contain complex mixes of different selectors, it is of great importance to determine how common and strong such interactions (in particular synergistic) are to correctly assess their selective potential.
Over all, these results indicate that, apart from exposure in treated humans and animals, selection can occur in many ex vivo environments where bacteria are exposed to sub-MIC levels of combinations of antimicrobial drugs due to their excretion from treated subjects (for example, effluent from hospitals), the use of antimicrobials in farming and aquaculture, the natural presence of heavy metals in soils, and pollution from antimicrobial production plants. Thus, it is likely that common weak selection pressures are important contributors to resistance selection and enrichment. However, at present we cannot estimate their relative importance as compared to that of the usually (above MIC) strong selection pressures reached in humans and animals that are treated for infections. Furthermore, with the introduction of MSC determinations as a tool to measure the selection potential of antibiotics, it might also be prudent to reexamine the maximum residue limits (MRLs) set for antibiotics in food. Based on the MSCs, our calculations for fluoroquinolones and tetracyclines (59) and the MRLs set for meat (70) suggest that a normal (100-g) daily consumption of meat that is contaminated with these particular antibiotic residues just below the MRL would still be high enough to cause selection for resistant bacteria (Andersson, unpublished data), implying that these MRLs ought to be reduced.
Other Selectors
Apart from different antimicrobials, there are other mechanisms that could enrich for and maintain resistance in a bacterial population. With regard to plasmids they might encode, apart from antibiotic resistances, virulence factors or growth-promoting factors. In addition, many plasmids have stability systems that ensure plasmid maintenance even without direct selection. The successful epidemic plasmids associated with the widespread clones of E. coli ST131 and K. pneumoniae ST258 appear to owe their success partly to the presence of highly stable (due to multiple partitioning and postsegregational killing systems) IncF-type plasmids encoding several antibiotic resistance genes and virulence factors (71). Furthermore, even though generally plasmids will reduce bacterial fitness in the absence of antibiotics, it has also been demonstrated that certain plasmids might enhance host fitness (growth rate) even in the absence of a selective pressure (72), indicating that plasmids can be maintained and enriched also without direct selection (73–77). At present, the mechanisms of such fitness increases are unclear.
Reducing Selection
With regard to our possibilities to reduce the emergence and frequency of resistance, a reduction in the selective pressure is our most effective and realistic approach. Considering the unwise way antibiotics have been and still are being used, there is considerable room for improvement when it comes to antibiotic stewardship. The required changes are manyfold and are, for example, associated with reducing economic incentives for antibiotic use, educating prescribers and patients, tightening regulation of antibiotic prescriptions, and discontinuing the use of antibiotics as growth promoters in animal farming. Similarly, the potential impact of biocides on enrichment of antibiotic resistance needs much further study and evaluation, which possibly might result in reduced biocide use.
Reduced antibiotic use will reduce the rate of emergence and enrichment of antibiotic-resistant bacteria, but whether the already existing problem of frequent resistance is reversible is questionable. As has been discussed in many papers, we find it likely that once a resistance problem has emerged the rate of reversibility will generally be slow after discontinued/reduced antibiotic use and that a reversibility strategy is unlikely to succeed (for a review, see reference 54).
FACTORS INFLUENCING THE RATE OF EMERGENCE AND SUCCESS OF RESISTANT CLONES
At a given selective pressure, the rate of emergence of resistant mutants, the steady-state frequency of resistance, and the potential for reversibility will be determined by a complex interplay between a number of biological parameters that are intrinsic to the specific combination of bacterial species and antibiotic. Thus, mutation/HGT rates, population sizes, fitness costs of resistance, the rate and efficiency of compensatory evolution, and potential epistatic interactions between drugs and/or combinations of resistance mutations all affect resistance evolution. As several recent reviews have covered this subject, we will not discuss this further here (54, 63, 78) except for a few brief comments on how we could potentially utilize these factors to our benefit.
One general conclusion is that the emergence of resistance in a population is predicted to be slowed by high fitness costs of resistance, especially if compensatory evolution is inefficient. Unfortunately, our ability to influence these parameters is limited, except during drug development, where one could specifically focus on drugs and drug targets in which the above requirements are fulfilled. At present, it is not possible to predict which types of targets are the least prone to resistance evolution, so identification of such targets and drugs remains a purely experimental question. However, a few generalities have emerged from such studies, suggesting that drugs with multiple targets, targets that are encoded by several identical genes (e.g., rRNA), and targets that when mutated show reduced fitness due to pleiotropic physiological effects are less prone to resistance evolution (79). Furthermore, drugs that target complex essential metabolites (e.g., vancomycin, which targets a precursor of peptidoglycans) are expected to be less prone to resistance development because of the difficulties for the cell to mutationally alter the target while maintaining functionality (but obviously resistance could still appear by drug inactivation, reduced uptake, increased efflux, etc.). Thus, an interesting avenue for drug development would be to specifically search for novel drugs that target metabolites outside of the inner membrane (e.g., metabolites involved in lipopolysaccharide, teichoic acid, outer membrane, and peptidoglycan synthesis) since problems with cellular uptake (no need for uptake over the inner membrane) would be less and the propensity for resistance probably lower.
Other possible strategies could be to (i) identify drugs that reduce mutation rates, for example, by inhibiting the SOS response (80–82); (ii) identify drugs that reduce conjugation rates (82, 83); or (iii) use drug combinations during treatment and in particular to utilize the concept of collateral sensitivity. In our opinion, it is unlikely that drug developers will have the luxury of choosing between alternative development candidates with different effects on mutation rate, or that drugs inhibiting bacterial conjugation would significantly contribute to breaking resistance transmission. Hovever, the third possibility, drug combinations, has precedent in clinical treatment of, for example, tuberculosis and HIV to suppress the rate of resistance evolution. Of particular interest is collateral sensitivity, in which resistance to one drug confers increased susceptibility to a second drug (84–86), allowing two (or more) drugs to be alternated to select against resistance to either drug. Experimental data suggest that this approach can slow the rate of resistance development (84, 85), but until it has been shown in treated patients to reduce resistance evolution, the concept remains hypothetical.
TRANSMISSION OF RESISTANCE GENES
Many of the most problematic resistance problems today are directly associated with HGT of resistance genes. Examples include plasmid-borne resistance to β-lactams, macrolides, aminoglycosides, and fluoroquinolones (87), and most recently plasmid-borne resistance to colistin (88). In addition, HGT is the cause of resistant mosaic genes in pneumococci and Neisseria species (89, 90), and the source of most of the virulence genes found on pathogenicity islands (91). In this section, we discuss how antibiotics influence rates of HGT, and ongoing efforts to discover small-molecule inhibitors of HGT.
Mechanisms of HGT of Antibiotic Resistance Genes
The various mechanisms of HGT of resistance genes (24, 92) and the importance of HGT for mobilizing different reservoirs of resistance genes (26) have been extensively reviewed by others and are only outlined here. Briefly, the process of HGT involves two stages: firstly, getting resistance genes into a recipient cell, and secondly, ensuring that they can be stably maintained. Selection is expected to act at several levels of HGT (Fig. 2). The actual DNA transfer process can occur by direct cell-to-cell contact, for example, involving conjugative plasmids or conjugative transposons that mediate the transfer resistance genes from one bacterial cell into another, including across species boundaries. HGT of resistance genes can also be mediated in the absence of direct cell-to-cell contact, with bacteriophages acting as vectors, or the DNA can be taken up directly from the environment (e.g., after release from dead cells) by transformation into a recipient cell. The rates of transfer in the case of conjugation and transformation are expected to be strongly influenced by the population density of donor and recipient cells. Thus, bacteria that, for example, share a common environment such as the human intestine or nasal passages are potentially much more likely to share DNA by processes involving direct contact than bacteria that normally live in, for example, marine environments. In such enclosed environments conjugation (and transformation) are likely to be the dominant mechanisms of HGT. Bacteriophages, however, may play a more important role in mediating the transfer of DNA between bacterial species that occupy different environmental niches, because they are by their nature more widely spread and the DNA they contain is more likely to survive long periods than naked DNA in the environment. Thus, depending on the particular environment and the source of the genetic material involved, each of these mechanisms may play an important role in the HGT of resistance genes.
FIGURE 2.
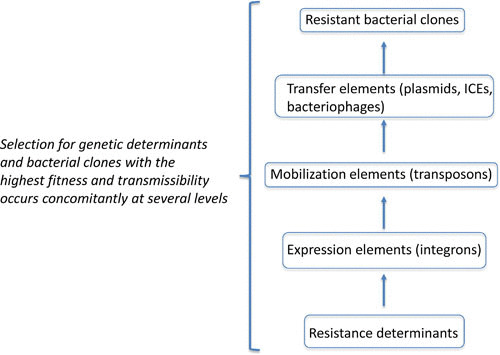
Selection for antibiotic resistance occurs at several levels of complexity to generate a successful resistant clone.
