

Abstract
The adenosine antagonist 9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazolin-5-amine (CGS 15943) binds nonselectively to human A1, A2A, and A3 receptors with high affinity. Acylated derivatives and one alkyl derivative of the 5-amino group and other modifications were prepared in an effort to enhance A2B or A3 subtype potency. In general, distal modifications of the N5-substituent were highly modulatory to potency and selectivity at adenosine receptors, as determined in radioligand binding assays at rat brain A1 and A2A receptors and at recombinant human A3 receptors. In Chinese hamster ovary cells stably transfected with human A2B receptor cDNA, inhibition of agonist-induced cyclic AMP production was measured. An N5-(2-iodophenyl)acetyl derivative was highly selective for A2A receptors. An (R)-N5-α-methyl-(phenylacetyl) derivative was the most potent derivative at A3 receptors, with a Ki value of 0.36 nM. A bulky N5-diphenylacetyl derivative, 13, displayed a Ki value of 0.59 nM at human A3 receptors and was moderately selective for that subtype. Thus, a large, nondiscriminating hydrophobic region occurs in the A3 receptor in proximity to the N5-substituent. A series of straight-chain N5-aminoalkylacyl derivatives demonstrated that for A2B receptors the optimal chain length occurs with three methylene groups, i.e., the N5-γ-aminobutyryl derivative 27 which had a pA2 value of 8.0 but was not selective for A2B receptors. At A1, A2A, and A3 receptors however the optimum occurs with four methylene groups. An N5-pivaloyl derivative, which was less potent than 27 at A1, A2A, and A3 receptors, retained moderate potency at A2B receptors. A molecular model of the 27–A2B receptor complex based on the structure of rhodopsin utilizing a “cross-docking” procedure was developed in order to visualize the environment of the ligand binding site.
Introduction
Of the four known subtypes of adenosine receptors, A1, A2A, A2B, and A3 receptors, ligand development at the first two subtypes has been well-explored for both selective agonists and antagonists.1–3 Radioligands selective for A1 receptors have been known since the early 1980s,4 and radioligands selective for A2A receptors were first introduced in the late 1980s.5 These tools have aided greatly in the identification of numerous ligands for the A1 and A2A adenosine receptor subtypes. Such selective agonists and antagonists are potentially useful in the treatment of disorders of the CNS6–8 and the renal system9 and also cardiovascular disorders.10
Several classes of agonists and antagonists selective for the most recently cloned adenosine receptor, the A3 subtype, have also been reported.11 These agents have been used pharmacologically to demonstrate both cerebroprotective12 (chronic agonist or acute antagonist) and cardioprotective13 (agonist) effects. A3 receptor agonists at high concentration may also induce apoptosis.14 Four diverse chemical classes have been identified as promising leads for A3 antagonists: flavonoids15 (phenolics found in all vascular plants), pyridines,16 dihydropyridines17–19 (structurally similar to Ca2+ channel antagonists), and triazoloquinazolines,20 such as 1a (9-chloro-2-(2-furyl)[1,2,4]triazolo[1,5-c]quinazolin-5-amine, CGS 15943; Chart 1). Chemical optimization of the leads has resulted in antagonists having subnanomolar affinity, for example, 5-(phenylacetylamino)-9-chloro-2-(2-furyl)[1,2,4]triazolo[1,5-c]quinazoline (MRS 1220, 1b), which had a Ki value in binding of 0.65 nM at human A3 receptors. 1,4-Dihydropyridine derivatives 2 in some cases displayed >30000-fold selectivity for human A3 receptors (e.g., MRS 1334, 2b). Antagonist radioligands for the A3 receptor are still lacking.
Chart 1.
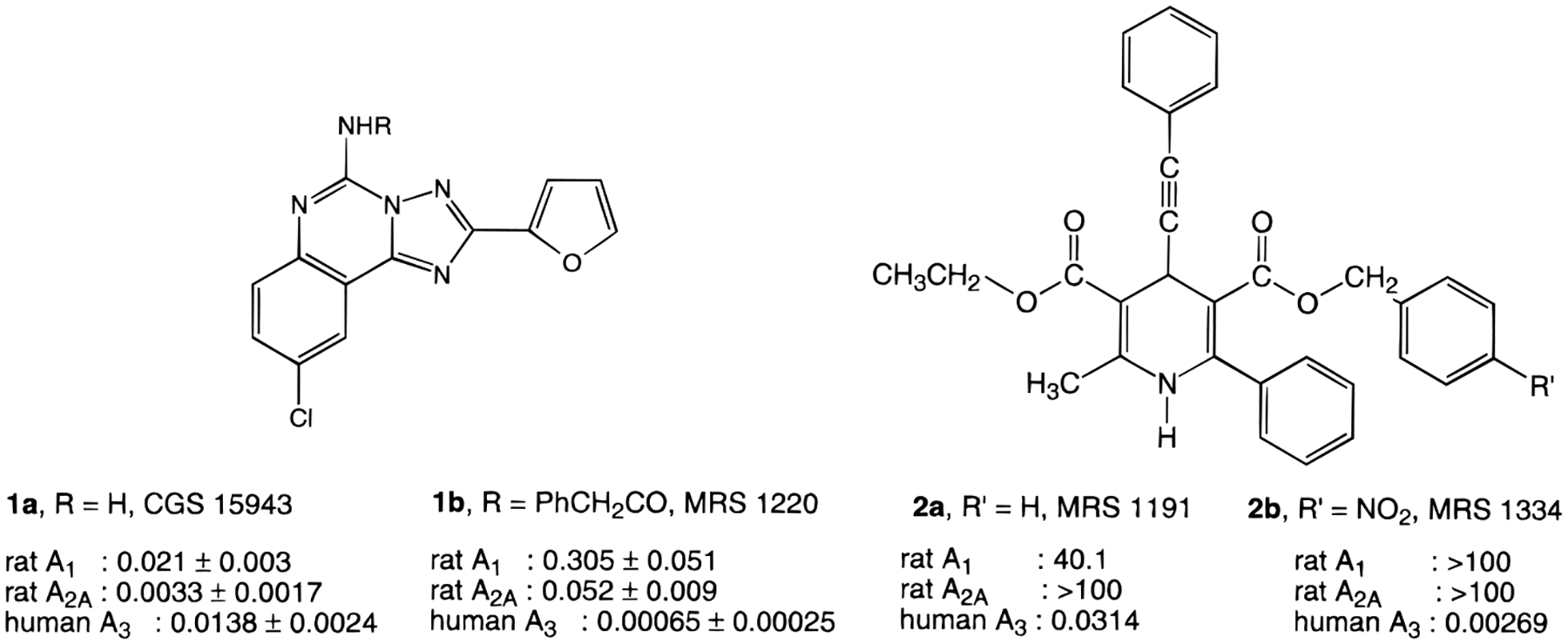
Structures of High-Affinity A3 Receptor Antagonists (Ki Values in μM).
The adenosine A2B receptor21 is the poorest characterized subtype of adenosine receptors. Whereas for the other subtypes selective agonists, antagonists, and radiolabeled ligands are available, no such compounds are known for the A2B receptor. For pharmacological studies, both selective agonists and antagonists are needed. An A2B selective antagonist may prove useful in the treatment of asthma.43
As an approach to find both selective antagonists for the A2B receptor and more potent antagonists for the A3 receptor, we have synthesized and screened a series of triazoloquinazolines derived from the nonselective A1/A2A receptor antagonist 1a. In this study we have further explored the pharmacophore region in the vicinity of the N5 position of 1a and the acyl group of 1b in binding to the human A3 receptor and have defined the previously unknown structure–activity relationship (SAR) at the human A2B receptor.
Results and Discussion
Synthesis.
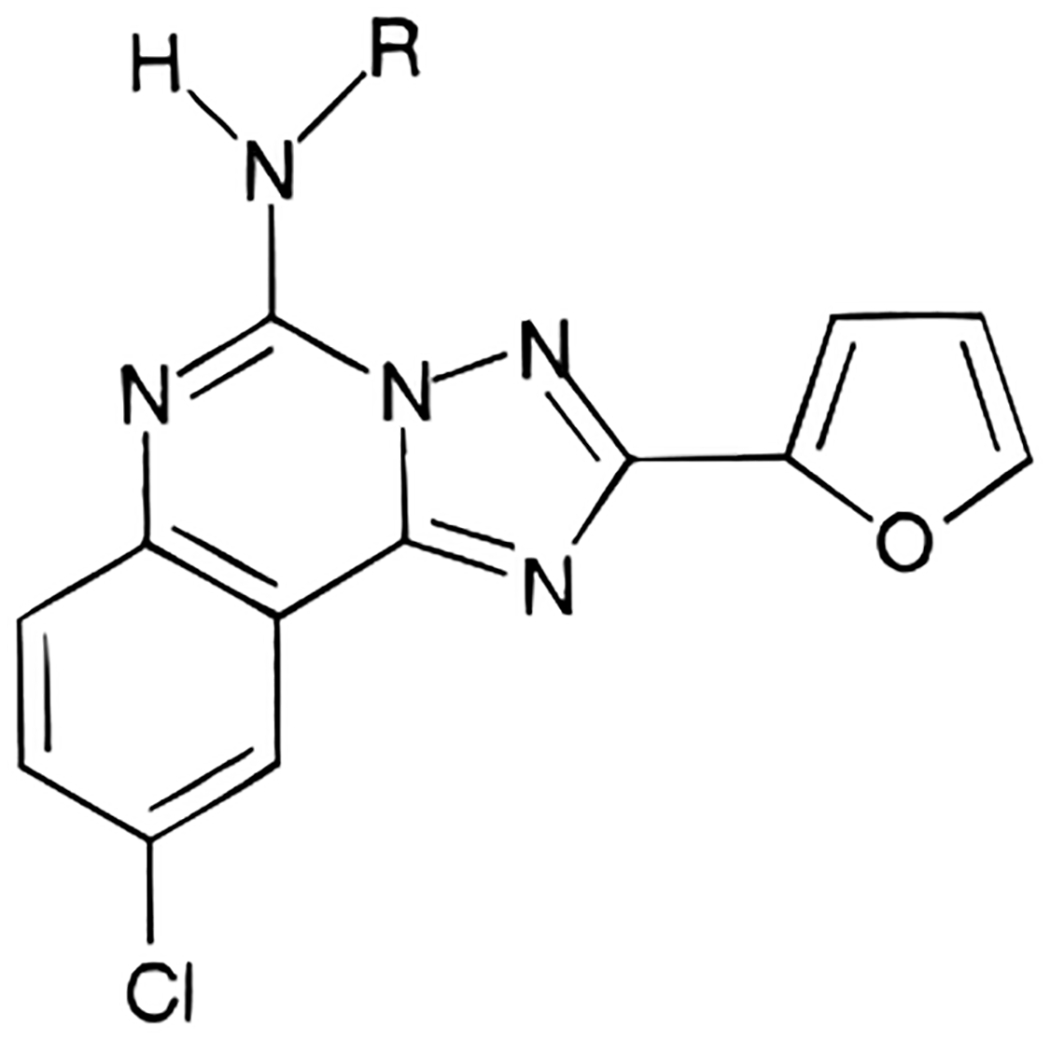
The structures of the triazoloquinazoline derivatives tested for affinity in radioligand binding assays at adenosine receptors are shown in Table 1. A few derivatives, e.g., the phenylacetyl derivative 1b and the 5-aminoacyl derivatives 20 and 27 in Table 1 and compounds 35-46 in Table 3, had been reported by us previously.20 The 5-benzyl derivative 3 was prepared through an alkylation reaction using potassium hydroxide and benzyl bromide. The 4-amino-3-iodophenylacetyl derivative 5 was synthesized by iodination22 (using iodine and calcium carbonate) of the 4-aminophenylacetyl derivative 6 which was prepared from catalytic reduction of the corresponding 4-nitrophenylacetyl derivative. The other various acyl derivatives were synthesized using standard acylation methods described in the Experimental Section, except for the 4-carboxybutanoyl congener 34 which was prepared by heating with glutaric anhydride at 80 °C without base or coupling agents. The R- and S-enantiomeric pairs (11 and 14; 17 and 18) were prepared from the corresponding optically pure carboxylic acids. The yields and chemical characterization of these compounds are reported in Table 2.
