

Abstract
Triplophysa bombifrons, a species of bony fish localized in China, has largely been understudied genetically, with limited data available beyond its mitochondrial genome. This study introduces a chromosome-level genome assembly for T. bombifrons, achieved through the integration of PacBio long-read sequencing and Hi-C chromatin interaction mapping. The assembly reveals a genome structure comprising 25 chromosomes and an overall size of 655.95 Mb. The quality of this assembly is demonstrated by a scaffold N50 length of 24.25 Mb, and a genome completeness evaluation via BUSCO, which shows that 97.4% of the Vertebrata Benchmarking Universal Single-Copy Orthologs are present. Gene prediction methods including de novo, homology-based, and transcript-based, identified 34,211 genes, with 34,151 being functionally annotated. These findings may provide valuable resources in conservation, functional genomics, and molecular breeding of T. bombifrons, as well as the molecular phylogenetics and evolutionary patterns in Triplophysa.
Subject terms: Genome, Water resources
Background & Summary
Triplophysa bombifrons (Herzenstein1), a species of bony fish within the Nemacheilidae family, was first identified by Russian scientist Herzenstein in 1888 and originally named Nemachilus bombifrons Herzenstein1. This fish has a limited distribution in China, predominantly inhabiting the Tarim and Aksu Rivers2. Over recent decades, the wild population of T. bombifrons has experienced significant declines due to environmental disturbances such as pollution and habitat destruction. As a result, it has been classified as critically endangered in the Red List of China’s Vertebrates (2016)3. Belonging to the genus Triplophysa, which is part of the Cobitoidea superfamily and includes over 141 species, T. bombifrons is a key component of the ichthyofauna on the Qinghai-Tibetan Plateau (QTP). Despite its significance, the phylogenetic position of Triplophysa remains unresolved due to limited molecular data. To date, 28 complete mitochondrial genomes (mitogenomes) of Triplophysa species have been documented, using mitochondrial genes or complete mitogenome data to explore phylogenetic relationships within the genus2,4–19. However, the specific phylogenetic links between T. bombifrons and other species within Triplophysa are still unclear20,21. Although sequencing technologies have advanced significantly and reduced sequencing costs, only five chromosome-level genomes from this genus have been sequenced: T. tibetana, T. rosa, T. bleekeri, T. dalaica, and T. siluroides. Unfortunately, T. bombifrons still lacks a reference genome, limiting comprehensive genetic and evolutionary studies22–26.
Sequencing the genome of T. bombifrons could provide essential data for analyzing its genetic diversity and population history. Such genomic information is crucial for understanding the genetic adaptations and molecular mechanisms behind the species’ endangerment, aiding in the development of more effective management and conservation strategies.
In this research, we successfully compiled and annotated a chromosome-level genome of T. bombifrons using PacBio single-molecular DNA sequencing complemented by high-throughput chromatin conformation capture (Hi-C) technology. The chromosome-level genome assembly of T. bombifrons measured 655.95 Mb, with a contig N50 length of 14.6 Mb and a scaffold N50 of 24.25 Mb. Approximately 628.24 Mb of sequences were anchored onto 25 chromosomes, covering 99.38% of the total assembly. A chromosome-scale genome assembly of of T. bombifrons was 628.24 Mb with the scaffold N50 length of 24.25 Mb. The sequence continuity of T. bombifrons is confirmed through the high N50 lengths, which align with those found in similar species. Our analysis revealed that the genome is notably complete, as indicated by the presence of over 97.4% of BUSCO genes. We identified 34,211 protein-coding genes in the T. bombifrons genome, of which 99% were successfully annotated against public protein databases. This high-quality genomic resource will support future studies in comparative genomics, phylogenomics, and evolutionary research within the Nemacheilidae family. The research establishes a high-quality genome resource that will underpin future investigations into the population dynamics, conservation strategies, and environmental adaptations of fish in extreme habitats.