The second stage of HGT, ensuring the maintenance of the acquired genes, is equally important. HGT by plasmids is the most efficient because conjugative plasmids typically have inbuilt mechanisms to ensure their stable maintenance in a recipient bacterial cell. Genes that enter a recipient genome after transfer on a conjugative transposon typically depend on the recombination activity of an encoded integrase enzyme. DNA that enters a recipient by bacteriophage transduction or by transformation could also be recombined into the chromosome by an integrase enzyme if one is encoded on the transferred DNA, but otherwise will depend on homologous recombination. The ability of plasmids to replicate independently of the host chromosome (not depending on recombination to ensure maintenance) explains their very close relationship with HGT resistance genes. The contribution of bacteriophages was thought to be limited by the requirement that they attach to a specific host surface receptor to deliver DNA into a recipient, but recent evidence suggests that many bacteriophages may actually have very broad host range. The other factor that restricts the relative contribution of bacteriophages and transformation to HGT is the requirement that the delivered DNA can be successfully integrated into the recipient genome. This requirement explains the observed close connection between HGT and genes encoding integrases, but in some clinically important examples of resistance (e.g., penicillin resistance in streptococci and neisseriae), the integration of foreign DNA is primarily by homologous recombinations because donor and recipient are related species living in close proximity (89).
Antibiotics Can Influence Rates of HGT
As described above, factors such as the concentration and physical proximity of donor and recipient cells or DNA, the recipient spectrum of transducing phages, and the probability that an integrase enzyme successfully recombines foreign DNA into a genome are each expected to have a significant impact on the overall probability of HGT of antibiotic resistance genes. In addition to these factors, exposure of bacteria to antibiotics can in itself influence the rate of HGT by several mechanisms connected with induction of the SOS response (Fig. 3). The SOS system is a set of bacterial genes involved in the repair of DNA damage. The genes of the SOS system are coregulated by a repressor protein, LexA, and the system is induced in response to DNA damage when activated RecA protein interacts with LexA, catalyzing autocleavage and relieving repression (93). Exposure of bacteria to sublethal doses of several important classes of antibiotics, including fluoroquinolones such as ciprofloxacin (94), β-lactams such as ampicillin (95), and trimethoprim (96), has been shown to induce the SOS system (97). Antibiotic-induced induction of SOS is associated with several downstream effects that promote HGT of antibiotic resistance genes and genes encoding virulence factors. Examples include induction of integrating conjugative elements (ICEs) such as SXT in Vibrio cholerae, which encodes resistance to chloramphenicol, sulfamethoxazole, trimethoprim, and streptomycin (98); induction of several different temperate phages that package and transfer a pathogenicity island encoding virulence factors in staphylococci (99, 100); induction of a prophage encoding Shiga toxin in E. coli (101); and induction of phages that package and transduce an antibiotic resistance plasmid from Salmonella (102). There are also reported examples where sublethal antibiotic exposure promotes HGT of conjugative transposons (103, 104) and plasmids (105), independently of SOS induction. Another example of an SOS-independent effect on HGT involves transcriptional activation of the com regulon by of sublethal antibiotic exposure in Streptococcus pneumoniae, resulting in an increased frequency of transformation (106).
FIGURE 3.

Effects of antibiotics on HGT and potential inhibition points.
Besides increasing rates of HGT as described above, sublethal exposure of bacteria to antibiotics has also been shown to increase the rate of homologous recombination between divergent sequences (107), increasing the probability of successfully recombining HGT genes into a recipient genome. SOS induction by antibiotic exposure has also been shown to increase cassette rearrangements within integrons (108), and in one case an antibiotic-induced rearrangement was shown to result in increased expression of a β-lactamase in a clinical isolate of Pseudomonas aeruginosa (109).
Efforts To Interfere with HGT of Resistance Genes
In the sections above we have presented evidence that a major source of antibiotic resistance is through acquisition of resistance genes in HGT events, sometimes stimulated by the presence of antibiotics in the environment. This raises the question of whether it is possible to interfere with HGT rates, and if so, how and where this should be done. Different antiresistance drug strategies have been suggested, some directed against plasmid-borne resistance and others aimed at developing drugs to suppress the SOS response. The direct antiplasmid approach includes the discovery of small molecules that interfere with plasmid replication, that activate plasmid-borne toxin genes, or that interfere with plasmid-mediated conjugation (110, 111). The heterogeneity of plasmid replication systems and of toxin-antitoxin systems argues that it might be very difficult to identify drugs with a useful spectrum of activity against these targets. Currently there is ongoing research into small-molecule drugs or antibodies that can interfere with plasmid conjugation systems. Unsaturated fatty acids (oleic and linoleic acids) were discovered to inhibit bacterial conjugation, acting on a clinically interesting range of Gram-negative bacteria including Escherichia, Salmonella, Pseudomonas, and Acinetobacter species (83, 112), by targeting a type IV secretion traffic ATPase (113). Recently, after screening a library of bioactive compounds from aquatic organisms, tanzawaic acids (fungal polyketides) were identified as inhibitors of IncW and IncFII conjugation systems (114). Because each of these conjugation-inhibiting compounds is a relatively nontoxic natural product, they may have uses in natural environments to reduce conjugation frequencies, or in therapy in combination with antibiotics. A very interesting recent finding is that bacterial conjugation efficiency is reduced by coculture with human intestinal cells, with the data suggesting that a secreted protein or peptide is responsible for the effect (115). This discovery opens up the possibility that a drug therapy could be developed to modulate conjugation of multidrug resistance plasmids in the human gut.
The second approach to control HGT of resistance genes is to investigate the possibility of developing drugs that reduce induction of the bacterial SOS response. It was previously shown that the accumulation of chromosomal mutations conferring resistance to fluoroquinolones or rifampin in E. coli in an in vivo infection model was strongly dependent on the bacteria having an active SOS response (116). This suggested that drugs capable of altering the bacterial SOS response would be generally useful in reducing the evolution of resistance because they could reduce mutation rates to resistance, reduce rates of HGT of resistance genes, and possibly act as adjuvants to DNA-damaging antibiotics (117). Screening programs have already identified several small-molecule inhibitors that prevent or reduce the ciprofloxacin-induced SOS response in E. coli (118), Staphylococcus aureus (119), or Mycobacterium tuberculosis (120), raising the hope that some compounds might be developed further into clinically useful drugs.
TRANSMISSION OF RESISTANT CLONES
In the previous section, we discussed HGT of resistance genes. However, for a resistant gene to be a clinical problem it must be associated with a genome that is successful in terms of its ability to promote bacterial transmission from the environment to a human host, or from host to host. In this section, we discuss some of the important routes and modes of transmission and the role played by successful epidemic clones in spreading multidrug resistance globally. Finally, we discuss some of the measures that might be implemented to reduce rates of transmission.
Reservoirs of Resistant Bacteria and Modes of Transmission
In recent years several reviews have been published describing and discussing the ecological resevoirs of resistance genes and resistant bacterial pathogens, as well as the major routes of transmission (46, 121–125). The most important conclusion from these reviews is that there is currently very little evidence to quantify either the magnitude or the direction of transmission of resistance genes or pathogens between humans and any of these reservoirs. Here we limit ourselves to briefly summarizing some of the main points and conclusions from these reviews, before considering in more detail the significance of successful bacterial clones in the global dissemination of multidrug resistance. There is a great need for systematic, well-designed, long-term research, using metagenomic sequencing techniques, to map and quantify the routes and directions of transmission of both mobile resistance genes and resistant bacterial pathogens. At the moment, the only robust conclusion that can be made is that the anthropogenic use of antibiotics has almost certainly led to the contamination of many environments with antibiotic-resistant bacteria. What is not clear is the degree to which this contamination is responsible for the resistance problems within human populations. Acquiring high-quality information to quantitatively answer this question, using molecular typing on human, animal, and other environmental isolates, is essential for designing appropriate intervention strategies in the future.
The Role of Successful (Epidemic) Clones
Antibiotic resistance, whether arising due to mutation or by HGT, could in principle be selected in any of the bacteria within a pathogen population. However, whether a particular resistant clone subsequently becomes the founder of a bacterial population that causes a significant clinical problem depends on many factors, in particular its relative fitness in a range of appropriate environments and its transmissibility (Fig. 4). With the increasing use of whole-genome sequencing to analyze antibiotic-resistant bacterial pathogens, it has become clear that many of the major resistance problems are associated with a few successful bacterial clones within a species. Here we shall examine the evidence for clonal success, focusing on two of the most problematic multidrug-resistant Gram-negative pathogens, E. coli ST131 and K. pneumoniae ST258.
FIGURE 4.

Generic scheme for the creation and spread of globally successful antibiotic-resistant clones. In all environments (human, animal, and the wider environment), there are bacterial variants with resistance plasmids, resistance mutations, resistance genes, virulence genes, genes that increase transmission, etc. The mechanisms of HGT, coupled with selection by use of antibiotics, can select for combinations of these elements in one clone. When a clone arises that combines clinical resistance with high fitness and transmissibility, such a clone can spread through the global human population and become a dominant successful clone such as ST131 or ST258.