Table 1.
Affinities or Antagonistic Activities of Triazoloquinazoline Derivatives in Radioligand Binding Assays at A1, A2A, and A3 Receptorsa–c
| ||||||
|---|---|---|---|---|---|---|
| compd | R | Ki or IC50 (nM) | ||||
| rA1 | rA2A | hA3 | rA1/hA3 | rA2A/hA3 | ||
| 1a (CGS 15943) | H | 21 ± 3.0d | 3.3 ± 1.7d | 13.8 ± 2.4 | ||
| 1b | COCH2-Ph | 52.7 ± 11.8e | 10.3 ± 3.7e | 0.65 ± 0.25 | 81 | 16 |
| 3 | CH2-Ph | 1200 ± 15 | 200 ± 34 | 42.5 ± 6.91 | 28 | 4.7 |
| 4 | COCH2-(4-CH3O-Ph) | 30.2 ± 6.3 | 28.0 ± 4.8 | 14.4 ± 3.2 | 2.1 | 1.9 |
| 5 | COCH2-(4-NH2-3-I-Ph) | 217 ± 65 | 10.4 ± 2.2 | 49.3 ± 17.9 | 4.4 | 0.21 |
| 6 | COCH2-(4-NH2-Ph) | 24.9 ± 7.7 | 6.97 ± 1.13 | 3.56 ± 1.24 | 7.0 | 2.0 |
| 7 | COCH2-(2-I-Ph) | 2300 ± 590 | 17.2 ± 4.0 | >10000 | <1 | ≪1 |
| 8 | COCH2-(3-I-Ph) | 45.4 ± 8.7 | 9.67 ± 1.74 | 882 ± 242 | 0.051 | 0.011 |
| 9 | COCH2-(4-I-Ph) | 13.8 ± 4.6 | 9.93 ± 2.00 | 62.9 ± 13.0 | 0.22 | 0.16 |
| 10 | COCH2-(3-Cl-Ph) | 43.0 ± 6.4 | 9.43 ± 2.65 | 32.1 ± 11.3 | 1.3 | 0.29 |
| 11 | (R)-COCH(CH3)(Ph) | 46.2 ± 11.9 | 11.1 ± 1.3 | 0.362 ± 0.053 | 130 | 31 |
| 12 | (S)-COCH(CH3)(Ph) | 37.5 ± 8.5 | 20.1 ± 3.7 | 0.468 ± 0.111 | 80 | 43 |
| 13 | COCH(Ph)2 | 129 ± 43 | 12.1 ± 3.4 | 0.586 ± 0.196 | 220 | 21 |
| 14 | COC(CH3)(Ph)2 | 890 ± 131 | 453 ± 156 | 194 ± 42 | 4.6 | 2.3 |
| 15 | COCH2CH2-Ph | 45.2 ± 7.5 | 28.3 ± 10.3 | 23.6 ± 7.6 | 1.9 | 1.6 |
| 16 | COCH=CH-Ph (trans) | 282 ± 71 | 59.8 ± 13.4 | 72.1 ± 15.6 | 3.9 | 0.71 |
| 17 | d-COCH(CH3)(NH-Boc) | 48.6 ± 6.7 | 18.8 ± 5.3 | 46.3 ± 5.4 | 1.0 | 0.41 |
| 18 | L-COCH(CH3)(NH-Boc) | 54.7 ± 13.4 | 6.65 ± 1.56 | 82.9 ± 2.7 | 0.66 | 0.080 |
| 19 | CO(CH2)2-NH-Boc | 31.0 ± 2.2 | 7.58 ± 0.80 | 6.71 ± 0.67 | 4.6 | 1.1 |
| 20 | CO(CH2)3-NH-Boc | 45.9 ± 13.2 | 19.9 ± 4.6 | 32.9 | 1.4 | 0.60 |
| 21 | CO(CH2)4-NH-Boc | 30.1 ± 6.8 | 3.64 ± 0.34 | 22.0 ± 3.1 | 1.4 | 0.17 |
| 22 | CO(CH2)5-NH-Boc | 53.8 ± 12.7 | 33.7 ± 7.3 | 33.8 ± 10.2 | 1.6 | 1.0 |
| 23 | CO(CH2)6-NH-Boc | 38.2 ± 2.6 | 11.1 ± 1.9 | 53.7 ± 31.0 | 0.71 | 0.21 |
| 24 | d-COCH(CH3)(NH2) | 193 ± 17 | 29.7 ± 9.1 | 1140 ± 370 | 0.17 | 0.026 |
| 25 | l-COCH(CH3)(NH2) | 390 ± 127 | 143 ± 13 | 1200 ± 460 | 0.33 | 0.12 |
| 26 | CO(CH2)2-NH2 | 89.8 ± 23.3 | 7.58 ± 2.1 | 874 ± 4 | 0.10 | 0.0088 |
| 27 | CO(CH2)3-NH2 | 8.75 ± 2.28 | 1.38 ± 0.23 | 80.8 ± 7.4 | 0.11 | 0.017 |
| 28 | CO(CH2)4-NH2 | 6.99 ± 1.38 | 1.13 ± 0.41 | 57.9 ± 20.8 | 0.12 | 0.020 |
| 29 | CO(CH2)5-NH2 | 99.6 ± 6.7 | 10.3 ± 3.7 | 213 ± 27 | 0.47 | 0.048 |
| 30 | CO(CH2)6-NH2 | 114 ± 26 | 55.9 ± 5.5 | 346 ± 77 | 0.33 | 0.16 |
| 31 | CO(CH2)4-COOBn | 304 ± 126 | 16.7 ± 2.5 | 44.7 ± 14.1 | 6.8 | 0.37 |
| 32 | CO(CH2)2-COOCH3 | 71.8 ± 7.2 | 28.8 ± 10.7 | 55.1 ± 8.6 | 1.3 | 0.52 |
| 33 | CO(CH2)6-COOCH3 | 46.6 ± 10.5 | 7.43 ± 2.72 | 59.0 ± 18.1 | 0.79 | 0.13 |
| 34 | CO(CH2)3-COOH | 45 ± 4 | 1.15 ± 0.47 | 81.3 ± 11.0 | 0.55 | 0.014 |
Displacement of specific [3H]R-PIA binding in rat brain membranes, expressed as Ki ± SEM (n = 3–5).
Displacement of specific [3H]CGS 21680 binding in rat striatal membranes, expressed as Ki ± SEM (n = 3–6).
Displacement of specific [125I]AB-MECA binding at human A3 receptors expressed in HEK cells, in membranes, expressed as Ki ± SEM (n = 3–4).
IC50 values for 1a (see ref 20).
Previously reported Ki values were 305 and 52.0 nM for rat A1 and A2A receptors, respectively.20
Table 3.
Potency of Triazoloquinazoline Derivatives in a Functional Assay at Human A2B Receptors Stably Expressed in CHO Cells

| ||||
|---|---|---|---|---|
| compd | R1 or R3 | % inhibitiona,c | IC50 (μM)b,d | pA2d |
| 1a | H | 100 (100–100) | 1.20 ± 0.43 | 8.0 ± 0.3 |
| 1b | COCH2-Ph | 71 (73–69) | ||
| 3 | CH2-Ph | 21 (20–22) | ||
| 5 | COCH2(4-NH2-3-I-Ph) | 23 (18–27) | ||
| 6 | COCH2(4-NH2-Ph) | 90 (85–94) | 7.4 ± 4.5 | |
| 9 | COCH2-(4-I-Ph) | 42 (39–45) | ||
| 10 | COCH2-(3-Cl-Ph) | 45 (55–34) | ||
| 15 | COCH2CH2-Ph | 43 (50–36) | ||
| 16 | COCH=CH-Ph | 38 ± 14 | ||
| 20 | CO(CH2)3-NH-Boc | 47 (57–36) | ||
| 26 | CO(CH2)2-NH2 | 83 (85–80) | 13.3 ± 4.0 | |
| 27 | CO(CH2)3-NH2 | 100 (100–100) | 0.27 ± 0.04 | 8.0 ± 0.3 |
| 28 | CO(CH2)4-NH2 | 100 (100–100) | 1.77 ± 1.25 | |
| 29 | CO(CH2)5-NH2 | 79 (73–85) | ||
| 30 | CO(CH2)6-NH2 | 75 (74–75) | ||
| 34 | CO(CH2)3-COOH | 1.90 ± 0.13 | ||
| 35 | COCH3 | 92 (94–90) | 2.65 ± 1.00 | |
| 36 | COCH2CH3 | 66 (75–56) | ||
| 37 | CO(CH2)2CH3 | 66 (73–58) | ||
| 38 | CO(CH2)3CH3 | 79 (86–72) | ||
| 39 | COC(CH3)3 | 89 (92–85) | 2.40 ± 1.70 | |
| 40 | CO-OC(CH3)3 | 92 (94–90) | 2.41 ± 1.02 | |
| 41 | CO-Ph | 58 (69–47) | ||
| 42 | CO-(3-I-Ph) | 38 (51–25) | ||
| 43 | H, R2 = Br | 16 (24–9) | ||
| 44 | CO-Ph, R2 = Br | 0 (0–0) | ||
| 45 | H | 0 (0–0) | ||
| 46 | (CH2)2CH3 | 0 (0–0) | ||
Percentage inhibition by 50 μM of each antagonist of cyclic AMP production induced by 50 μM NECA.
Concentration of antagonist inhibiting 50% of cyclic AMP production induced by 50 μM NECA.
Values are the means of duplicate measurements (values of individual measurements between parentheses) or are the means of at least three experiments ± SEM.
Values are given ± SEM and are the means of at least three experiments.
Table 2.
