

Abstract
The synthesis, characterization, and reactivity of a NiOH core bearing a tridentate redox-active ligand capable of reaching three molecular oxidation states is presented in this paper. The reduced complex [LNiOH]2– was characterized by single-crystal X-ray diffraction analysis, depicting a square-planar NiOH core stabilized by intramolecular H-bonding interactions. Cyclic voltammetry measurements indicated that [LNiOH]2– can be reversibly oxidized to [LNiOH]− and [LNiOH] at very negative reduction potentials (−1.13 and −0.39 V vs ferrocene, respectively). The oxidation of [LNiOH]2– to [LNiOH]− and [LNiOH] was accomplished using 1 and 2 equiv of ferrocenium, respectively. Spectroscopic and computational characterization suggest that [LNiOH]2–, [LNiOH]−, and [LNiOH] are all NiII species in which the redox-active ligand adopts different oxidation states (catecholate-like, semiquinone-like, and quinone-like, respectively). The NiOH species were found to promote H-atom abstraction from organic substrates, with [LNiOH]− acting as a 1H+/1e– oxidant and [LNiOH] as a 2H+/2e– oxidant. Thermochemical analysis indicated that [LNiOH] was capable of abstracting H atoms from stronger O–H bonds than [LNiOH]−. However, the greater thermochemical tendency of [LNiOH] reactivity toward H atoms did not align with the kinetics of the PCET reaction, where [LNiOH]− reacted with H-atom donors much faster than [LNiOH]. The unique stereoelectronic structure of [LNiOH]− (radical character combined with a basic NiOH core) might account for its enhanced PCET reactivity.
Short abstract
Herein, we describe the synthesis, characterization, and reactivity of a NiOH core bearing a tridentate redox-active ligand capable of reaching three molecular oxidation states. Spectroscopic and computational characterization suggest that the NiOH complexes are all NiII species in which the redox-active ligand adopts different oxidation states (catecholate-like, semiquinone-like, and quinone-like, respectively). The unique stereoelectronic structure of the NiOH semiquinone-like species (radical character and basicity) is proposed to enhance its PCET reactivity.
Introduction
Mononuclear Ni hydroxide species (e.g., NiIIOH and NiIIIOH) are invoked as intermediates in multiple organic transformations, including CO2 fixation,1,2 coupling reactions,3 and C–H bond activation.4 Despite their relevance, only a handful of mononuclear NiOH complexes have been synthesized and characterized (see some selected examples in Figure 1A).5−7 It is believed that this is due to the tendency of the hydroxide ligand to bridge between late 3d transition metals and produce species of higher nuclearity [e.g., NiII2(OH)2 and CuII2(OH)2 cores].8 The formation of multinuclear species can be avoided by using pincer ligand scaffolds with bulky substituents and/or by utilizing ligands with H-bonding donors to stabilize mononuclear M–OH cores.9 With this in mind, we recently utilized a tridentate redox-active ligand containing ureanyl H-bond donors to generate a mononuclear CuOH complex capable of reaching three molecular oxidation states (Figure 1B).10,11 Structural and spectroscopic characterization of the CuOH species (namely, [LCuOH]2–, [LCuOH]−, and [LCuOH]) suggested that these are formulated as CuIIOH cores with the ligand adopting different oxidation states (catecholate-like, semiquinone-like and quinone-like, respectively). In this paper, we report the synthesis, characterization, and reactivity of the analogous NiOH system.
Figure 1.

(A) Selected examples of mononuclear NiOH cores stabilized by pincer ligands. (B) Mononuclear CuOH and NiOH complexes discussed in this paper.
The oxidative reactivity of metal oxo and metal hydroxide species usually entails a proton-coupled electron-transfer (PCET) key step, involving the transfer of a proton (PT) and an electron (ET) from an organic substrate (C–H, N–H, and O–H bonds) to the M–O(H) species (Figure 2A).12,13 Classically, it is proposed that these PCET processes can occur in a concerted fashion (coupled-proton electron transfer, CPET) or stepwise fashion (proton transfer followed by electron transfer, PT–ET, or electron transfer followed by proton transfer, ET–PT). Very recently, a third category of PCET transformation has been described (asynchronous CPET) that differs from the limiting extremes mentioned above, in which the one-step CPET transformation occurs via the formation of a sole transition state with more proton-transfer character (asynchronous basic CPET) or more electron-transfer character (asynchronous oxidative CPET).14,15
Figure 2.

Reaction pathways (A) and thermochemical analysis (B) of the PCET reactivity of metal hydroxide complexes.
The thermochemistry and kinetics of PCET reactions can be studied by determining the bond dissociation free energy (BDFE) of the bonds broken and formed during the reaction (Figure 2B).12 For example, the ability of a metal hydroxide species to accept H atoms is driven by the BDFE of the O–H bond of the resulting M-aqua complex. In general, the higher the difference between the BDFEO–H of the M-aqua complex and the BDFEX–H of the PCET reagent (BDFEO–H ≫ BDFEX–H), the higher the driving force of the PCET reaction will be, which usually correlates with faster reaction rates.13 However, several reports have shown that the kinetics of PCET reactions depend not only on the thermochemistry of the reaction but also on the spin state of the H-atom-abstracting metal species (e.g., two-state reactivity in nonheme Fe-oxo complexes) or the mechanism by which the metal species reacts (e.g., synchronous vs asynchronous CPET in Co-oxo species or PT–ET vs CPET in Mn-oxo).16,17
For the [LCuOH]n− system, we have shown that the CuOH species abstracted H atoms from organic substrates containing weak O–H and N–H bonds in a multiproton multielectron fashion (Figure 3A).10 Our studies also indicated that the thermochemical driving force of the 1H+/1e– reduction of the CuOH cores depended on the oxidation state of the ligand scaffold (i.e., the BDFE of the [LCuOH]/[LCuOH(H)] couple was higher than the BDFE of the [LCuOH]−/[LCuOH(H)]− couple; Figure 3B), which impacted the reactivity of the CuOH cores, as [LCuOH] reacted with phenols while [LCuOH]− did not. In this paper, we show that the PCET reactivity of the [LNiOH]n− species also depends on the oxidation state of the complex. The PCET reactions between TEMPOH and [LNiOH]− were substantially faster than the analogous reaction involving [LNiOH], despite the superior thermochemical driving force of [LNiOH] to accept H atom from organic substrates.18,19
Figure 3.

