

Abstract
Upon infection of murine trigeminal ganglia with herpes simplex virus type 1 (HSV-1), an immune response is initiated resulting in significant infiltration of CD8+ T cells. Previous investigators have observed a lack of apoptosis in HSV-1 trigeminal ganglia even in the presence of cytotoxic immune cells. To determine the role of the latency-associated transcript (LAT) in inhibiting apoptosis, we examined mice during acute and latent infection with HSV-1 (strain 17 or a LAT-negative deletion mutant strain 17 N/H) by terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) and fluorescence-activated cell sorting (FACS). FACS analysis revealed CD8+ T cells in the trigeminal ganglia by day 7, with more being present in 17- than 17 N/H-infected trigeminal ganglia (6.22% versus 3.5%) and a decrease in number through day 30 (2.7% to 1.2%). To detect apoptotic CD8+ T cells, sections were assayed by TUNEL and stained for CD8+ T cells. By day 7, ∼10% of CD8+ T cells in both 17- and 17 N/H-infected trigeminal ganglia had undergone apoptosis. By day 30, 58% and 74% of all T cells had undergone apoptosis in 17- and 17 N/H-infected trigeminal ganglia, respectively. Furthermore, no HSV strain 17-infected trigeminal ganglion neurons were apoptotic, but 0.087% of 17ΔSty and 0.98% of 17 N/H-infected neurons were apoptotic. We conclude that the antiapoptotic effect of LAT appears to require the LAT promoter, with most of the antiapoptotic effect mapping within the StyI (+447) to the HpaI (+1667) region and a minor contribution from the upstream StyI (+76) to StyI (+447) region.
Herpes simplex virus type 1 (HSV-1) infects epithelial cells and undergoes an active lytic infection. New viral progeny is released and infects neighboring sensory neurons. Upon infection of these sensory neurons, HSV-1 establishes a latent infection, which lasts for the life of the neuron, while maintaining the ability to reactivate. HSV-1 latency is characterized by a lack of viral gene expression except for latency-associated transcripts (LAT) (for a review, see references 10 and 38). The LAT is transcribed as an 8.3-kb primary transcript which is processed into a stable 2-kb LAT intron (8, 43) and a 6.3-kb poorly defined mRNA. The exact function of the LAT remains to be definitively established, but experiments with LAT-negative mutants seems to suggest a role for LAT in establishing latency, reactivation from latency, and antiapoptosis (1, 15, 16, 29, 30, 40).
Upon infection of neurons, HSV-1 is capable of both inducing and inhibiting apoptosis (3, 12). Several HSV-1 genes have been implicated in inhibiting apoptosis. During acute infection in Jurkat cells, apoptosis appears to be inhibited by the immediate-early genes Us3, ICP27, and Us5 (2, 3, 18, 22). Viral glycoproteins (US6 and US5) have also been shown to block apoptosis during a productive infection in SK-N-SH cells (45).
During the primary infection, immunity to neuronal infection is mostly innate in nature with the involvement of complement (6), natural killer cells (24), and interferon (44). Following viral replication in the mucosa, virus enters sensory nerve terminals and HSV virions are transported to the nerve ganglia by retrograde transport (4). In mice, infection of the trigeminal ganglia results in virus replication and neuronal cell death in some nerves (27). Because there is inflammation associated with HSV replication in mouse trigeminal ganglia, a T-cell-mediated response is mounted against HSV (24). The appearance of CD8+ T cells has been well documented at the site of infection on the mucosa, around infected sensory neurons, and in the central nervous system (7, 37). The importance of cytotoxic T cells in controlling HSV infections and pathology has been clearly demonstrated in CD8+ T-cell-depleted mice, with these immune-deficient mice having significant neuronal death in the trigeminal ganglia when infected with HSV-1 (25, 35).
The possible role of CD8+ T cells in HSV latency is less clear. Following infection by ocular scarification, HSV establishes a latent infection in the trigeminal ganglia of mice. By 3 days postinfection, CD8+ T cells begin infiltrating the trigeminal ganglia, peaking at 14 days and persisting there for months (20, 24, 39). However, even with the near-constant presence of CD8+ T cells, neurons in infected trigeminal ganglia evade eradication by cytotoxic T cells. HSV-1 has an array of defenses against the cytotoxic mechanisms of CD8+ T cells. Upon infection, ICP47 down regulates expression of antigen associated with major histocompatibility complex class I, thereby allowing infected cells to evade cell-mediated immunity (11). Even though ICP47 is much less effective in murine than human tissue (41), infection of mice with an ICP47-null mutant exhibited reduced neurovirulence in mice (13). HSV can also evade the immune system by infecting and eliminating antiviral T cells (31). Infected tissue can also inactivate CD8+ T cells through the expression of US3 (36).
Once CD8+ T cells successfully engage target cells, they are capable of inducing apoptosis by Fas ligand (14, 19, 33) and granzymes A and B (28). Even with the constant presence of antiviral CD8+ T cells, trigeminal ganglion neurons remain resistant to apoptosis during HSV latency (17, 21, 40). One viral factor that has been implicated in this protection from apoptosis is the LAT. The antiapoptotic qualities of LAT have been documented in vitro as well as in vivo (1, 16, 30). In this study, we investigate the contribution of LAT to protecting neurons from apoptosis using HSV-1 mutants containing a large deletion including the LAT promoter, exon 1, and the 5′ half of the 2-kb intron (17 N/H) (5) and a mutant containing a deletion in LAT exon 1 (17ΔSty) (26). We demonstrate that part of the HSV-1 LAT region protects trigeminal ganglion neurons from apoptosis during viral latency.