Yields and Chemical Characterization of Triazoloquinazoline Derivatives
| compd no. | % yield | mp (°C) | MS | formula | anal. |
|---|---|---|---|---|---|
| 3 | 29 | 184–185 | EI: 375 | C20H14N5O2Cl·0.81H2O | C,H,N |
| 4 | 12 | 258–260 | EI: 433 | C22H16N5O3Cl·0.53CH2Cl2 | C,H,N |
| 5 | 25 | 210–211 | CI: 545 | C21H14N6O2ClI | C,H,N |
| 6 | 45 | 197–199 | EI: 418 | C21H15N6O2Cl·0.64H2O | C,H,N |
| 7 | 22 | 280–282 | EI: 529 | C21H13N5O2ClI·0.94(CH3)2CO | C,H,N |
| 8 | 16 | 224–227 | CI: 530 | C21H13N5O2ClI·1.17(CH3)2CO | C,H,N |
| 9 | 51 | 260–262 dec | EI: 529 | C21H13N5O2ClI | C,H,N |
| 10 | 33 | 248–250 | CI: 438 | C21H13N5O2Cl2 | C,H,N |
| 11 | 38 | 207–209 dec | EI: 417 | C22H16N5O2Cl | C,H,N |
| 12 | 58 | 115–118 | EI: 417 | C22H16N5O2Cl | C,H,N |
| 13 | 57 | 205–207 | CI: 480 | C27H18N5O2Cl | C,H,N |
| 14 | 100 | 175–178 | EI: 493 | C28H20N5O2Cl | C,H,N |
| 15 | 65 | 253–255 | EI: 417 | C22H17N5O2Cl | C,H,N |
| 16 | 43 | 305–307 | CI: 416 | C21H15N5O2Cl | C,H,N |
| 17 | 13 | 131–134 | CI: 457 | C21H21N6O4Cl | C,H,N |
| 18 | 15 | 220 | CI: 457 | C21H21N6O4Cl·2.6CH3OH | C,H,N |
| 19 | 50 | 216–218 | CI: 457 | C21H21N6O4Cl | C,H,N |
| 21 | 53 | 189–192 | CI: 485 | C23H25N6O4Cl | C,H,N |
| 22 | 100 | 196–198 | CI: 499 | C24H27N6O4Cl | C,H,N |
| 23 | 100 | 156–157 | EI: 512 | C25H29N6O4Cl·0.91H2O | C,H,N |
| 24 | 56 | 175–176 | FAB: 357 | C16H13N6O2Cl | FAB+a |
| 25 | 71 | 278 | FAB: 357 | C16H13N6O2Cl | FAB+a |
| 26 | 72 | 137–139 | EI: 356 | C16H13N6O2Cl·CF3COOH | C,H,N |
| 28 | 64 | 231–232 | FAB: 385 | C18H17N6O2Cl | FAB+a |
| 29 | 80 | 153–155 | CI: 399 | C19H19N6O2Cl·CF3COOH·0.87CH3OH | C,H,N |
| 30 | 70 | 165–167 | EI: 412 | C20H21N6O2Cl | FAB+a |
| 31 | 71 | 149–151 | EI: 503 | C26H22N5O4Cl·CF3COOH | C,H,N |
| 32 | 54 | 185–187 | EI: 399 | C18H14N5O4Cl·0.3EtOAc | C,H,N |
| 33 | 62 | 165 | EI: 455 | C22H22N5O4Cl·0.37(C2H5)2O | C,H,N |
| 34 | 26 | 209–211 | FAB: 400 | C18H14N5O4Cl·0.56CH2Cl2 | C,H,N |
High-resolution mass in FAB+ mode (m/z) determined to be within acceptable limits. 24: calcd, 357.0867; found, 357.0879. 25: calcd, 357.0867; found, 357.0865. 28: calcd, 385.1180; found, 385.1168. 30: calcd, 413.1491; found, 413.1493.
Affinity at Adenosine Receptors. Binding Assays at A1, A2A, and A3 Receptors.
Ki values were determined in radioligand binding assays at rat cortical A1 receptors vs [3H]R-PIA ([3H]-(R)-N6-(phenylisopropyl)adenosine) and at rat striatal A2A receptors vs [3H]CGS 21680 ([3H]-2-[[4-(2-carboxyethyl)phenyl]ethylamino]-5′-(N-ethylcarbamoyl)adenosine).4,5 Affinity at cloned human A3 receptors expressed in HEK-293 cells23 was determined using [125I]AB-MECA (N6-(4-amino-3-[125I]iodobenzyl)-5′-(N-methylcarbamoyl)adenosine).24
N5-Alkylation in the N-benzyl derivative 3 decreased binding affinity at A1, A2A, and A3 receptors. Simple phenyl ring substitutions were made on the structure of the N5-phenylacetyl derivative 1b including: methoxy, 4; amino, 5 and 6; and halo, 7-10. None of these derivatives exceeded the affinity of 1b at human A3 receptors. The most potent of these derivatives was the 4-amino derivative 6 which was prepared as a potential substrate for radioiodination. The Ki value of 6 at human A3 receptors was 3.56 nM, which would have been satisfactory for a radioligand; however upon iodination to give compound 5, the affinity at human A3 receptors decreased by 14-fold. Thus, the receptor affinity is highly dependent on the substitution of the phenyl ring of 1b. The affinities of ring-iodinated isomers of 1b, 7-9, were compared. The N5-(2-iodophenyl)acetyl derivative 7 was highly selective for A2A receptors. The N5-(3-chlorophenyl)acetyl and N5-(3-iodophenyl)acetyl derivatives 10 and 8, respectively, differed in affinity only at the A3 receptor, such that 8 was 27-fold less potent in binding.
Substitution of the two prochiral methylene hydrogens of the acetyl group was carried out. The α-methyl substitutions, resulting in the R- and S-enantiomers 11 and 12, appeared to slightly enhance affinity in both cases. No significant stereoselectivity of binding to A1, A2A, and A3 receptors was evident at this chiral center. The most potent derivative at A3 receptors in the present study was the (R)-N5-α-methyl(phenylacetyl) derivative 11 with a Ki value of 0.36 nM. Curiously, even a phenyl substitution at the methylene group, compound 13, resulted in extremely high affinity at human A3 receptors, with a Ki value of 0.59 nM and selectivities of 220- and 21-fold vs rat A1 and A2A receptors, respectively. Thus, the binding region of the receptor surrounding the α-carbon of the acetyl group of 1b is not highly sterically restricted. Nevertheless, retention of at least one methylene hydrogen of the acetyl group was required for high affinity at the adenosine receptors, since the α-methyldiphenyl derivative 14 was 330-fold less potent than the α-diphenyl derivative 13 in binding to human A3 receptors. Homologation of 1b to give the phenylpropionyl derivative 15 reduced the affinity at human A3 receptors by 27-fold, and the corresponding styryl derivative 16 was even less potent at A3 receptors.
The γ-aminobutyric acid derivatives, both the Boc-protected form 20 and the free amino form 27, were reported in the previous study.20 The SAR of homologues of these derivatives was explored. Although nanomolar affinities at human A3 receptors were not obtained, there was a distinct dependence of affinity upon lengthening or shortening the methylene chain. A minimum in the Ki values at A1, A2A, and A3 receptors among the free amine derivatives was obtained at a chain length of four methylene groups, corresponding to compound 28 (Figure 1), which had a Ki value of 1.13 nM at A2A receptors. For free amine derivatives, the order of affinity was invariably: A2A > A1 > A3 receptors. Thus, the interactions responsible for this affinity enhancement must be common to these three receptor subtypes. For Boc-amine derivatives, however, a clear dependence of binding affinity on chain length was not evident at A1, A2A, and A3 adenosine receptor subtypes. In this series of amino acid derivatives, no stereoselectivity of binding to A1, A2A, and A3 receptors was evident at the α-carbon prochiral center, as seen from the affinities of the d- and l-Ala conjugates, 17 vs 18 and 24 vs 25. At A2A receptors the presence of the free amino group, e.g., 27-29, in general resulted in higher affinity, while at A3 receptors the Boc-protected form, e.g., 20-22, appeared to be preferred. This suggested that human A3 receptors contain a more highly hydrophobic pocket in this region than do rat A2A receptors. Furthermore, at A1 receptors the free amine is preferred over the Boc-amine for analogues containing three and four methylene groups.
Figure 1.
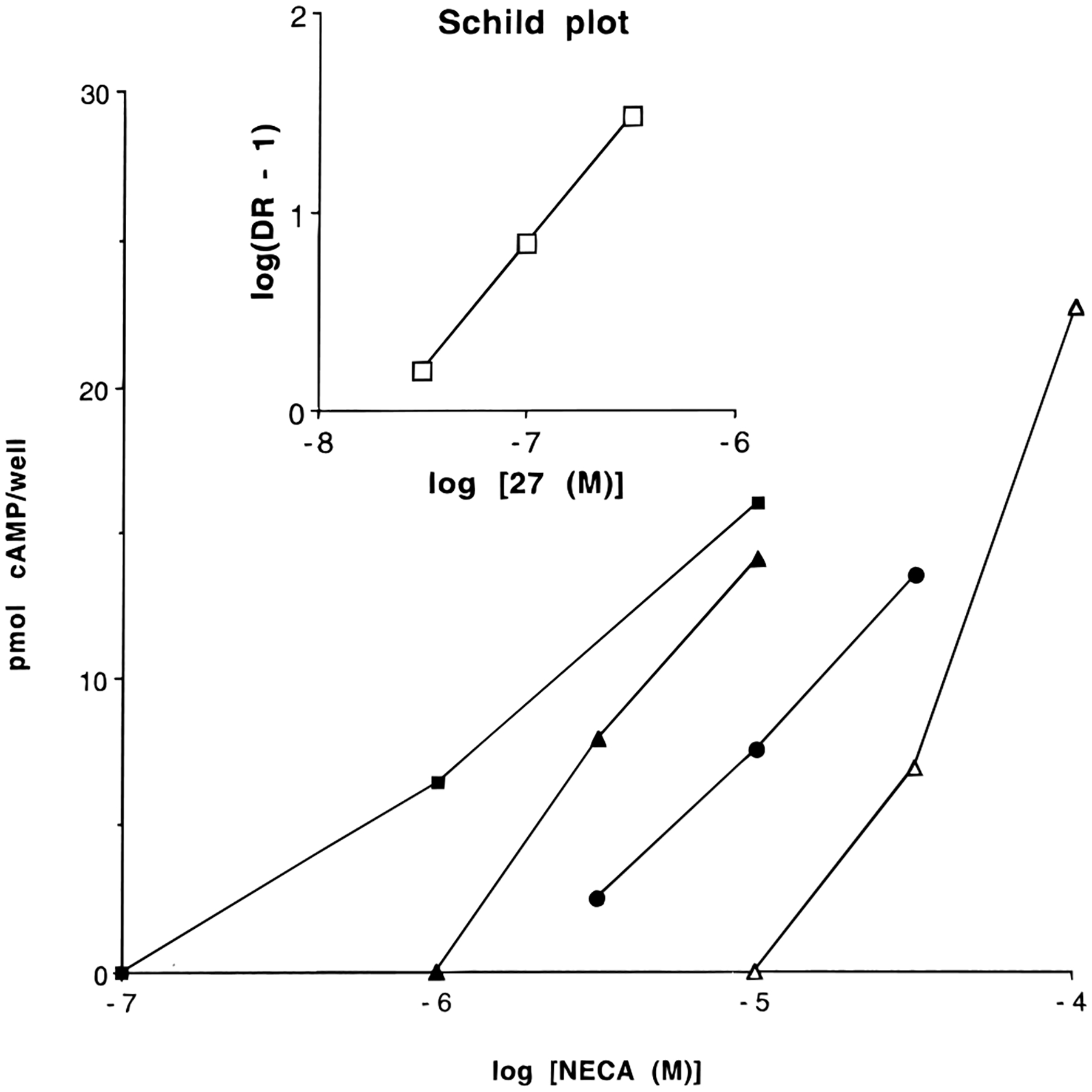
Determination of the pA2 value of compound 27 at human A2B receptors. Data were taken from a typical experiment. Dose-response curves of NECA were recorded in the absence and presence of three concentrations of the antagonist 27: ■, no antagonist added; ▲, 0.03 μM; ●, 0.1 μM; Δ, 0.3 μM. pA2 values were determined in a Schild plot (inset) with a slope of approximately unity.
The effects of ester, 31-33, and carboxylic acid, 34, groups on the N5-acyl substituent were also explored. The affinity at A3 receptors was not significantly affected by the presence of a negatively charged carboxylate group vs a neutral ester group.
Functional Assay at A2B Receptors.