Methods
Experimental sampling
A female T. bombifrons was captured using nets in the 2nd Yurungkax River, located at coordinates 37°6′39.6” E, 79°54′46.8” N, in the Hotan district of the Xinjiang Uygur Autonomous Region, China. The specimen was preserved and catalogued at Tarim University under the accession number GYQ2022030001, and is under the care of Xinyue Wang (Fig. 1). The species and sex were confirmed by Professors Adark and YongSong through dissection and examination of the gonads. Pectoral fin samples were collected, preserved in 75% ethanol, and stored at -80°C for subsequent DNA/RNA extraction. All procedures adhered to the relevant regulations on animal care and use.
Fig. 1.

Picture of T. bombifrons. Note: Photographs of vouchers was taken by Shengao Chen.
Library construction, and sequencing
Sequencing of the T. bombifrons genome was conducted using multiple platforms. For short-read sequencing, the sample was randomly fragmented into approximately 350 bp DNA fragments using an ultrasonic processor (Covaris S220; Woburn, MA, USA). The fragments were then prepared by performing end repair, adding a 3′ A tail, and ligating adapters. The resulting library was sequenced on a MGIDNBSEQ T7 platform (BENAGEN Company, Wuhan, China). Raw short reads were filtered using Fastp to eliminate adapters and low-quality reads, yielding a total of 85.1 Gb of clean data for T. bombifrons, providing an approximate coverage of 135 × for the T. bombifrons genome.
To construct the PacBio HiFi library, the DNA template was sheared to an average size of 20 kb using a g-TUBE (Covaris, Inc., MA, USA), and target DNA fragments were recovered through size selection using the BluePippin system (Sage Science, Inc., MA, USA). The SMRTbell library was prepared with the SMRTbell Express Template Prep kit 2.0 (Pacific Biosciences, California, USA) following the manufacturer’s instructions. Sequencing was performed on the PacBio Sequel II platform (Pacific Biosciences, Menlo Park, USA). We confirm that the genome sequencing was conducted in CCS (HiFi) mode, generating Circular Consensus Sequencing (CCS) reads (also referred to as HiFi reads). These HiFi reads were produced using the CCS software (https://github.com/pacificbiosciences/unanimity) with the parameter ‘-minPasses 3’, ensuring highly accurate consensus sequences. This process yielded approximately 22.47 Gb of data, with the average and N50 lengths of these reads being 14.404 kb and 49.465 kb, respectively, achieving approximately 30x coverage of the T. bombifrons genome (Table 1).
Table 1.
Sequencing data for the T. bombifrons genome assembly.
| Sequencing | libraries Insert size | Raw data (Gb) | Clean data (Gb) | Mean read length (bp) | Sequence coverage (×) |
|---|---|---|---|---|---|
| Illumina reads | 250 bp | 85.1 | 85.1 | 150 | 135 |
| Pacbio HiFi reads | 20 Kb | 393.52 | 22.47 | 14,404 | 35 |
| Hi-C reads | 250 bp | 70 | 67 | 150 | 106 |
| Iso-sedq reads | 20 Kb | 47.19 | 44.1 | 1,878 | 70 |
Fresh musscle tissue of T. bombifrons was used to construct a library for Hi-C analysis. The tissue was first cross-linked with formaldehyde, followed by cell lysis using Nuclear Isolation Buffer. Chromatin DNA was then digested with the restriction enzyme MboI, creating sticky ends at the cleavage sites. These sticky ends were biotinylated and proximity-ligated to form chimeric junctions, which were enriched. The DNA was purified, and impurities were removed before being randomly fragmented into 300–500 bp pieces for library construction. The purified DNA underwent blunt-end repair, A-tailing, and adaptor ligation, followed by biotin-streptavidin pull-down and PCR amplification. The Hi-C libraries were quantified and sequenced on the Illumina NovaSeq platform (Illumina, San Diego, CA, USA). The resulting 113.84 Gb of raw data provided 166 × coverage of the genome (Table 1).
