

Abstract
α-Actinin and vinculin orchestrate reorganization of the actin cytoskeleton following the formation of adhesion junctions. α-Actinin interacts with vinculin through the binding of an α-helix (αVBS) present within the R4 spectrin repeat of its central rod domain to vinculin's N-terminal seven-helical bundle domain (Vh1). The Vh1:αVBS structure suggests that αVBS first unravels from its buried location in the triple-helical R4 repeat to allow it to bind to vinculin. αVBS binding then induces novel conformational changes in the N-terminal helical bundle of Vh1, which disrupt its intramolecular association with vinculin's tail domain and which differ from the alterations in Vh1 provoked by the binding of talin. Surprisingly, αVBS binds to Vh1 in an inverted orientation compared to the binding of talin's VBSs to vinculin. Importantly, the binding of αVBS and talin's VBSs to vinculin's Vh1 domain appear to also trigger distinct conformational changes in full-length vinculin, opening up distant regions that are buried in the inactive molecule. The data suggest a model where vinculin's Vh1 domain acts as a molecular switch that undergoes distinct structural changes provoked by talin and α-actinin binding in focal adhesions versus adherens junctions, respectively.
α-Actinin is a ubiquitously expressed and essential cytoskeletal protein that cross-links actin filaments at sites of cell-cell (adherens junctions) and cell-matrix (focal adhesion) junctions and at the leading edges of the cell membranes of migrating cells. α-Actinin is a member of a family of structurally related proteins, including spectrin, dystrophin, and utrophin, which regulate the organization of the actin cytoskeleton in a cell-type-specific fashion (32). These proteins have a globular structure, with calponin homology (CH) domains at their N termini, a central rod domain containing α-helical spectrin repeats, and a C-terminal domain that contains calmodulin-like domains harboring EF-hand motifs (7).
α-Actinin is a rod-shaped antiparallel dimer of two 100-kDa monomers; this configuration positions its actin-binding CH motifs at either end of the rigid rod, an arrangement that allows α-actinin to efficiently cross-link actin filaments into tight bundles (12, 40). The central rod domain of α-actinin contains four spectrin repeats (R1 to R4), which are triple-helical coiled-coil bundles that are connected by helical linkers (7, 12). In α-actinin, these repeats are aligned in a symmetric fashion that allows for the formation of the rigid dimer through interactions of the R1 and R4 repeats and of the R2 and R3 repeats (12, 40). However, spectrin repeats can also form stable unfolded intermediates when subjected to mechanical stress (1), as occurs following the formation of adhesion complexes, and the spectrin repeats of α-actinin also harbor docking sites for a number of other cytoskeletal proteins (32), in particular vinculin (31).
α-Actinin directly binds to vinculin in solution (22, 26, 31) and colocalizes with vinculin in cells at sites of adherens junctions and/or focal adhesions (8, 30, 35). Each protomer within the α-actinin dimer binds to one vinculin molecule through an amphipathic α-helix (human residues 731 to 760) (26, 31) that, in α-actinin's native structure, is buried within the R4 spectrin repeat (40). Like α-actinin, vinculin has a globular structure with head (Vh) and tail (Vt) domains (10, 39), and the intramolecular interaction of a seven-helical bundle domain within vinculin's head (Vh1, residues 1 to 258) with the five-helical bundle that comprises the Vt domain (residues 879 to 1066) (3, 19) locks the protein in a closed, inactive conformation. The crystal structure of the 117-kDa monomeric vinculin molecule in its resting state (2, 5) established that the protein also contains three additional α-helical bundle domains that are similar in their structure to either Vh1 (Vh2 and Vh3) or Vt (Vt2) (5).
α-Actinin also binds to the cytoplasmic tails of cadherin receptors in adherens junctions and of β-integrin receptors at sites of focal adhesion (32). The interaction of α-actinin with these receptors thus provides a structural link for vinculin with cell surface adhesion receptors. However, vinculin must transition from its inactive, closed conformation to its open and activated state to link adhesion complexes to the actin cytoskeleton (16, 23). Activation of vinculin requires severing of the Vh1-Vt interaction (23), and this was thought to be directed by the binding of phosphatidylinositol-4,5-bisphosphate (PIP2) (16), which can bind to and alter the conformation of Vt (3). However, the binding site for PIP2 is partially occluded in the full-length structure of inactive vinculin (2, 5). Thus, other triggers, in particular the vinculin binding sites (VBSs) of talin and α-actinin, may also contribute to vinculin activation. Indeed, their binding displaces Vt from preexisting Vh1:Vt complexes, and talin's VBSs provoke dramatic conformational changes in the structure of Vh1 (15, 19, 20). Surprisingly, all of these VBSs are buried in the native rod structures of talin (15, 33) and α-actinin (40).
Since spectrin repeats can form stable unfolded intermediates (1), we reasoned that the structure of the Vh1-αVBS complex might reveal insights into the structural dynamics that might allow α-actinin to bind to vinculin and how αVBS activates vinculin. Here we report the crystal structure of the Vh1-αVBS complex to 1.8-Å resolution, which reveals that αVBS binds to vinculin in an orientation that is inverted with respect to the binding of talin's VBSs. Our Vh1-αVBS structure further suggests that activation of vinculin by α-actinin involves a sequence of structural alterations that are initiated by unraveling of αVBS from within its buried location in the triple-helical R4 spectrin repeat. Finally, biochemical studies suggest that αVBS alone appears sufficient to alter the conformation of full-length vinculin and indicate that these changes differ from those provoked by the binding of talin's VBSs. These findings establish that vinculin's Vh1 domain is dynamic and undergoes unique conformational changes when bound by its partners, and they suggest that these changes may, in turn, cause differences in the overall structure of vinculin that would endow it with the ability to selectively interact with its other binding partners.
MATERIALS AND METHODS
Protein expression and purification.
The N-terminally hexahistidine-tagged human Vh1 (residues 1 to 258) and Vt (residues 879 to 1066) expression constructs, the C-terminally octahistidine-tagged human full-length vinculin (residues 1 to 1066) expression construct, and the purification of Vh1, Vt, and full-length vinculin have been previously described (5, 19). An N-terminally tagged recombinant form of human vinculin containing nearly the entire head domain but lacking the proline-rich hinge region and the Vt domain (VH, residues 1 to 780) was cloned into the pET3 expression vector (Novagen). Protein was expressed in Escherichia coli BL21(DE3), and cells were lysed in Tris-HCl (pH 8), 0.5 M NaCl, and phenylmethylsulfonyl fluoride. The expressed VH protein was purified using a chelating nickel affinity column (Amersham) and eluted over a gradient to 0.5 M imidazole. VH protein was then further purified using an anion-exchange column (Q Sepharose; Amersham). To form the VH-Vt complex, the two proteins were incubated and purified over a Superdex-200 column. All proteins were dialyzed into Tris-acetate (pH 7.6), 10 mM dithiothreitol, and 1 mM EDTA. All proteins were concentrated to 10 mg/ml, aliquoted, and stored at −20°C.
Crystallization of the Vh1-αVBS complex.
Human α-actinin (residues 731 to 760; αVBS) was synthesized and high-pressure liquid chromatography purified in our in-house facility. Initial crystallization conditions were identified at the Hauptman-Woodward Institute (Buffalo, NY). Vh1-αVBS crystals were obtained by equilibrating 2 moles of αVBS per mole of Vh1 against a reservoir of 14% polyethylene glycol 3000, 100 mM sodium phosphate buffer (pH 6.0), and 10 mM BaCl2. Crystals appeared within 1 week at room temperature. These crystals belong to space group P21, with one heterodimer in the asymmetric unit, a solvent content of 42%, and a volume-to-mass ratio of 2.13 Å3/Da. The Vh1:αVBS crystals were cryoprotected in 30% polyethylene glycol 400 and 100 mM phosphate buffer (pH 6.2).