Compound 1a and selected N5-acyl-substituted analogues (Table 3) were screened by measuring the percentage inhibition by the analogue (50 μM) of the cyclic AMP production induced by 50 μM 5′-(N-ethylcarboxamido)adenosine (NECA) in a Chinese hamster ovary cell line stably transfected with human A2B receptor cDNA. For derivatives that displayed ≥80% inhibition, IC50 values were determined on cyclic AMP production induced by 50 μM NECA. Finally, pA2 values were determined for two of the most active derivatives, 1a and 27. For this purpose, NECA concentration–response curves were recorded in the absence and presence of three increasing concentrations of antagonist. pA2 values were derived from Schild plots (Figure 1), which displayed slopes of approximately unity.
Compound 1a had an IC50 value of 1.20 μM. In the same system a pA2 value of 8.0 ± 0.3 was measured. This agrees with earlier findings of Alexander et al., who have measured a value of 7.8 in a similar assay, using another CHO cell line expressing human A2B receptors.42 The γ-aminobutyryl-substituted derivative 27 displayed an elevated potency at the A2B receptor (IC50 value of 0.27 μM). However, its pA2 value was similar to that of 1a. Concentration–response curves of NECA in the presence and absence of 27 are shown in Figure 2. There was no gain in selectivity toward the other receptor subtypes.
Figure 2.

Minimized energy conformations of 27, showing two conformers (cis and trans) of the aminoalkyl side chain.
Derivatives with a 2-(5-bromofuryl) substituent (43, 44) and derivatives that lack the 5-amino function (45, 46) were weak or inactive as A2B receptor antagonists. Diminished affinities of these analogues for the other subtypes have been reported previously.20
Among the amino acid conjugates a minimum in the IC50 values in A2B receptor antagonism was observed with a chain length of three methylene groups (27), while at A1, A2A, and A3 receptors aminoalkyl derivatives having three and four methylene groups were nearly equipotent. At A2B receptors the Boc-protected γ-aminobutyryl derivative 20 had a much lower activity than the corresponding free amino derivative 27 (only 47% inhibition at 50 μM).
The γ-carboxybutyryl derivative 34 showed a high potency (1.9 μM), but it was less active than the γ-aminobutyryl derivative 27. Apparently, a positively charged amino group results in a higher activity than a negatively charged carboxylate function.
In the N5-phenylacetyl-substituted series only the 4-amino derivative 6 displayed a moderate potency (7.4 μM). A phenylacetyl group appears to be not well-tolerated in the A2B receptor. Increase of potency by substitution with an amino function was also observed in propionyl, 36, vs 3-aminopropionyl, 26, and butyryl, 37, vs 4-aminobutyryl, 27, not only at the A2B receptor (Table 3) but also at A1 and A2A receptors (refer to binding data of 36 and 37 in ref 20).
In the series of compounds with N5-acyl chain substituents, the N5-acetyl (35) and N5-pivaloyl (39) derivatives were most active. Apparently, there is room in the A2B receptor for a bulky tert-butyl group. The N5-Boc-substituted derivative 40, with a tert-butyl group too, showed a similar activity. Remarkably, for maximal activity, the N5-acetyl group should either be unsubstituted as in 35 or methyl-trisubstituted as in 39, but not methyl-monosubstituted as in the propionyl derivative 36.
Although the N5-pivaloyl derivative 39 had a slightly decreased activity (IC50 value 2.4 μM), it showed largely diminished affinities for A1, A2A, and A3 receptors in the previous study (Ki A1 = 205 ± 20 nM; Ki A2A = 88.8 ± 20.5 nM; Ki A3 = 244 ± 6 nM).20 The N5-Boc-substituted derivative 40 with an IC50 value of 2.41 ± 1.02 μM behaved similarly (Ki A1 = 190 ± 16 nM; Ki A2A = 92 ± 8 nM; Ki A3 = 82.5 ± 23.3 nM).20 Thus, N5-pivaloyl or N5-Boc substitution might lead to A2B receptor selectivity.
Molecular Modeling.
A rhodopsin-based molecular model for the human A2A receptor has been published previously.25 As an aid in interpreting the present results at the human A2B receptor, we provide the first description of ligand–A2B receptor interactions, using 27 as a ligand to probe the A2B receptor binding pocket. We have recently introduced the cross-docking procedure26 (see the Experimental Section) which can be considered a further refinement in building the putative antagonist binding site in the human A2B receptor. As with other G protein-coupled receptor models,27 the length of the transmembrane region is approximately 40 Å. The interhelical distance between pairs of adjacent helical axes is roughly 10 Å, consistent with a common interhelical contact distance.28 The interhelical angles, measured between the principal axes of adjacent helices, are around −150–170° for antiparallel and 10–25° for the parallel helices. This is typical of a 3–4 type helix–helix contact associated with optimal interactions between nearly parallel-aligned helices.28 Each helix maintained almost the same position and tilting found in the published rhodopsin 2D electron density map.29,30
In all our calculations, we used the γ-amino-protonated form of compound 27, consistent with protonation at physiological pH. Since 27 has high conformational flexibility in the region of the γ-aminobutyryl side chain, a complete random search conformational analysis was performed. After sampling and minimization procedures, two energetically important conformers have been selected, one of these with the amide bond in the cis conformation and the other one in which the amide bond was in the trans conformation (see Figure 2). RHF/AM1 semiempirical calculations were conducted to obtain the optimized geometries. Only the cis conformer may be docked in an energetically favorable manner inside the TM bundle. The trans isomer would force the aminobutyl side chain toward the transmembrane helices leading to an overall distortion of the receptor architecture.
Moreover, in the cross-docked model TM3, TM4, TM5, TM6, and TM7 were rotated clockwise by 15°, 10°, 5°, 10°, and 5°, respectively, about their transmembrane axes with respect to the ligand–free receptor model. The energy of the 27–A2B receptor complex structure was lowered 55 kcal/mol through the cross-docking procedure. Consistent with the proposal of Gouldson et al.,31 rotations and translations of the TM domains are crucial factors in the ligand recognition process in different GPCRs. Consequently, our approach to docking is designed to mimic the natural domain movement within a receptor.
One of the most important limitations in the correct identification of the ligand binding regions of the A2B receptor is that no site-directed mutagenesis studies are available. Using both the A2A/A2B sequence homology and the site-directed mutagenesis results previously obtained in our laboratory for the human A2A receptor,25 we were able to identify a hypothetical binding site for 27 in the human A2B receptor. Figure 3 shows the 3D structural models of the human A2B receptor after the application of cross-docking with 27.
Figure 3.
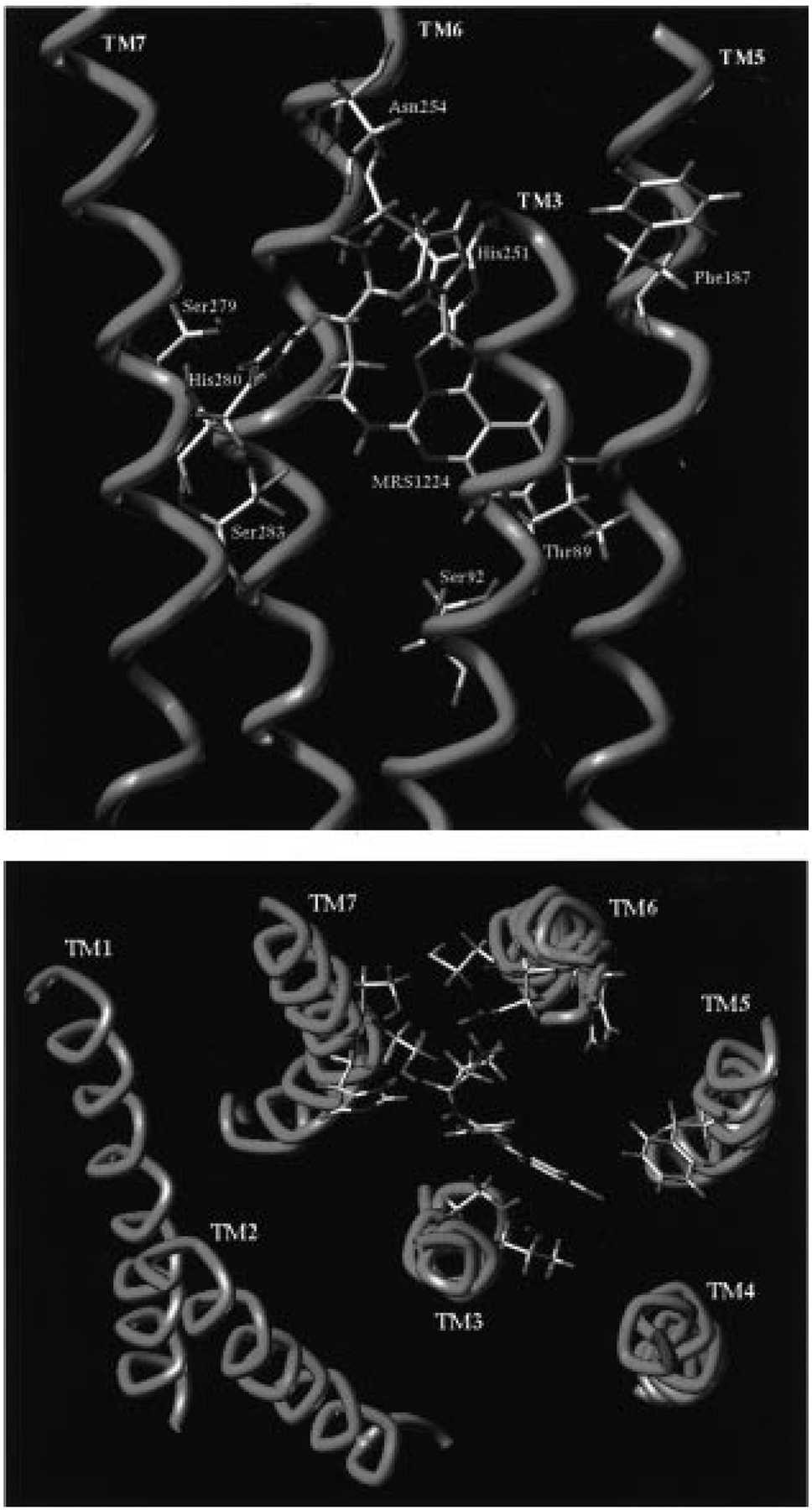
Model of the cis conformation of 27 docked in the putative binding site of the human A2B receptor, as viewed from the plane of the plasma membrane (top) and from the extracellular side (bottom).
We identified the hypothetical binding site of 27 in proximity to TMs 3, 5, 6, and 7, with the furan ring pointing toward the extracellular environment. The γ-aminobutyryl side chain is located midway between His251 (TM6) and His280 (TM7). According to the previously reported site-directed mutagenesis results for A1 and A2A receptors,32,25 these two histidine residues (conserved in the same positions of TM6 and TM7 helices for both receptors) were found crucial for the binding of both agonists and antagonists.
The oxygen atom of Ser283 (TM7) is within hydrogen-bonding distance (2.6 Å) of the carbonyl group of the amide moiety at the 5-position of the triazoloquinazoline structure. The amide group is surrounded by four polar amino acids: His251 (TM6), Ser279 (TM7), His280, and Ser283 (TM7). This region seems to be very important for the recognition of the antagonist structures. In particular, our side-directed mutagenesis results for human A2A receptor suggested that this serine (Ser281) was crucial for binding of both agonists and antagonists.25
Another important interaction (1.9 Å) is possible between the amino group of Asn254 (TM6) and the protonated nitrogen of the γ-aminobutyryl side chain. Also this Asn, conserved among all adenosine receptor subtypes, was found important for ligand binding. This interaction can explain the common trend observed between the different adenosine receptor subtypes, of the affinity values upon lengthening or shortening of the methylene chain. A minimum in the Ki values at A1, A2A, A2B, and A3 among the free amine derivatives was obtained at chain lengths of three or four methylene groups, indicating that there is a common anchor point inside the receptor binding cavity. We speculate that Asn254 (TM6) corresponds to this anchor point in the human A2B receptor.