MATERIALS AND METHODS
Infection of mice and isolation of trigeminal ganglia.
Six- to eight-week-old female BALB/c mice (Jackson Laboratories) were anesthetized with an intraperitoneal injection of ketamine (87 mg/kg of body weight)-xylazine (13 mg/kg of body weight) and inoculated with 2.5 × 104 PFU per eye of HSV-1 17+, 17 N/H (5), and 17ΔSty (26) (Fig. 1). At 7, 15, and 30 days postinfection, mock-, 17+-, and 17 N/H-infected mice were sacrificed by cervical dislocation and trigeminal ganglia were removed. Mice infected with 17ΔSty had their trigeminal ganglia extracted and processed only at day 30.
FIG. 1.

Map of HSV-1 genome and LAT deletion mutants. (A) Linear map of the HSV-1 genome with its unique long (UL) and unique short (US) regions flanked by inverted repeat (IR) elements. (B) LAT region of the HSV-1 genome. The LAT region is enlarged to show the latency-active promoter (LAP1) and the 2-kb LAT intron. (C) LAT deletion region in 17 N/H. (D) Exon 1 deletion region in 17ΔSty. (E) LAT deletion region in 17Δ A/H.
Trigeminal ganglia (TG) were processed for sectioning by fixing them in 4% paraformaldehyde for 4 h. TG were then incubated in 100% ethanol overnight followed by 70% ethanol overnight. TG were then embedded in paraffin and sectioned.
TG were processed for fluorescence-activated cell sorter (FACS) analysis by placing them in phosphate-buffered saline (PBS) containing collagenase type XI (1 mg/ml; Sigma, St. Louis, MO) and incubating them at 37°C for 90 min with trituration every 15 min. Samples of cells were removed every 15 min and checked by microscope to gauge cellular dissociation. Cells were washed twice with PBS and used for immunohistochemical staining or terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) analysis.
In situ hybridization.
A LAT probe was made using a digoxigenin-nick translation system (Roche, Manheim, Germany). Briefly, a vector containing a BstEII-BstEII subfragment of the BamHI B fragment (119195 to 120092) was linearized with HindIII and labeled with digoxigenin-11-dUTP using the above kit. The labeled probe was purified using a Sephadex G-50 spin column. Trigeminal ganglion tissue sections were fixed and mounted on slides, and in situ hybridization was performed as described previously (23, 42).
Immunohistochemical staining.
Tissue sections on slides were deparaffinized and rehydrated. The tissue was incubated in 5% bovine serum albumin in PBS at room temperature for 1 h followed by incubation with an anti-CD8 primary monoclonal antibody (BD Pharmingen) at a dilution of 1:5,000 in PBS at 4°C overnight. The slides were then incubated with a fluorescein-conjugated anti-rat secondary antibody (Sigma) at a dilution of 1:2,000 at room temperature for 2 h. Slides were mounted with 50% glycerol and viewed with a Leica DMRE fluorescent microscope connected to a Hamamatsu digital cameral. Microscope images are captured and processed using Improvision Open Lab version 3.1.6 software.
TUNEL analysis.
TUNEL analysis on tissue sections was performed by first permeabilizing the tissue with permeabilization solution (0.1% Triton X-100, 0.1% sodium citrate) for 2 min on ice. Following permeabilization, the slides were washed with PBS and the TUNEL reaction was done using the fluorescein in situ cell death detection kit (Roche, Germany). Slides were viewed and analyzed using the microscope and image analysis software detailed above.
FACS analysis.
Dissociated cells were washed twice in PBS. Cells were then resuspended in 500 μl of ice-cold PBS and fixed with 5 ml of cold ethanol, which was added drop by drop while vortexing the mixture. Fixed cells were counted, and 2.5 × 105 cells were added to a 1.5-ml Eppendorf tube in which they were washed twice with PBS. TUNEL analysis was done with the in situ cell death detection kit (Roche, Germany). DNA in apoptotic nuclei incorporated fluorescein-labeled nucleotides and was analyzed by FACS (excitation, 488 nm; emission, 560 nm) at the Wistar Institute Flow Cytometry Facility.
Trigeminal ganglion neurons were disassociated and fixed as described above. Cells were counted and stained for CD8 using a phycoerythrin (PE)-conjugated anti-mouse CD8a monoclonal antibody (BD Pharmingen) at a dilution of 1:250 in FACS buffer (1× PBS, 1% bovine serum albumin). Cells were incubated on ice for 1 h and washed three times in FACS buffer.
RESULTS
C57BL/6 mice were infected with the HSV-1 LAT-null mutant 17 N/H and its parent virus, 17+. The deletion spans from the NotI site 5′ of the LAT promoter (118443) to the HpaI site halfway into the 2-kb LAT intron (120463) (Fig. 1c). A 400-bp piece of λ DNA was inserted into this deleted site (5). Because essential promoter elements were deleted, no LATs are made. Apoptosis experiments were also performed on 17ΔSty-infected trigeminal ganglia. 17ΔSty contains a 370-bp (11880 to 119250) deletion in exon 1 (Fig. 1d) (26). This deletion removes just over half of the exon 1 sequence.