Stoichiometry (A) and thermochemical analysis (B) of the multi-PCET transformations promoted by the [LCuOH]n− systems.
Results and Discussion
Synthesis and Characterization of the NiOH Complex
The [LNiOH]2– complex was obtained following a protocol similar to the one used for the synthesis of [LCuOH]2– (Figure 4). In the glovebox, 3 equiv of KH was added to a DMF solution of the tridentate ligand (LH3), after which 1 equiv of Ni(OAc)2 and 2 equiv of NMe4OH·5H2O were stirred to form the complex [LNiOH](NMe4)2 [see the Supporting Information (SI) for further details].
Figure 4.

Synthesis and SC-XRD characterization of [LNiOH]2–. Note that the NMe4+ countercations, lattice DMF solvent molecules, and most H atoms were omitted for clarity.
Crystalline material suitable for single-crystal X-ray diffraction (SC-XRD) analysis was obtained by layering a DMF solution of [LNiOH]2– with diethyl ether. The Ni complex exhibited a slightly distorted square-planar geometry around the nickel center, typical of d8 4-coordinated NiII complexes (τ4 value of 0.15; for square-planar structures, a τ4 value of 0.0 is expected, whereas for tetrahedral structures, a τ4 value of 1.0 is expected20). Compared to the Cu complex (τ4 value of 0.2), the Ni system adopts a higher degree of square-planarity. The Ni–N bond distances averaged 1.91 Å, whereas the Cu–N bond averaged 1.98 Å. With respect to the metal hydroxide bond, the Ni–O bond was found to be 0.14 Å shorter than the Cu–O bond (1.88 and 2.02 Å respectively). These bond lengths, in tandem with shorter intramolecular H-bond interactions (1.80 Å for [LNiOH]2– and 1.90 Å for [LCuOH]2–), indicated the formation of a more compact square-planar structure for the NiOH complex.
Analysis of the CAr–Nα and CAr–CAr distances (∼1.42 and ∼1.40 Å, respectively) indicated that the ligand scaffold in [LNiOH]2– adopted the catecholate-like form.10,211H NMR and EPR measurements confirmed the diamagnetic nature of [LNiOH]2– (S = 0), which was formulated as a low-spin NiIIOH core bound by the fully reduced trianionic form of the ligand (see the SI).11
Electrochemistry
The hallmark of metal complexes bearing redox-active ligands is the propensity for these complexes to reach multiple oxidation states via ligand and/or metal reduction/oxidation events.22−24 In 2020, we reported three reversible, ligand-based redox events in [LCuOH]2–, where the ligand was reversibly oxidized from the catecholate-like (L3–) form to the semiquinone-like (L•2–), and quinone-like (L–) forms, and the metal center remained a CuII ion.10 In the current Ni system, seven oxidation states are possible, ranging from a Ni0 complexed with the catecholate-like form of the ligand ([LNiOH]4–) to a NiIV complexed by the quinone-like form of the ligand ([LNiOH]2+; Figure 5). However, cyclic voltammetry experiments showed three observable states, where [LNiOH]2– can be reversibly oxidized to [LNiOH]− and [LNiOH] at −1.13 and −0.39 V, respectively (V vs ferrocene, Fc0/+). Both of these redox events are comparatively more reductive than the Cu system, which undergoes redox events at −1.05 and −0.28 V, correspondingly.
Figure 5.

(A) Possible oxidation states that can be reached in the oxidation/reduction of the [LNiOH]2– complex. (B) Electrochemical and spectroscopic characterization of [LNiOH]2–, [LNiOH]−, and [LNiOH]. Note: Only 3 of the 7 possible oxidation states ([LNiOH]2–, [LNiOH]−, and [LNiOH]) were observed by CV. See the SI for further details.
The oxidation of [LNiOH]2– to the “high-valent” oxidation states [LNiOH]− and [LNiOH] can also be accomplished by stoichiometric additions of an oxidant such as ferrocenium hexafluorophosphate (FcPF6; Figure 5B). The addition of 1 equiv of FcPF6 to [LNiOH]2– (650 nm, ε = 600 M–1 cm–1) generated a new species that absorbed at 655 nm (ε = 5600 M–1 cm–1), which was assigned as [LNiOH]−. This species was stable at room temperature (t1/2 > 24 h). However, attempts to crystallize this complex were futile at both room and low temperatures. The subsequent addition of 1 equiv of FcPF6 generated a species with absorbances at 440 nm (ε = 7200 M–1 cm–1) and 950 nm (ε = 8000 M–1 cm–1), which was assigned as [LNiOH]. This species was metastable at room temperature (t1/2 ∼ 40 min). After the formation of [LNiOH] from FcPF6, stoichiometric additions of the 1e– reductant cobaltocene reduced the complex to regenerate [LNiOH]− and [LNiOH]2–, respectively (see the SI).
Due to the ambiguous electronic nature of the metal complexes bound by redox-active ligands (noninnocent ligands), [LNiOH]− can be formulated as a NiII ion bound by the semiquinone-like form of the ligand or a NiIII bound by the catecholate-like form, among other possibilities. Evidence for the formulation of [LNiOH]− as a semiquinone-like NiIIOH species was obtained by EPR, in which a spectrum characteristic of a species containing a ligand-centered radical (i.e., semiquinone-like ligand) was observed (Figure 5B). These results contrast with the EPR spectra of known NiIII complexes.25 The magnetic susceptibility of [LNiOH]− was measured at room temperature using the Evans method (see the SI). The μeff obtained (1.77 μB) was consistent with the formation of a low-spin S = 1/2 complex, in agreement with the EPR measurements.