A hydrophobic region, delimited by three apolar amino acids such as Val85 (TM3), Phe187 (TM5), and Val191 (TM5), is also present in the binding site model. The corresponding amino acids Leu90 (TM3) and Phe182 (TM5) were essential for binding of both agonists and antagonists at human A2A receptors.25 The chlorophenyl moiety of the triazoloquinazoline structure of 27 is located within this region. No direct interactions are predicted between the two polar amino acids, Thr89 (TM3) and Ser92 (TM3), and the antagonist structure. As previously reported, these two amino acids are important only for the coordination of the agonist derivatives at A1 and A2A receptors, respectively.25
Another unresolved question is why there are great differences in ligand affinities between A2A and A2B receptors. A comparison of the nature of the critical amino acids in the TM regions involved in the recognition of the ligand at the A2A and A2B subtypes fails to provide a clear explanation. In fact, there is a very good correspondence between the important residues found using site-directed mutagenesis of the human A2A receptor and those present in the A2B receptor. We speculate that there are two important differences between the molecular structures of A2A and A2B receptors: (i) the replacement of two amino acids, Leu267 (hA2A) with Lys269 (hA2B) and Tyr270 (hA2A) with Asn273 (hA2B), at the top of TM7 that could be involved in the movement of the ligand into the binding site and (ii) the different primary and secondary structures of the second extracellular loops that seem to be involved in the extracellular recognition process of the ligand.33 The second extracellular loop has been shown to be important for both agonist and antagonist binding at the human A2A receptor and could represent one of the strategic keys of the different selectivities between the different receptor subtypes.
Experimental Section
Computational Methods.
The human A2B receptor model was built and optimized using SYBYL 6.334 and MacroModel5.035 modeling packages, respectively, based on the approaches described by van Rhee et al.36 and Moro et al.26 All calculations were performed on a Silicon Graphics Indigo2 R8000 workstation. Briefly, transmembrane domains were identified with the aid of Kyte–Doolittle hydrophobicity37 and Emini37 surface probability parameters. Transmembrane helices were built from the sequences and minimized individually. The minimized helices were then grouped together to form a helical bundle matching the overall characteristics of the electron density map of rhodopsin. The helical bundle was minimized using the Amber38 all-atom force field, until the rms value of the conjugate gradient (CG) was <0.1 kcal/mol/Å. A fixed dielectric constant = 4.0 was used throughout these calculations.
A model of 5-[(6-aminohexanoyl)amino]-9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazoline (27) was constructed using the Sketch Molecule module of SYBYL. Conformational analysis of 27 was performed on all rotatable bonds of the aminohexanoyl side chain, using the random search procedure of SYBYL. The optimized geometries of the resulting conformers were calculated using MOPAC software (RHF method and AM1 Hamiltonian, keywords: PREC, GNORM=0.1, EF).
The fully minimized conformers were rigidly docked into the helical bundle using graphical manipulation coupled to continuous energy monitoring (Dock module of SYBYL). When a final position was reached, consistent with a local energy minimum, the complexes of receptor and ligand were subjected to an additional CG minimization run of 300 steps. Partial atomic charges for the ligands were imported from the MOPAC output files.
Recently, we have introduced a “cross-docking” procedure to obtain energetically refined structures of ligand–P2Y1 receptor complexes.26 We applied the same technique to obtain the structure of the 27–A2B receptor complex. This is a way to explore possible ligand-induced rearrangements of 7TM bundle by sampling 7TM conformations in the presence of the docked ligands. Cross-docking was carried out using the Dock module of SYBYL. Each helix was separated from the ligand–receptor complex structure, and its relative position was changed until a new lower-energy geometry was obtained. These adjustments consisted of small translations and rotations of the principal axis of the helix with respect to its original position. When a new final position was reached, consistent with the lowest-local-energy minimum, the separated helix was merged again into the ligand–receptor complex. The hydropathy profile for the new oriented helix was checked using the Kyte–Doolittle method.37 The new complex was subjected to an additional CG minimization run of 300 steps. This procedure was repeated for TM3, TM4, TM5, TM6, and TM7. The manual adjustments were followed by 25 ps of molecular dynamics (MD module of MacroModel) performed at a constant temperature of 300 K using a time step of 0.001 ps and a dielectric constant = 4.0. This procedure was followed by another sequence of CG energy minimization to a gradient threshold of < 0.1 kcal/mol/Å. Energy minimization of the complex was performed using the AMBER all-atom (Macro-Model) force field.
Materials.
Compound 1a, R-PIA, and 2-chloroadenosine were purchased from Research Biochemicals International (Natick, MA). All acylating agents were obtained from Aldrich (St. Louis, MO).
Synthesis.
Proton nuclear magnetic resonance spectroscopy was performed on a Varian GEMINI-300 spectrometer, and spectra were taken in DMSO-d6 or CDCl3. Unless noted, chemical shifts are expressed as ppm downfield from tetramethylsilane. Chemical-ionization (CI) mass spectrometry was performed with a Finnigan 4600 mass spectrometer and electron-impact (EI) mass spectrometry with a VG7070F mass spectrometer at 6 kV. High-resolution FAB (fast atom bombardment) mass spectrometry was performed with a JEOL SX102 spectrometer using 6-kV Xe atoms. The compounds were previously desorbed from glycerol or magic bullet matrix. Optical rotation was measured using a Perkin-Elmer polarimeter 341. Elemental analysis (± 0.4% acceptable) was performed by Atlantic Microlab Inc. (Norcross, GA) or Galbraith Laboratories, Inc. (Knoxville, TN). All melting points were determined with a Unimelt capillary melting point apparatus (Arthur H. Thomas Co., PA) and were uncorrected. All triazoloquinazoline derivatives showed one spot on TLC (MK6F silica, 0.25 mm, glass-backed; Whatman Inc., Clifton, NJ). Where needed, evaluation of purity was done on a Hewlett-Packard 1090 HPLC system using an OD-5–60 C18 analytical column (150 mm × 4.6 mm; Separation Methods Technologies, Inc., Newark, DE) in two different linear gradient solvent systems. One solvent system (A) was 0.1 M TEAA (pH = 5.0)/CH3CN, 50:50 to 10:90, in 20 min with flow rate 1 mL/min. The other (B) was H2O/MeOH, 40:60 to 10:90, in 20 min with flow rate 1 mL/min. Peaks were detected by UV absorption using a diode array detector.
General Procedure for the Preparation of 5-N-Acyl Derivatives of 1a. Method A (Acid Chloride).
To a stirred solution of 1a (20 mg, 0.07 mmol) and anhydrous pyridine (80 μL, 1.0 mmol) in 2 mL of anhydrous CH2Cl2 was added the desired acyl chloride (0.21 mmol), prepared from the acid and excess thionyl chloride (unless commercially available). The mixture was stirred at room temperature for 24 h and then evaporated to dryness under reduced pressure. The residue was purified by preparative silica gel TLC (CH2Cl2–MeOH, 50:1–75:1, or Hex:CHCl3:MeOH, 1:1:0.1) to afford the desired compounds (4, 7-16, 32, 33).
Method B (Carbodiimide).
A solution of 1a (20 mg, 0.07 mmol), the desired carboxylic acid compound (0.42 mmol), EDAC (82 mg, 0.42 mmol), DMAP (4 mg, 0.032 mmol), and triethylamine (0.146 mL, 1.05 mmol) in 3 mL of anhydrous DMF/CH2Cl2 (1:1 v/v) was stirred at room temperature for 48h. The mixture was treated with the same procedure as method A for purification of the desired compounds (17-23, 31).
Method C (Deprotection of Boc Group).
To a solution of 0.02 mmol of Boc-protected amino acyl derivatives of 1a in 1 mL of CH2Cl2 was added 50 μL of TFA, and the mixture stirred at room temperature for 10 min. The mixture was evaporated under reduced pressure, and white solids were obtained from ether or CH2Cl2/MeOH as TFA salts (24-30).
9-Chloro-2-(2-furanyl)-5-(benzylamino)[1,2,4]triazolo[1,5-c]quinazoline (3).
To a solution of 1a (24.0 mg, 0.08 mmol) in 2 mL of anhydrous DMSO was added powdered potassium hydroxide (67.2 mg, 1.20 mmol) followed by benzyl bromide (28.8 mg, 0.17 mmol). The mixture was stirred at room temperature overnight and then diluted with 2 mL of water. The reaction product was extracted with CHCl3 (10 mL × 3). The fractions were combined, neutralized with 1 N HCl, washed with water (25 mL × 2), and dried over sodium sulfate. The product was isolated×from preparative silica gel TLC (hexanes–ethyl acetate, 4:1) (29%): 1H NMR (CDCl3) δ 4.92 (2H, d, J = 5.86 Hz, −CH2), 6.55 (1H, t, J = 5.86 Hz, NH), 6.61 (1H, t, J = 1.95 Hz, H-4′), 7.28 (1H, s, H-3′), 7.36–7.49 (4H, m, aromatic-H), 7.61–7.64 (2H, m, H-8 and H-5′), 7.70 (1H, d, J = 9.77 Hz, H-7), 8.41 (1H, d, J = 1.95 Hz, H-10).
9-Chloro-2-(2-furanyl)-5-[(4-methoxyphenylacetyl)-amino][1,2,4]triazolo[1,5-c]quinazoline (4):
1H NMR (DMSO-d6) δ 3.74 (3H, s, −OCH3), 3.98 (2H, s, −CH2CO), 6.78–6.80 (1H, m, H-4′), 6.93 (2H, d, J = 8.7 Hz, aromatic-H), 7.32–7.35 (3H, m, H-3′ and aromatic-H), 7.92 (2H, s, H-8 and H-7), 8.03 (1H, s, H-5′), 8.39 (1H, s, H-10), 11.24 (1H, s, NH).
9-Chloro-2-(2-furanyl)-5-[(4-amino-3-iodophenylacetyl)-amino][1,2,4]triazolo[1,5-c]quinazoline (5).
To a solution of 6 (10 mg, 0.024 mmol) in 1 mL of CH2Cl2/MeOH was added solid I2 (6 mg, 0.024 mmol) followed by solid CaCO3 (3 mg, 0.028 mmol). The reaction mixture was stirred for 12 h at room temperature. The solution was evaporated with a nitrogen stream and partitioned between NaHSO3/EtOAc. EtOAc layer was dried over anhydrous Na2SO4 and evaporated to dryness under reduced pressure. The residue was purified by preparative silica gel TLC (CHCl3–MeOH, 30:1) to afford 3.3 mg of 5 as a brown solid (25%):
1H NMR (CDCl3) δ 4.13 (NH2, s), 4.22 (2H, s, −CH2CO), 6.63–6.64 (1H, m, H-4′), 6.76 (1H, d, J = 7.9 Hz, aromatic-H), 7.20 (1H, d, J = 7.9 Hz, aromatic-H), 7.28–7.29 (2H, m, H-3′ and aromatic-H), 7.69 (1H, s, H-5′), 7.75 (1H, dd, J = 8.7, 2.9 Hz, H-8), 7.94 (1H, d, J = 8.7 Hz, H-7), 8.50 (1H, d, J = 2.9 Hz, H-10), 9.08 (1H, s, NH).
9-Chloro-2-(2-furanyl)-5-[(4-aminophenylacetyl)amino][1,2,4]triazolo[1,5-c]quinazoline (6).