RNA sequencing
RNA samples were extracted from muscle, liver, and brain tissues using standard Trizol reagent (Invitrogen, CA, USA) and then combined in equal proportions for sequencing. The purity and integrity of the RNA were assessed using a NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA). RNA contamination was also checked using 1.5% agarose gel electrophoresis. Full-length cDNA was synthesized using the Clontech SMARTer PCR cDNA Synthesis Kit (Takara Biotechnology, China), followed by SMRTbell library construction with the Pacific Biosciences SMRTbell Template Prep Kit (Pacific Biosciences, USA). The transcriptome was then sequenced using the PacBio Iso-Seq protocol in CCS mode on the PacBio Sequel II platform by Frasergen Bioinformatics Co., Ltd. (Wuhan, China). Importantly, no Illumina-based RNA sequencing was performed in this study. After removing adaptors from the polymerase reads, a total of 47.19 Gb of Iso-Seq subreads were obtained (Table 1).
Quality control of sequencing data For the PacBio sequencing data (including both genome assembly HiFi CCS reads and Iso-Seq RNA-seq reads), any reads with a read quality value (RQ-value) below 0.99 were discarded to ensure high accuracy. For the DNA paired-end (PE) sequencing data, which included only genomic short-insert and Hi-C reads, raw data were filtered using Fastp (version 0.23.2)27 to remove adapter sequences and low-quality reads (those containing more than 10% unknown bases or more than 40 low-quality bases). No RNA-seq data were generated using this DNA PE sequencing approach. All remaining high-quality sequencing data from each platform were retained for subsequent analyses.
Genome estimate and assembly
The estimated sizes of the predicted genomes were determined in silico using JELLYFISH (version 2.2.3) and Genomescope (http://qb.cshl.edu/genomescope/), which analyzed the frequency distribution of 27-mers28,29. K-mer analysis suggests that the T. bombifrons genome spans approximately 510.674 Mb, with 217 Mb consisting of repeated sequences and a heterozygosity rate estimated at 0.849% (Fig. 2).
Fig. 2.
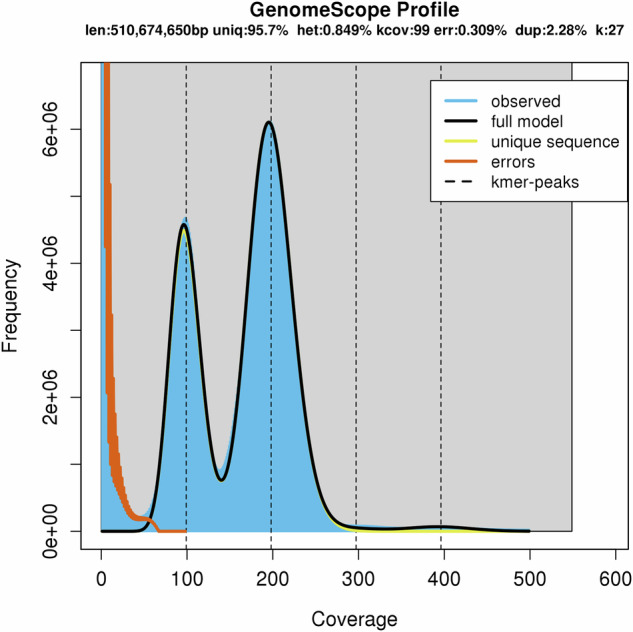
Genome size estimates for T. bombifrons using Kmer-based method.
To create a high-quality genome assembly for T. bombifrons, we employed a hierarchical strategy utilizing various sequencing data types—PacBio long reads, genomic short-insert reads, and Hi-C reads. Initially, PacBio HiFi reads were assembled into contigs using Hifiasm (version 0.17.4) with default settings30. Subsequently, high-accuracy short-read Illumina data were aligned to these contigs using BWA (version 0.7.12), and the draft assembly was refined using Pilon (version 1.21)31,32. Following this, valid Hi-C paired-end reads were processed and a contact matrix with 100 kb resolution was generated using HiC-Pro33. This tool also filtered out low-quality, unmapped, and invalid paired reads, and constructed a contact matrix based on interaction frequencies. After filtering, uniquely mapped sequences from valid paired-end reads were used for additional assembly steps. The genome was then assembled using the Hi-C data with Lachesis, which managed clustering, ordering, and orientation of sequences. A heat map was employed to evaluate the assembly quality. Using PacBio long reads, the T. bombifrons genome was assembled into a structure measuring 655.95 Mb, comprising 609 contigs, with an N50 length of 14.6 Mb. The longest contig measured 26.65 Mb (Table 2).