Complex [LNiOH] was also characterized by UV–vis, EPR, and NMR spectroscopy. The absorption spectrum of [LNiOH] was similar to the one observed for [LCuOH], which is characteristic of metal complexes bound by tridentate quinone-like ligands.21 The formulation of [LNiOH] as a paramagnetic NiII ion bound by a quinone-like ligand was also supported by EPR (no signal in standard perpendicular mode, S = 1) and NMR (paramagnetic spectrum; see the SI). Magnetic susceptibility measurements using the Evans method were also consistent with the formulation of [LNiOH] as a high-spin triplet paramagnetic species (2.81 μB).
DFT Calculations
Computations provided additional evidence on the geometry and electronic structure of the [LNiOH]n− complexes (Figure 6). For each species, we optimized the geometry of the complex in two possible spin states and computed the difference in energy between the two isomers. For example, [LNiOH]2– could adopt two different spin states, a diamagnetic isomer (S = 0) or a paramagnetic isomer (S = 1). Computations on [LNiOH]2– suggested that the isomer in the singlet spin state (S = 0) was ∼8 kcal/mol lower than the isomer in the triplet state (S = 1). Hence, DFT calculations agreed with the experimental evidence (NMR, magnetic moment measurements) that formulated [LNiOH]2– as a diamagnetic species (see the sections above). Computation of the d-orbital occupancy in [LNiOH]2– indicated that the Ni ion was a d8 metal (8 of out of 10 of the electrons in the d orbitals had a higher occupancy than 0.726), implying that the ligand scaffold was in the catecholate-like form.
Figure 6.

DFT calculations on the electronic structure of the [LNiOH]n− systems. Note: see the SI for details on the DFT computations, including the structure, spin density plots, and geometric parameters of the other spin isomers of the complexes (e.g., the isomer in the triplet spin state of [LNiOH]2–).
Computations on [LNiOH]− found that the isomer in the doublet spin state (S = 1/2) was ∼13 kcal/mol lower than the isomer in the quadruplet spin state (S = 3/2). Like in [LNiOH]2–, the d-orbital occupancy calculations on [LNiOH]− (S = 1/2) indicated that the Ni ion is a d8 metal, suggesting that the ligand scaffold was in the semiquinone-like form and in agreement with the EPR measurements. This hypothesis was further confirmed with the spin density plot (visualization of the “electron density of unpaired electrons”), in which substantial spin in the N and C atoms of the redox-active ligand was observed (Figure 6).
For [LNiOH], our DFT calculations suggested that the isomer in the singlet spin state (S = 0) was ∼4.5 kcal/mol lower in energy than the isomer in the triplet spin state (S = 1). The small energy gap between the two isomers is somewhat in agreement with paramagnetism recorded, suggesting that the triplet spin state is significantly populated at room temperature. Based on the d-orbital occupancy, the singlet spin state isomer was formulated as a NiII complex (d8) bound by the quinone-like form of the ligand. Computations on the isomer in the triplet spin state were consistent with a NiIIIOH core bound by a semiquinone-like ligand, in which the unpaired metal-based electron is ferromagnetically coupled with the ligand-centered radical. As previously stated, UV–vis and NMR measurements suggest that [LNiOH] is formulated as a quinone-like NiII paramagnetic complex. The origin for the discrepancy between the experiments and the computations is unclear, but it could be due to a change in the geometry of the Ni ion (e.g., formation of tetrahedral Ni species, which could favor the high-spin configuration27), solvent coordination (inducing the formation of a pseudo-octahedral NiII paramagnetic complex28), or other changes (e.g., ureanyl reorientation to bind with either NH or carbonyl or keto–enol urea isomerization29). Calculations on [LNiOH] coordinated by two molecules of DMF found that both isomers (singlet and triplet spin state) were formulated as NiIIOH quinone-like species, in agreement with the spectroscopic data (see the SI).
Our computations indicate that the oxidation of the ligand from the catecholate-like form in [LNiOH]2– to the semiquinone-like form in [LNiOH]− and quinone-like form in [LNiOH] entailed shortening of the C···Nα bond (from 1.38 to 1.36 Å). The sequential oxidation of [LNiOH]2– to [LNiOH]− and [LNiOH] also led to shortening of the Ni–O bond (from 1.97 to 1.82 Å), elongation of the intramolecular H-bonding interactions (Hα′···O, from 1.71 to 1.98 Å), and a slight increase of the τ4 value (from 0.14 to 0.23).
PCET Reactivity of the [LNiOH]n− Complexes: Scope and Stoichiometry
The H-atom-abstraction reactivity of [LNiOH]− and [LNiOH] with PCET reagents was studied in detail (Figure 7). Similar to the CuOH system, none of the NiOH complexes were able to react with organic substrates with weak C–H bonds (e.g., dihydroanthracene, DHA). The NiOH systems can, however, perform H-atom abstractions on substrates containing weak O–H and N–H bonds. [LNiOH]− reacted with 1-hydroxy-2,2,6,6-tetramethylpiperidine (TEMPOH; BDFEO–H = 65.7 kcal/mol) and with diphenylhydrazine (PhNHNHPh; BDFEavg = 65.2 kcal/mol) but not with 4-substituted 2,6-di-tert-butylphenols such as 4-methoxy-2,6-tert-butylphenol (4-MeO-2,6-DTBP; BDFEO–H = 71.9 kcal/mol) and 2,4,6-tri-tert-butylphenol (2,4,6-TTBP; BDFEO–H = 75.1 kcal/mol). [LNiOH], on the other hand, reacted with TEMPOH, PhNHNHPh, and the above-mentioned phenols (note that the BDFE values of the substrates and complexes discussed in this and the following section have been determined experimentally with an uncertainty of 1 kcal/mol unless stated12,30).
Figure 7.