A mixture of PtO2 (5 mg) and 9-chloro-2-(2-furanyl)-5-[(4-nitrophenylacetyl)amino][1,2,4]triazolo[1,5-c]quinazoline (10 mg, 0.022 mmol), which was prepared using method A, in 3 mL of CH2Cl2/MeOH, was stirred under hydrogen atmosphere for 3 h at room temperature. The reaction mixture was filtered through a Celite bed, and the filtrate was concentrated and purified by preparative silica gel TLC (Hex:CHCl3:MeOH, 1:1:0.1) to afford 4.1 mg of 6 as a brown solid (45%): 1H NMR (CDCl3) δ 3.74 (NH2, s), 4.16 (2H, s, −CH2CO), 6.61–6.62 (1H, m, H-4′), 6.73 (2H, d, J = 7.8 Hz, aromatic-H), 7.21 (2H, d, J = 7.8 Hz, aromatic-H), 7.24 (1H, m, H-3′), 7.66 (1H, s, H-5′), 7.72 (1H, dd, J = 8.8, 2.9 Hz, H-8), 7.92 (1H, d, J = 8.8 Hz, H-7), 8.45 (1H, d, J = 2.9 Hz, H-10), 9.11 (1H, bs, NH).
9-Chloro-2-(2-furanyl)-5-[(2-iodophenylacetyl)amino][1,2,4]triazolo[1,5-c]quinazoline (7):
1H NMR (CDCl3) δ 4.56 (2H, s, −CH2CO), 6.64–6.65 (1H, m, H-4′), 7.03–7.09 (1H, m, aromatic-H), 7.29 (1H, m, H-3′), 7.41–7.43 (2H, m, aromatic-H), 7.69 (1H, s, H-5′), 7.75 (1H, dd, J = 8.8, 2.9 Hz, H-8), 7.92–7.96 (2H, m, aromatic-H and H-7), 8.51 (1H, d, J = 2.9 Hz, H-10), 9.19 (1H, bs, NH).
9-Chloro-2-(2-furanyl)-5-[(3-iodophenylacetyl)amino][1,2,4]triazolo[1,5-c]quinazoline (8):
1H NMR (CDCl3) δ 4.38 (2H, s, −CH2CO), 6.63–6.65 (1H, m, H-4′), 7.10–7.19 (1H, m, aromatic-H), 7.30–7.31 (1H, m, H-3′), 7.40 (1H, d, J = 7.8 Hz, aromatic-H), 7.64 (1H, d, J = 7.8 Hz, aromatic-H), 7.69 (1H, s, H-5′), 7.75 (1H, dd, J = 8.8, 1.9 Hz, H-8), 7.82 (1H, s, aromatic-H), 7.91 (1H, d, J = 8.8 Hz, H-7), 8.50 (1H, d, J = 1.9 Hz, H-10), 9.10 (1H, bs, NH).
9-Chloro-2-(2-furanyl)-5-[(4-iodophenylacetyl)amino][1,2,4]triazolo[1,5-c]quinazoline (9):
1H NMR (CDCl3) δ 4.37 (2H, s, −CH2CO), 6.64 (1H, s, H-4′), 7.17 (2H, d, J = 7.8 Hz, aromatic-H), 7.28–7.29 (1H, m, H-3′), 7.69–7.76 (4H, m, H-5′, H8 and aromatic-H), 7.90 (1H, d, J = 8.8 Hz, H7), 8.03 (1H, s, H-5′), 8.50 (1H, s, H-10), 9.08 (1H, s, NH).
9-Chloro-2-(2-furanyl)-5-[(3-chlorophenylacetyl)amino][1,2,4]triazolo[1,5-c]quinazoline (10):
1H NMR (CDCl3) δ 4.42 (2H, s, −CH2CO), 6.64–6.65 (1H, m, H-4′), 7.28–7.32 (4H, m, aromatic-H and H-3′), 7.45 (1H, s, aromatic-H), 7.68 (1H, s, H-5′), 7.76 (1H, dd, J = 8.8, 2.9 Hz, H-8), 7.91 (1H, d, J = 8.8 Hz, H-7), 8.50 (1H, d, J = 2.9 Hz, H-10), 9.10 (1H, bs, NH).
9-Chloro-2-(2-furanyl)-5-[[(2R)-phenylpropionyl]amino][1,2,4]triazolo[1,5-c]quinazoline (11):
1H NMR (CDCl3) δ 1.69 (3H, d, J = 6.8 Hz, −CH3), 4.54 (1H, q, J = 6.8 Hz, −CHCO), 6.60–6.62 (1H, m, H-4′), 7.20–7.21 (1H, m, H-3′), 7.29–7.50 (5H, m, aromatic-H), 7.65 (1H, s, H-5′), 7.72 (1H, dd, J = 8.8, 2.9 Hz, H-8), 7.94 (1H, d, J = 8.8 Hz, H-7), 8.44 (1H, d, J = 2.9 Hz, H-10), 9.03 (1H, bs, NH); [α]D20 = +1.04 (c = 0.23, CHCl3).
9-Chloro-2-(2-furanyl)-5-[[(2S)-phenylpropionyl]amino][1,2,4]triazolo[1,5-c]quinazoline (12):
1H NMR (CDCl3) identical to 11, [α]D20 = −2.26 (c = 0.38, CHCl3).
9-Chloro-2-(2-furanyl)-5-(diphenylacetylamino)[1,2,4]triazolo[1,5-c]quinazoline (13):
1H NMR (CDCl3) δ 6.09 (1H, s, −CHCO), 6.61–6.64 (1H, m, H-4′), 7.05–7.45 (11H, m, H-3′ and aromatic-H), 7.65 (1H, s, H-5′), 7.73 (1H, dd, J = 8.8, 2.9 Hz, H-8), 7.92 (1H, d, J = 8.8 Hz, H-7), 8.46 (1H, d, J = 2.9 Hz, H-10), 9.26 (1H, bs, NH).
9-Chloro-2-(2-furanyl)-5-[(2,2-diphenylpropionyl)amino][1,2,4]triazolo[1,5-c]quinazoline (14):
1H NMR (CDCl3) δ 2.18 (3H, s, −CH3), 6.57–6.59 (1H, m, H-4′), 7.06 (1H, d, J = 3.9 Hz, H-3′), 7.29–7.46 (10H, m, aromatic-H), 7.63 (1H, s, H-5′), 7.71 (1H, dd, J = 8.8, 1.9 Hz, H-8), 8.01 (1H, d, J = 8.8 Hz, H-7), 8.45 (1H, d, J = 1.9 Hz, H-10), 9.33 (1H, bs, NH).
9-Chloro-2-(2-furanyl)-5-[(3-phenylpropionyl)amino][1,2,4]triazolo[1,5-c]quinazoline (15):
1H NMR (CDCl3) δ 3.16 (2H, t, J = 7.8 Hz, −CH2), 3.43 (2H, t, J = 7.8 Hz, −CH2), 6.64 (1H, s, H-4′), 7.22–7.25 (1H, m, H-3′), 7.30–7.33 (5H, m, aromatic-H), 7.70 (1H, s, H-5′), 7.74 (1H, dd, J = 8.8, 1.9 Hz, H-8), 7.85 (1H, d, J = 8.8 Hz, H-7), 8.49 (1H, d, J = 1.9 Hz, H-10), 8.98 (1H, bs, NH).
9-Chloro-2-(2-furanyl)-5-(cinnamoylamino)[1,2,4]triazolo[1,5-c]quinazoline (16):
1H NMR (CDCl3) δ 6.66 (1H, m, H-4′), 7.35 (1H, d, J = 3.9 Hz, H-3′), 7.48–7.49 (3H, m, aromatic-H), 7.68–7.72 (3H, m, H-5′ and aromatic-H), 7.75–7.78 (2H, m, H-8 and olefinic-H), 7.97 (1H, d, J = 8.7 Hz, H-7), 8.02 (1H, d, J = 16.6 Hz, olefinic-H), 8.53 (1H, d, J = 1.9 Hz, H-10), 9.10 (1H, bs, NH).
5-[[N-(tert-Butoxycarbonyl)-d-alanyl]amino]-9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazoline (17) and the corresponding l-enantiomer (18):
1H NMR (CDCl3) δ 1.50 (9H, s, −Boc), 1.58 (3H, d, J = 5.8 Hz, −CH3), 4.95 (1H, m, H-α), 5.13 (1H, bs, NH), 6.64 (1H, m, H-4′), 7.31 (1H, d, J = 2.9 Hz, H-3′), 7.67 (1H, s, H-5′), 7.75 (2H, dd, J = 8.7, 1.9 Hz, H-8), 7.97 (1H, d, J = 8.7 Hz, H-7), 8.51 (1H, d, J = 1.9 Hz, H-10), 9.92 (1H, bs, NH).
5-[[3-[(tert-Butoxycarbonyl)amino]propionyl]amino]-9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazoline (19):
1H NMR (CDCl3) δ 1.44 (9H, s, −Boc), 3.32 (2H, t, J = 5.8 Hz, −CH2), 3.62 (2H, q, J = 5.8 Hz, −CH2), 5.28 (1H, bs, NH), 6.64 (1H, m, H-4′), 7.31 (1H, d, J = 3.8 Hz, H-3′), 7.68 (1H, s, H-5′), 7.73 (2H, dd, J = 8.8, 1.9 Hz, H-8), 7.87 (1H, d, J = 8.8 Hz, H-7), 8.47 (1H, d, J = 1.9 Hz, H-10), 9.06 (1H, bs, NH).
5-[[5-[(tert-Butoxycarbonyl)amino]pentanoyl]amino]-9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazoline (21):
1H NMR (CDCl3) δ 1.44 (9H, s, −Boc), 1.63–1.73 (2H, m, −CH2), 1.81–1.92 (2H, m, −CH2), 3.10 (2H, t, J = 6.8 Hz, −CH2), 3.22 (2H, q, J = 6.8 Hz, −CH2), 4.67 (1H, bs, NH), 6.63 (1H, m, H-4′), 7.30 (1H, d, J = 3.9 Hz, H-3′), 7.68 (1H, s, H-5′), 7.72 (2H, dd, J = 8.8, 1.9 Hz, H-8), 7.87 (1H, d, J = 8.8 Hz, H-7), 8.46 (1H, d, J = 1.9 Hz, H-10), 9.01 (1H, bs, NH).
5-[[6-[(tert-Butoxycarbonyl)amino]hexanoyl]amino]-9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazoline (22):
1H NMR (CDCl3) δ 1.45 (9H, s, −Boc), 1.50–1.61 (4H, m, 2 × −CH2), 1.80–1.90 (2H, m, −CH2), 3.05 (2H, t, J = 6.8 Hz, −CH2), 3.14–3.18 (2H, m, −CH2), 4.66 (1H, bs, NH), 6.62 (1H, m, H-4′), 7.29 (1H, m, H-3′), 7.67 (1H, s, H-5′), 7.70 (2H, dd, J = 8.8, 1.9 Hz, H-8), 7.84 (1H, d, J = 8.8 Hz, H-7), 8.42 (1H, d, J = 1.9 Hz, H-10), 9.01 (1H, bs, NH).
5-[[7-[(tert-Butoxycarbonyl)amino]heptanoyl]amino]-9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazoline (23):
1H NMR (CDCl3) δ 1.45 (9H, s, −Boc), 1.50–1.61 (6H, m, 3 × −CH2), 1.86 (2H, m, J = 6.85 Hz, −CH2), 3.03 (2H, t, J = 7.81 × Hz, −CH2), 3.13 (2H, m, −CH2), 4.59 (1H, bs, NH), 6.63 (1H, m, J = 1.94 Hz, H-4′), 7.30 (1H, s, H-3′), 7.67 (1H, s, H-5′), 7.72 (1H, m, H-8), 7.86 (1H, d, J = 8.79 Hz, H-7), 8.45 (1H, d, J = 2.94 Hz, H-10), 9.01 (1H, s, NH).