X-ray data collection and processing.
The Vh1-αVBS data set was collected at the Advanced Photon Source (SBC-CAT ID beamline), integrated using MOSFLM (9), and scaled and merged with SCALA (14). The estimation of overloads in MOSFLM was crucial to achieving good completeness at low resolution, which turned out to be essential for the success of subsequent molecular replacement calculations. Data statistics are provided in Table 1.
TABLE 1.
Data reduction statistics of the Vh1-αVBS data set
| Parameter | Value |
|---|---|
| Cell dimensions | |
| a (Å) | 42.8 |
| b (Å) | 54.3 |
| c (Å) | 63.6 |
| β (°) | 107.6 |
| Resolution range (Å) | 40.5-1.8 |
| Last shell (Å) | 1.85-1.8 |
| Rmeas0a | 0.106 |
| Rmeas0a (last shell) | 0.325 |
| Total data | 99,804 |
| Total data (last shell) | 2,968 |
| Unique data | 24,098 |
| Unique data (last shell) | 1,140 |
| Avg F2/o(F2) | 14.1 |
| Avg F2/o(F2) (last shell) | 4.8 |
| Completeness | 0.929 |
| Completeness (last shell) | 0.605 |
| Redundancy | 4.1 |
| Redundancy (last shell) | 2.6 |
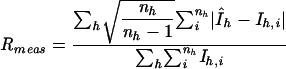 with
with 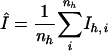 . For Rmeas I+ and I− are treated separately, whereas for Rmeas0 they are assumed to be equivalent (11).
. For Rmeas I+ and I− are treated separately, whereas for Rmeas0 they are assumed to be equivalent (11).
Structure determination and refinement of the Vh1-αVBS complex.
The C-terminal helical bundle of Vh1 (PDBID 1RKE, chain A, residues 130 to 258) was placed by molecular replacement and the N-terminal bundle (PDBID 1SYQ, residues 1 to 129) was subsequently placed by two-body molecular replacement, both using MOLREP (37). Attempts at using the whole Vh1 domain as a search model were unsuccessful because of the elongated shape of the molecule. The resulting map was solvent flattened with the program DM (9) and completely traced with the program ARP/wARP (9). The model was improved by extensive rebuilding with the O interactive graphics program (24). The final stages involved interactive model building of the αVBS peptide and the α1-α2 loop of Vh1. Positional and B-factor refinement was carried out with BUSTER/TNT (6, 36). Refinement statistics are provided in Table 2.
TABLE 2.
Crystallographic refinement statistics for the Vh1-αVBS structure
| Parameter | Value |
|---|---|
| Resolution range (Å) | 40.5-1.8 |
| Last shell (Å) | 1.91-1.8 |
| No. of reflections (working set) | 22,894 |
| No. of reflections (test set) | 1,192 |
| R factora (overall) | 0.171 |
| R factor (last shell) | 0.187 |
| Rfreeb (overall) | 0.232 |
| Rfree (last shell) | 0.225 |
| No. of amino acid residues | 294 |
| No. of protein atoms | 2,337 |
| No. of solvent molecules | 489 |
| Avg B factor (main chain) (Å2) | 19.7 |
| Avg B factor (side chain) (Å2) | 30.6 |
| Avg B factor (solvent) (Å2) | 37.2 |
| RMSDc from ideal geometry | |
| Covalent bond lengths (Å) | 0.01 |
| Bond angles (°) | 1.12 |
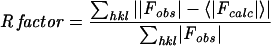 , where 〈|Fcalc|〉is the expectation of |Fcalc| under the error model used in maximum-likelihood refinement.
, where 〈|Fcalc|〉is the expectation of |Fcalc| under the error model used in maximum-likelihood refinement.
The free R factor is a cross-validation residual calculated by using 5% of the native data, which were randomly chosen and excluded from the refinement.
RMSD, root mean square deviation.
αVBS binding assays.
αVBS, talin-VBS1, or talin-VBS3 peptide was added to VH:Vt at a molar ratio of 4:1 and allowed to incubate for 20 min. Complexes were then resolved by native polyacrylamide gel electrophoresis. As controls, unbound VH and the talin-VBS1-, talin-VBS3-, and αVBS-bound VH complex were also analyzed by native polyacrylamide gel electrophoresis. The identity of the complexes in native gels was confirmed by cutting out the bands and analysis on sodium dodecyl sulfate (SDS)-polyacrylamide gels (not shown).
Surface plasmon resonance assays.
Binding studies were performed on a Biacore 2000 surface plasmon resonance (SPR) instrument. All experiments were performed using HBS-P (10 mM HEPES at pH 7.4, 150 mM NaCl, 0.1 mg/ml bovine serum albumin, 0.005% p-20) as running buffer at room temperature. Vh1 or full-length vinculin proteins were covalently attached to a carboxymethyldextran-coated gold surface (CM-5 chip; Biacore). The carboxymethyl groups on the chip were activated with N-ethyl-N′-dimethylaminopropylcarbodiimide and N-hydroxysuccinimide to form the N-hydroxysuccinimide ester of carboxymethyldextran. Vh1 and full-length vinculin proteins were attached to this activated surface by reaction of the carboxyl groups of dextran with the primary amines of Vh1 and full-length protein to form an amide linkage. Any remaining reactive sites on the surface were blocked by reaction with ethanolamine. Reference cells were prepared similarly except that no Vh1 or full-length vinculin proteins were added. Binding was measured by flowing the αVBS peptide at a flow rate of 20 μl/min through the reference and Vh1- or vinculin-containing flow cells in sequence. Blanks consisting of only buffer were also run. After the injection, release of the bound αVBS peptide was measured by flowing only buffer through the flow cells. Data reported are the difference in SPR signal between the flow cells containing Vh1 or vinculin and the reference cells. Any contribution to the signal was removed by subtraction of the blank (buffer) injection from the reference-subtracted signals.
Protease sensitivity assays.
Talin-VBS1 and -VBS3 peptides have been previously described (19, 20). Vinculin (1 μg in 20 mM Tris [pH 7.6], 1.0 mM EDTA, 1.0 mM EGTA, and 10 mM dithiothreitol) was left untreated or was incubated with a 10-fold molar excess of αVBS, talin-VBS1, or talin-VBS3 peptides at room temperature for 15 min. Papain, staphylococcal V8 protease, or chymotrypsin (1:1,000 [wt/wt]) was then added to each sample, and digestion was allowed to proceed at room temperature for 5 or 15 min. The reactions were then immediately quenched by the addition of boiling SDS lysis buffer, and the samples were then further heated at 97°C for 1 min.
Papain-digested peptides were resolved on 12% SDS-polyacrylamide gels, and peptide bands were visualized by silver staining or were blotted onto polyvinylidene difluoride membranes and stained with Coomassie blue. Peptides were directly sequenced on the polyvinylidene difluoride membranes by automated Edman degradation using a PE Applied Biosystems 494cLC protein sequencer. Immunoblotting confirmed the identities of several of these peptides (data not shown). In addition, the identities of these peptides were also determined using combined mass spectrometry-capillary liquid chromatography/tandem mass spectrometry (28) of papain-digested (15 min, quenched by lowering the pH) unbound vinculin or of αVBS-, talin-VBS1-, or talin-VBS3-bound vinculin. These analyses established the identities of all the peptides and confirmed that all were major papain digestion products of vinculin.
As a control, all three of the VBS peptides were also treated with papain, V8, or chymotrypsin and shown not to be substrates of these proteases. Furthermore, these VBS peptides did not affect self-cleavage of these proteases (data shown only for papain).