E. coli ST131
Although E. coli lives as a commensal of the gastrointestinal tract of humans and other animals, some genetic variants have developed into extraintestinal pathogens (126). Within the past 20 years a clone of the sequence type ST131 has become the predominant multidrug-resistant extraintestinal E. coli human pathogen globally (127). Its phenotype includes resistance to fluoroquinolones and extended-spectrum cephalosporins, primarily associated with CTX-M-15 (128, 129). Genome-wide sequence analysis of historical isolates by several groups has provided evidence supporting the hypothesis that ST131 is clonal and delineating its recent evolutionary history. An allele of fimH (encoding the type I fimbrial adhesin gene), designated fimH30, was identified as a signature of the globally dominant clone of ST131 (130). Fluoroquinolone resistance apparently evolved in the early 2000s, in a single ancestor within the H30 lineage of ST131, referred to as H30-R (131). ST131 isolates can carry plasmids with different β-lactamase genes, but most frequently carry CTX-M-15 (127, 132–137). More than 90% of the CTX-M-15-producing isolates of ST131 form a single-ancestor subclone (associated with three additional single nucleotide polymorphisms) within the fluoroquinolone resistance H30 lineage, referred to as H30-Rx (138). Together these data strongly support the hypothesis that ST131 is a successsful multidrug-resistant clone that has spread globally in the recent past, raising the question of which features make ST131 such a successful pathogen. It is apparently associated with a high level of host-to-host transmissibility within household and hospital settings (137, 139, 140) and is more virulent than non-H30 ST131 isolates (141), but the responsible virulence factors have not yet been identified (127). Recent comprehensive genomic analysis of ST131 isolates from varied sources provides strong evidence that not only are there multiple subtypes of ST131 circulating but that a key feature of their success has been the accumulation of compensatory mutations in regulatory regions in response to acquisition of multidrug resistance plasmids (72). ST131 has apparently not reached the end of its drug resistance evolutionary trajectory because there are increasingly frequent reports of carbapenemase resistance in ST131 isolates from around the world (142–149).
K. pneumoniae ST258
K. pneumoniae is a major cause of hospital-acquired pneumonias and bloodstream infections. Antibiotic resistance is a major problem, and it has become much worse in recent years as resistance to carbapenems, the preferred last-resort agents, has increased globally (150). Resistance to carbapenems in K. pneumoniae is due to plasmid-borne carbapenemases, KPC-2 and KPC-3 (151, 152), mainly associated with a global pandemic strain, ST258 (71). ST258 (clade II) originated after a major recombination event between the chromosomes of two different K. pneumoniae strains, ST11 and ST442, together with acquisition of an ICE (ICEKp258.2) encoding a type IV pilus and a type III restriction-modification system (153). In a subsequent recombination event, the capsular polysaccharide region of the chromosome was replaced with the equivalent region from K. pneumoniae strain ST42 to create ST258 (clade I) (153). The success of both ST258 clades may be closely connected with the presence of ICEKp258.2, which facilitates the carriage of KPC genes on narrow-host-range IncF plasmids (154). In contrast, its ancestor ST11 (which lacks ICEKp258.2) is associated with different carbapenemases carried on broad-host-range plasmids (155, 156).
Comparing E. coli ST131 and K. pneumoniae ST258 can provide some insights into how globally successful clones can originate. ST131 may be an example of an accident waiting to happen: a preexisting successful clone (with good virulence properties) that was initially selected to global prominence by exposure to fluoroquinolones (spontaneous resistance mutations), and capable of acquiring and maintaining IncF plasmids encoding CTX-M β-lactamases, providing resistance to cephalosporins (157). It has been speculated that the fluoroquinolone resistance mutations might also contribute by reducing the carriage cost of IncF CTX-M plasmids (71), although recent evidence suggests that a very important role was played by the accumulation of compensatory mutations affecting gene regulation (72). Accordingly, ST131 emerged from a favorable genotype in a series of relatively high-probability steps under selection by human use of antibiotics. In contrast to ST131, ST258 had its origin in a chromosomal recombination event, which together with the integration of ICEKp258.2 into the chromosome created a unique hybrid strain (153). Because we don’t know how unlikely this series of recombination events is, it is difficult to say whether the creation of ST258 was a very unfortunate accident or a frequent event waiting to be selected. This creation of the chromosomal recombinant provided a suitable environment for the acquisition and maintenance of multidrug-resistant IncF plasmids that also carry KPC-2 or KPC-3 enzymes (154). Whether, or which, additional features of the ST258 genome are critical for its global success is not currently known. ST131 and ST258 are not the only examples of successful antibiotic-resistant clones. Other high-risk resistant clones include Salmonella enterica serovar Typhimurium DT104, whose creation involved acquisition of the Salmonella genomic island I (158); P. aeruginosa ST146, which is highly transmissible in cystic fibrosis patients (159, 160); penicillin-nonsuseptible vaccine-escape strains of S. pneumoniae (161, 162); and the M1T1 clonal serotype of Streptococcus pyogenes, which evolved by multiple acquisitions of DNA encoding virulence factors (163).
Strategies To Reduce Transmission
Although the rapid evolution and global spread of ST131, ST258, and these other clones is alarming, their very clonality holds out the hope that a deep understanding of the genetics of these individual strains might provide clues for the design of targeted interventions, leading, for example, to the application of novel antivirulence drugs or vaccines that, if successful, would significantly reduce the burden on health care systems globally (157, 162, 164). Additional research and investment is required to understand more fully the epidemiology of these successful clones to determine which measures should be implemented to most successfully reduce transmission. Several obvious strategies are frequently suggested to reduce the reservoirs of antibiotic resistance and the transmission of resistance genes or resistant bacteria. Such measures include reducing the release of antibiotics into the environment, reducing unnecessary antibiotic use in medicine, reducing antibiotic use in animal husbandry and aquaculture, treating wastewater plants to destroy antibiotic residues, improving hospital hygiene, increasing patient isolation for particular indications, and developing vaccine treatments to replace antibiotic use where possible (121–124). Information is available on the frequencies of clinically relevant pathogens in environments as varied as wastewater treatment plants, sewage sludge, and farm animals. However, there is very little data on the rates and directions of transmission of these resistant bacteria and genes between humans and any of these environments (121, 123, 125). What is required to tackle the transmission problem is a well-planned and long-term surveillance program. Without such a program there is a real danger that the major investment required could be misdirected. The surveillance must be based on detailed genetic and genomic analysis to identify bacterial variants and genes with the aim of quantifying accurately and in detail the major sources and directions of transmission of resistance genes and resistant bacteria between different environmental and biological compartments. Only with this information will it be possible to evaluate the effectiveness of alternative measures designed to reduce the burden of resistance on human health care.
REFERENCES
- 1.Carlet J, Jarlier V, Harbarth S, Voss A, Goossens H, Pittet D, Participants of the 3rd World Healthcare-Associated Infections Forum. 2012. Ready for a world without antibiotics? The Pensières Antibiotic Resistance Call to Action. Antimicrob Resist Infect Control 1:11. 10.1186/2047-2994-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wittekamp BH, Bonten MJ. 2012. Antibiotic prophylaxis in the era of multidrug-resistant bacteria. Expert Opin Investig Drugs 21:767–772. 10.1517/13543784.2012.681642. [DOI] [PubMed] [Google Scholar]