Table 2.
Assembly and annotation statistics of the T. bombifrons genome.
| Total length (Mb) | N_contig | Contig N50 (Mb) | N_scaffold | Scaffold N50 (Mb) | |
|---|---|---|---|---|---|
| PacBio sequencing | 655.95 | 609 | 12.65 | ||
| Hi-c sequencing | 628.24 | 133 | 24.25 | ||
| Genome annotation | |||||
| Protein-coding gene | 34, 211 (34, 151 annotated,99%) | ||||
| Repeatitive sequence | 61.77% | ||||
| GC content | 39% | ||||
Note: N_contig and N_scaffold denote number of contig and number of scaffold respectively.
Hi-C technology utilizes interaction information between different chromosomal regions, predicated on the notion that interactions are more frequent among proximal regions than distant ones. In this study, we generated 113.84 Gb of sequencing data through Hi-C library sequencing. Utilizing this interaction data, we achieved a chromosome assembly totaling 656.20 Gb with a scaffold N50 length of 24.25 Mb. Remarkably, over 628 Mb of sequences were successfully anchored across 25 chromosomes, demonstrating a high chromosome anchoring rate of 99.38% at the base level (Fig. 3). The integrity of the assembled genome was assessed using BUSCO (v5.5.0) with the Vertebrata odb10 database34.
Fig. 3.

The Hi-C contact map of the T. bombifrons genome. chr 01–25 represented for the 25 pseudo-chromosomes. The color bar showed the contact density from white (low) to red (high).
Gene prediction and annotation
We employed two strategies—homology-based and de novo prediction—to identify repetitive content in the T. bombifrons genome. In the homology-based approach, known transposable elements (TEs) within the T. bombifrons genome were identified using RepeatMasker (version 4.0.7) and the Repbase TE library35,36. For the de novo prediction, we developed a repeat library specifically for the T. bombifrons genome using RepeatModeler (version 2.0.6) (http://www.repeatmasker.org/RepeatModeler/). This tool automatically engages two core de novo repeat-finding programs, RECON (version 1.08) and RepeatScout (version 1.0.5), to generate, refine, and classify consensus models of potential interspersed repeats37. Additionally, we conducted a de novo search for long terminal repeat (LTR) retrotransposons within the genome sequences using LTR_FINDER (version 1.0.7), LTR_harvest (version 1.5.11), and LTR_retriever (version 2.7)38–40. Tandem repeats were identified using the Tandem Repeat Finder (TRF) package, and simple sequence repeats (SSRs) were detected using MISA (version 1.0)41. Subsequently, we merged the library files from both methods and used RepeatMasker to catalog the repeat contents. Our annotation pipeline revealed that 228.3 Mb of the genome sequences, representing 34.60% of the genome, were annotated as repetitive elements in the T. bombifrons genome (Tab.4). Specifically, these included 2.63% DNA transposons (17.2 Mb), 3.19% long interspersed nuclear elements (LINEs) (20.931 Mb), and 5.58% long terminal repeats (LTRs) (36.63 Mb) (Table 3).
Table 3.