Accession number.
The atomic coordinates have been deposited in the Protein Data Bank under accession code 1YDI.
RESULTS
α-Actinin provokes novel alterations in vinculin structure.
α-Actinin binds to vinculin, through the agency of an α-helix (αVBS) present within the R4 spectrin repeat of its central rod domain, to vinculin's N-terminal seven-helical bundle domain Vh1 (19, 31). We solved the crystal structure of the Vh1-αVBS complex to 1.8-Å resolution by molecular replacement using the crystal structure of the Vh1 domain as the search model (see Materials and Methods). The structure was refined to 1.8-Å resolution with an R factor of 17% and an Rfree of 23% (Tables 1 and 2).
The crystal structure of the Vh1-αVBS complex demonstrated that one molecule of αVBS binds in an intimate fashion between helices α1 and α2 of the N-terminal helical bundle of Vh1 (Fig. 1, top left). The interactions of αVBS with this bundle are extensive, burying 48% of αVBS's solvent-accessible surface area. Binding of αVBS occurs largely through van der Waals interactions, where the hydrophobic face of αVBS interacts with the hydrophobic core of the Vh1 N-terminal helical bundle (Fig. 1, top right). As a consequence of this interaction, αVBS binding provokes striking and novel alterations in the conformation of the Vh1 N-terminal helical bundle compared to its structure as found in inactive full-length vinculin (see Fig. S1A in the supplemental material). In particular, the insertion of αVBS pushes helix α1 of Vh1 into the Vh1-Vt interface (see Video S1 in the supplemental material) and thus provokes steric hindrance of α1 with Vt; this alteration thus resolves how αVBS displaces Vt from preexisting Vh1-Vt complexes (19). Despite the dramatic changes produced by αVBS in the structure of the N-terminal helical bundle of Vh1, the structure of the C-terminal helical bundle of Vh1 remains essentially unchanged compared to its structure in vinculin's resting state, supporting the notion that this helical bundle is a rigid structure that serves as a scaffold for the dynamic structural alterations that occur in the N-terminal helical bundle of Vh1 (19).
FIG. 1.

Helical bundle conversion of vinculin's Vh1 domain by α-actinin. (Top left) Ribbon representation of vinculin's Vh1 domain (pink) in complex with αVBS (α-actinin residues 737 to 760; cyan). The seven α-helices of the Vh1 domain are labeled (α1 to α7). The N termini of α-helices are marked with a plus sign, while their C termini are indicated with a minus sign, in agreement with the helix dipole moment. The N- and C-terminal helical bundles are labeled (Vh1 N and Vh1 C, respectively). (Top right) Final omit electron density map for αVBS (gray bonds, blue labels) bound to helices α1 and α2 of Vh1 (pink bonds, black labels). The contour level of the electron density map is 2σ, and the resolution is 1.8 Å. αVBS refined to a temperature factor of 36.2 Å2. (Bottom left) Top panel, Cα-trace superposition of helices α1 and α2 of the Vh1-talin-VBS1 structure (gray) onto the Vh1:αVBS structure (pink). Talin-VBS1 bonds are shown in black, and αVBS bonds are shown in cyan. Some Cα positions are numbered, and specific peptide residues are labeled (talin-VBS1 in black and αVBS in blue). While the 2.7-Å Vh1-talin-VBS3 (19) structure is very similar to our human 2.4-Å Vh1-talin-VBS1 structure (20) and to the chicken 2-Å Vh1-talin-VBS1 structure (33), for clarity only the higher-resolution Vh1-talin-VBS1 structure is superimposed onto our Vh1-αVBS structure, but similar results are obtained when superimposing all known Vh1-talin-VBS structures. Bottom panel, Structure-based amino acid sequence alignment of αVBS in its reverse (C- to N-terminal) orientation onto talin-VBS1. Identical residues are in red, and similar residues are in green.
The alterations in Vh1 caused by αVBS binding suggested that they might be similar to those induced by talin's VBSs, which also provoke dramatic alterations in the N-terminal bundle of Vh1, by a process termed helical bundle conversion (19, 20). We therefore directly compared the structures of the Vh1-αVBS complex with those of the Vh1 domain in vinculin's resting state and when in complex with talin-VBS1 (20) and talin-VBS3 (19). Interestingly, superposition of the Vh1-αVBS structure with these structures established that the orientation of the new five-helical bundle provoked by the binding of αVBS relative to the C-terminal bundle is in fact very different from that of the five-helical bundle provoked by the binding of talin-VBS1 (see Fig. S1B in the supplemental material) or VBS3 (see Fig. S1C in the supplemental material). Indeed, the relative orientation of the N- and C-terminal bundles of the novel Vh1-αVBS structure is actually more similar to that of the Vh1 structure as seen in inactive full-length vinculin (see Fig. S1A in the supplemental material). Nonetheless, the solvent-exposed surface area buried upon binding the α-helix of αVBS (residues 738 to 760) is of the same magnitude as that which is buried upon binding talin's VBS1 (approximately 1,200 Å2) (20).
α-Actinin binds to vinculin in an inverted orientation relative to talin.
The structure of the Vh1-αVBS complex also revealed, quite surprisingly, that the N- to C-terminal orientation of the αVBS peptide is the opposite of that observed for talin-VBS1 (20, 33), talin-VBS2 (15), or talin-VBS3 (19) when αVBS inserts into the N-terminal helical bundle of Vh1. Indeed, the structure indicates that the α-helix of αVBS is antiparallel to α1 and parallel to α2 of Vh1, while talin's VBSs lie parallel to α1 and antiparallel to α2 (Fig. 1, bottom left). A sequence alignment of human talin-VBS1 (residues 607 to 627) and talin-VBS3 (residues 1949 to 1969) reveals six identical amino acids and eight similar residues. Only two of these residues (Leu-608 and Ala-612 in VBS1), however, are conserved in talin-VBS2 (residues 855 to 875) (20, 31), which binds to Vh1 with lower affinity than VBS1 or VBS3 (Kd of 32 nM for talin-VBS2 versus 15 nM and 3 nM for VBS1 and VBS3, respectively) (20). Alignment of all three of talin's VBSs with αVBS in its conventional N- to C-terminal orientation (residues 741 to 762 with an insertion at 758; alignment not shown) shows that these leucine and alanine residues are also conserved in αVBS, and there are an additional five residues that show strong similarity. However, alignment of the primary sequence of αVBS in its reverse orientation with talin-VBS1 generates the identical structure-based alignment obtained by superposition of Vh1 helices α1 and α2 (Fig. 1, bottom left). Indeed, inverting the direction of αVBS results in a much better alignment with talin-VBS1 (three identical residues and eight similar residues) (Fig. 1, bottom left) than when αVBS is positioned in an N- to C-terminal orientation (two identical and five similar residues).