- 3.Woodford N, Livermore DM. 2009. Infections caused by Gram-positive bacteria: a review of the global challenge. J Infect 59(Suppl 1):S4–S16. 10.1016/S0163-4453(09)60003-7. [PubMed] [DOI] [PubMed] [Google Scholar]
- 4.Procópio RE, Silva IR, Martins MK, Azevedo JL, Araújo JM. 2012. Antibiotics produced by Streptomyces. Braz J Infect Dis 16:466–471. 10.1016/j.bjid.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 5.Hasani A, Kariminik A, Issazadeh K. 2014. Streptomycetes: characteristics and their antimicrobial activities. Int J Adv Biol Biomed Res 2:63–75. [Google Scholar]
- 6.Hall BG, Barlow M. 2004. Evolution of the serine β-lactamases: past, present and future. Drug Resist Updat 7:111–123. 10.1016/j.drup.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Bhullar K, Waglechner N, Pawlowski A, Koteva K, Banks ED, Johnston MD, Barton HA, Wright GD. 2012. Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS One 7:e34953. 10.1371/journal.pone.0034953. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D’Costa VM, King CE, Kalan L, Morar M, Sung WW, Schwarz C, Froese D, Zazula G, Calmels F, Debruyne R, Golding GB, Poinar HN, Wright GD. 2011. Antibiotic resistance is ancient. Nature 477:457–461. 10.1038/nature10388. [DOI] [PubMed] [Google Scholar]
- 9.Petrova M, Gorlenko Z, Mindlin S. 2011. Tn5045, a novel integron-containing antibiotic and chromate resistance transposon isolated from a permafrost bacterium. Res Microbiol 162:337–345. 10.1016/j.resmic.2011.01.003. [PubMed] [DOI] [PubMed] [Google Scholar]
- 10.Waksman SA, Woodruff HB. 1940. The soil as a source of microorganisms antagonistic to disease-producing bacteria. J Bacteriol 40:581–600. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinez JL, Fajardo A, Garmendia L, Hernandez A, Linares JF, Martínez-Solano L, Sánchez MB. 2009. A global view of antibiotic resistance. FEMS Microbiol Rev 33:44–65. 10.1111/j.1574-6976.2008.00142.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 12.Abrudan MI, Smakman F, Grimbergen AJ, Westhoff S, Miller EL, van Wezel GP, Rozen DE. 2015. Socially mediated induction and suppression of antibiosis during bacterial coexistence. Proc Natl Acad Sci U S A 112:11054–11059. 10.1073/pnas.1504076112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruiz J, Pons MJ, Gomes C. 2012. Transferable mechanisms of quinolone resistance. Int J Antimicrob Agents 40:196–203. 10.1016/j.ijantimicag.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 14.Robicsek A, Jacoby GA, Hooper DC. 2006. The worldwide emergence of plasmid-mediated quinolone resistance. Lancet Infect Dis 6:629–640. 10.1016/S1473-3099(06)70599-0. [DOI] [PubMed] [Google Scholar]
- 15.Robicsek A, Strahilevitz J, Jacoby GA, Macielag M, Abbanat D, Park CH, Bush K, Hooper DC. 2006. Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nat Med 12:83–88. 10.1038/nm1347. [DOI] [PubMed] [Google Scholar]
- 16.Favrot L, Blanchard JS, Vergnolle O. 2016. Bacterial GCN5-related N-acetyltransferases: from resistance to regulation. Biochemistry 55:989–1002. 10.1021/acs.biochem.5b01269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelly JA, Dideberg O, Charlier P, Wery JP, Libert M, Moews PC, Knox JR, Duez C, Fraipont C, Joris B, Dusart J, Frere JM, Ghuysen JM. 1986. On the origin of bacterial resistance to penicillin: comparison of a beta-lactamase and a penicillin target. Science 231:1429–1431. 10.1126/science.3082007. [DOI] [PubMed] [Google Scholar]
- 18.Massova I, Mobashery S. 1998. Kinship and diversification of bacterial penicillin-binding proteins and β-lactamases. Antimicrob Agents Chemother 42:1–17. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chesnel L, Zapun A, Mouz N, Dideberg O, Vernet T. 2002. Increase of the deacylation rate of PBP2x from Streptococcus pneumoniae by single point mutations mimicking the class A β-lactamases. Eur J Biochem 269:1678–1683. 10.1046/j.1432-1327.2002.02815.x. [DOI] [PubMed] [Google Scholar]
- 20.Peimbert M, Segovia L. 2003. Evolutionary engineering of a β-lactamase activity on a d-Ala d-Ala transpeptidase fold. Protein Eng 16:27–35. 10.1093/proeng/gzg008. [DOI] [PubMed] [Google Scholar]
- 21.Sun S, Selmer M, Andersson DI. 2014. Resistance to β-lactam antibiotics conferred by point mutations in penicillin-binding proteins PBP3, PBP4 and PBP6 in Salmonella enterica. PLoS One 9:e97202. 10.1371/journal.pone.0097202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang W, Moore IF, Koteva KP, Bareich DC, Hughes DW, Wright GD. 2004. TetX is a flavin-dependent monooxygenase conferring resistance to tetracycline antibiotics. J Biol Chem 279:52346–52352. 10.1074/jbc.M409573200. [DOI] [PubMed] [Google Scholar]
- 23.Linkevicius M, Sandegren L, Andersson DI. 2015. Potential of tetracycline resistance proteins to evolve tigecycline resistance. Antimicrob Agents Chemother 60:789–796. 10.1128/AAC.02465-15. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soucy SM, Huang J, Gogarten JP. 2015. Horizontal gene transfer: building the web of life. Nat Rev Genet 16:472–482. 10.1038/nrg3962. [DOI] [PubMed] [Google Scholar]
- 25.Wright GD. 2010. Antibiotic resistance in the environment: a link to the clinic? Curr Opin Microbiol 13:589–594. 10.1016/j.mib.2010.08.005. [PubMed] [DOI] [PubMed] [Google Scholar]
- 26.Perry JA, Westman EL, Wright GD. 2014. The antibiotic resistome: what’s new? Curr Opin Microbiol 21:45–50. 10.1016/j.mib.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 27.Poirel L, Kämpfer P, Nordmann P. 2002. Chromosome-encoded Ambler class A β-lactamase of Kluyvera georgiana, a probable progenitor of a subgroup of CTX-M extended-spectrum β-lactamases. Antimicrob Agents Chemother 46:4038–4040. 10.1128/AAC.46.12.4038-4040.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Humeniuk C, Arlet G, Gautier V, Grimont P, Labia R, Philippon A. 2002. β-Lactamases of Kluyvera ascorbata, probable progenitors of some plasmid-encoded CTX-M types. Antimicrob Agents Chemother 46:3045–3049. 10.1128/AAC.46.9.3045-3049.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Decousser JW, Poirel L, Nordmann P. 2001. Characterization of a chromosomally encoded extended-spectrum class A β-lactamase from Kluyvera cryocrescens. Antimicrob Agents Chemother 45:3595–3598. 10.1128/AAC.45.12.3595-3598.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poirel L, Lartigue MF, Decousser JW, Nordmann P. 2005. ISEcp1B-mediated transposition of blaCTX-M in Escherichia coli. Antimicrob Agents Chemother 49:447–450. 10.1128/AAC.49.1.447-450.2005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poirel L, Rodriguez-Martinez JM, Mammeri H, Liard A, Nordmann P. 2005. Origin of plasmid-mediated quinolone resistance determinant QnrA. Antimicrob Agents Chemother 49:3523–3525. 10.1128/AAC.49.8.3523-3525.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poirel L, Héritier C, Nordmann P. 2004. Chromosome-encoded Ambler class D β-lactamase of Shewanella oneidensis as a progenitor of carbapenem-hydrolyzing oxacillinase. Antimicrob Agents Chemother 48:348–351. 10.1128/AAC.48.1.348-351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guiney DG, Jr, Hasegawa P, Davis CE. 1984. Expression in Escherichia coli of cryptic tetracycline resistance genes from bacteroides R plasmids. Plasmid 11:248–252. 10.1016/0147-619X(84)90031-3. [DOI] [PubMed] [Google Scholar]
- 34.Park BH, Levy SB. 1988. The cryptic tetracycline resistance determinant on Tn4400 mediates tetracycline degradation as well as tetracycline efflux. Antimicrob Agents Chemother 32:1797–1800. 10.1128/AAC.32.12.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Speer BS, Salyers AA. 1988. Characterization of a novel tetracycline resistance that functions only in aerobically grown Escherichia coli.J Bacteriol 170:1423–1429. 10.1128/jb.170.4.1423-1429.1988. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bartha NA, Sóki J, Urbán E, Nagy E. 2011. Investigation of the prevalence of tetQ, tetX and tetX1 genes in Bacteroides strains with elevated tigecycline minimum inhibitory concentrations. Int J Antimicrob Agents 38:522–525. 10.1016/j.ijantimicag.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 37.de Vries LE, Vallès Y, Agersø Y, Vaishampayan PA, García-Montaner A, Kuehl JV, Christensen H, Barlow M, Francino MP. 2011. The gut as reservoir of antibiotic resistance: microbial diversity of tetracycline resistance in mother and infant. PLoS One 6:e21644. 10.1371/journal.pone.0021644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghosh S, Sadowsky MJ, Roberts MC, Gralnick JA, LaPara TM. 2009. Sphingobacterium sp. strain PM2-P1-29 harbours a functional tet(X) gene encoding for the degradation of tetracycline. J Appl Microbiol 106:1336–1342. 10.1111/j.1365-2672.2008.04101.x. [DOI] [PubMed] [Google Scholar]