Following corneal scarification and infection with HSV-1 (2 × 104 PFU/eye), mice were sacrificed at 7, 15, and 30 days postinfection. Initial experiments were focused on the effect of LAT on apoptosis of neurons during latency. Since 30 days postinfection is widely accepted as a point in time by which latency has been established, the trigeminal ganglia of mice from this time point were used for our apoptosis studies. In situ hybridization for the LAT was performed using a riboprobe directed against the HSV-1 LAT intron (see Materials and Methods). In Fig. 2a, there are 2 LAT-positive neurons in a section of 17+-infected trigeminal ganglia. The TUNEL assay done on the same section shows a number of apoptotic nuclei (Fig. 2b). Careful visual inspection shows that none of the apoptotic nuclei colocalized with trigeminal ganglion neurons. Because the probe that was used is located in the deletion found in 17 N/H, no LAT signal is seen in 17 N/H sections (Fig. 2c). The TUNEL assay on the same section shows a large number of apoptotic nuclei as seen with 17+-infected trigeminal ganglia. A more careful and thorough survey of 17 N/H sections reveals apoptotic neurons. Figure 3a shows phase pictures of a 17 N/H-infected section with a well-defined neuronal cell body (see also Fig. 9a). The TUNEL assay done on the same section clearly shows an apoptotic nucleus in that neuron (Fig. 3b; see Fig. 9c). Mock-infected trigeminal ganglia had no apoptotic cells when subjected to TUNEL assay (data not shown).
FIG. 2.

Detection of LAT expression and apoptosis in 17+- and 17 N/H-infected trigeminal ganglia. BALB/c mice were infected with 17+ (A, B) and 17 N/H (C, D). At 30 days postinfection, mice were sacrificed, trigeminal ganglia were extracted and fixed, and sections were cut and processed for in situ hybridization for LAT message (A, C) and apoptosis (B, D) using the fluorescein in situ cell death detection kit (Roche). Sections were visualized by light microscopy and fluorescent microscopy using a fluorescein filter.
FIG. 3.

Detection of apoptotic neurons in 17 N/H-infected trigeminal ganglia. Trigeminal ganglia were explanted 30 days postinfection from 17 N/H-infected BALB/c mice. Neuronal apoptosis was detected using the fluorescein in situ cell death detection kit (Roche). Sections were visualized by light microscopy (A) and fluorescent microscopy (B).
FIG. 9.

Localization of CD8+ T cells and apoptosis in 17 N/H-infected trigeminal ganglia. BALB/c mice were infected by corneal scarification with 2.5 × 104 PFU of 17 N/H virus in each eye. At 30 days postinfection, mice were sacrificed and trigeminal ganglia were extracted and sectioned for slides. Slides were stained for CD8a antigen and apoptosis using TUNEL. Sections were viewed by light microscopy (A), UV microscopy using a rhodamine filter to view CD8a staining (B), and UV microscopy using a fluorescein filter to view TUNEL staining (C). (D) Fields B and C were merged for colocalization of CD8 cells and apoptosis.
To measure the incidence of neuronal apoptosis, a large number of neurons in 17+-, 17ΔSty-, 17 N/H-, and mock-infected trigeminal ganglia were identified by morphology and apoptotic neurons were counted (Fig. 4). Three independent experiments were performed, and approximately 1,000 neurons were identified and scored for apoptosis in each experiment. These neurons were derived from 15 mice (5 mice per experiment), and every attempt was made to ensure that the neurons counted were from as many different trigeminal ganglia as possible to provide a wide distribution of neurons. For example, 10 or more slides were counted from each experiment, and only nonsequential slides were used to ensure that the same neurons were not counted two or more times. Standard deviations were calculated from the data from the three experimental groups. Of the neurons in trigeminal ganglia infected with 17 N/H, 0.98% were apoptotic. HSV-1 17+-infected trigeminal ganglia had no apoptotic neurons, and almost no apoptotic neurons were observed in mock-infected (0.009%) and 17ΔSty-infected (0.087%) trigeminal ganglia. Statistical analysis (t tests) of the above data shows that there are significantly more apoptotic neurons in trigeminal ganglia infected with 17 N/H than in trigeminal ganglia infected with 17ΔSty or mock-infected trigeminal ganglia (P < 0.05).
FIG. 4.

Percentage of neurons in 17+-, 17ΔSty-, 17 N/H-, and mock-infected trigeminal ganglia that are apoptotic. 17+-, 17ΔSty-, 17 N/H-, and mock-infected BALB/c mice were sacrificed 30 days postinfection, and trigeminal ganglia were explanted and sectioned. Neurons were identified by morphology, and apoptosis was determined by the presence of fluorescent nuclei using the fluorescein in situ cell death detection kit (Roche). The asterisk indicates statistically significant differences (P < 0.05) between 17 N/H- and 17ΔSty-infected trigeminal ganglia and between mock- and 17+-infected trigeminal ganglia.