Structures comprising two subdomains that move relative to one other can, of course, be superimposed in several ways. When comparing the structures of the inactive Vh1 domain with those of its activated forms, we chose to superimpose the α-helices of the C-terminal helical bundle of Vh1 (19, 20), which appears to act like an anchor as it does not move in these structures and which does not contribute to the binding of Vt or of talin's or α-actinin's VBSs. Further, it is the N-terminal helical bundle of Vh1 that undergoes helical bundle conversion, which alters this structure from a four-helical bundle (as seen in vinculin's inactive conformation) to a five-helical bundle (when vinculin is in its activated forms). In this scenario, one can envision the C-terminal helical bundle of Vh1 as the wrist of a hand and the N-terminal helical bundle of Vh1 as the fingers that grab onto the amphipathic α-helices of either talin's or α-actinin's VBSs. This superposition also takes into account the fact that vinculin is a protein of five domains, rather than just considering an isolated small portion of the molecule. However, to focus on the localized changes and the specific differences in the interactions of Vh1 with talin-VBS1 versus αVBS binding in its inverted orientation, independent of vinculin's additional four domains, we superimposed the helices α1 and α2 of these two structures (Fig. 1, bottom left; see Fig. S1 in the supplemental material). Indeed, when superimposed in this fashion, the α1 and α2 helices of Vh1 that sandwich either talin-VBS1 or αVBS open up in a similar fashion to accommodate these binding partners, and talin-VBS1 or αVBS also binds in a similar manner. However, not only does αVBS run in the opposite direction compared to talin-VBS1, but the αVBS α-helix is also shifted by about 2.2 Å compared to the position of the talin-VBS1 α-helix when bound to Vh1 (Fig. 1, bottom left). This relative movement of the α-helix of αVBS thus causes the unique structural differences seen by its binding to Vh1 versus those triggered by the binding of talin's VBSs.
These structural comparisons are in agreement with our native gel analyses of the Vh1-Vt, Vh1-VBS1, Vh1-VBS3, and Vh1-αVBS complexes. Specifically, all of these VBSs displace Vt from preexisting Vh1-Vt complexes to generate uniquely migrating Vh1-containing complexes (19, 20), and all of the interactions of these VBSs with Vh1 are mutually exclusive, including that of αVBS versus talin's VBSs (references 19 and 20 and data not shown). Our supplemental videos model and underscore the very distinct movements that we propose occur upon displacement of Vt by either αVBS (see Video S1 in the supplemental material) or talin-VBS1 (see Video S2 in the supplemental material). Specifically, the binding of these two VBS peptides triggers very distinct movements that distort the Vh1-Vt interface in unique ways to generate unique five-helical bundle domains.
The VBS of α-actinin has high affinity for vinculin.
The structure of the Vh1-αVBS complex revealed that a substantial portion (48%) of the total available surface area of αVBS is buried upon its insertion between helices α1 and α2 of vinculin's Vh1 domain (Fig. 1, top right). The intimate nature of these hydrophobic interactions was very reminiscent of those seen with the interactions of talin's VBSs with the Vh1 domain (15, 19, 20, 33), which bind to vinculin's Vh1 domain with high affinity, with Kds that range from ∼3 nM (VBS3) to ∼30 nM (VBS2) (20). To determine the affinity of the interaction between αVBS and vinculin's Vh1 domain, we performed surface plasmon resonance assays. As expected from the structure of the Vh1-αVBS complex, these analyses demonstrated a single, high-affinity binding site for VBS1 in the Vh1 domain, with a Kd of 1.7 nM (Fig. 2, top).
FIG. 2.

Binding affinities of αVBS for vinculin. Biacore surface plasmon resonance was used to measure the affinity of αVBS. Biotinylated Vh1 or full-length human vinculin was captured on a carboxymethyldextran-coated gold surface (CM-5 chip; Biacore). αVBS peptide was injected over the reference and Vh1- or vinculin-immobilized cells in sequence, and release of bound αVBS peptide was determined. Responses were referenced for reference flow cell and blank injections of buffer. Representative sensorgrams are shown. (Top) Affinity of αVBS for vinculin's Vh1 domain (residues 1 to 258). Binding responses were fit using a mass transport model, and the calculated Kd is shown in the inset. (Bottom) Affinity of αVBS for full-length human vinculin (residues 1 to 1066). Binding responses were fit using a simple model and the calculated Kd is shown in the inset. The signal appears noisier for full-length vinculin compared to Vh1 because of the lower levels of vinculin on the surface, resulting in lower binding capacity.
Vinculin activation has been suggested to require two or more rather low-affinity ligands to collectively provide the free energy necessary to break the nanomolar intramolecular interactions of vinculin's Vt domain with those in its head, which principally involve hydrophobic interactions with Vh1 and several electrostatic interactions with the Vh3 seven-helical bundle domain (5). Measurement of the affinity of the interaction of full-length, native α-actinin with full-length, native vinculin by low-speed equilibrium centrifugation has indicated a Kd of only 1.3 × 10−5 (31). However, this assay measures the affinity of these two partners only in their inactive states, whereas the αVBS-Vh1 complex captures the activated states of both of these partners (see below). The intramolecular hydrophobic interactions of vinculin's Vh1 and Vt domains occur at a distance (19) from the surface that mediates the interaction of αVBS with helices α1 and α2 of Vh1 (Fig. 1; see Video S1 in the supplemental material). This finding suggested that, when in its activated state, the VBS of α-actinin might also have a high affinity for full-length vinculin. Remarkably, when measured by surface plasmon resonance, the Kd of the interaction of αVBS with full-length vinculin was essentially equivalent to that for vinculin's Vh1 domain (with a Kd of 1.78 nM) (Fig. 2, bottom). Therefore, the VBS of α-actinin binds to vinculin with an affinity that is comparable to and likely higher than that of the intramolecular interaction of vinculin's Vh1 and Vt domains (4).
The αVBS of α-actinin activates vinculin.
The high-affinity interaction of αVBS for full-length vinculin suggested that it would efficiently disrupt intramolecular interactions that hold vinculin in its inactive state (2, 5). The crystal structure of full-length inactive human vinculin demonstrated that Vt is clamped by its interactions with the Vh1 and Vh3 domains (5). To recapture the essential components and regulation of vinculin's intramolecular interaction and to test whether αVBS, and talin's VBSs were indeed alone sufficient to disrupt the extensive interactions of Vt with these domains in the context of a near-full-length molecule, we generated a recombinant human vinculin protein that contains most of the vinculin head (termed VH, residues 1 to 780, comprising Vh1, Vh2, Vh3, and most of Vt2) yet lacks the proline-rich hinge region and Vt. The VH protein was then tested for its ability to form complexes with the Vt domain and with the VBSs of α-actinin and talin. As expected, both Vt and all three VBSs bound avidly to VH, as was evident by the formation of distinct VH-Vt, VH-VBS1, VH-VBS3, and VH-αVBS complexes on native polyacrylamide gels (Fig. 3), which were confirmed by SDS-polyacrylamide gel electrophoresis of these complexes (data not shown). Importantly, the addition of αVBS, talin-VBS1, or talin-VBS3 peptides (at a fourfold molar excess) to preexisting VH-Vt complexes demonstrated that all three of these VBSs were sufficient to displace Vt from preexisting VH-Vt complexes (Fig. 3). Therefore, in solution, the VBSs of α-actinin or talin are alone sufficient to efficiently disrupt the nanomolar intramolecular interactions of Vt with vinculin's Vh1 and Vh3 domains, which normally hold vinculin in its inactive state.
FIG. 3.

Binding of talin-VBS1, talin-VBS3, or αVBS peptides to vinculin's head domain (VH, human residues 1 to 780) is sufficient to displace Vt from preexisting VH-Vt complexes. Native polyacrylamide gel electrophoresis analysis of free VH (lane 1) and of VH bound to talin-VBS1 (lane 2), talin-VBS3 (lane3), αVBS (lane 4), or Vt (human vinculin residues 879 to 1066; lane 5) is shown. Free VH is indicated with an asterisk. Talin-VBS1, talin-VBS3, and αVBS were then added to preexisting VH-Vt complexes (circle) at a fourfold molar excess and displaced Vt to form the respective VH-talin-VBS1 (lane 6), VH-talin-VBS3 (lane 7), or VH-αVBS (lane 8). Free Vt is not visible in native gels because of its basic pI.
αVBS and talin's VBSs alter the conformation of full-length vinculin.