- 39.Heuer H, Kopmann C, Binh CTT, Top EM, Smalla K. 2009. Spreading antibiotic resistance through spread manure: characteristics of a novel plasmid type with low %G+C content. Environ Microbiol 11:937–949. 10.1111/j.1462-2920.2008.01819.x. [DOI] [PubMed] [Google Scholar]
- 40.Zhang XX, Zhang T. 2011. Occurrence, abundance, and diversity of tetracycline resistance genes in 15 sewage treatment plants across China and other global locations. Environ Sci Technol 45:2598–2604. 10.1021/es103672x. [DOI] [PubMed] [Google Scholar]
- 41.Leski TA, Bangura U, Jimmy DH, Ansumana R, Lizewski SE, Stenger DA, Taitt CR, Vora GJ. 2013. Multidrug-resistant tet(X)-containing hospital isolates in Sierra Leone. Int J Antimicrob Agents 42:83–86. 10.1016/j.ijantimicag.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 42.Knapp CW, Dolfing J, Ehlert PAI, Graham DW. 2010. Evidence of increasing antibiotic resistance gene abundances in archived soils since 1940. Environ Sci Technol 44:580–587. 10.1021/es901221x. [DOI] [PubMed] [Google Scholar]
- 43.Hughes VM, Datta N. 1983. Conjugative plasmids in bacteria of the ‘pre-antibiotic’ era. Nature 302:725–726. 10.1038/302725a0. [DOI] [PubMed] [Google Scholar]
- 44.Bellanger X, Guilloteau H, Bonot S, Merlin C. 2014. Demonstrating plasmid-based horizontal gene transfer in complex environmental matrices: a practical approach for a critical review. Sci Total Environ 493:872–882. 10.1016/j.scitotenv.2014.06.070. [DOI] [PubMed] [Google Scholar]
- 45.Michael I, Rizzo L, McArdell CS, Manaia CM, Merlin C, Schwartz T, Dagot C, Fatta-Kassinos D. 2013. Urban wastewater treatment plants as hotspots for the release of antibiotics in the environment: a review. Water Res 47:957–995. 10.1016/j.watres.2012.11.027. [DOI] [PubMed] [Google Scholar]
- 46.Marti E, Variatza E, Balcazar JL. 2014. The role of aquatic ecosystems as reservoirs of antibiotic resistance. Trends Microbiol 22:36–41. 10.1016/j.tim.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 47.Bouki C, Venieri D, Diamadopoulos E. 2013. Detection and fate of antibiotic resistant bacteria in wastewater treatment plants: a review. Ecotoxicol Environ Saf 91:1–9. 10.1016/j.ecoenv.2013.01.016. [DOI] [PubMed] [Google Scholar]
- 48.Zhang XX, Zhang T, Fang HH. 2009. Antibiotic resistance genes in water environment. Appl Microbiol Biotechnol 82:397–414. 10.1007/s00253-008-1829-z. [DOI] [PubMed] [Google Scholar]
- 49.Pei J, Yao H, Wang H, Ren J, Yu X. 2016. Comparison of ozone and thermal hydrolysis combined with anaerobic digestion for municipal and pharmaceutical waste sludge with tetracycline resistance genes. Water Res 99:122–128. 10.1016/j.watres.2016.04.058. [DOI] [PubMed] [Google Scholar]
- 50.Oh J, Salcedo DE, Medriano CA, Kim S. 2014. Comparison of different disinfection processes in the effective removal of antibiotic-resistant bacteria and genes. J Environ Sci (China) 26:1238–1242. 10.1016/S1001-0742(13)60594-X. [DOI] [PubMed] [Google Scholar]
- 51.Kimura M, Ohta T. 1969. The average number of generations until fixation of a mutant gene in a finite population. Genetics 61:763–771. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Charlesworth B. 2009. Fundamental concepts in genetics: effective population size and patterns of molecular evolution and variation. Nat Rev Genet 10:195–205. 10.1038/nrg2526. [PubMed] [DOI] [PubMed] [Google Scholar]
- 53.Berg OG. 1996. Selection intensity for codon bias and the effective population size of Escherichia coli. Genetics 142:1379–1382. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Andersson DI, Hughes D. 2011. Persistence of antibiotic resistance in bacterial populations. FEMS Microbiol Rev 35:901–911. 10.1111/j.1574-6976.2011.00289.x. [DOI] [PubMed] [Google Scholar]
- 55.van de Sande-Bruinsma N, Grundmann H, Verloo D, Tiemersma E, Monen J, Goossens H, Ferech M, European Antimicrobial Resistance Surveillance System Group, European Surveillance of Antimicrobial Consumption Project Group. 2008. Antimicrobial drug use and resistance in Europe. Emerg Infect Dis 14:1722–1730. 10.3201/eid1411.070467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bergman M, Nyberg ST, Huovinen P, Paakkari P, Hakanen AJ, Finnish Study Group for Antimicrobial Resistance. 2009. Association between antimicrobial consumption and resistance in Escherichia coli. Antimicrob Agents Chemother 53:912–917. 10.1128/AAC.00856-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goossens H. 2009. Antibiotic consumption and link to resistance. Clin Microbiol Infect 15(Suppl 3):12–15. 10.1111/j.1469-0691.2009.02725.x. [DOI] [PubMed] [Google Scholar]
- 58.Drlica K, Zhao X. 2007. Mutant selection window hypothesis updated. Clin Infect Dis 44:681–688. 10.1086/511642. [PubMed] [DOI] [PubMed] [Google Scholar]
- 59.Gullberg E, Cao S, Berg OG, Ilbäck C, Sandegren L, Hughes D, Andersson DI. 2011. Selection of resistant bacteria at very low antibiotic concentrations. PLoS Pathog 7:e1002158. 10.1371/journal.ppat.1002158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu A, Fong A, Becket E, Yuan J, Tamae C, Medrano L, Maiz M, Wahba C, Lee C, Lee K, Tran KP, Yang H, Hoffman RM, Salih A, Miller JH. 2011. Selective advantage of resistant strains at trace levels of antibiotics: a simple and ultrasensitive color test for detection of antibiotics and genotoxic agents. Antimicrob Agents Chemother 55:1204–1210. 10.1128/AAC.01182-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gullberg E, Albrecht LM, Karlsson C, Sandegren L, Andersson DI. 2014. Selection of a multidrug resistance plasmid by sublethal levels of antibiotics and heavy metals. mBio 5:e01918-e14. 10.1128/mBio.01918-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baquero F, Coque TM. 2014. Widening the spaces of selection: evolution along sublethal antimicrobial gradients. mBio 5:e02270. 10.1128/mBio.02270-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Andersson DI, Hughes D. 2014. Microbiological effects of sublethal levels of antibiotics. Nat Rev Microbiol 12:465–478. 10.1038/nrmicro3270. [DOI] [PubMed] [Google Scholar]
- 64.Scientific Committee on Emerging and Newly Identified Health Risks (SCENHIR). 2009. Assessment of the Antibiotic Resistance Effects of Biocides. European Union, Brussels, Belgium. http://ec.europa.eu/health/ph_risk/committees/04_scenihr/docs/scenihr_o_021.pdf. Accessed 19 July 2017. [Google Scholar]
- 65.European Chemicals Agency (ECHA). Biocidal Products Regulation. ECHA, Helsinki, Finland. https://echa.europa.eu/regulations/biocidal-products-regulation. Accessed 19 July 2017. [Google Scholar]
- 66.Webber MA, Whitehead RN, Mount M, Loman NJ, Pallen MJ, Piddock LJ. 2015. Parallel evolutionary pathways to antibiotic resistance selected by biocide exposure. J Antimicrob Chemother 70:2241–2248. 10.1093/jac/dkv109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Webber MA, Randall LP, Cooles S, Woodward MJ, Piddock LJ. 2008. Triclosan resistance in Salmonella enterica serovar Typhimurium. J Antimicrob Chemother 62:83–91. 10.1093/jac/dkn137. [DOI] [PubMed] [Google Scholar]
- 68.Sivaraman S, Zwahlen J, Bell AF, Hedstrom L, Tonge PJ. 2003. Structure-activity studies of the inhibition of FabI, the enoyl reductase from Escherichia coli, by triclosan: kinetic analysis of mutant FabIs. Biochemistry 42:4406–4413. 10.1021/bi0300229. [DOI] [PubMed] [Google Scholar]
- 69.Pal C, Bengtsson-Palme J, Kristiansson E, Larsson DG. 2015. Co-occurrence of resistance genes to antibiotics, biocides and metals reveals novel insights into their co-selection potential. BMC Genomics 16:964. 10.1186/s12864-015-2153-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.U.S. Department of Agriculture (USDA). Maximum Residue Limits (MRL) Database. USDA, Washington, DC. http://www.fas.usda.gov/maximum-residue-limits-mrl-database. Accessed 19 July 2017. [Google Scholar]