Genome quality assessment statistics of the T. bombifrons genome.
| TYPE | Number | Percent (%) |
|---|---|---|
| Total BUSCO groups searched | 3354 | |
| Complete BUSCOs (C) | 3264 | 97.4 |
| Complete and single-copy BUSCOs (S) | 3212 | 95.8 |
| Complete and duplicated BUSCOs (D) | 52 | 1.6 |
| Fragmented BUSCO (F) | 17 | 0.5 |
| Missing BUSCO (M) | 73 | 2.1 |
The protein-coding genes of the T. bombifrons genome were predicted using three distinct methods: ab initio gene prediction, homology-based gene prediction, and RNA-Seq-guided gene prediction. Initially, the assembled T. bombifrons genome was processed for both hard and soft masking using RepeatMasker (version 4.1.6). For the ab initio gene prediction, we utilized Augustus (version 3.3.3), training models on a set of high-quality proteins derived from our RNA-Seq dataset42. Homology-based gene prediction was conducted using MAKER (version 2.31.10). This involved aligning protein sequences and transcript sequences to the genome assembly, and predicting coding genes with default parameters in MAKER43. For RNA-Seq-guided gene prediction, we first mapped clean RNA-Seq reads to the genome using Minimap2 (version 2.24). Gene structures were then deduced using Transdecoder (version 5.5) and MAKER244. Ultimately, the results from all three methods were consolidated into a comprehensive gene set using EVidenceModeler (version 1.1.1)45.
Functional annotations for the predicted protein-coding genes were determined by aligning their sequences to several public databases. These databases include Gene Ontology (http://geneontology.org/), the Integrated Resource of Protein Domains and Functional Sites (InterPro: https://www.ebi.ac.uk/interpro/), Kyoto Encyclopedia of Genes and Genomes (KEGG: https://www.kegg.jp/), Clusters of Orthologous Groups of Proteins (COG: https://www.ncbi.nlm.nih.gov/COG/), Swiss-Prot (www.uniprot.org), TrEMBL (www.uniprot.org), and the nonredundant proteins database (NR: https://ftp.ncbi.nlm.nih.gov/blast/db). Additionally, four types of non-coding RNAs—microRNAs, transfer RNAs (tRNA), ribosomal RNAs (rRNA), and small nuclear RNAs (snRNA)—were predicted within the T. bombifrons genome. These predictions were performed using tRNAscan-SE (version 1.3.1) for tRNAs, and Infernal (version 1.1.3) with the Rfam database for the remaining RNAs46,47.The efforts encompassed protein and non-coding gene prediction as well as functional annotation Table 4.
Table 4.
Statistics of repetitive sequences in the T. bombifrons genome.
| Types | Number of elements | Length | Percentage occupied of sequence |
|---|---|---|---|
| SINEs | 0 | 0 bp | 0.00% |
| ALUs | 0 | 0 bp | 0.00% |
| MIRs | 0 | 0 bp | 0.00% |
| LINEs | 40763 | 20939947 bp | 3.19% |
| LINE1 | 80 | 38293 bp | 0.01% |
| LINE2 | 32403 | 14044271 bp | 2.14% |
| L3/CR1 | 349 | 151062 bp | 0.02% |
| LTR elements | 68297 | 36625328 bp | 5.58% |
| ERVL | 0 | 0 bp | 0.00% |
| ERVL-MaLRs | 0 | 0 bp | 0.00% |
| ERV_classI | 1284 | 870386 bp | 0.13% |
| ERV_classII | 0 | 0 bp | 0.00% |
| DNA elements | 42683 | 17227146 bp | 2.63% |
| hAT-Charlie | 8585 | 1306322 bp | 0.20% |
| TcMar-Tigger | 0 | 0 bp | 0.00% |
| Unclassified | 687759 | 142724487 bp | 21.75% |
| Total interspersed repeats | 217516908 bp | 33.15% | |
| Small RNA | 0 | 0 bp | 0.00% |
| Satellites | 0 | 0 bp | 0.00% |
| Simple repeats | 142044 | 10548685 bp | 1.61% |
| Low complexity | 18015 | 1102503 bp | 0.17% |
Using de novo, homology, and RNA-seq data methods, a total of 34,211 protein-coding genes were predicted in the T. bombifrons genome. Among these genes, approximately 99%, 95%, and 68% exhibited homologous sequences in the NCBI NR, TrEMBL, and Swiss-Prot databases, respectively. Furthermore, 70% of these genes were associated with Pfam domains, while 45%, 40%, and 50% were assigned Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Clusters of Orthologous Groups (KOG) terms, respectively (Table 5). Overall, 34,151 of the protein-coding genes, representing 99% of the total, received functional annotations.