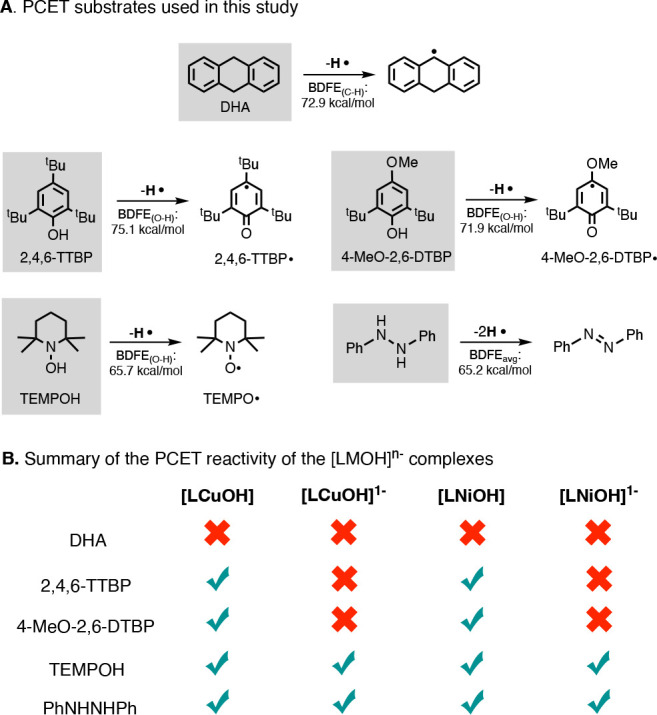
Reactivity of [LMOH]n−, including the substrates utilized (A) and variations on the reactivity depending on the oxidation state of the complex (B).
The stoichiometry of the reactions between [LNiOH]− and [LNiOH] with PCET reagents was determined by UV–vis, NMR, and EPR (note: see the overall proposed stoichiometry in Figure 10). The UV–vis spectra of the reaction of [LNiOH]− (0.25 mM) with 20 equiv of TEMPOH at room temperature showed fast decay of the features associated with [LNiOH]− (λmax = 655 nm; Abs ∼1.5) to produce a Ni complex with features that resembled [LNiOH]2– (λmax = 640 nm; Abs ∼0.2), suggesting that [LNiOH]− underwent a 1H+/1e– reduction (Figure 8A). EPR analysis of the reaction between [LNiOH]− and TEMPOH confirmed the formation of 1 equiv of TEMPO radical (yield ∼80%; see the SI). The addition of 1 equiv of a strong acid (DMF·CF3SO3H) to [LNiOH]2– produced the same UV–vis spectrum (Figure 8B), suggesting that the final product of the protonation/reduction of [LNiOH]− (namely, [LNiOH(H)]−) was a NiII complex with a catecholate-like form of the ligand (Figure 8B; note that one of the possible structures of [LNiOH(H)]− is depicted in Figure 10; see also the SI). Additional DMF·CF3SO3H led to a decrease of the UV–vis features of [LNiOH(H)]−, suggesting that the catecholate-like complex can be further protonated. NMR analysis of the protonation of [LNiOH]− indicated that the addition of 1 equiv of acid produced the diamagnetic species [LNiOH(H)]−, which reacts with additional acid to form 1 equiv of ligand catLH3.
Figure 10.

Summary of the stoichiometry and species formed in the reductive protonation of [LNiOH] and [LNiOH]−. Note that the structures of INT390, INT517, and [LNiOH(H)]− are ambiguous. One proposed structure is shown in this figure. See the SI for additional structures.
Figure 8.
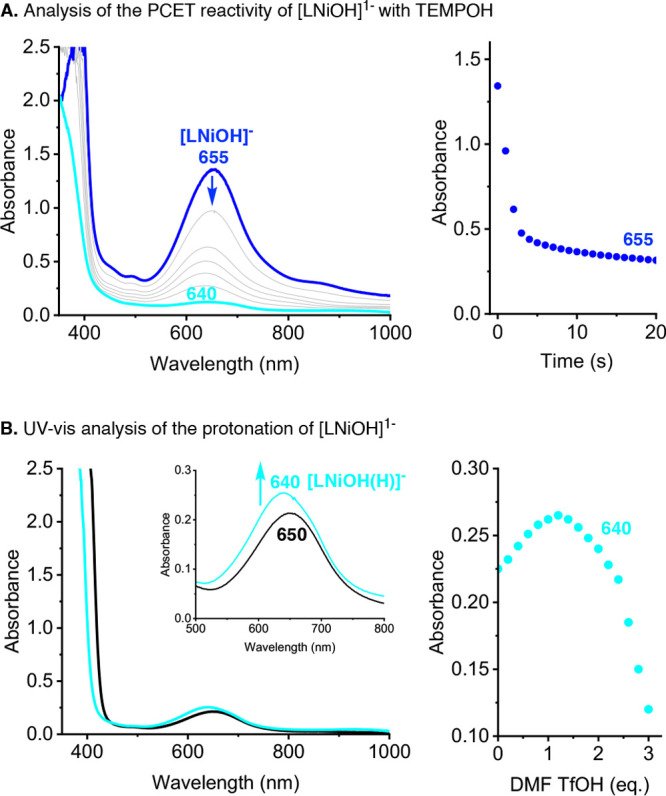
UV–vis analysis of the reaction between [LNiOH]− and TEMPOH (A) and protonation of [LNiOH]2– (B).
The reaction of [LNiOH]− with PhNHNHPh was followed by UV–vis and NMR (see the SI). Like in the reaction with TEMPOH, the addition of PhNHNHPh to [LNiOH]− caused fast decay of its UV–vis features to produce [LNiOH(H)]−. The 1H NMR spectrum of the reaction between [LNiOH]− and PhNHNHPh confirmed that [LNiOH]− acted as a 1H+/1e– acceptor, producing 0.5 equiv of PhN=NPh and the diamagnetic catecholate-like complex [LNiOH(H)]−.
The reaction of [LNiOH] with TEMPOH was followed by UV–vis and EPR (see Figure 9 and the SI). The addition of excess amounts of TEMPOH to [LNiOH] caused spectral changes characteristic of multistep reactivity. The UV–vis features of [LNiOH] (λmax = 950 nm; Abs ∼2.0) decayed to produce two reaction intermediates (INT390, λmax = 390 nm; INT517, λmax = 517 nm), which sequentially formed and decayed to generate a species with weak absorption (λmax = 640 nm). EPR analysis of the reaction between [LNiOH] with TEMPOH confirmed the formation of 2 equiv of TEMPO radical, suggesting that [LNiOH] acted as a 2H+/2e– acceptor. The reaction between [LNiOH] with PhNHNHPh was followed by UV–vis and NMR (see the SI). The UV–vis spectra recorded were similar to that of the stepwise reaction between [LNiOH] with TEMPOH (i.e., formation and decay of intermediate species with λmax = 517 nm; see the SI). NMR analysis indicated that [LNiOH] reacted as the 2H+/2e– acceptor, producing 1 equiv of PhN=NPh and 1 equiv of ligand (catLH3).