- 71.Mathers AJ, Peirano G, Pitout JD. 2015. The role of epidemic resistance plasmids and international high-risk clones in the spread of multidrug-resistant Enterobacteriaceae. Clin Microbiol Rev 28:565–591. 10.1128/CMR.00116-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McNally A, Oren Y, Kelly D, Pascoe B, Dunn S, Sreecharan T, Vehkala M, Välimäki N, Prentice MB, Ashour A, Avram O, Pupko T, Dobrindt U, Literak I, Guenther S, Schaufler K, Wieler LH, Zhiyong Z, Sheppard SK, McInerney JO, Corander J. 2016. Combined analysis of variation in core, accessory and regulatory genome regions provides a super-resolution view into the evolution of bacterial populations. PLoS Genet 12:e1006280. 10.1371/journal.pgen.1006280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Enne VI, Bennett PM, Livermore DM, Hall LM. 2004. Enhancement of host fitness by the sul2-coding plasmid p9123 in the absence of selective pressure. J Antimicrob Chemother 53:958–963. 10.1093/jac/dkh217. [DOI] [PubMed] [Google Scholar]
- 74.Dionisio F, Conceição IC, Marques AC, Fernandes L, Gordo I. 2005. The evolution of a conjugative plasmid and its ability to increase bacterial fitness. Biol Lett 1:250–252. 10.1098/rsbl.2004.0275. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yates CM, Shaw DJ, Roe AJ, Woolhouse ME, Amyes SG. 2006. Enhancement of bacterial competitive fitness by apramycin resistance plasmids from non-pathogenic Escherichia coli. Biol Lett 2:463–465. 10.1098/rsbl.2006.0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bouma JE, Lenski RE. 1988. Evolution of a bacteria/plasmid association. Nature 335:351–352. 10.1038/335351a0. [DOI] [PubMed] [Google Scholar]
- 77.Starikova I, Al-Haroni M, Werner G, Roberts AP, Sørum V, Nielsen KM, Johnsen PJ. 2013. Fitness costs of various mobile genetic elements in Enterococcus faecium and Enterococcus faecalis. J Antimicrob Chemother 68:2755–2765. 10.1093/jac/dkt270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Andersson DI, Hughes D. 2010. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol 8:260–271. 10.1038/nrmicro2319. [DOI] [PubMed] [Google Scholar]
- 79.Hughes D, Andersson DI. 2015. Evolutionary consequences of drug resistance: shared principles across diverse targets and organisms. Nat Rev Genet 16:459–471. 10.1038/nrg3922. [DOI] [PubMed] [Google Scholar]
- 80.Cirz RT, Romesberg FE. 2006. Induction and inhibition of ciprofloxacin resistance-conferring mutations in hypermutator bacteria. Antimicrob Agents Chemother 50:220–225. 10.1128/AAC.50.1.220-225.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alam MK, Alhhazmi A, DeCoteau JF, Luo Y, Geyer CR. 2016. RecA inhibitors potentiate antibiotic activity and block evolution of antibiotic resistance. Cell Chem Biol 23:381–391. 10.1016/j.chembiol.2016.02.010. [PubMed] [DOI] [PubMed] [Google Scholar]
- 82.Culyba MJ, Mo CY, Kohli RM. 2015. Targets for combating the evolution of acquired antibiotic resistance. Biochemistry 54:3573–3582. 10.1021/acs.biochem.5b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Getino M, Sanabria-Ríos DJ, Fernández-López R, Campos-Gómez J, Sánchez-López JM, Fernández A, Carballeira NM, de la Cruz F. 2015. Synthetic fatty acids prevent plasmid-mediated horizontal gene transfer. mBio 6:e01032-e15. 10.1128/mBio.01032-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim S, Lieberman TD, Kishony R. 2014. Alternating antibiotic treatments constrain evolutionary paths to multidrug resistance. Proc Natl Acad Sci U S A 111:14494–14499. 10.1073/pnas.1409800111. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Imamovic L, Sommer MO. 2013. Use of collateral sensitivity networks to design drug cycling protocols that avoid resistance development. Sci Transl Med 5:204ra132. 10.1126/scitranslmed.3006609. [DOI] [PubMed] [Google Scholar]
- 86.Lázár V, Nagy I, Spohn R, Csörgő B, Györkei Á, Nyerges Á, Horváth B, Vörös A, Busa-Fekete R, Hrtyan M, Bogos B, Méhi O, Fekete G, Szappanos B, Kégl B, Papp B, Pál C. 2014. Genome-wide analysis captures the determinants of the antibiotic cross-resistance interaction network. Nat Commun 5:4352. 10.1038/ncomms5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Blair JM, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJ. 2015. Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol 13:42–51. 10.1038/nrmicro3380. [DOI] [PubMed] [Google Scholar]
- 88.Liu YY, Wang Y, Walsh TR, Yi LX, Zhang R, Spencer J, Doi Y, Tian G, Dong B, Huang X, Yu LF, Gu D, Ren H, Chen X, Lv L, He D, Zhou H, Liang Z, Liu JH, Shen J. 2016. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis 16:161–168. 10.1016/S1473-3099(15)00424-7. [DOI] [PubMed] [Google Scholar]
- 89.Hakenbeck R, Brückner R, Denapaite D, Maurer P. 2012. Molecular mechanisms of β-lactam resistance in Streptococcus pneumoniae. Future Microbiol 7:395–410. 10.2217/fmb.12.2. [DOI] [PubMed] [Google Scholar]
- 90.Tapsall JW. 2009. Neisseria gonorrhoeae and emerging resistance to extended spectrum cephalosporins. Curr Opin Infect Dis 22:87–91. 10.1097/QCO.0b013e328320a836. [DOI] [PubMed] [Google Scholar]
- 91.Finlay BB, Falkow S. 1997. Common themes in microbial pathogenicity revisited. Microbiol Mol Biol Rev 61:136–169. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.von Wintersdorff CJ, Penders J, van Niekerk JM, Mills ND, Majumder S, van Alphen LB, Savelkoul PH, Wolffs PF. 2016. Dissemination of antimicrobial resistance in microbial ecosystems through horizontal gene transfer. Front Microbiol 7:173. 10.3389/fmicb.2016.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Butala M, Zgur-Bertok D, Busby SJ. 2009. The bacterial LexA transcriptional repressor. Cell Mol Life Sci 66:82–93. 10.1007/s00018-008-8378-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Drlica K, Zhao X. 1997. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev 61:377–392. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Maiques E, Ubeda C, Campoy S, Salvador N, Lasa I, Novick RP, Barbé J, Penadés JR. 2006. β-Lactam antibiotics induce the SOS response and horizontal transfer of virulence factors in Staphylococcus aureus. J Bacteriol 188:2726–2729. 10.1128/JB.188.7.2726-2729.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lewin CS, Amyes SG. 1991. The role of the SOS response in bacteria exposed to zidovudine or trimethoprim. J Med Microbiol 34:329–332. 10.1099/00222615-34-6-329. [DOI] [PubMed] [Google Scholar]
- 97.Baharoglu Z, Mazel D. 2011. Vibrio cholerae triggers SOS and mutagenesis in response to a wide range of antibiotics: a route towards multiresistance. Antimicrob Agents Chemother 55:2438–2441. 10.1128/AAC.01549-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Beaber JW, Hochhut B, Waldor MK. 2004. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature 427:72–74. 10.1038/nature02241. [DOI] [PubMed] [Google Scholar]
- 99.Ubeda C, Maiques E, Knecht E, Lasa I, Novick RP, Penadés JR. 2005. Antibiotic-induced SOS response promotes horizontal dissemination of pathogenicity island-encoded virulence factors in staphylococci. Mol Microbiol 56:836–844. 10.1111/j.1365-2958.2005.04584.x. [DOI] [PubMed] [Google Scholar]
- 100.Chen J, Novick RP. 2009. Phage-mediated intergeneric transfer of toxin genes. Science 323:139–141. 10.1126/science.1164783. [PubMed] [DOI] [PubMed] [Google Scholar]
- 101.Zhang X, McDaniel AD, Wolf LE, Keusch GT, Waldor MK, Acheson DW. 2000. Quinolone antibiotics induce Shiga toxin-encoding bacteriophages, toxin production, and death in mice. J Infect Dis 181:664–670. 10.1086/315239. [DOI] [PubMed] [Google Scholar]
- 102.Bearson BL, Brunelle BW. 2015. Fluoroquinolone induction of phage-mediated gene transfer in multidrug-resistant Salmonella. Int J Antimicrob Agents 46:201–204. 10.1016/j.ijantimicag.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 103.Torres OR, Korman RZ, Zahler SA, Dunny GM. 1991. The conjugative transposon Tn925: enhancement of conjugal transfer by tetracycline in Enterococcus faecalis and mobilization of chromosomal genes in Bacillus subtilis and E. faecalis. Mol Gen Genet 225:395–400. 10.1007/BF00261679. [DOI] [PubMed] [Google Scholar]
- 104.Stevens AM, Shoemaker NB, Li LY, Salyers AA. 1993. Tetracycline regulation of genes on Bacteroides conjugative transposons. J Bacteriol 175:6134–6141. 10.1128/jb.175.19.6134-6141.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Barr V, Barr K, Millar MR, Lacey RW. 1986. Beta-lactam antibiotics increase the frequency of plasmid transfer in Staphylococcus aureus. J Antimicrob Chemother 17:409–413. 10.1093/jac/17.4.409. [PubMed] [DOI] [PubMed] [Google Scholar]
- 106.Prudhomme M, Attaiech L, Sanchez G, Martin B, Claverys JP. 2006. Antibiotic stress induces genetic transformability in the human pathogen Streptococcus pneumoniae. Science 313:89–92. 10.1126/science.1127912. [DOI] [PubMed] [Google Scholar]