Given the low incidence of apoptotic neurons in 17+- and 17 N/H-infected trigeminal ganglia, we decided to try to identify the apoptotic cells observed in Fig. 2b and d. Hematoxylin and eosin staining of trigeminal ganglion sections shows a large number of infiltrating cells in 17+- and 17 N/H-infected trigeminal ganglia (Fig. 5b and c) but not in mock-infected trigeminal ganglia (Fig. 5a). Several investigators have proposed that immune cells infiltrate HSV-1-infected trigeminal ganglia and may undergo apoptosis (20, 40). To determine whether the infiltrating cells seen in hematoxylin- and eosin-stained trigeminal ganglion sections (Fig. 5) were cytotoxic T cells, sections were stained with an anti-CD8 antibody followed by a fluorescein-conjugated secondary antibody. In Fig. 6, we see CD8+ T cells entering 17+-infected trigeminal ganglia by day 7 (Fig. 6a) and remaining there at day 15 and 30 (Fig. 6b and c, respectively). A similar pattern of CD8+ T-cell infiltration was observed in 17 N/H-infected trigeminal ganglia (6d, e, and f). No CD8+ T cells were observed in mock-infected trigeminal ganglia (Fig. 6g, h, and i).
FIG. 5.

Trigeminal ganglion sections stained with hematoxylin and eosin. Trigeminal ganglia were explanted at day 15 postinfection from mock (A)-, 17+ (B)-, and 17 N/H (C)-infected BALB/c mice.
FIG. 6.

Detection of CD8+ T cells in trigeminal ganglia. Trigeminal ganglia of mice infected with 17+ (A, B, C) or 17 N/H (D, E, F) or mock infected (G, H, I) are shown at day 7 (A, D, G), 15 (B, E, H), and 30 (C, F, I). Trigeminal ganglia were sectioned and stained with a monoclonal antibody against the antigen CD8a followed by a fluorescein-conjugated secondary antibody. Sections were visualized by fluorescence microscopy.
CD8+ T-cell infiltration was quantitated by FACS analysis (Fig. 7). Disassociated trigeminal ganglia were stained for CD8a antigen with a PE-conjugated antibody. Values of the percentage of total trigeminal ganglion cells that are CD8+ T cells were graphed in Fig. 8. A significant number of CD8+ T cells enter 17+- and 17 N/H-infected trigeminal ganglia by day 7 (Fig. 8) (8.8% and 8.07% of total cells, respectively). In 17+-infected trigeminal ganglia, CD8+ T-cell levels peak at day 15 (Fig. 8) (12.9%). In 17 N/H-infected trigeminal ganglia, CD8+ T-cell levels decrease from day 7 levels at day 15 (8.8% to 5.12%). Despite decreasing in number, there are a substantial number of CD8+ T cells left in 17+- and 17 N/H-infected trigeminal ganglia (Fig. 8) (8.3% and 4.2%, respectively) at day 30.
FIG. 7.

FACS analysis of infiltrating CD8+ T cells. 17+ (A, B, C)- and 17 N/H (D, E, F)-infected BALB/c mice were sacrificed on day 7 (A, D), 15 (B, E), and 30 (C, F) postinfection. Trigeminal ganglia were explanted, dissociated by collagenase, and stained with a PE-conjugated anti-CD8a monoclonal antibody. Cells were analyzed by flow cytometry.
FIG. 8.

CD8+ T-cell infiltration into 17+- and 17 N/H-infected trigeminal ganglia. Trigeminal ganglia from 17+ (white bars)- and 17 N/H (black bars)-infected mice were explanted at 7, 15, and 30 days and analyzed by flow cytometry (Fig. 7). Values for the number of CD8+ T cells detected in trigeminal ganglia relative to the total number of cells counted were graphed.
To determine whether the apoptotic cells seen in infected trigeminal ganglia are CD8+ T cells, sections of 17+- and 17 N/H-infected ganglia were stained with anti-CD8 antibody and subjected to TUNEL analysis. Fields were recorded for CD8 staining (PE filter) and TUNEL (fluorescein filter) and overlaid using Improvision Open Lab version 3.1.6 software. In Fig. 9, this analysis was done on 30-day-postinfection 17 N/H-infected trigeminal ganglia. Higher-magnification pictures of these sections shows apoptotic CD8+ T cells and neurons. Phase pictures clearly shows 3 cell bodies of neurons (Fig. 9a). CD8 staining (Fig. 9b) shows a number of cytotoxic T cells in the field. TUNEL analysis of the same section shows 2 apoptotic neurons (a rare event) and a number of other apoptotic cells. Overlaying the fields (Fig. 9d) reveals that all of the CD8+ cells in Fig. 9b are undergoing apoptosis. These apoptotic CD8+ T cells appear to be surrounding apoptotic neurons.
To quantitate how many CD8+ T cells are undergoing apoptosis, disassociated trigeminal ganglion cells were stained with an anti-CD8 PE-conjugated antibody and assayed by TUNEL before quantification by FACS analysis (Fig. 10). FACS analysis shows that in 17+- and 17 N/H-infected trigeminal ganglia, initial infiltrating CD8+ cells undergo apoptosis at a very low rate (Fig. 11) (6.3% and 10%, respectively). As infiltrating CD8+ T cells continue to enter 17+- and 17 N/H-infected trigeminal ganglia, they undergo apoptosis at a much higher rate at 15 days (Fig. 11) (53.4% and 59.9%, respectively) and at 30 days (Fig. 11) (57.8% and 75.7%, respectively).