Figure 9.
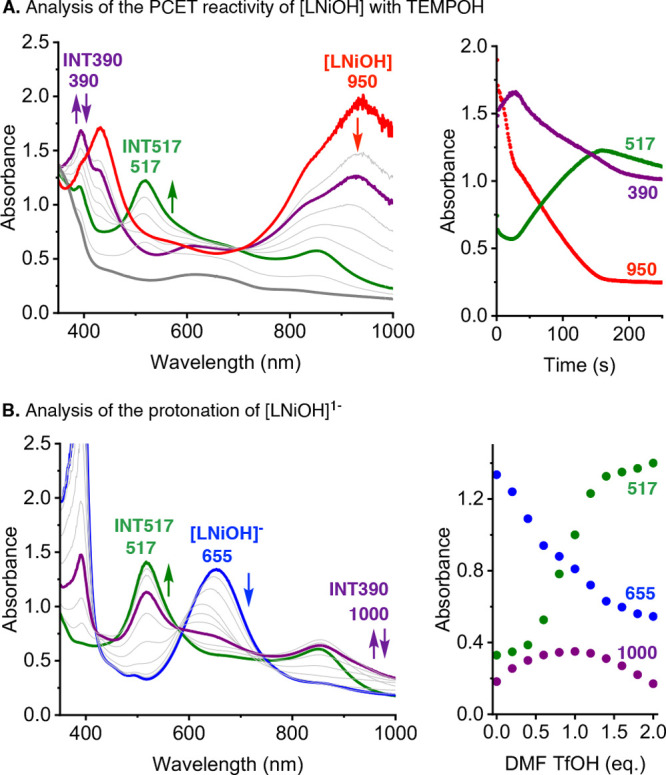
UV–vis analysis of the reaction between [LNiOH] and TEMPOH (A) and protonation of [LNiOH]− (B). See the SI for details.
Further evidence on the identity of the intermediates formed in the reductive protonation of [LNiOH] was obtained via protonation of [LNiOH]− (Figure 9B). The reaction was followed by UV–vis, NMR, and EPR (see the SI for details). The addition of stoichiometric amounts of DMF·CF3SO3H (from 0.2 to 0.4 equiv) led to a slight shift in the UV–vis band associated with [LNiOH] and the formation of a new band at ∼1000 nm, consistent with the formation of INT390. Further addition of acid (from 0.4 to 1 equiv) led to an increase on the UV–vis features associated with the formation of INT517. Full decay of INT390 and full formation of INT517 species were accomplished by adding an additional 1 equiv of acid (total of 2 equiv of DMF·CF3SO3H; Figure 9B). After protonation of [LNiOH]− with 2 equiv of acid, the addition of excess amounts of strong base did not lead to regeneration of [LNiOH]−, which suggests that this process is irreversible. In a separate UV–vis experiment, we reacted [LNiOH]− with 2 equiv of acid (added all at once), and we observed the initial formation of INT390, which evolved to form INT517 (see the SI).
NMR analysis of the protonation of [LNiOH]− with 1 equiv of acid showed the formation of ∼0.5 equiv of free ligand and a diamagnetic Ni species (see the SI). NMR analysis of the protonation of [LNiOH]− with 2 equiv of acid showed a similar spectrum, with slightly more ligand formed. EPR analysis of the reaction after addition of 1 equiv of acid led to a substantial decrease in [LNiOH]−(see the SI). The addition of 2 equiv of acid led to quenching of all of the EPR signals, suggesting the formation of an EPR-silent species.
On the basis of all of this evidence, we propose that the 1H+/1e– reductive protonation of [LNiOH] and the protonation of [LNiOH]− produce a metastable mononuclear Ni-semiquinone complex (INT390) that undergoes an irreversible disproportionation reaction to generate 0.5 equiv of a catecholate-like complex ([(catL3–)NiOH2]−) and 0.5 equiv of a quinone-like complex (INT517; Figure 10). Our data suggest that once formed, the catecholate-like NiII species is irreversibly protonated to generate ligand (catLH3) and free NiII in solution, which explains the fact that 2 equiv of acid is required to fully protonate [LNiOH]−. In the presence of substrate (e.g., TEMPOH), the diamagnetic quinone-like complex (INT517) undergoes reductive protonation, which also leads to the formation of catLH3 and free NiII in solution. To corroborate this hypothesis, we carried out the protonation of [LNiOH]−, followed by the addition of PCET reagents (see the experimental details and spectra in the SI). EPR analysis of the reaction between [LNiOH]−, 2 equiv of acid, and TEMPOH confirmed the formation of 1 equiv of TEMPO radical (∼80%). NMR analysis of the reaction between [LNiOH]−, 2 equiv of acid, and PhNHNHPh led to the formation of 0.5 equiv of PhN=NPh and 1 equiv of ligand (catLH3). Evidence on the formation of free NiII in solution after protonation of [LNiOH]− and protonation of [LNiOH]2– was obtained by adding bipyridine ligand, which produced the paramagnetic [NiII(bpy)3]2+ complex that could be observed by NMR (see the SI).