5-[(d-Alanyl)amino]-9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazoline (24):
1H NMR (DMSO-d6) δ 1.14 (3H, m, −CH3), 4.25 (1H, bs, H-R), 5.26 (2H, bs, NH2), 6.73 (1H, m, H-4′), 7.12 (1H, m, H-3′), 7.64 (2H, m, H-8 and H-7), 7.93 (1H, m, H-5′), 8.17 (1H, m, H-10), 11.63 (1H, bs, NH); [α]D20 = +6.00 (c = 0.1, CHCl3/CH3OH, 10:1). HPLC retention time: 4.11 min (>95% purity) using solvent system A, 3.91 min (>95% purity) using solvent system B.
5-[(l-Alanyl)amino]-9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazoline (25):
[α]D20 = −7.33 (c = 0.09, CHCl3/CH3OH, 10:1). HPLC retention time: 4.09 min (>95% purity) using solvent system A, 4.13 min (>95% purity) using solvent system B.
5-[(3-Aminopropionyl)amino]-9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazoline (26):
1H NMR (DMSO-d6) δ 3.13–3.15 (4H, m, 2 × −CH2), 6.80–6.81 (1H, m, H-4′), 7.36 (1H, d, J = 2.9 Hz, H-3′), 7.83 (2H, bs, NH2), 7.90 (1H, d, J = 8.8 Hz, H-7), 7.95 (1H, dd, J = 8.8, 1.9 Hz, H-8), 8.03 (1H, s, H-5′), 8.42 (1H, d, J = 1.9 Hz, H-10), 11.10 (1H, s, NH).
5-[(5-Aminopentanoyl)amino]-9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazoline (28):
1H NMR (DMSO-d6) δ 1.68 (4H, bs, 2 × −CH2), 2.73–2.76 (2H, m, −CH2), 2.85 (2H, bs, −CH2), 6.78–6.79 (1H, m, H-4′), 7.35 (1H, d, J = 2.9 Hz, H-3′), 7.70 (2H, bs, NH2), 7.92 (2H, s, H-8 and H-7), 8.02 (1H, s, H-5′), 8.40 (1H, s, H-10), 11.07 (1H, s, NH). HPLC retention time: 6.3 min (>95% purity) using solvent system A, 8.7 min (>95% purity) using solvent system B.
5-[(6-Aminohexanoyl)amino]-9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazoline (29):
1H NMR (DMSO-d6) δ 1.42–1.70 (6H, m, 3 × −CH2), 2.72 (2H, t, J = 6.8 Hz, −CH2), 2.81 (2H, t, J = 6.8 Hz, −CH2), 6.78–6.79 (1H, m, H-4′), 7.34 (1H, d, J = 3.9, H-3′), 7.70 (2H, bs, NH2), 7.92 (2H, d, J = 1.9 Hz, H-8 and H-7), 8.01 (1H, s, H-5′), 8.40 (1H, s, H-10), 11.04 (1H, s, NH).
5-[(7-Aminoheptanoyl)amino]-9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazoline (30):
1H NMR (DMSO-d6) δ 1.24 (2H, m, −CH2), 1.39 (2H, m, −CH2), 1.53 (2H, m, −CH2), 1.66 (2H, m, −CH2), 2.71 (2H, m, −CH2), 2.78 (2H, m, −CH2), 6.79 (1H, s, H-4′), 7.35 (1H, s, H-3′), 7.64 (2H, bs, NH2), 7.93 (2H, s, H-7 and H-8), 8.02 (1H, s, H-5′), 8.40 (1H, s, H-10), 11.04 (1H, s, NH). HPLC retention time: 4.6 min (>95% purity) using solvent system A, 8.5 min (>95% purity) using solvent system B.
5-[[5-(Benzyloxycarbonyl)pentanoyl]amino]-9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazoline (31).
The title compound was synthesized using method B with monobenzyl adipate, which was prepared according to the literature:39 1H NMR (CDCl3) δ 1.82–1.88 (4H, m, 3 −CH2), 2.48 (2H, t, J = 6.8 Hz, −CH2), 3.09 (2H, t, J = 6.8 × Hz, −CH2), 5.13 (2H, s, −CH2), 6.62–6.63 (1H, m, H-4′), 7.28–7.36 (6H, m, H-3′ and aromatic-H), 7.67–7.72 (2H, m, H-8 and H-5′), 7.84 (1H, d, J = 8.8 Hz, H-7), 8.44 (1H, d, J = 1.9 Hz, H-10), 8.97 (1H, s, NH).
5-[[3-(Methoxycarbonyl)propionyl]amino]-9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazoline (32):
1H NMR (CDCl3) δ 2.86 (2H, t, J = 6.8 Hz, −CH2), 3.44 (2H, t, J = 6.8 Hz, −CH2), 3.75 (3H, s, −OCH3), 6.62–6.64 (1H, m, H-4′), 7.28–7.31 (1H, m, H-3′), 7.68 (1H, s, H-5′), 7.72 (1H, d, J = 8.8, 1.9 Hz, H-8), 7.86 (1H, d, J = 8.8 Hz, H-7), 8.46 (1H, d, J = 1.9 Hz, H-10), 9.12 (1H, s, NH).
5-[[7-(Methoxycarbonyl)heptanoyl]amino]-9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazoline (33):
1H NMR (CDCl3) δ 1.47 (2H, m, −CH2), 1.68 (2H, t, J = 6.83 Hz, −CH2), 1.85 (2H, t, J = 6.83 Hz, −CH2), 2.34 (2H, t, J = 7.81 Hz, −CH2), 3.05 (2H, t, J = 7.81 Hz, −CH2), 3.67 (3H, s, −OCH3), 6.65 (1H, s, H-4′), 7.33 (1H, d, J = 3.91 Hz, H-3′), 7.69 (1H, s, H-5′), 7.76 (1H, d, J = 6.84 Hz, H-8), 7.89 (1H, d, J = 9.96 Hz, H-7), 8.50 (1H, d, J = 1.95 Hz, H-10), 8.99 (1H, s, NH).
5-[(4-Carboxylbutanoyl)amino]-9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazoline (34).
A solution of 30 mg (0.105 mmol) of 1a and 60 mg (0.525 mmol) of glutaric anhydride in 2 mL of acetone was refluxed at 85 °C for 2 days. After cooling, the mixture was purified by preparative silica gel TLC (CHCl3:MeOH, 10:1) and crystallization from CH2Cl2 to afford 23 mg of 34 as a white solid: 1H NMR (DMSO-d6) δ 1.87 (2H, t, J = 6.8 Hz, −CH2), 2.35 (2H, t, J = 6.8 Hz, −CH2), 2.73 (2H, t, J = 6.8 Hz, −CH2), 6.77 (1H, s, H-4′), 7.33 (1H, s, H-3′), 7.89 (2H, s, H-7 and H-8), 8.00 (1H, s, H-5′), 8.37 (1H, s, H-10).
Pharmacology. Radioligand Binding Studies.
Binding of [3H]-(R)-N6-(phenylisopropyl)adenosine ([3H]R-PIA; Amersham, Chicago, IL) to A1 receptors from rat cerebral cortical membranes and of [3H]CGS 21680 (DuPont NEN, Boston, MA) to A2A receptors from rat striatal membranes was performed as described previously.4,5 Adenosine deaminase (3 units/mL) was present during the preparation of the brain membranes, in a preincubation of 30 min at 30 °C, and during the incubation with the radioligands.
Binding of [125I]-N6-(4-amino-3-iodobenzyl)-5′-(N-methylcarbamoyl)adenosine ([125I]AB-MECA; Amersham) in membranes prepared from HEK-293 cells stably expressing the human A3 receptor (Receptor Biology, Inc., Baltimore, MD) was as described.24 The assay medium consisted of a buffer containing 50 mM Tris, 10 mM Mg2+, and 1 mM EDTA, at pH 8.0. The glass incubation tubes contained 100 μL of the membrane suspension (0.3 mg of protein/mL, stored at −80 °C in the same buffer), 50 μL of [125I]AB-MECA (final concentration 0.3 nM), and 50 μL of a solution of the proposed antagonist. Nonspecific binding was determined in the presence of 200 μM NECA.
All nonradioactive compounds were initially dissolved in DMSO and diluted with buffer to the final concentration, where the amount of DMSO never exceeded 2%.
Incubations were terminated by rapid filtration over Whatman GF/B filters, using a Brandell cell harvester (Brandell, Gaithersburg, MD). The tubes were rinsed three times with 3 mL of buffer each.
At least five different concentrations of competitor, spanning 3 orders of magnitude adjusted appropriately for the IC50 of each compound, were used. IC50 values, calculated with the nonlinear regression method implemented in the InPlot program (Graph-PAD, San Diego, CA), were converted to apparent Ki values using the Cheng–Prusoff equation40 and Kd values of 1.0 nM ([3H]R-PIA), 14 nM ([3H]CGS 21680), and 0.59 nM ([125I]AB-MECA at human A3 receptors). Hill coefficients of the tested compounds were in the range of 0.8–1.1.
Cyclic AMP Assay of Antagonist Potency at the Human A2B Receptor:
1. Cell Culture.
CHO-A2B cells were grown under 5% CO2/95% O2 humidified atmosphere at a temperature of 37 °C in DMEM supplemented with Hams F12 nutrient mixture (1/1), 10% newborn calf serum, 2 mM glutamine, and containing 50 IU/mL penicillin, 50 μg/mL streptomycin, and 0.2 mg/mL Geneticin (G418, Boehringer Mannheim). Cells were cultured in 10-cm diameter round plates and subcultured when grown confluent (approximately after 72 h). PBS/EDTA containing 0.25% trypsin was used for detaching the cells from the plates. Experimental cultures were grown overnight as a monolayer in 24-well tissue culture plates (400 μL/well; 0.8 × 106 cells/well). CHO-A2B cells were kindly provided by Dr. K.-N. Klotz (University of Würzburg, Germany).
2. Cyclic AMP Generation.
Cyclic AMP generation was performed in DMEM/HEPES buffer (DMEM containing 50 mM HEPES, pH 7.4, 37 °C). To each well, washed twice with DMEM/HEPES buffer, were added 100 μL of adenosine deaminase (final concentration 10 IU/mL) and 100 μL of rolipram/cilostamide (final concentration 10 μM/10 μM), followed by 50 μL of test compound (appropriate concentration) or buffer. After incubation for 40 min at 37 °C, 100 μL of NECA was added (final concentration 50 μM for compound screening and IC50 determination, appropriate concentrations for recording dose–response curves for pA2 calculations). After 15 min, incubation at 37 °C was terminated by removing the medium and adding 200 μL of 0.1 M HCl. Wells were stored at −20 °C until assay.
3. Cyclic AMP Determination.
The amounts of cyclic AMP were determined after a protocol with cAMP binding protein (PKA)41 with the following minor modifications. As a buffer was used 150 mM K2HPO4/10 mM EDTA/0.2% BSA FV at pH 7.5. Samples (20 μL) were incubated for 90 min at 0 °C. Incubates were filtered over GF/C glass microfiber filters in a Brandel M-24 cell harvester. The filters were additionally rinsed with 4 × 2 mL of 150 mM K2HPO4/10 mM EDTA (pH 7.5, 4 °C). Punched filters were counted in Packard emulsifier safe scintillation fluid after 2 h of extraction.