The ability of αVBS and talin's VBSs to displace Vt from preexisting Vh1-Vt complexes (19) and the novel structures of the N-terminal helical bundle of the Vh1 domain when bound by αVBS versus talin's VBSs (Fig. 1; see Videos S1 and S2 in the supplemental material) suggested that αVBS and talin's VBSs might also alter the conformation of full-length vinculin. To test if αVBS or talin's VBSs could indeed affect the flexibility or conformation of vinculin, we performed limited papain proteolysis of full-length vinculin and of full-length vinculin bound to αVBS, talin-VBS1, or talin-VBS3 (Fig. 4). Papain did not cleave any of the VBSs alone, and these peptides did not influence the overall activity of papain, as judged by self-cleavage of the protease (Fig. 4, top right). Importantly, αVBS-, talin-VBS1-, and talin-VBS3-bound vinculin all displayed accelerated rates of proteolysis as judged by the appearance of the 90-kDa head of vinculin (Fig. 4, top left), which is linked to Vt by the proline-rich hinge region, suggesting an overall increase in flexibility in their structures that made them more susceptible to cleavage.
FIG. 4.

αVBS and talin's VBSs provoke unique conformational alterations in full-length vinculin. (Top left) αVBS and talin's VBS1 and VBS3 alter the sensitivity of human full-length vinculin to papain cleavage. Full-length human vinculin protein was left untreated or was bound with αVBS, talin-VBS1, or talin-VBS3 peptides for 15 min. The proteins were then treated with papain, and at the indicated intervals digestion products of vinculin were analyzed by polyacrylamide gel electrophoresis and silver staining of the gels. αVBS-, talin-VBS1-, and talin-VBS3-bound vinculin were much more susceptible to papain cleavage at Met-350 and Ala-613, indicating that binding of all of these VBSs exposes regions buried in the inactive vinculin structure (5). The arrow indicates full-length (uncleaved) vinculin. (Top right) αVBS and talin's VBS1 and VBS3 are not substrates of papain. αVBS, talin-VBS1, or talin-VBS3 peptides were treated with papain and after 15 min were analyzed by polyacrylamide gel electrophoresis and silver staining of the gels. αVBS, talin-VBS1, and talin-VBS3 were not cleaved by papain. Further, there were no obvious effects of these VBSs on papain activity, as levels of self-digestion products of papain (which were very minor) were essentially unaffected. (Middle) Densitometric scans of papain cleavage products of unbound vinculin or of αVBS- or talin-VBS3-bound vinculin. The cleavage sites were identified by mass spectrometry, N-terminal sequencing, and immunoblotting with anti-His tag antibody (data not shown). The scans of talin-VBS1- andtalin-VBS3-bound vinculin were essentially equivalent (data not shown). By contrast, note the very distinct patterns of papain digestion products of αVBS- versus talin-VBS3-bound vinculin, in particular at Ala-490, which resides in the interface of the Vh2-Vt interaction in inactive vinculin (5) and which is especially prominent in talin-VBS3-bound vinculin. (Bottom) Schematic representation of the locations of the papain cleavage sites and their cleavage efficiency in inactive and αVBS- or talin-VBS3-bound vinculin. Plus signs indicate the relative efficiency of papain cleavage at the indicated sites (e.g., at Met-350 and Ala-613, papain cleavage was 16-fold more efficient in αVBS-bound vinculin than in inactive vinculin). The results shown are representative of those from eight independent experiments.
Interestingly, papain digestion of αVBS-, talin-VBS1-, or talin-VBS3-bound vinculin also revealed that distinct regions of the protein were much more susceptible to proteolytic cleavage than inactive vinculin (Fig. 4, middle). Identification of the cleavage sites, by mass spectrometry and N-terminal sequencing of these peptide fragments (data not shown), revealed that inactive vinculin was really only significantly susceptible to cleavage at two residues (Leu-854 and Asp-856), both within the flexible proline-rich region of the molecule which is disordered in the structure of full-length inactive vinculin (5). In contrast, cleavage of αVBS-, talin-VBS1-, or talin-VBS3-bound vinculin revealed shared cleavage sites at Met-350, which resides at the C terminus of helix α3 of the Vh2 domain, and at Ala-613, which lies within the four-helical N-terminal bundle of the Vh3 domain (Fig. 4, bottom). Significantly, in the closed conformation of vinculin Ala-613 resides on α-helix α4, which is buried in the hydrophobic core of this helical bundle, where it engages in intradomain van der Waals interactions with Phe-626, Ala-679, and Leu-682 (5). Thus, binding to either αVBS or talin's VBSs opens up the Vh3 helical bundle.
The protease cleavage patterns of talin-VBS1- and talin-VBS3-bound vinculin were essentially equivalent (Fig. 4, top left), in agreement with their nearly identical effects on the structure of the Vh1 domain (19, 20). However, obvious differences were evident in talin- versus α-actinin-bound vinculin when the peptide fragments of talin-VBS1- or talin-VBS3-bound vinculin were compared to those generated in αVBS-bound vinculin. Specifically, Ala-490, which resides in the loop between the Vh2 and Vh3 domains, was much more susceptible to cleavage in talin-VBS3-bound vinculin than in αVBS-bound vinculin (Fig. 4, middle). In inactive vinculin, Ala-490 resides at the interface of the Vh2-Vt interdomain interaction (5), and thus displacement of Vt by the binding of talin's VBSs to the Vh1 domain must also disrupt the Vh2-Vt interface, but this does not occur to a significant extent in αVBS-bound vinculin. Therefore, the unique structural alterations in the Vh1 domain caused by the binding of talin's VBSs versus αVBS (Fig. 1) appear to also cause unique conformational changes in far-distant regions of the entire molecule.
To address whether differences in the conformation of αVBS- versus talin-VBS-bound vinculin were also evident with other proteases, we also treated vinculin bound by αVBS, talin-VBS1, or talin-VBS3 with the proteases V8 and chymotrypsin. The V8 protease of Staphylococcus aureus initially cleaves vinculin (twice) within its proline-rich hinge region that links Vt to the reminder of the molecule, and indeed this cleavage first defined the intramolecular interactions of vinculin's head and tail domain (23). As judged by the appearance of the 90-kDa head fragment, talin-VBS1- and talin-VBS3-bound vinculin had accelerated rates of cleavage compared to αVBS-bound vinculin (see Fig. S2A in the supplemental material). Furthermore, αVBS-bound vinculin displayed some unique chymotrypsin cleavage products compared to talin-VBS-bound vinculin (see Fig. S2B in the supplemental material). Collectively, the protease data are consistent with the notions that binding of talin's or α-actinin's VBSs appears to alter the overall conformation of full-length vinculin and, further, that their binding appears to alter far-distant regions of the molecule in unique ways.
The activated conformation of α-actinin.
An obvious paradox presented by our Vh1-αVBS structure was that dynamic changes in structure appear to also occur on the α-actinin end, which would allow the amphipathic α-helix of αVBS to bind to vinculin. In their native structure the R1-R4 spectrin repeats contain three helices, “A,” “B,” and “C,” which are connected by two loops that have been predicted to serve as hinges that open up to unfold the triple helix (1). The αVBS helix within the R4 spectrin repeat represents the “C” helix, and in native α-actinin the αVBS helix makes intramolecular contacts with α-helices A and B of the R4 repeat and intermolecular contacts with helices B and C in the R1 helical bundle that dimerizes with R4 (Fig. 5, top). Strikingly, the seven amino-terminal residues of αVBS present in native α-actinin are not in a helical conformation in the Vh1-αVBS structure, and this alteration in the length and structure of the αVBS helix prevents steric hindrance of bound αVBS with the α1-α2 loop of Vh1 (not shown). Indeed, superposition of αVBS as seen in the α-actinin rod structure onto αVBS when bound to vinculin places the α1 helix of Vh1 on the R4 C helix, whereas the R4 B helix is placed between Vh1 helices α3 and α4 but more proximal to α4 (not shown).