FIG. 10.

FACS analysis of CD8+ T cells and apoptosis. 17+ (A, B, C)- and 17 N/H (D, E, F,)-infected BALB/c mice were sacrificed on day 7 (A, D), 15 (B, E), and 30 (C, F) postinfection. Trigeminal ganglia were explanted, dissociated by collagenase, and stained for CD8+ T cells with a PE-conjugated anti-CD8a monoclonal antibody and TUNEL using fluorescein-conjugated nucleotides. Cells were analyzed by flow cytometry.
FIG. 11.
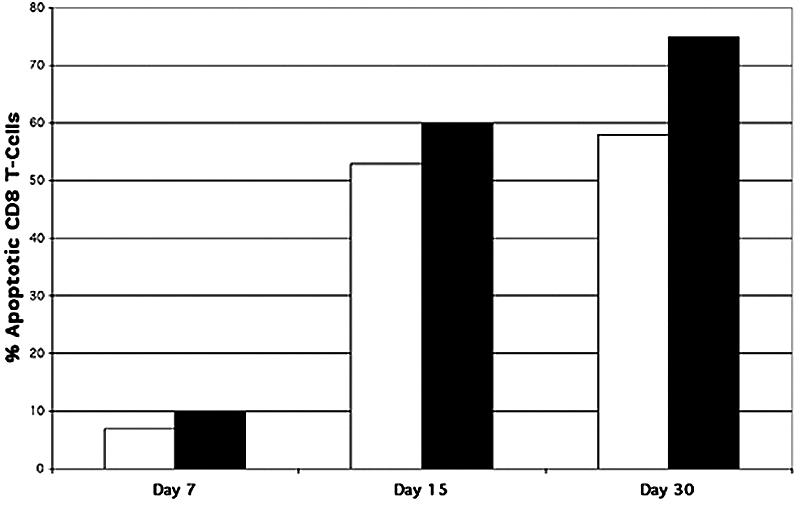
Apoptosis of CD8+ T cells infiltrating trigeminal ganglia. BALB/c mice were infected with 17+ (white bars) and 17 N/H (black bars) by corneal scarification and sacrificed at 30 days postinfection. Explanted trigeminal ganglia were digested with collagenase, and dissociated cells were stained for CD8+ T cells and apoptosis using TUNEL. Cells were analyzed by FACS, and values were graphed.
DISCUSSION
HSV-1 establishes latency in the peripheral nervous system, with LAT being the only detectable transcript (10, 32). While LAT expression has been implicated in the establishment of latency and reactivation from latency, several investigators have demonstrated an antiapoptotic effect in vivo and in vitro (1, 29). Transfection of expression vectors containing the LAT sequence into tissue culture cells protect those cells from apoptotic agents such as ceramide, etoposide, and anti-Fas antibody (1, 29, 30). Investigators have mapped this in vitro antiapoptotic effect to exon 1 (1) or the first 1.5 kb of the LAT (1, 29, 30).
This antiapoptotic effect is more difficult to detect in vivo. The trigeminal ganglia of rabbits infected with the LAT-negative mutant dLAT2903 (−161 to +1667; 7 days postinfection) have been shown to have a large number of TUNEL-positive apoptotic neurons (30). Few apoptotic neurons were observed in the trigeminal ganglia infected with the LAT-positive parental virus or a rescuant of dLAT2903. Apoptosis in these neurons was further confirmed by staining for the apoptotic protein PARP (a caspase-3 cleavage product of poly-ADP-ribose polymerase) (30).
In this study, mice were inoculated with a LAT-negative mutant (17 N/H), a partial exon 1 deletion (371 bp deleted from the 660 bp in exon 1) mutant (17ΔSty), and the parent strain (17+). Once latency is established at 30 days postinfection, trigeminal ganglia were excised, fixed, and stained for apoptosis by TUNEL. We observed no apoptotic neurons in 17+-infected trigeminal ganglia, and only 0.087% of all neurons in 17ΔSty-infected trigeminal ganglia were apoptotic. 17 N/H-infected trigeminal ganglia had significantly more apoptotic neurons (0.98%; P < 0.05). Based on our results with 17ΔSty and 17 N/H, we conclude that the in vivo antiapoptotic effect of the LAT is located within the a fragment of the 3′ end of exon 1, which was not part of the 17ΔSty deletion (+447 to +661), and the 5′ end of the LAT intron (+661 to +1667). This mapping is in agreement with that of Inman et al. (16).
Even though the LAT deletion in our mutant is similar to that used by other investigators, we observed significantly more apoptotic neurons. Other investigators observed that infection of mouse trigeminal ganglia with the LAT-null mutant 17ΔA/H resulted in a decrease in survivability in trigeminal ganglion neurons (40). However, they observed (contrary to our results) practically no evidence of apoptosis in the eradicated neurons. One possible explanation for this discrepancy is the deletion difference between their LAT-null mutant 17ΔA/H (−1136 to +828) (Fig. 1D) and our mutant 17ΔN/H (−359 to +1667).