Table 5.
Functional annotation of protein-coding genes for T. bombifrons.
| Data_base | annotated_number | annotated_ratio |
|---|---|---|
| GO | 15643 | 45% |
| kegg | 13990 | 40% |
| KOG | 17216 | 50% |
| nr | 34145 | 99% |
| pfam | 24262 | 70% |
| swiss_prot | 23603 | 68% |
| TrEMBL | 32679 | 95% |
| Total | 34151 | 99% |
Data Records
The DNA and RNA sequencing data were submitted to the NCBI Sequence Read Archive (SRA) database under the SRA IDs: SRR22343166,SRR22343167,SRR22343168 and SRR22343165, which is associated with the BioProject accession number PRJNA90322748. The assembled genome of Triplophysa bombifrons have been deposited at the NCBI GenBank (https://identifiers.org/ncbi/insdc.gca:GCA_029783895.1)49.The annotation results of repeated sequences, gene structure and functional prediction have been deposited at the Figshare database (10.6084/m9.figshare.27054901)50.
Technical Validation
Evaluation of the genome assembly
To assess the integrity of the assembly, short reads were mapped to the genomes using BWA v0.7.17, giving a mapping rate of 96.6% and a genome coverage of 99.70%. The completeness and accuracy of the final genome assembly were checked by Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.5.0 with vertebrata_odb10. The BUSCO analysis revealed that 97.4% of the complete genes were retrieved in the genome, with 95.8% being single-copy and 1.6% duplicated. Only 0.5% and 2.1% of BUSCO genes were fragmented and missing, respectively(Table 3).
Acknowledgements
This work was supported by the Integrated recycling of saline-alkali soil and water and ecological improvement fishery model (2023YFD2401000), the Third Xinjiang Scientific Expedition Project (2021xjkk0604), the Tianshan Talent Training Project of Xinjiang (2023TSYCCX0128), the Corps Science and Technology Bureau Project (2017DB003, 2022DB019).
Author contributions
Shengao Chen conceived and designed the study; Chengxin Wang, Yong Song and Xinyue Wang collected the samples; Site Luo and Liting Yang performed the bioinformatics analysis, including genome size estimation, genome assembly, annotation, and gene prediction; Chengxin Wang and Site Luo wrote the manuscript. Shengao Chen revised the manuscript. All authors read and approved the final manuscript for submission.
Code availability
The software versions, settings and parameters used are described below. No custom code was used during this study for the curation and/or validation of the dataset. CCS v6.4.0–min-rq 0.99. Fastp: -q 20 -u 40. Jellyfish v2.2.3: default. Hifiasm v0.17.4: default. Bwa v0.7.17: default. Pilon:–threads 40. Lachesis v1.21: default. Busco v5.5.0 -l vertebrata_odb10 -m genome. LTR_FINDER v1.0.7: default. LTR_harvest v1.5.11: default. LTR_retriever v2.7: default. MISA v1.0: default. tRNAscan-SE v1.3.1: default. Infernal v1.1.3: default. RepeatModeler v2.0.6: default. RECON v 1.08: default. RepeatScout v1.0.5: default. RepeatMasker v4.1.6:default. Augustus v3.3.3: default. MAKER v2.31.10: default. Minimap2 v 5.5: default. EVidenceModeler v1.1.1:–genome final_assembly.fasta–weights weights.txt–gene_predictions gene_predictions.gff3–transcripts transcripts.gff3–proteins proteins.gff3.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Chengxin Wang, Site Luo.
References
- 1.Herzenstein, S. Wissenschaftliche resultate der von NM przewalski nach central-asien unternommenen reisen (scientific results of von NM przewalski while travelling through central asia. In german). Zoologischer Theil3 (1888).