FIG. 5.

Proposed movements of α-actinin's VBS that allow binding to vinculin. (Top) Superposition of α-actinin's vinculin binding site as seen in the native α-actinin rod crystal structure (40) (PDBID 1HCI; gray and yellow) onto αVBS as when bound to the N-terminal helical bundle of Vh1 (cyan). In the dimer of the α-actinin rod, the C-terminal spectrin repeat that contains αVBS (R4, yellow) of one protomer interacts with the N-terminal spectrin repeat (R1, gray) of the other protomer. Residues involved in binding of αVBS to vinculin are shown. In α-actinin's native state, these residues are oriented to the inside of the hydrophobic R4 repeat. The proposed movement that occurs to allow the binding of αVBS to Vh1 is indicated. (Bottom) Model for the binding of the full-length α-actinin dimer (ribbon drawing) to two full-length vinculin molecules (surface representation). The αVBS helix (red) within the R4 spectrin repeat of α-actinin's rod (gray) swings out from its packed position in the α-actinin dimer to expose its hydrophobic face and interact with the N-terminal helical bundle of the Vh1 domain of vinculin (pink). Based on the full-length α-actinin cryo-electron microscopy structure (29) (PDBID 1SJJ), the proposed movements of αVBS also move the C-terminal EF hand domain (black) without steric hindrance. αVBS binding displaces the five-helical bundle of Vt (cyan), which is connected to a four-helical bundle, Vt2 (dark blue), by a proline-rich linker (gray). The two other seven-helical bundles of vinculin, Vh2 (yellow) and Vh3 (orange), are also shown.
Electron microscopy has shown that monomers of vinculin are bound at the ends of the rod region of the antiparallel α-actinin dimer, and this interaction site was mapped, in solution, to the R4 repeat (31). This observation, together with the crystal structures of the α-actinin rod (40) and our Vh1-αVBS complex (Fig. 1), therefore suggests that the αVBS helix swings out from its packed position within the R4 spectrin repeat to expose its hydrophobic face to interact with vinculin. Further, this movement would be predicted to occur without disturbing the α-actinin dimer, as it leaves the intermolecular interactions of the R2 and R3 repeats of the rod structurally intact (Fig. 5, bottom) and since α-actinin exists as a dimer in the vinculin-α-actinin complex (31). In support of the proposed independent movement of the R4 repeat of α-actinin when bound to vinculin without affecting the dimerization of its R2 and R3 repeats, it has been recently shown that the spectrin repeats of avian α-spectrin are capable of independent movements without affecting the dimerization of adjacent repeats (27).
In spectrin, the formation of stable unfolding intermediates, provoked by mechanical stress, involves the induction of a kink in helix B at a conserved proline residue that essentially splits this helix in two and which generates a loop that might facilitate the unfolding of the triple helix and/or stabilize the unfolded intermediate. In addition, the lengths of the α-helices in the unfolded intermediates of spectrin are dynamic (1). We propose that a similar model may apply to the α-actinin-vinculin interaction, where alterations in the structure of the α-actinin rod caused by, for example, mechanical stress might provide the free energy necessary to induce the αVBS helix to break and swing free of its hydrophobic contacts to then allow it to bind to vinculin (Fig. 5). This event may also generate an unfolded intermediate having a shorter αVBS helix, which could then bind in an unconstrained manner to the α1 and α2 helices of Vh1. In support of this notion, we note the presence of a conserved proline residue (Pro-683) in the B helix of R4, which could contribute to the formation of stable unfolded intermediates in α-actinin in a fashion akin to the conserved proline residue of the B helix of spectrin (1).
DISCUSSION
Vinculin is activated by α-actinin and talin in adhesion signaling.
Collectively, these studies and our previous findings (5, 19, 20) suggest a role for both α-actinin and talin in triggering vinculin activation in signaling pathways induced by adherens junctions (α-actinin) and focal adhesions (both α-actinin and talin). Interestingly, while our structures reveal that the VBSs of both α-actinin and talin insert between helices α1 and α2 of the N-terminal helical bundle of vinculin's Vh1 domain, they also suggest that α-actinin and talin may provoke distinct structural changes in the overall conformation of vinculin, and in part this could be due to the inverted orientation of their VBSs when they bind to vinculin and the unique structural alterations that they induce when bound to vinculin's Vh1 domain (Fig. 1, bottom left). Nonetheless, the binding of talin's and α-actinin's VBSs does result in a shared outcome, the activation of vinculin by displacing Vt from a distance. These findings establish the remarkable flexibility of the Vh1 domain, which is now shown to take on distinct identities when bound to different partners. Further, given the apparent differences in the conformation of full-length vinculin when bound by talin's VBSs versus αVBS, we suggest that these specific alterations may bestow on vinculin the ability to bind distinct partners in adherens junctions versus focal adhesions.
The key molecular event in vinculin activation is generally agreed to be the severing of the Vh1-Vt interaction (16, 23), which directs the most extensive interdomain contacts within the inactive full-length protein (5). PIP2, αVBS, and talin's VBSs are all individually capable of disrupting this interaction in vitro (16, 19, 20), but which ligand(s) activates vinculin under physiological conditions remains to be resolved (38). The binding site for PIP2 is partially occluded in the structure of inactive full-length vinculin, suggesting that binding of PIP2 would logically follow that of other partners that initially open up the protein (2). We suspect, based on our findings, that the VBSs of α-actinin and talin may be mediators of this initial response. First, their binding to Vh1 displaces Vt from preexisting Vh1-Vt complexes (19, 20), and they also displace Vt's interaction with the head domain (Fig. 3), indicating that they alone are sufficient to sever the Vh1-Vt interaction. Second, all of these VBSs provoke extraordinary changes in the structure of the N-terminal helical bundle of Vh1, which sterically displace Vt from afar by altering the structures of α-helices within this four-helical bundle to create entirely new five-helical bundle structures. Third, although these VBSs are predicted to have relatively low affinity for Vh1 when buried in their full-length rod structures (2), when free, as predicted in their activated states (Fig. 5, bottom) (15, 19, 20, 33), the binding affinities of these VBSs for Vh1 are indeed higher (20) (Fig. 2) than that of Vt (16). Finally, αVBS and talin's VBSs binding alone also appears to alter the conformation of full-length vinculin in vitro (Fig. 4; see Fig. S2 in the supplemental material). Nonetheless, in its native state the Vh1 and Vt domains are covalently linked, a condition that should increase the affinity of Vh1-Vt interaction. Thus, it has been proposed that activation of vinculin may be allosteric, where the binding of two or more ligands, for example, αVBS and PIP2, would be required in concert to generate the free energy necessary to break the high-affinity Vh1-Vt interaction (2, 38). While more studies are clearly required to resolve this debate, we suggest, given the ability of αVBS or talin's VBSs to alter the sensitivity of full-length vinculin to various proteases, that these VBSs, once unraveled from their buried locations within the helical bundles of α-actinin's or talin's rods, may trigger initial changes in vinculin structure that allow it to bind to other ligands such as PIP2. This, in turn, might then confer the ability of vinculin to interact with other partners that reorganize the actin cytoskeleton in adherens junctions and focal adhesions.
Structural alterations as effectors in adhesion signaling.