Although neither LAT mutant expresses detectable levels of LAT, it is clear that 17ΔA/H is similar to strain 17 in terms of protecting neurons from apoptosis, even though 17ΔN/H has a significant sequence difference. Thus, the antiapoptotic effect could be from a small transcript originating at the LAT promoter and mapping in the region from +828 to +1667. It is formally possible that it may be due to the effect of the DNA sequence in this region. Our data support the observations made by Perng et al. (30) that the LAT has an antiapoptotic effect. Data obtained in this study with the 17ΔSty mutant support the mapping of the antiapoptotic function to a region including at least a portion of exon 1. A prior study showed by in situ hybridization that 17ΔSty expresses a LAT (+399 to +1296) (26).
We also show that CD8+ T cells infiltrate trigeminal ganglia during the early stages of HSV-1 infection and remain there past the establishment of latency. These observations concur with those of other investigators who showed a population of CD8+ T cells in trigeminal ganglia after 84 days (20). During the lytic phase of infection, significant levels of viral antigens are present which stimulate the recruitment of cytotoxic cells to the trigeminal ganglia. However, upon establishment of latency in neurons, viral genomes are transcriptionally silent except for LAT expression. Feldman et al. (9), using in situ hybridization, demonstrated that a small percentage of neurons express viral transcripts, such as ICP4, during the latent phase in mice. It was thought that these cells were very rare reactivating neurons. Despite their low frequency (less than 1 positive cell detected per ganglion), it is possible that they contribute to the CD8+ infiltrate.
Once cytotoxic T cells are recruited to an infected area, they can eradicate target cells by inducing apoptosis by engaging the target cell's Fas receptor with Fas ligand, found on the T cell's surface (14, 33), and subsequently releasing lytic granules (28). The documented in vivo antiapoptotic effect of LAT may protect infected neurons in trigeminal ganglia from cytotoxic T cells. HSV-1 can inhibit apoptosis by cytotoxic T cells 2 h postinfection during the lytic phase of infection (16). It was postulated that the HSV-1 protein kinase Us3 is responsible for lytic-phase apoptosis protection. We believe that the HSV LAT may act as an inhibitor of cytotoxic-T-lymphocyte-directed apoptosis during latency, since we observe a large number of apoptotic neurons in 17 N/H-infected trigeminal ganglia and none in 17+-infected trigeminal ganglia. These CD8+ T cells may be recruited into the infected trigeminal ganglia by the low-level expression of viral transcriptional activators like ICP4, as described by Feldman et al. (9).
As we see in Fig. 2, 9, and 10, there are a number of nonneuronal apoptotic cells. By colocalizing CD8 staining and TUNEL staining, we see that up to 80% of apoptotic cells are cytotoxic T cells (data not shown). There are several possible reasons why CD8+ T cells recruited to infected trigeminal ganglia undergo apoptosis. One such phenomenon is called activation-induced T-cell death, in which activation through CD3 T-cell molecules in the absence of an inflammatory response activates pathways that lead to caspase activation and cell apoptosis (34). This would correspond with our observation that significant CD8+ T-cell apoptosis occurs at days 15 and 30, which would be a point where lytic infection is ending and inflammation is resolving.
Another theory concerning cytotoxic T-cell apoptosis is that HSV-1 infection of cytotoxic T cells leaves them vulnerable to apoptosis from other virus-specific cytotoxic T cells. This process is called fratricide and may, in effect, be a mechanism of viral immune evasion (31). We have identified HSV-1-infected apoptotic T cells during the early phase of infection (7 days postinfection) but not at latent time points (30 days postinfection).
In conclusion, we show that neurons in mouse trigeminal ganglia, infected with a LAT-expressing HSV-1, are protected from apoptosis once they become latent. We also show a constant presence of CD8+ T cells in the trigeminal ganglia of infected mice. A possible source of antigen for this recruitment of CD8+ T cells may be the low-level expression of viral transcriptional transactivators observed by Feldman et al. (9). Even in the continual presence of CD8+ T cells, latent neurons infected with a LAT-expressing HSV-1 remain protected from apoptosis, possibly because of the expression of LAT.