PCET Reactivity of the [LNiOH]n− Complexes: Thermochemistry
In the reaction between [LNiOH] and 2,4,6-TTBP, we observed that the addition of excess amounts of phenolic substrate caused only partial decay of the UV–vis features of [LNiOH]. This behavior is usually observed when species involved in the PCET reaction have similar BDFEs, which leads to a PCET equilibrium. Given the BDFEO–H of 2,4,6-TTBP (75.1 kcal/mol in DMF) and the fact that excess 2,4,6-TTBP (e.g., 100 equiv) is required for [LNiOH] to partially react, we can calculate that the BDFE for the 1H+/1e– reductive protonation of [LNiOH] is ∼71 kcal/mol (see the SI for details). As we discussed in the Introduction, the thermochemical driving force (BDFE) for proton–electron acceptors (e.g., metal hydroxide) can also be determined using the Bordwell equation and the corresponding E1/2 and pKa values.12 A partial protonation of [LNiOH]− can be accomplished by 4-NO2–PhOH (pKa = 12.6 in DMF31). Thus, the pKa of the protonation of [LNiOH]− is calculated to be ∼10 (see the SI for details). By the Bordwell equation, this results in a BDFE of ∼72 kcal/mol, in agreement with the reactivity between [LNiOH] and 2,4,6-TTBP (Figure 11).
Figure 11.
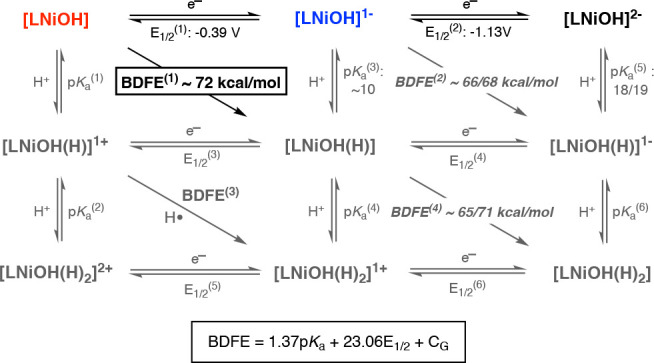
Summary of the BDFE analysis of the species involved in the PCET reactivity of the [LNiOH]n− complexes.
Because [LNiOH]− reacts with TEMPOH (BDFEO–H = 65.7 kcal/mol) but not with 4-MeO-2,6-DTBP (BDFEO–H = 71.9 kcal/mol), we can estimate the BDFE for the 1H+/1e– reductive protonation [LNiOH]− to be between 65 and 72 kcal/mol. [LNiOH]2– was found to be protonated by 4-NO2-2,6-DTBP (BDFE = 79 kcal/mol; pKa = 8.3 in DMF31) and partially protonated with excess 2,6-DTBP (BDFE = 76.7 kcal/mol; pKa = 18.2 in DMF31) and 4-MeO-2,6-DTBP (BDFE = 71.9 kcal/mol; pKa = 19.0 in DMF31), suggesting that the pKa value for protonating [LNiOH]2– is ∼18–19. Considering this estimation and the E1/2 of [LNiOH]−/[LNiOH]2– couple (−1.13 V vs Fc0/+), we can narrow down the BDFE for the reductive protonation of [LNiOH]− to be between 66 and 68 kcal/mol (Figure 11).
Our data suggest that the BDFE for the reductive protonation of [LNiOH] was slightly lower than that for [LCuOH] (72 vs 75 kcal/mol). In fact, [LCuOH] was fully reduced by 2,4,6-TTBP, while [LNiOH] reacted partially with this PCET substrate. This could be attributed to the lower redox potential of the [LNiOH]/[LNiOH]− couple (−0.39 V vs Fc0/+) when compared to the analogous Cu couple (−0.28 V vs Fc0/+) because the pKa values for the protonation of [LNiOH]− and [LCuOH]− are comparable (∼10). Similarly, the BDFE for the reductive protonation of [LNiOH]− was also slightly lower than that for the Cu counterpart (69–71 vs 66–68 kcal/mol). This could also be explained by the lower E1/2 of the [LNiOH]−/[LNiOH]2– couple (−1.13 V) when compared to Cu (−1.05 V), where both [LMOH]2– systems have similar pKa values (∼18/19).
PCET Reactivity of the [LNiOH]n− Complexes: Kinetics
The kinetics of the reaction between the [LNiOH]n− complexes and TEMPOH was analyzed at −40 °C in DMF under pseudo-first-order conditions ([Ni] = 0.25 mM; [TEMPOH] = 2.5–12.5 mM; see the SI). For [LNiOH]−, we observed a fast decay of the UV–vis features of the Ni-semiquinone species, which were fitted to an exponential decay that allowed for calculating the pseudo-first-order kinetic constant kobs (Figure 12). Variations on the concentration of TEMPOH led to a linear increase of the reaction rates, allowing us to obtain a second-order rate constant (k2 = 26.2 M–1 s–1). The kinetics for the reaction of [LNiOH] with TEMPOH were also evaluated at −40 °C. The initial decay of UV–vis features of [LNiOH] was fitted to a linear function, by the method of the initial rates, to determine the pseudo-first-order kinetic constant. The second-order rate constant for the reaction between [LNiOH] and TEMPOH was significantly lower than the k2 observed for [LNiOH]− (0.1 vs 26.2 M–1 s–1). The kinetics of the reaction between the Cu analogues and TEMPOH were also studied (see the SI). Our results indicated that the reaction between TEMPOH and [LCuOH] was significantly faster than the reaction with [LCuOH]− (k2 = 5.3 vs 0.05 M–1 s–1).
Figure 12.

Kinetic analysis of the reaction between [LMOH]n− and TEMPOH, including the kinetic traces (A), rate constants at different TEMPOH(D) concentrations (B), Eyring plots (C), and summary table (D). See the SI for further details.
Considering the BDFE of the 1H+/1e– reductive protonation of [LNiOH], [LNiOH]−, [LCuOH], and [LCuOH]−, we expected that the complexes with higher thermodynamic driving force (e.g., [LNiOH] and [LCuOH]) would react with TEMPOH faster than the complexes with lower driving forces ([LNiOH]− and [LCuOH]−). Hence, we assumed that the k2 for the reaction between TEMPOH and the [LMOH]n− complexes would follow the trend [LCuOH] > [LNiOH] ≫ [LCuOH]− > [LNiOH]−. However, the reaction rates observed for [LNiOH]− were unexpectedly high. Analysis of the kinetics at different temperatures allowed for obtaining the activation parameters (i.e., ΔH⧧, ΔS⧧, and ΔG⧧233 K) for the PCET transformations (Figure 12C). For all of the complexes, similar ΔS⧧ values were obtained (ΔS⧧ between −20 and −30 cal/mol·K) and the low ΔG⧧233 K of [LNiOH]− can be attributed to its low ΔH⧧ when compared to the other complexes.