- 2.Ming Han, M. et al. Complete mitochondrial genome of the Triplophysa bombifrons and Triplophysa strauchii. Mitochondrial DNA Part A27, 4710–4711 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Jiang, Z. et al. Red list of china’s vertebrates. Biodiversity Science24, 500 (2016). [Google Scholar]
- 4.Wang, C. et al. Complete mitochondrial DNA genome of Triplophysa venusta (cypriniformes: Cobitida). Mitochondrial DNA Part A27, 4617–4619 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Yang, X., Wen, H., Luo, T. & Zhou, J. Complete mitochondrial genome of Triplophysa nasobarbatula. Mitochondrial DNA Part B5, 3771–3772 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan, P., Li, J., Ma, Q., Deng, Y. & Song, Z. Complete mitochondrial genome of Triplophysa robusta (Teleostei: Cypriniformes: Balitoridae). Mitochondrial DNA Part A27, 1715–1716 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Ning, X. et al. The complete mitochondrial DNA sequence of Kashgarian loach (Triplophysa yarkandensis) from Bosten Lake. Mitochondrial DNA Part B5, 821–823 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang, Y. et al. The complete mitochondrial genome of a cave-dwelling loach Triplophysa baotianensis (Teleostei: Nemacheilidae). Mitochondrial DNA Part B6, 1209–1211 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, I.-S., Liu, G.-D. & Prokofiev, A. M. The complete mitochondrial genome of giant stone loach Triplophysa siluroides (Cypriniformes: Balitoridae). Mitochondrial DNA Part A27, 998–1000 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Feng, X., Chen, Y., Sui, X. & Chen, Y. The complete mitochondrial genome of Triplophysa cuneicephala (Cypriniformes: Balitoridae) with phylogenetic consideration. Mitochondrial DNA Part B4, 1239–1240 (2019). [Google Scholar]
- 11.Jing, H., Yan, P., Li, W., Li, X. & Song, Z. The complete mitochondrial genome of Triplophysa lixianensis (Teleostei: Cypriniformes: Balitoridae) with phylogenetic consideration. Biochemical Systematics and Ecology66, 254–264 (2016). [Google Scholar]
- 12.Liu, T. & You, P. The complete mitochondrial genome of Triplophysa sp.(Teleostei: Cypriniformes: Balitoridae). Mitochondrial DNA Part A27, 4557–4558 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Wang, J. et al. The complete mitochondrial genome of Triplophysa tibetana. Mitochondrial DNA Part B4, 1411–1412 (2019). [Google Scholar]
- 14.Que, Y. et al. The complete mitochondrial genome sequence of Triplophysa anterodorsalis (Teleostei, Balitoridae, Nemacheilinae). Mitochondrial DNA Part A27, 937–938 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Tang, Q. et al. The complete mitochondrial genome sequence of Triplophysa bleekeri (Teleostei, Balitoridae, Nemacheilinae). Mitochondrial DNA24, 25–27 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Yan, Y. & Luo, D. The complete mitochondrial genome sequence of Triplophysa stenura (Teleostei, Cypriniformes): genome characterization and phylogenetic analysis. Mitochondrial DNA Part B1, 607–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang, X., Cao, L. & Zhang, E. The complete mitochondrial genome sequence of Triplophysa xiangxiensis (Teleostei: Nemacheilidae). Mitochondrial DNA Part A28, 171–172 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Wang, J. et al. The complete mitogenome sequence of a cave loach Triplophysa rosa (Teleostei, Balitoridae, Nemacheilinae). Mitochondrial DNA23, 366–368 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Lei, D. et al. The complete mtDNA genome of Triplophysa dorsalis (Cypriniformes, Balitoridae, Cobitoidea): genome characterization and phylogenetic analysis. Mitochondrial DNA Part A27, 3745–3746 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Yang, X. et al. Haplotype-resolved chinese male genome assembly based on high-fidelity sequencing. Fundamental Research2(6), 946–953 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li, X. et al. First annotated genome of a mandibulate moth, Neomicropteryx cornuta, generated using PacBio HiFi sequencing. Genome biology and evolution13, evab229 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang, L. et al. A chromosome-scale reference assembly of a Tibetan Loach, Triplophysa siluroides. Frontiers in genetics10, 991 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan, D. et al. Chromosomal genome of Triplophysa bleekeri provides insights