REFERENCES
- 1.Ahmed, M., M. Lock, C. G. Miller, and N. W. Fraser. 2002. Regions of the herpes virus type 1 latency-associated transcript that protect cells from apoptosis in vitro and protect neuronal cells in vivo. J. Virol. 76:717-729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asano, S., T. Honda, F. Goshima, D. Watanabe, Y. Miyake, Y. Sugiura, and Y. Nishiyama. 1999. US3 protein kinase of herpes simplex virus type 2 plays a role in protecting corneal epithelial cells from apoptosis in infected mice. J. Gen. Virol. 80:51-56. [DOI] [PubMed] [Google Scholar]
- 3.Aubert, M., and J. A. Blaho. 1999. The herpes simplex virus type 1 regulatory protein ICP27 is required for the prevention of apoptosis in infected human cells. J. Virol. 73:2803-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bak, I. J., C. H. Markham, M. L. Cook, and J. G. Stevens. 1977. Intraaxonal transport of herpes simplex virus in the rat central nervous system. Brain Res. 136:415-429. [DOI] [PubMed] [Google Scholar]
- 5.Block, T. M., S. Deshmane, J. Masonis, J. Maggioncalda, T. Valyi-Nagi, and N. M. Fraser. 1993. An HSV LAT null mutant reactivates slowly from latent infection and makes small plaques on CV-1 monolayers. Virology 192:618-630. [DOI] [PubMed] [Google Scholar]
- 6.Da Costa, X. J., M. A. Brockman, E. Alicot, M. Ma, M. B. Fischer, X. Zhou, M. Knipe, and M. C. Carroll. 1999. Humoral response to herpes simplex virus is complement-dependent. Proc. Natl. Acad. Sci. USA 96:12708-12712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esiri, M. M. 1982. Herpes simplex encephalitis. An immunohistological study of the distribution of viral antigen within the brain. J. Neurol. Sci. 54:209-226. [DOI] [PubMed] [Google Scholar]
- 8.Farrell, M. J., A. T. Dobson, and L. T. Feldman. 1991. Herpes simplex virus latency-associated transcript is a stable intron. Proc. Natl. Acad. Sci. USA 88:790-794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feldman, L. T., A. R. Ellison, C. C. Voytek, L. Yang, P. Krause, and T. P. Margolis. 2002. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. Proc. Natl. Acad. Sci. USA 99:978-983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fraser, N. W., T. M. Block, and J. G. Spivack. 1992. The latency-associated transcripts of herpes simplex virus: RNA in search of function. Virology 191:1-8. [DOI] [PubMed] [Google Scholar]
- 11.Fruh, K., K. Ahn, H. Djaballah, P. Sempe, P. M. Van Endert, R. Tampe, P. A. Peterson, and Y. Yang. 1995. A viral inhibitor of peptide transporters for antigen presentation. Nature 375:415-418. [DOI] [PubMed] [Google Scholar]
- 12.Galvan, V., and B. Roizman. 1998. Herpes simplex virus 1 induces and blocks apoptosis at multiple steps during infection and protects cells from exogenous inducers in a cell-type-dependent manner. Proc. Natl. Acad. Sci. USA 95:3931-3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldsmith, K., W. Chen, D. C. Johnson, and R. L. Hendricks. 1998. Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J. Exp. Med. 187:341-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Golstein, P. 1995. Fas-based T cell-mediated cytotoxicity. Curr. Top. Microbiol. Immunol. 198:25-37. [DOI] [PubMed] [Google Scholar]
- 15.Hill, J. M., F. Sedarati, R. T. Javier, E. K. Wagner, and J. G. Stevens. 1990. Herpes simplex virus latent phase transcription facilitates in vivo reactivation. Virology 174:117-125. [DOI] [PubMed] [Google Scholar]
- 16.Inman, M., G. C. Perng, G. Henderson, H. Ghiasi, A. B. Nesburn, S. L. Wechsler, and C. Jones. 2001. Region of herpes simplex virus type 1 latency-associated transcript sufficient for wild-type spontaneous reactivation promotes cell survival in tissue culture. J. Virol. 75:3636-3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jerome, K. R., J. F. Tait, D. M. Koelle, and L. Corey. 1998. Herpes simplex virus type 1 renders infected cells resistant to cytotoxic T-lymphocyte-induced apoptosis. J. Virol. 72:436-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jerome, K. R., R. Fox, Z. Chen, A. E. Sears, H. Lee, and L. Corey. 1999. Herpes simplex virus inhibits apoptosis through the action of 2 genes, Us5 and Us3. J. Virol. 73:8950-8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kagi, D., F. Vignaux, and B. E. A. Ledermann. 1994. Fas and perforin pathways as major mechanisms of T cell-mediated cytotoxicity. Science 265:528-533. [DOI] [PubMed] [Google Scholar]
- 20.Khanna, K. M., R. H. Bonneau, P. R. Kinchington, and R. L. Hendricks. 2003. Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity 18:593-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koyama, A. H., and Y. Miwa. 1997. Suppression of apoptotic DNA fragmentation in herpes simplex virus type 1-infected cells. J. Virol. 71:2567-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leopardi, R., C. Van Sant, and B. Roizman. 1997. The herpes simplex virus 1 protein kinase US3 is required for protection from apoptosis induced by the virus. Proc. Natl. Acad. Sci. USA 94:7891-7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levine, E. M., M. Passini, P. F. Hitchcock, E. Glasgow, and N. Schechter. 1997. Vsx-1 and Vsx-2: two Chx10-like homeobox genes expressed in overlapping domains in the adult goldfish retina. J. Comp. Neurol. 387:439-448. [PubMed] [Google Scholar]