A striking consequence of the structure of the Vh1-αVBS complex is that it suggests that alterations in α-actinin structure first occur to allow it to bind to vinculin (Fig. 5, bottom). Comparisons of our Vh1-αVBS structure to that of the twisted antiparallel α-actinin rod (13, 29, 40) suggest that the αVBS α-helix found in one protomer of the R4 spectrin repeat (a three-helical bundle) of the α-actinin dimer first unravels from its buried location within the triple helix to allow it to bind between helices α1 and α2 of the N-terminal helical bundle of Vh1, where it then becomes part of a new five-helical bundle structure. This is proposed to occur without affecting the overall structure of the α-actinin dimer (Fig. 5, bottom), as vinculin monomers are bound at the R4 repeat of the rod of the antiparallel α-actinin dimer (31). Interestingly, cryoelectron microscopy has recently revealed that the molecular ends of the α-actinin containing the actin-binding and calmodulin-like domains have different structures and orientations (29). Specifically, the two calponin homology domains, CH1 and CH2, comprising the actin-binding domains, have a closed conformation on one amino terminus and an open conformation on the other, due to domain swapping. This results in different orientations of the EF hand domains that follow the αVBS α-helix in the R4 spectrin repeat on either carboxyl terminus. The linker between αVBS and the EF hand domains together with its flexibility, due to free rotations in the CH1 and CH2 domains and the observed rotations of the EF hand domains (29), would then facilitate the binding of αVBS to vinculin's Vh1 domain, without steric hindrance of the CH domains in either their closed or open conformations (Fig. 5, bottom). Such a scenario would preserve the ability of the α-actinin rod to direct cross-linking of actin filaments, while also providing direct links to vinculin and to cadherin and integrin receptors.
What signals may trigger the proposed unraveling of αVBS from its buried location in the R4 spectrin repeat of α-actinin are not resolved. One possibility is that vinculin itself plays an active role in this response, where binding of the N-terminal helical bundle of the Vh1 domain could conceivably provoke changes in the structure of the R4 repeat that allows the αVBS to swing out from its buried location. We feel that this scenario is unlikely, as vinculin activation requires that the intramolecular Vh1-Vt interaction must first be severed to allow Vh1 (and Vt) to bind to its partners. More likely triggers that might facilitate unraveling of the αVBS could include modifications of α-actinin itself, as phosphorylations of α-actinin (18) and/or binding of polyphosphoinositides (17) both occur following the formation of adhesion junctions. Alternatively, the signal could be mechanical stress, as this is sufficient to unravel the highly related repeats of spectrin, which can then form stable intermediates (1). Indeed, from their structure (e.g., the conserved proline residue in the B helix of R4), the spectrin repeats of α-actinin are predicted to form similar unfolded intermediates in response to sheer stress, and α-actinin binds to cadherin and integrin receptors in adherens junctions and focal adhesions, respectively, where mechanical stress plays a major role in activation of these receptors (21, 25, 34). If this scenario applies here, mechanical stress caused by ligation of these receptors would be transferred to bound α-actinin, and this stress might then provoke αVBS in the R4 repeat to swing out and allow its binding to vinculin.
We also propose that these findings, and previous biochemical studies (19, 20), suggest an order and mechanism for how α-actinin acts as a signaling effector to activate vinculin in adhesion pathways. Specifically, we suggest that structural alterations in α-actinin's rod are an initiating event that allows αVBS to swing free from its buried location to bind to the Vh1 domain of vinculin. A similar scenario has recently been proposed in the activation of talin-VBS1 and talin-VBS2 in focal adhesions, which are also buried in α-helical bundle domains (15, 33). However, at least for talin-VBS1, the rules that apply appear to be quite different, as the structure of VBS1 in its native five-helical bundle is remarkably similar to its structure in the five-helical bundle created when in complex with the N-terminal helical bundle of Vh1 (33). In contrast, in the α-actinin-vinculin interaction the αVBS α-helix undergoes fairly drastic changes in its helical architecture and length, to shift from being buried within the three-helical bundle of α-actinin's R4 repeat to then create an entirely new five-helical bundle Vh1 structure.
αVBS binding provokes dramatic conformational changes in Vh1 that displace Vt, which in the context of full-length vinculin would be predicted to release its five domains and allow vinculin to be free to interact with other binding partners. Thus, collectively, these findings suggest a domino effect of structural alterations in adhesion signaling, whereby alterations in the structure of one protein (α-actinin) might then provoke changes in the structure and function of another (vinculin), in effect a chain reaction of structural signals.
Supplementary Material
Acknowledgments
We are indebted to John Cleveland for many helpful discussions and for critical review of the manuscript, to David Myszka for surface plasmon resonance assays, and to Gerard Bricogne for continuous advice. We thank our in-house Hartwell Center for peptide synthesis, mass spectrometry, and N-terminal sequencing; the Hauptman-Woodward Institute for determining the initial crystallization conditions; Charles Ross for maintaining the computing facilities; Christina Rush for technical assistance; and the staff at the Advanced Photon Source (APS SBC-CAT ID beamline) for synchrotron support.
This work was supported in part by National Institutes of Health grant GM071596, Cancer Center Support (CORE) grant CA21765, and the American Lebanese Syrian Associated Charities (ALSAC). P.R.J.B. is a Van Vleet Fellow.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Altmann, S. M., R. G. Grunberg, P. F. Lenne, J. Ylanne, A. Raae, K. Herbert, M. Saraste, M. Nilges, and J. K. Horber. 2002. Pathways and intermediates in forced unfolding of spectrin repeats. Structure 10:1085-1096. [DOI] [PubMed] [Google Scholar]
- 2.Bakolitsa, C., D. M. Cohen, L. A. Bankston, A. A. Bobkov, G. W. Cadwell, L. Jennings, D. R. Critchley, S. W. Craig, and R. C. Liddington. 2004. Structural basis for vinculin activation at sites of cell adhesion. Nature 430:583-586. [DOI] [PubMed] [Google Scholar]
- 3.Bakolitsa, C., J. M. de Pereda, C. R. Bagshaw, D. R. Critchley, and R. C. Liddington. 1999. Crystal structure of the vinculin tail suggests a pathway for activation. Cell 99:603-613. [DOI] [PubMed] [Google Scholar]
- 4.Bass, M. D., B. J. Smith, S. A. Prigent, and D. R. Critchley. 1999. Talin contains three similar vinculin-binding sites predicted to form an amphipathic helix. Biochem. J. 341:257-263. [PMC free article] [PubMed] [Google Scholar]
- 5.Borgon, R. A., C. Vonrhein, G. Bricogne, P. R. J. Bois, and T. Izard. 2004. Crystal structure of human vinculin. Structure 12:1189-1197. [DOI] [PubMed] [Google Scholar]
- 6.Bricogne, G. 1997. The Bayesian viewpoint of crystallography: basic concepts and applications. Methods Enzymol. 276:361-423. [DOI] [PubMed] [Google Scholar]
- 7.Broderick, M. J. F., and S. J. Windes. 2002. Towards a complete atomic structure of spectrin family proteins. J. Struct. Biol. 137:184-193. [DOI] [PubMed] [Google Scholar]
- 8.Bubeck, P., S. Pistor, J. Wehland, and B. M. Jockusch. 1997. Ligand recruitment by vinculin domains in transfected cells. J. Cell Sci. 110:1361-1371. [DOI] [PubMed] [Google Scholar]
- 9.Collaborative Computational Project. 1994. The CCP4 suite: programs for protein crystallography. Collaborative Computational Project, no. 4. Acta Crystallogr. D50:760-763. [DOI] [PubMed] [Google Scholar]
- 10.Critchley, D. R. 2000. Focal adhesions—the cytoskeletal connection. Curr. Opin. Cell Biol. 12:133-139. [DOI] [PubMed] [Google Scholar]
- 11.Diedrichs, K., and A. Karplus. 1997. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 4:269-275. [DOI] [PubMed] [Google Scholar]
- 12.Djinovic-Carugo, K., M. Gautel, J. Ylanne, and P. Young. 2002. The spectrin repeat: a structural platform for cytoskeletal protein assemblies. FEBS Lett. 513:119-123. [DOI] [PubMed] [Google Scholar]
- 13.Djinovic-Carugo, K., P. Young, M. Gautel, and M. Saraste. 1999. Structure of the α-actinin rod: molecular basis for cross-linking of actin filaments. Cell 98:537-546. [DOI] [PubMed] [Google Scholar]
- 14.Evans, P. R. 1987. Postrefinement of oscillation camera data, p. 58-66. In J. R. Helliwell, P. A. Machin, and M. Z. Papiz (ed.), Proceedings of the Daresbury Study Weekend at Daresbury Laboratory, 23-24 January. Daresbury Laboratory, Warrington, United Kingdom.