Analysis of the kinetics of the reaction between the [LMOH]n− complexes and TEMPOD helped to elucidate the kinetic isotope effect (KIE) of the PCET transformations. A primary KIE was observed for all of the complexes, which is consistent with O–H cleavage of the substrate during the rate-determining step of the reaction and suggestive of one-step CPET processes.32,33 While modest primary KIEs were found for [LCuOH], [LNiOH], and [LCuOH]− (KIE between 2.3 and 4.4), the KIE for [LNiOH]− was very large (KIE = 17.8), suggestive of H-atom tunneling.34
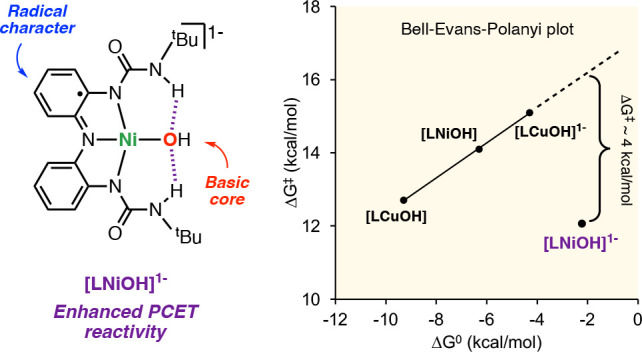
Overall, the kinetic evidence seems to indicate that the reaction mechanism by which [LNiOH]− abstracts H atoms from TEMPOH differs from the other [LMOH]n− complexes. In the Bell–Evans–Polanyi (BEP) plot for the reaction between the [LMOH]n− complexes (ΔG⧧ vs ΔG0), we observed that [LCuOH], [LNiOH], and [LCuOH]− produced a linear regression with a slope ∼0.5, characteristic of synchronous CPET transformations (Figure 13). The BEP plot clearly shows the deviation of [LNiOH]− from the trend, also suggesting that the H-atom transfer for this NiOH complex may be driven by additional factors (see discussion below).
Figure 13.
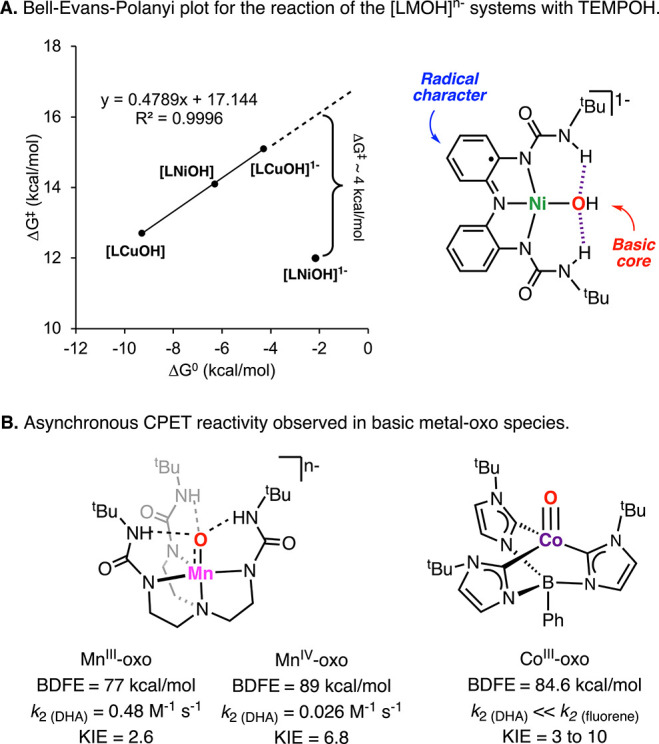
BEP plot for the reaction between [LMOH]n− and TEMPOH (A) and selected examples of complexes where higher thermodynamic driving forces do not correlate to faster rates (B).
Contextualization of Our Findings
In 2009, Borovik and co-workers described the H-atom-abstraction reactivity of a MnIII-oxo and MnIV-oxo toward DHA (Figure 13B).35 The authors found that the reaction rates for the MnIII-oxo species were substantially higher than the one for the MnIV-oxo, despite the latter having a higher thermochemical driving force for PCET (77 vs 89 kcal/mol). Initially, the authors suggested a change in the PCET mechanism due to the high basicity of the MnIII-oxo, with the MnIV-oxo species reacting via a concerted CPET and the MnIII-oxo reacting via a stepwise PT–ET mechanism. However, recent publications from the same research group suggest that both Mn-oxo complexes react in an asynchronous CPET fashion in which the activation energy of the PCET transformation is highly affected by the basicity of the Mn-oxo species and the pKa of the substrate oxidized.16,36 In a seminal research report, Anderson and co-workers found that the reactivity of a terminal CoIII-oxo complex toward C–H bonds was also dictated by the basicity of the metal-oxo complex (Figure 13B).17 The authors reported that the reaction rates for the oxidation of substrates with similar BDFE (e.g., DHA and 9-fluorene) were highly dependent on the pKa of the substrate (higher acidity of 9-fluorene led to higher reaction rates). In a subsequent study, the authors described that this asynchronous CPET transformation had also very high KIEs, suggesting that the tunneling effect is not exclusive to synchronous CPET transformations.34
Theoretical investigations by Srnec and co-workers have recently shown that some of the deviations on the BEP plots can be quantified using the nonclassical thermodynamic factors asynchronicity (η) and frustration (σ), which arise from differences in the reduction potentials and pKa of all of the species depicted in the square scheme of a PCET transformation.14,15 However, the authors noted that their model did not fully account for nonthermodynamic factors that also affect the activation energy of PCET transformations, such as sterics, adiabaticity, H-atom tunneling, and the involvement of different spin states and excited states. The fact that the thermochemical parameters for the 1H+/1e– reductive protonation [LNiOH]− and [LCuOH]− are similar (e.g., pKa for protonation of [LNiOH]− and [LCuOH]− are ∼10 in both cases and E1/2 for the reduction of [LNiOH]− and [LCuOH]− only differ by 80 mV) suggest that the marked difference in the kinetics for PCET might arise from nonthermodynamic factors. We believe that the unique stereoelectronic structure of [LNiOH]−, the only complex that combines the high basicity of the hydroxide core and the radical character of the semiquinone-like ligand, might be a contributor to lowering the activation energy of the PCET reaction. The radical character of metal-oxo complexes (“oxyl character”) explains the high reactivity of some biological and synthetic oxidants, including Ni-oxyl complexes.37 Similarly, NiII-iminyl radicals (as opposed to NiIII-imino species) have also been invoked in challenging Ni-mediated C–H amination reactions.38
Conclusions