- 107.López E, Elez M, Matic I, Blázquez J. 2007. Antibiotic-mediated recombination: ciprofloxacin stimulates SOS-independent recombination of divergent sequences in Escherichia coli. Mol Microbiol 64:83–93. 10.1111/j.1365-2958.2007.05642.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 108.Guerin E, Cambray G, Sanchez-Alberola N, Campoy S, Erill I, Da Re S, Gonzalez-Zorn B, Barbé J, Ploy MC, Mazel D. 2009. The SOS response controls integron recombination. Science 324:1034. 10.1126/science.1172914. [DOI] [PubMed] [Google Scholar]
- 109.Hocquet D, Llanes C, Thouverez M, Kulasekara HD, Bertrand X, Plésiat P, Mazel D, Miller SI. 2012. Evidence for induction of integron-based antibiotic resistance by the SOS response in a clinical setting. PLoS Pathog 8:e1002778. 10.1371/journal.ppat.1002778. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Williams JJ, Hergenrother PJ. 2008. Exposing plasmids as the Achilles’ heel of drug-resistant bacteria. Curr Opin Chem Biol 12:389–399. 10.1016/j.cbpa.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Spengler G, Molnár A, Schelz Z, Amaral L, Sharples D, Molnár J. 2006. The mechanism of plasmid curing in bacteria. Curr Drug Targets 7:823–841. 10.2174/138945006777709601. [PubMed] [DOI] [PubMed] [Google Scholar]
- 112.Fernandez-Lopez R, Machón C, Longshaw CM, Martin S, Molin S, Zechner EL, Espinosa M, Lanka E, de la Cruz F. 2005. Unsaturated fatty acids are inhibitors of bacterial conjugation. Microbiology 151:3517–3526. 10.1099/mic.0.28216-0. [DOI] [PubMed] [Google Scholar]
- 113.Ripoll-Rozada J, García-Cazorla Y, Getino M, Machón C, Sanabria-Ríos D, de la Cruz F, Cabezón E, Arechaga I. 2016. Type IV traffic ATPase TrwD as molecular target to inhibit bacterial conjugation. Mol Microbiol 100:912–921. 10.1111/mmi.13359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Getino M, Fernández-López R, Palencia-Gándara C, Campos-Gómez J, Sánchez-López JM, Martínez M, Fernández A, de la Cruz F. 2016. Tanzawaic acids, a chemically novel set of bacterial conjugation inhibitors. PLoS One 11:e0148098. 10.1371/journal.pone.0148098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Machado AM, Sommer MO. 2014. Human intestinal cells modulate conjugational transfer of multidrug resistance plasmids between clinical Escherichia coli isolates. PLoS One 9:e100739. 10.1371/journal.pone.0100739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cirz RT, Chin JK, Andes DR, de Crécy-Lagard V, Craig WA, Romesberg FE. 2005. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol 3:e176. 10.1371/journal.pbio.0030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mo CY, Manning SA, Roggiani M, Culyba MJ, Samuels AN, Sniegowski PD, Goulian M, Kohli RM. 2016. Systematically altering bacterial SOS activity under stress reveals therapeutic strategies for potentiating antibiotics. mSphere 1:e00163-16. 10.1128/mSphere.00163-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wigle TJ, Sexton JZ, Gromova AV, Hadimani MB, Hughes MA, Smith GR, Yeh LA, Singleton SF. 2009. Inhibitors of RecA activity discovered by high-throughput screening: cell-permeable small molecules attenuate the SOS response in Escherichia coli. J Biomol Screen 14:1092–1101. 10.1177/1087057109342126. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Peng Q, Zhou S, Yao F, Hou B, Huang Y, Hua D, Zheng Y, Qian Y. 2011. Baicalein suppresses the SOS response system of Staphylococcus aureus induced by ciprofloxacin. Cell Physiol Biochem 28:1045–1050. 10.1159/000335791. [DOI] [PubMed] [Google Scholar]
- 120.Nautiyal A, Patil KN, Muniyappa K. 2014. Suramin is a potent and selective inhibitor of Mycobacterium tuberculosis RecA protein and the SOS response: RecA as a potential target for antibacterial drug discovery. J Antimicrob Chemother 69:1834–1843. 10.1093/jac/dku080. [DOI] [PubMed] [Google Scholar]
- 121.Holmes AH, Moore LS, Sundsfjord A, Steinbakk M, Regmi S, Karkey A, Guerin PJ, Piddock LJ. 2016. Understanding the mechanisms and drivers of antimicrobial resistance. Lancet 387:176–187. 10.1016/S0140-6736(15)00473-0. [DOI] [PubMed] [Google Scholar]
- 122.Yates TA, Khan PY, Knight GM, Taylor JG, McHugh TD, Lipman M, White RG, Cohen T, Cobelens FG, Wood R, Moore DA, Abubakar I. 2016. The transmission of Mycobacterium tuberculosis in high burden settings. Lancet Infect Dis 16:227–238. 10.1016/S1473-3099(15)00499-5. [DOI] [PubMed] [Google Scholar]
- 123.Aarestrup FM. 2015. The livestock reservoir for antimicrobial resistance: a personal view on changing patterns of risks, effects of interventions and the way forward. Philos Trans R Soc Lond B Biol Sci 370:20140085. 10.1098/rstb.2014.0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Huijbers PM, Blaak H, de Jong MC, Graat EA, Vandenbroucke-Grauls CM, de Roda Husman AM. 2015. Role of the environment in the transmission of antimicrobial resistance to humans: a review. Environ Sci Technol 49:11993–12004. 10.1021/acs.est.5b02566. [DOI] [PubMed] [Google Scholar]
- 125.Arnold KE, Williams NJ, Bennett M. 2016. ‘Disperse abroad in the land’: the role of wildlife in the dissemination of antimicrobial resistance. Biol Lett 12:12. 10.1098/rsbl.2016.0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Croxen MA, Finlay BB. 2010. Molecular mechanisms of Escherichia coli pathogenicity. Nat Rev Microbiol 8:26–38. 10.1038/nrmicro2265. [PubMed] [DOI] [PubMed] [Google Scholar]
- 127.Nicolas-Chanoine MH, Bertrand X, Madec JY. 2014. Escherichia coli ST131, an intriguing clonal group. Clin Microbiol Rev 27:543–574. 10.1128/CMR.00125-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nicolas-Chanoine MH, Blanco J, Leflon-Guibout V, Demarty R, Alonso MP, Caniça MM, Park YJ, Lavigne JP, Pitout J, Johnson JR. 2008. Intercontinental emergence of Escherichia coli clone O25:H4-ST131 producing CTX-M-15. J Antimicrob Chemother 61:273–281. 10.1093/jac/dkm464. [DOI] [PubMed] [Google Scholar]
- 129.Coque TM, Novais A, Carattoli A, Poirel L, Pitout J, Peixe L, Baquero F, Cantón R, Nordmann P. 2008. Dissemination of clonally related Escherichia coli strains expressing extended-spectrum β-lactamase CTX-M-15. Emerg Infect Dis 14:195–200. 10.3201/eid1402.070350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Johnson JR, Tchesnokova V, Johnston B, Clabots C, Roberts PL, Billig M, Riddell K, Rogers P, Qin X, Butler-Wu S, Price LB, Aziz M, Nicolas-Chanoine MH, Debroy C, Robicsek A, Hansen G, Urban C, Platell J, Trott DJ, Zhanel G, Weissman SJ, Cookson BT, Fang FC, Limaye AP, Scholes D, Chattopadhyay S, Hooper DC, Sokurenko EV. 2013. Abrupt emergence of a single dominant multidrug-resistant strain of Escherichia coli. J Infect Dis 207:919–928. 10.1093/infdis/jis933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Price LB, Johnson JR, Aziz M, Clabots C, Johnston B, Tchesnokova V, Nordstrom L, Billig M, Chattopadhyay S, Stegger M, Andersen PS, Pearson T, Riddell K, Rogers P, Scholes D, Kahl B, Keim P, Sokurenko EV. 2013. The epidemic of extended-spectrum-β-lactamase-producing Escherichia coli ST131 is driven by a single highly pathogenic subclone, H30-Rx. mBio 4:e00377-e13. 10.1128/mBio.00377-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Woodford N, Carattoli A, Karisik E, Underwood A, Ellington MJ, Livermore DM. 2009. Complete nucleotide sequences of plasmids pEK204, pEK499, and pEK516, encoding CTX-M enzymes in three major Escherichia coli lineages from the United Kingdom, all belonging to the international O25:H4-ST131 clone. Antimicrob Agents Chemother 53:4472–4482. 10.1128/AAC.00688-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Naseer U, Haldorsen B, Tofteland S, Hegstad K, Scheutz F, Simonsen GS, Sundsfjord A, Norwegian ESBL Study Group. 2009. Molecular characterization of CTX-M-15-producing clinical isolates of Escherichia coli reveals the spread of multidrug-resistant ST131 (O25:H4) and ST964 (O102:H6) strains in Norway. APMIS 117:526–536. 10.1111/j.1600-0463.2009.02465.x. [DOI] [PubMed] [Google Scholar]