into its evolution and environmental adaptation. GigaScience9, giaa132 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang, X. et al. Chromosome-level genome assembly of Triplophysa tibetana, a fish adapted to the harsh high-altitude environment of the Tibetan Plateau. Molecular ecology resources19, 1027–1036 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Zhao, Q., Shao, F., Li, Y., Yi, S. V. & Peng, Z. Novel genome sequence of Chinese cavefish (Triplophysa rosa) reveals pervasive relaxation of natural selection in cavefish genomes. Molecular Ecology31, 5831–5845 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou, C. et al. The chromosome-level genome of Triplophysa dalaica (Cypriniformes: Cobitidae) provides insights into its survival in extremely alkaline environment. Genome biology and evolution13, evab153 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics34, i884–i890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marcais, G. & Kingsford, C. Jellyfish: A fast k-mer counter. Tutorialis e Manuais1, 1–8 (2012). [Google Scholar]
- 29.Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nature communications11, 1–10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature methods18, 170–175 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walker, B. J. et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLOS ONE9, e112963 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-bio] 10.6084/M9.FIGSHARE.963153.V1 (2013).
- 33.Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology16, 259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics31, 3210–3212 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics5, 4–10 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile Dna6, 1–6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics21, i351–i358 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant physiology176, 1410–1422 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC bioinformatics9, 1–14 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res35, 265–268 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beier, S., Thiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics33, 2583–2585 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic acids research34, 435–439 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campbell, M. S., Holt, C., Moore, B. & Yandell, M. Genome annotation and curation using MAKER and MAKER-P. Current protocols in bioinformatics48, 4–11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics12, 491 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome biology9, 1–22 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: Improved Detection and Functional Classification of Transfer RNA Genes. bioRxiv 614032 (2019). [DOI] [PMC free article] [PubMed]
- 47.Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics29, 2933–2935 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sequence Read Archivehttps://identifiers.org/ncbi/insdc.sra:SRP408683.
- 49.Triplophysa bombifrons breed wild isolate L2, whole genome shotgun sequencing project. Genbankhttps://identifiers.org/ncbi/insdc.gca:GCA_029783895.1 (2023).
- 50.Wang, C. et al. The protein-coding annotation file of Triplophysa bombifrons genome. Figshare. 10.6084/m9.figshare.27054901 (2024).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Triplophysa bombifrons breed wild isolate L2, whole genome shotgun sequencing project. Genbankhttps://identifiers.org/ncbi/insdc.gca:GCA_029783895.1 (2023).
Data Availability Statement
The software versions, settings and parameters used are described below. No custom code was used during this study for the curation and/or validation of the dataset. CCS v6.4.0–min-rq 0.99. Fastp: -q 20 -u 40. Jellyfish v2.2.3: default. Hifiasm v0.17.4: default. Bwa v0.7.17: default. Pilon:–threads 40. Lachesis v1.21: default. Busco v5.5.0 -l vertebrata_odb10 -m genome. LTR_FINDER v1.0.7: default. LTR_harvest v1.5.11: default. LTR_retriever v2.7: default. MISA v1.0: default. tRNAscan-SE v1.3.1: default. Infernal v1.1.3: default. RepeatModeler v2.0.6: default. RECON v 1.08: default. RepeatScout v1.0.5: default. RepeatMasker v4.1.6:default. Augustus v3.3.3: default. MAKER v2.31.10: default. Minimap2 v 5.5: default. EVidenceModeler v1.1.1:–genome final_assembly.fasta–weights weights.txt–gene_predictions gene_predictions.gff3–transcripts transcripts.gff3–proteins proteins.gff3.