- 24.Liu, T., Q. Tang, and R. L. Hendricks. 1996. Inflammatory infiltration of the trigeminal ganglion after herpes simplex virus type 1 corneal infection. J. Virol. 70:264-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lopes, C., R. Ryshke, and M. Bennet. 1980. Marrow-dependent cells depleted by 89Sr mediate genetic resistance to herpes simplex virus type 1 infection in mice. Infect. Immun. 28:1028-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maggioncalda, J., A. Mehta, Y. H. Su, N. W. Fraser, and T. Block. 1996. Correlation between herpes simplex virus type 1 rate of reactivation from latent infection and the number of infected neurons in trigeminal ganglia. Virology 225:72-81. [DOI] [PubMed] [Google Scholar]
- 27.Margolis, T. P., F. Sedarati, A. T. Dobson, L. T. Feldman, and J. G. Stevens. 1992. Pathways of viral gene expression during acute neuronal infection with HSV-1. Virology 189:150-160. [DOI] [PubMed] [Google Scholar]
- 28.Medema, J. P., R. E. Toes, C. Scaffidi, T. S. Zheng, R. A. Flavell, C. J. Melief, M. E. Peter, R. Offringa, and P. H. Krammer. 1997. Cleavage of FLICE (caspase-8) by granzyme B during cytotoxic T lymphocyte-induced apoptosis. Eur. J. Immunol. 27:3492-3498. [DOI] [PubMed] [Google Scholar]
- 29.Perng, G. C., S. M. Slanina, A. Yukht, H. Ghiasi, A. B. Nesburn, and S. L. Wechsler. 2000. The latency-associated transcript gene enhances establishment of herpes simplex virus type 1 latency in rabbits. J. Virol. 74:1885-1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perng, G. C., C. Jones, J. Ciacci-Zanella, M. Stone, G. Henderson, A. Yukht, S. M. Slanina, F. M. Hofman, H. Ghiasi, A. B. Nesburn, and S. L. Wechsler. 2000. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 287:1500-1503. [DOI] [PubMed] [Google Scholar]
- 31.Raftery, M. J., C. K. Behrens, A. Muller, P. H. Krammer, H. Walczak, and G. Schonrich. 1999. Herpes simplex virus type 1 infection of activated cytotoxic T cells: induction of fratricide as a mechanism of viral immune evasion. J. Exp. Med. 190:1103-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rock, D. L., A. B. Nesburn, H. Ghiasi, J. Ong, T. L. Lewis, J. R. Lokensgard, and S. L. Wechsler. 1987. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J. Virol. 61:3820-3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rouvier, E., M. F. Luciani, and P. Goldstein. 1993. Fas involvement in Ca(2+)-independent T cell-mediated cytotoxicity. J. Exp. Med. 177:195-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi, Y. F., B. M. Sahai, and D. R. Green. 1989. Cyclosporin A inhibits activation-induced cell death in T-cell hybridomas and thymocytes. Nature 339:625-626. [DOI] [PubMed] [Google Scholar]
- 35.Simmons, A., and D. C. Tscharke. 1992. Anti-CD8 impairs clearance of herpes simplex virus from the nervous system: implications for the fate of virally infected neurons. J. Exp. Med. 175:1337-1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sloan, D. D., G. Zahariadis, C. M. Posavad, N. T. Pate, S. J. Kussick, and K. R. Jerome. 2003. CTL are inactivated by herpes simplex virus-infected cells expressing a viral protein kinase. J. Immunol. 171:6733-6741. [DOI] [PubMed] [Google Scholar]
- 37.Sobel, R. A., A. B. Collins, R. B. Colvin, and A. K. Bhan. 1986. The in situ cellular immune response in acute herpes simplex encephalitis. Am. J. Pathol. 125:332-338. [PMC free article] [PubMed] [Google Scholar]
- 38.Stevens, J. G. 1989. Human herpesviruses: a consideration of the latent state. Microbiol. Rev. 53:318-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Theil, D., T. Derfuss, I. Paripovic, S. Herberger, E. Meinl, O. Schueler, M. Strupp, V. Abrusow, and T. Brandt. 2003. Latent herpesvirus infection in human trigeminal ganglia causes chronic immune response. Am. J. Pathol. 163:2179-2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thompson, R. L., and N. M. Sawtell. 2001. Herpes simplex type 1 latency-associated transcript gene promotes neuronal survival. J. Virol. 75:6660-6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tomazin, R., N. E. van Schoot, K. Goldsmith, P. Jugovic, P. Sempe, K. Fruh, and D. C. Johnson. 1998. Herpes simplex virus type 2 ICP47 inhibits human TAP but not mouse TAP. J. Virol. 72:2560-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valyi-Nagy, T., R. M. Gesser, B. Raengsakulrach, S. L. Deshmane, B. P. Randazzo, A. J. Dillner, and N. W. Fraser. 1994. A thymidine kinase-negative HSV-1 strain establishes a persistent infection in SCID mice that features uncontrolled peripheral replication but only marginal nervous system involvement. Virology 199:484-490. [DOI] [PubMed] [Google Scholar]
- 43.Wagner, E. K., W. M. Flanagan, G. Devi-Rao, Y. F. Zhang, J. M. Hill, K. P. Anderson, and J. G. Stevens. 1988. The herpes simplex virus latency-associated transcript is spliced during the latent phase of infection. J. Virol. 62:4577-4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zawatzky, R., I. Gresser, E. DeMaeyer, and H. Kirchner. 1982. The role of interferon in the resistance of C57BL/6 mice to various doses of herpes simplex virus type 1. J. Infect. Dis. 146:405-410. [DOI] [PubMed] [Google Scholar]
- 45.Zhou, G., V. Galvan, G. Campedelli-Fuime, and B. Roizman. 2000. Glycoprotein D or J delivered in trans blocks apoptosis in SK-N-SH cells induced by a herpes simplex virus 1 mutant lacking intact genes expressing both glycoproteins. J. Virol. 74:11782-11791. [DOI] [PMC free article] [PubMed] [Google Scholar]