- 134.Novais Â, Viana D, Baquero F, Martínez-Botas J, Cantón R, Coque TM. 2012. Contribution of IncFII and broad-host IncA/C and IncN plasmids to the local expansion and diversification of phylogroup B2 Escherichia coli ST131 clones carrying blaCTX-M-15 and qnrS1 genes. Antimicrob Agents Chemother 56:2763–2766. 10.1128/AAC.06001-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Partridge SR, Ellem JA, Tetu SG, Zong Z, Paulsen IT, Iredell JR. 2011. Complete sequence of pJIE143, a pir-type plasmid carrying ISEcp1-blaCTX-M-15 from an Escherichia coli ST131 isolate. Antimicrob Agents Chemother 55:5933–5935. 10.1128/AAC.00639-11. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bonnin RA, Poirel L, Carattoli A, Nordmann P. 2012. Characterization of an IncFII plasmid encoding NDM-1 from Escherichia coli ST131. PLoS One 7:e34752. 10.1371/journal.pone.0034752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mathers AJ, Peirano G, Pitout JD. 2015. Escherichia coli ST131: the quintessential example of an international multiresistant high-risk clone. Adv Appl Microbiol 90:109–154. 10.1016/bs.aambs.2014.09.002. [PubMed] [DOI] [PubMed] [Google Scholar]
- 138.Banerjee R, Johnson JR. 2014. A new clone sweeps clean: the enigmatic emergence of Escherichia coli sequence type 131. Antimicrob Agents Chemother 58:4997–5004. 10.1128/AAC.02824-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hilty M, Betsch BY, Bögli-Stuber K, Heiniger N, Stadler M, Küffer M, Kronenberg A, Rohrer C, Aebi S, Endimiani A, Droz S, Mühlemann K. 2012. Transmission dynamics of extended-spectrum β-lactamase-producing Enterobacteriaceae in the tertiary care hospital and the household setting. Clin Infect Dis 55:967–975. 10.1093/cid/cis581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Johnson JR, Miller S, Johnston B, Clabots C, Debroy C. 2009. Sharing of Escherichia coli sequence type ST131 and other multidrug-resistant and urovirulent E. coli strains among dogs and cats within a household. J Clin Microbiol 47:3721–3725. 10.1128/JCM.01581-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Banerjee R, Robicsek A, Kuskowski MA, Porter S, Johnston BD, Sokurenko E, Tchesnokova V, Price LB, Johnson JR. 2013. Molecular epidemiology of Escherichia coli sequence type 131 and its H30 and H30-Rx subclones among extended-spectrum-β-lactamase-positive and -negative E. coli clinical isolates from the Chicago region, 2007 to 2010. Antimicrob Agents Chemother 57:6385–6388. 10.1128/AAC.01604-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Johnson TJ, Hargreaves M, Shaw K, Snippes P, Lynfield R, Aziz M, Price LB. 2015. Complete genome sequence of a carbapenem-resistant extraintestinal pathogenic Escherichia coli strain belonging to the sequence type 131 H30R subclade. Genome Announc 3:e00272-15. 10.1128/genomeA.00272-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Accogli M, Giani T, Monaco M, Giufrè M, García-Fernández A, Conte V, D’Ancona F, Pantosti A, Rossolini GM, Cerquetti M. 2014. Emergence of Escherichia coli ST131 sub-clone H30 producing VIM-1 and KPC-3 carbapenemases, Italy. J Antimicrob Chemother 69:2293–2296. 10.1093/jac/dku132. [DOI] [PubMed] [Google Scholar]
- 144.Cai JC, Zhang R, Hu YY, Zhou HW, Chen GX. 2014. Emergence of Escherichia coli sequence type 131 isolates producing KPC-2 carbapenemase in China. Antimicrob Agents Chemother 58:1146–1152. 10.1128/AAC.00912-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Naas T, Cuzon G, Gaillot O, Courcol R, Nordmann P. 2011. When carbapenem-hydrolyzing β-lactamase Kpc meets Escherichia coli ST131 in France. Antimicrob Agents Chemother 55:4933–4934. 10.1128/AAC.00719-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Stoesser N, Sheppard AE, Peirano G, Sebra RP, Lynch T, Anson LW, Kasarskis A, Motyl MR, Crook DW, Pitout JD. 2016. First report of blaIMP-14 on a plasmid harboring multiple drug resistance genes in Escherichia coli sequence type 131. Antimicrob Agents Chemother 60:5068–5071. 10.1128/AAC.00840-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ortega A, Sáez D, Bautista V, Fernández-Romero S, Lara N, Aracil B, Pérez-Vázquez M, Campos J, Oteo J, Spanish Collaborating Group for the Antibiotic Resistance Surveillance Programme. 2016. Carbapenemase-producing Escherichia coli is becoming more prevalent in Spain mainly because of the polyclonal dissemination of OXA-48. J Antimicrob Chemother 71:2131–2138. 10.1093/jac/dkw148. [DOI] [PubMed] [Google Scholar]
- 148.O’Hara JA, Hu F, Ahn C, Nelson J, Rivera JI, Pasculle AW, Doi Y. 2014. Molecular epidemiology of KPC-producing Escherichia coli: occurrence of ST131-fimH30 subclone harboring pKpQIL-like IncFIIk plasmid. Antimicrob Agents Chemother 58:4234–4237. 10.1128/AAC.02182-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Peirano G, Bradford PA, Kazmierczak KM, Badal RE, Hackel M, Hoban DJ, Pitout JD. 2014. Global incidence of carbapenemase-producing Escherichia coli ST131. Emerg Infect Dis 20:1928–1931. 10.3201/eid2011.141388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.World Health Organization (WHO). 2014. Antimicrobial Resistance: Global Report on Surveillance 2014. WHO, Geneva, Switzerland. [Google Scholar]
- 151.Walther-Rasmussen J, Høiby N. 2007. Class A carbapenemases. J Antimicrob Chemother 60:470–482. 10.1093/jac/dkm226. [PubMed] [DOI] [PubMed] [Google Scholar]
- 152.Nordmann P, Cuzon G, Naas T. 2009. The real threat of Klebsiella pneumoniae carbapenemase-producing bacteria. Lancet Infect Dis 9:228–236. 10.1016/S1473-3099(09)70054-4. [DOI] [PubMed] [Google Scholar]
- 153.Chen L, Mathema B, Pitout JD, DeLeo FR, Kreiswirth BN. 2014. Epidemic Klebsiella pneumoniae ST258 is a hybrid strain. mBio 5:e01355-e14. 10.1128/mBio.01355-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chen L, Mathema B, Chavda KD, DeLeo FR, Bonomo RA, Kreiswirth BN. 2014. Carbapenemase-producing Klebsiella pneumoniae: molecular and genetic decoding. Trends Microbiol 22:686–696. 10.1016/j.tim.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Liu Y, Wan LG, Deng Q, Cao XW, Yu Y, Xu QF. 2015. First description of NDM-1-, KPC-2-, VIM-2- and IMP-4-producing Klebsiella pneumoniae strains in a single Chinese teaching hospital. Epidemiol Infect 143:376–384. 10.1017/S0950268814000995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Voulgari E, Gartzonika C, Vrioni G, Politi L, Priavali E, Levidiotou-Stefanou S, Tsakris A. 2014. The Balkan region: NDM-1-producing Klebsiella pneumoniae ST11 clonal strain causing outbreaks in Greece. J Antimicrob Chemother 69:2091–2097. 10.1093/jac/dku105. [DOI] [PubMed] [Google Scholar]
- 157.Stoesser N, Sheppard AE, Pankhurst L, De Maio N, Moore CE, Sebra R, Turner P, Anson LW, Kasarskis A, Batty EM, Kos V, Wilson DJ, Phetsouvanh R, Wyllie D, Sokurenko E, Manges AR, Johnson TJ, Price LB, Peto TE, Johnson JR, Didelot X, Walker AS, Crook DW, Modernizing Medical Microbiology Informatics Group (MMMIG). 2016. Evolutionary history of the global emergence of the Escherichia coli epidemic clone ST131. mBio 7:e02162. 10.1128/mBio.02162-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Leekitcharoenphon P, Hendriksen RS, Le Hello S, Weill FX, Baggesen DL, Jun SR, Ussery DW, Lund O, Crook DW, Wilson DJ, Aarestrup FM. 2016. Global genomic epidemiology of Salmonella enterica serovar Typhimurium DT104. Appl Environ Microbiol 82:2516–2526. 10.1128/AEM.03821-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fothergill JL, Walshaw MJ, Winstanley C. 2012. Transmissible strains of Pseudomonas aeruginosa in cystic fibrosis lung infections. Eur Respir J 40:227–238. 10.1183/09031936.00204411. [DOI] [PubMed] [Google Scholar]
- 160.McCallum SJ, Gallagher MJ, Corkill JE, Hart CA, Ledson MJ, Walshaw MJ. 2002. Spread of an epidemic Pseudomonas aeruginosa strain from a patient with cystic fibrosis (CF) to non-CF relatives. Thorax 57:559–560. 10.1136/thorax.57.6.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Brueggemann AB, Pai R, Crook DW, Beall B. 2007. Vaccine escape recombinants emerge after pneumococcal vaccination in the United States. PLoS Pathog 3:e168. 10.1371/journal.ppat.0030168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Henriques-Normark B, Blomberg C, Dagerhamn J, Bättig P, Normark S. 2008. The rise and fall of bacterial clones: Streptococcus pneumoniae. Nat Rev Microbiol 6:827–837. 10.1038/nrmicro2011. [DOI] [PubMed] [Google Scholar]
- 163.Nasser W, Beres SB, Olsen RJ, Dean MA, Rice KA, Long SW, Kristinsson KG, Gottfredsson M, Vuopio J, Raisanen K, Caugant DA, Steinbakk M, Low DE, McGeer A, Darenberg J, Henriques-Normark B, Van Beneden CA, Hoffmann S, Musser JM. 2014. Evolutionary pathway to increased virulence and epidemic group A Streptococcus disease derived from 3,615 genome sequences. Proc Natl Acad Sci U S A 111:E1768–E1776. 10.1073/pnas.1403138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ruer S, Pinotsis N, Steadman D, Waksman G, Remaut H. 2015. Virulence-targeted antibacterials: concept, promise, and susceptibility to resistance mechanisms. Chem Biol Drug Des 86:379–399. 10.1111/cbdd.12517. [DOI] [PubMed] [Google Scholar]