Abbreviations:
- AcOH
acetic acid
- Boc
tert-butoxycarbonyl
- CGS 21680
2-[[4-(2-carboxyethyl)phenyl]ethylamino]-5′-(N-ethylcarbamoyl)adenosine
- CGS 15943
9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazolin-5-amine
- CHO cells
Chinese hamster ovary cells
- DMAP
4-(dimethylamino)pyridine
- EDAC
3-ethyl-1-(3-(dimethylamino)propyl)carbodiimide
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- EDTA
ethylenediaminetetraacetate
- EI
electric ionization
- CI
chemical ionization
- FAB
fast atom bombardment
- HEK cells
human embryonic kidney cells
- HMPA
hexamethylphosphotriamide
- [125I]AB-MECA
[125I]-N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide
- K i
equilibrium inhibition constant
- MS
mass spectrum
- NECA
5′-(N-ethylcarbamoyl)adenosine
- R-PIA
(R)-N6-(phenylisopropyl)adenosine
- SAR
structure–activity relationship
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TLC
thin-layer chromatography
- Tris
tris(hydroxymethyl)aminomethane
- TEAA
triethylammonium acetate
References
- (1).Müller CE A1-adenosine receptor antagonists. Exp. Opin. Ther. Patents 1997, 7, 419–440. [Google Scholar]
- (2).Baraldi PG; Cacciari B; Spalluto G; Borioni A; Viziano M; Dionisotti S; Ongini E Current developments of A2A adenosine receptor antagonists. Curr. Med. Chem 1995, 2, 707–722. [Google Scholar]
- (3).Müller CE; Stein B Adenosine receptor antagonists – structures and potential therapeutic applications. Curr. Pharm. Des 1996, 2, 501–530. [Google Scholar]
- (4).Schwabe U; Trost T Characterization of adenosine receptors in rat brain by (−) [3H]N6-phenylisopropyladenosine. Naunyn-Schmiedeberg’s Arch. Pharmacol 1980, 313, 179–187. [DOI] [PubMed] [Google Scholar]
- (5).Jarvis MF; Schutz R; Hutchison AJ; Do E; Sills MA; Williams M [3H]CGS 21680, an A2 selective adenosine receptor agonist directly labels A2 receptors in rat brain tissue. J. Pharmacol. Exp. Ther 1989, 251, 888–893. [PubMed] [Google Scholar]
- (6).Fredholm BB Adenosine and neuroprotection. Intl. Rev. Neurobiol 1997, 40, 259–280. [PubMed] [Google Scholar]
- (7).Ongini E; Dionisotti S; Morelli M; Ferre S; Svenningsson P; Fuxe K; Fredholm BB Neuropharmacology of the adenosine A2A receptors. Drug Dev. Res 1996, 39, 450–460. [Google Scholar]
- (8).Latini S; Pazzagli M; Pepeu G; Pedata F A2 adenosine receptors - their presence and neuromodulatory role in the central-nervous-system. Gen. Pharmacol 1996, 27, 925–933. [DOI] [PubMed] [Google Scholar]
- (9).Mehra A; Cohen G; Johnson JV; Elkayam U Effect of adenosine on the renal circulation in patients with chronic heart-failure. J. Am. Coll. Cardiol 1997, 29, 7834–7834. [Google Scholar]
- (10).Rongen GA; Floras JS; Lenders J; Thien T; Smits P Cardiovascular pharmacology of purines. Clin. Sci 1997, 92, 13–24. [DOI] [PubMed] [Google Scholar]
- (11).Jacobson KA; Suzuki F Recent developments in selective agonists and antagonists acting at purine and pyrimidine receptors. Drug Dev. Res 1996, 39, 289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Jacobson KA; von Lubitz DKJE; Daly JW; Fredholm BB Adenosine receptor ligands – differences with acute versus chronic treatment. Trends Pharmacol. Sci 1996, 17, 108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tracey WR; Magee W; Masamune H; Kennedy SP; Knight DR; Buchholz RA; Hill RJ Selective adenosine A3 receptor stimulation reduces ischemic myocardial injury in the rabbit heart. Cardiovasc. Res 1997, 33, 410–415. [DOI] [PubMed] [Google Scholar]
- (14).Kohno Y; Sei Y; Koshiba M; Kim HO; Jacobson KA Induction of apoptosis in HL-60 human promyelocytic leukemia-cells by adenosine A3 receptor agonists. Biochem. Biophys. Res. Commun 1996, 221, 849–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Karton Y; Jiang JL; Ji XD; Melman N; Olah ME; Stiles GL; Jacobson KA Synthesis and biological-activities of flavonoid derivatives as as adenosine receptor antagonists. J. Med. Chem 1996, 39, 2293–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Jacobson MA; Chakravarty PK; Johnson RG; Norton R Novel nonxanthine selective A3 adenosine receptor antagonists. Drug. Dev. Res 1996, 37, 131–131. [Google Scholar]
- (17).van Rhee AM; Jiang JL; Melman N; Olah ME; Stiles GL; Jacobson KA Interaction of 1,4-dihydropyridine and pyridine-derivatives with adenosine receptors – selectivity for A3 receptors. J. Med. Chem 1996, 39, 2980–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Jiang JL; Van Rhee AM; Melman N; Ji XD; Jacobson KA 6-phenyl-1,4-dihydropyridine derivatives as potent and selective A3 adenosine receptor antagonists. J. Med. Chem 1996, 39, 4667–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Jiang JL; Van Rhee AM; Chang L; Patchornik A; Ji XD; Evans P; Melman N; Jacobson KA Structure–activity-relationships of 4-(phenylethynyl)-6-phenyl-1,4-dihydropyridines as highly selective A3 adenosine receptor antagonists. J. Med. Chem 1997, 40, 2596–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kim YC; Ji XD; Jacobson KA Derivatives of the triazoloquinazoline adenosine antagonist (CGS15943) are selective for the human A3 receptor subtype. J. Med. Chem 1996, 39, 4142–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Jacobson MA; Johnson RG; Luneau CJ; Salvatore CA Cloning and chromosomal localization of the human A2B adenosine receptor gene (ADORA2B) and its pseudogene. Genomics 1995, 27, 374–376. [DOI] [PubMed] [Google Scholar]
- (22).Gallo-Rodriguez C; Ji XD; Melman N; Siegman BD; Sanders LH; Orlina J; Fischer B; Pu QL; Olah ME; van Galen PJM; Stiles GL; Jacobson KA Structure–activity-relationships of N-6-benzyladenosine-5′-uronamides as A3-selective adenosine agonists. J. Med. Chem 1994, 37, 636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Salvatore CA; Jacobson MA; Taylor HE; Linden J; Johnson RG Molecular cloning and characterization of the human A3 adenosine receptor. Proc. Natl. Acad. Sci. U.S.A 1993, 90, 10365–10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Olah ME; Gallo-Rodriguez C; Jacobson KA; Stiles GL [125I]AB-MECA, a high affinity radioligand for the rat A3 adenosine receptor. Mol. Pharmacol 1994, 45, 978–982. [PMC free article] [PubMed] [Google Scholar]
- (25).Kim J; Wess J; van Rhee AM; Schoneberg T; Jacobson KA Site-directed mutagenesis identifies residues involved in ligand recognition in the human A2a adenosine receptor. J. Biol. Chem 1995, 270, 13987–13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Moro S; Guo D; Camaioni E; Boyer JL; Harden K; Jacobson KA Human P2Y1 receptor: molecular modeling and site-directed mutagenesis as a tools to identify agonist and antagonist recognition sites. J. Med. Chem 1998, 41, 1456–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lin Z; Shenker A; Pearlstein R A model of the lutropin/choriogonadotropin receptor: insights into the structural and functional effects of constitutively activating mutations. Protein Eng. 1997, 10, 501–510. [DOI] [PubMed] [Google Scholar]
- (28).Chiota C; Levitt M; Richardson D Helix to helix packing in proteins. J. Mol. Biol 1981, 145, 215–250. [DOI] [PubMed] [Google Scholar]
- (29).Schertler GF; Villa C; Henderson R Projection structure of rhodopsin. Nature 1993, 362, 770–772. [DOI] [PubMed] [Google Scholar]
- (30).Urger VM; Hargrave PA; Baldwin JM; Schertler GFX Arrangement of rhodopsin transmembrane α-helices. Nature 1997, 389, 203–206. [DOI] [PubMed] [Google Scholar]
- (31).Gouldson PR; Snell CR; Reynolds CA A new approach to docking in the β2-adrenergic receptor that exploits the domain structure of G-protein-coupled receptors. J. Med. Chem 1997, 40, 3871–3886. [DOI] [PubMed] [Google Scholar]
- (32).Olah ME; Ren HZ; Ostrowski J; Jacobson KA; Stiles GL Cloning, expression, and characterization of the unique bovine A1 adenosine receptor. Studies on the ligand binding site by site-directed mutagenesis. J. Biol. Chem 1992, 267, 10764–10770. [PMC free article] [PubMed] [Google Scholar]
- (33).Kim J; Jiang Q; Glashofer M; Yehle S; Wess J; Jacobson KA Glutamate residues in the second extracellular loop of the human A2a adenosine receptor are required for ligand recognition. Mol. Pharmacol 1996, 49, 683–691. [PMC free article] [PubMed] [Google Scholar]
- (34).The program SYBYL 6.3 is available from TRIPOS Associates, St. Louis, MO; 1993. [Google Scholar]
- (35).Mohamadi F; Richards NGJ; Guida WC; Liskamp R; Lipton M; Caufield C; Chang G; Hendrickson T; Still WC MacroModel-An Integrated Software System for Modeling Organic and Bioorganic Molecules using Molecular Mechanics. J. Comput. Chem 1990, 11, 440–450. [Google Scholar]
- (36).van Rhee AM; Fischer B; van Galen PJM; Jacobson KA Modeling the P2Y purinoceptor using rhodopsin as template. Drug Des. Discov 1995, 13, 133–154. [PMC free article] [PubMed] [Google Scholar]
- (37).Kyte J; Doolittle RF A simple method for displaying the hydrophobic character of a protein. J. Mol. Biol 1982, 157, 105–132. [DOI] [PubMed] [Google Scholar]
- (38).Weiner SJ; Kollman PA; Nguyen DT; Case DA An all-atom force field for simulation of protein and nucleic acids. J. Comput. Chem 1986, 7, 230–252. [DOI] [PubMed] [Google Scholar]
- (39).English AR; Girard D; Jasys VJ; Martingano RJ; Kellogg MS Orally Effective Acid Prodrugs of the β-Lactamase Inhibitor Sulbactam. J. Med. Chem 1990, 33, 344–347. [DOI] [PubMed] [Google Scholar]
- (40).Cheng YC; Prusoff WH Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50% inhibition (IC50) of an enzyme reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- (41).van der Wenden EM; Hartog-Witte HR; Roelen HCPF; von Frijtag Drabbe Künzel JK; Pirovano IM; Mathôt RAA; Danhof M; van Aerschot A; Lidaks MJ; IJzerman AP; Soudijn W. 8-Substituted adenosine and theophylline-7-riboside analogues as potential partial agonists for the adenosine A1 receptor. Eur. J. Pharmacol.-Mol. Pharmacol. Sect 1995, 290, 189–199. [DOI] [PubMed] [Google Scholar]
- (42).Alexander SPH; Cooper J; Shine J; Hill SJ Characterization of the human brain putative A2B adenosine receptor expressed in Chinese hamster ovary (CHO.A2B.4) cells. Br. J. Pharmacol 1996, 119, 1286–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Feoktistov I; Biaggioni I Adenosine A2B receptors. Pharmacol. Rev 1997, 49, 381–402. [PubMed] [Google Scholar]