- 15.Fillingham, I., A. R. Gingras, E. Papagrigoriou, B. Patel, J. Emsley, D. R. Critchley, G. C. Roberts, and I. L. Barsukov. 2005. A vinculin binding domain from the talin rod unfolds to form a complex with the vinculin head. Structure 13:65-74. [DOI] [PubMed] [Google Scholar]
- 16.Gilmore, A. P., and K. Burridge. 1996. Regulation of vinculin binding to talin and actin by phosphatidyl-inositol-4-5-bisphosphate. Nature 381:531-535. [DOI] [PubMed] [Google Scholar]
- 17.Greenwood, J. A., A. B. Theibert, G. D. Prestwich, and J. E. Murphy-Ullrich. 2000. Restructuring of focal adhesion plaques by PI 3-kinase. Regulation by PtdIns (3,4,5)-p(3) binding to alpha-actinin. J. Cell Biol. 150:627-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Izaguirre, G., L. Aguirre, P. Ji, B. Aneskievich, and B. Haimovich. 1999. Tyrosine phosphorylation of alpha-actinin in activated platelets. J. Biol. Chem. 274:37012-37020. [DOI] [PubMed] [Google Scholar]
- 19.Izard, T., G. Evans, R. A. Borgon, C. L. Rush, G. Bricogne, and P. R. Bois. 2004. Vinculin activation by talin through helical bundle conversion. Nature 427:171-175. [DOI] [PubMed] [Google Scholar]
- 20.Izard, T., and C. Vonrhein. 2004. Structural basis for amplifying vinculin activation by talin. J. Biol. Chem. 279:27667-27678. [DOI] [PubMed] [Google Scholar]
- 21.Jiang, G., G. Giannone, D. R. Critchley, E. Fukumoto, and M. P. Sheetz. 2003. Two-piconewton slip bond between fibronectin and the cytoskeleton depends on talin. Nature 424:334-337. [DOI] [PubMed] [Google Scholar]
- 22.Jockusch, B. M., P. Bubeck, K. Giehl, M. Kroemker, J. Moschner, M. Rothkegel, M. Rudiger, K. Schluter, G. Stanke, and J. Winkler. 1995. The molecular architecture of focal adhesions. Annu. Rev. Cell Dev. Biol. 11:379-416. [DOI] [PubMed] [Google Scholar]
- 23.Johnson, R. P., and S. W. Craig. 1995. F-actin binding site masked by the intramolecular association of vinculin head and tail domains. Nature 373:261-264. [DOI] [PubMed] [Google Scholar]
- 24.Jones, T. A., J.-Y. Zou, S. W. Cowan, and M. Kjeldgaard. 1991. Improved methods for building models into electron density maps and the location of errors in these models. Acta Crystallogr. A 47:110-119. [DOI] [PubMed] [Google Scholar]
- 25.Kim, M., C. V. Carman, and T. A. Springer. 2003. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science 301:1720-1725. [DOI] [PubMed] [Google Scholar]
- 26.Kroemker, M., A. H. Rudiger, B. M. Jockusch, and M. Rudiger. 1994. Intramolecular interactions in vinculin control α-actinin binding to the vinculin head. FEBS Lett. 355:259-262. [DOI] [PubMed] [Google Scholar]
- 27.Kusunoki, H., G. Minasov, R. I. Macdonald, and A. Mondragon. 2004. Independent movement, dimerization and stability of tandem repeats of chicken brain alpha-spectrin. J. Mol. Biol. 344:495-511. [DOI] [PubMed] [Google Scholar]
- 28.Link, A. J., J. Eng, D. M. Schieltz, E. Carmack, G. J. Mize, D. R. Morris, B. M. Garvik, and J. R. Yates III. 1999. Direct analysis of protein complexes using mass spectrometry. Nat. Biotechnol. 17:676-682. [DOI] [PubMed] [Google Scholar]
- 29.Liu, J., D. W. Taylor, and K. A. Taylor. 2004. A 3-D reconstruction of smooth muscle α-actinin by CryoEM reveals two different conformations at the actin-binding region. J. Mol. Biol. 338:115-125. [DOI] [PubMed] [Google Scholar]
- 30.Lu, M. H., C. DiLullo, T. Schultheiss, S. Holtzer, M. Murray, J. Choi, D. A. Fischman, and H. Holtzer. 1992. The vinculin/sarcomeric-alpha-actinin/alpha-actin nexus in cultured cardiac myocytes. J. Cell Biol. 117:1007-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McGregor, A., A. D. Blanchard, A. J. Rowe, and D. R. Critchley. 1994. Identification of the vinculin-binding site in the cytoskeletal protein α-actinin. Biochem. J. 301:225-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otey, C. A., and O. Carpen. 2004. α-Actinin revisited: a fresh look at an old player. Cell Motil. Cytoskel. 58:104-111. [DOI] [PubMed] [Google Scholar]
- 33.Papagrigoriou, E., A. R. Gingras, I. L. Barsukov, N. Bate, I. J. Fillingham, B. Patel, R. Frank, W. H. Ziegler, G. C. Roberts, D. R. Critchley, and J. Emsley. 2004. Activation of a vinculin-binding site in the talin rod involves rearrangement of a five-helix bundle. EMBO J. 23:2942-2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ridley, A. J., M. A. Schwartz, K. Burridge, R. A. Firtel, M. H. Ginsberg, G. Borisy, J. T. Parsons, and A. R. Horwitz. 2003. Cell migration: integrating signals from front to back. Science 302:1704-1709. [DOI] [PubMed] [Google Scholar]
- 35.Takubo, T., and N. Tatsumi. 1999. Microscopic co-distributions of myosin, actin, alpha-actinin and vinculin in human neutrophils during movement. Haematologia (Budapest) 29:285-294. [PubMed] [Google Scholar]
- 36.Tronrud, D. E., L. F. T. Eyck, and B. W. Matthews. 1987. An efficient general purpose least-squares refinement program for macromolecular structures. Acta Crystallogr. A 43:489-501. [Google Scholar]
- 37.Vagin, A., and A. Teplyakov. 1997. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30:1022-1025. [Google Scholar]
- 38.Weis, W. I. 2004. How to build a cell junction. Nature 430:513-515. [DOI] [PubMed] [Google Scholar]
- 39.Winkler, J., H. Lunsdorf, and B. M. Jockusch. 1996. The ultrastructure of chicken gizzard vinculin as visualized by high-resolution electron microscopy. J. Struct. Biol. 116:270-277. [DOI] [PubMed] [Google Scholar]
- 40.Ylanne, J., K. Scheffzek, P. Young, and M. Saraste. 2001. Crystal structure of the α-actinin rod reveals an extensive torsional twist. Structure 9:597-604. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.