In this research paper, we described the synthesis and characterization of a mononuclear NiOH core stabilized by a tridentate redox-active ligand containing H-bonding donors. The NiOH complex was found to reach three molecular oxidation states (namely, [LNiOH]2–, [LNiOH]−, and [LNiOH]), all of which are described as NiIIOH species bound by different oxidation states of the ligand. The PCET reactivity of the [LNiOH]n− complexes was studied in detail. The quinone-like complex [LNiOH] was found to act as a 2H+/2e– oxidant, capable of performing H-atom abstraction from O–H and N–H substrates with BDFEs lower than 72 kcal/mol. Conversely, the semiquinone-like species [LNiOH]− reacted as a 1H+/1e– oxidant toward substrates with lower BDFE (∼66 kcal/mol). Unexpectedly, the reaction rates for the 1H+/1e– dehydrogenation of TEMPOH were substantially faster for [LNiOH]− (∼250 times) despite [LNiOH] being a stronger 1H+/1e– acceptor (ΔBDFE ∼ 5 kcal/mol). This anomalous behavior, which might be a manifestation of asynchronous PCET, is hypothesized to be derived from the unique electronic properties of [LNiOH]−, which combines a noncoupled ligand-based radical (putative electron acceptor) with a highly basic NiOH core (putative proton acceptor).
Experimental Section
Materials
All reagents were purchased from commercial suppliers and used as received except as noted. [LCuOH]2– was prepared as previously reported.10 DMF·CF3SO3H was prepared by combining equimolar amounts of DMF and CF3SO3H in DCM for 30 min at 0 °C. All solvents were purchased at the highest level of purity and further purified and dried by passing through an activated alumina solvent purification system (MB SPS-7, MBRAUN INERTGAS-SYSTEME, Garching, Germany). Dimethylformamide (DMF) was distilled under partial vacuum before use. Deuterated solvents were purchased from Cambridge Isotope Laboratories (Tewksbury, MA) and used as received.
Physical Methods
Air-free handling of the copper and nickel complexes was performed inside a MBRAUN UNIlab Pro SP glovebox system with N2 working gas. Electrochemical measurements were performed using a CH Instruments 620E Electrochemical Workstation (CH Instruments, Austin, TX). UV–vis spectra were collected using a Hewlett-Packard 8454 diode-array spectrophotometer with a 1-cm-path-length quartz cell. The spectrometer was equipped with Agilent UV–visible ChemStation software (ver. B.05.02 [16], Agilent Technologies, Santa Clara, CA) and a Unisoku CoolSpeK UV cryostat (UNISOKU Co., Hirakata, Japan). NMR spectra were on a 500 MHz NMR spectrometer (NEO 500 or Avance III, Bruker Corp., Billerica, MA) to acquire spectra with 16 cumulative scans. Evans method experiments were performed using a 7 in., 5-mm-o.d. NMR tube with a smaller 3-mm-o.d. NMR tube inserted inside. The outer tube contained the analyte dissolved in a deuterated solvent with a dichloromethane (DCM) internal standard. The smaller inner tube contained the same deuterated solvent and DCM internal standard solution (without analyte). X-band EPR spectra of frozen solutions were recorded on a Bruker ELEXSYS spectrometer equipped with an Oxford liquid-helium cryostat and a Bruker bimodal cavity. The quantification of all signals was measured relative to a CuEDTA spin standard prepared from a copper atomic absorption standard (Sigma-Aldrich, St. Louis, MO). The spectra were recorded under nonsaturating power conditions. The microwave frequency was calibrated with a frequency counter, and the magnetic field was measured with an NMR gaussmeter. The sample temperature was calibrated against a calibrated CX-1050 Cernox sensor (Lake Shore Cryotronics, Westerville, OH) mounted inside an EPR tube. A modulation amplitude of 1 mT and frequency of 100 kHz was used for all EPR spectra. CHN analysis was performed by Midwest Micro Lab (Indianapolis, IN). A PerkinElmer Frontier FT-IR spectrometer with an attenuated-total-reflectance attachment containing a germanium crystal was used. Spectra were obtained over a range of 4000–700 cm–1 with 0.4 cm–1 resolution.
Synthesis and Characterization of [LNiOH]2–
This complex was prepared using a method adapted from our previous publication.1 In the glovebox, ligand LH3 (100 mg, 0.25 mmol) was dissolved in DMF (1 mL) in an 8 mL vial with a stir bar. KH (30 mg, 0.75 mmol) was added to the solution and stirred for 30 min or until hydrogen evolution ceased. Ni(OAc)2 (45 mg, 0.25 mmol) was added as a solid and allowed to stir for 3 h. Me4NOH·5H2O (90 mg, 0.50 mmol) was added as a solid and allowed to react for 6 h to yield a dark-green solution. Et2O (20 mL) was used to precipitate the complex as a green powder. The precipitate was dried, filtered, redissolved in DMF (1 mL), and recrystallized by vapor diffusion with Et2O. Green crystals of [LNiOH]2– were obtained in 40% yield (65 mg).
Elemental analysis: (C30H53N7NiO3). Experimental C: 58.06%, H: 9.12%, N: 15.87%. Calculated C: 58.26%, H: 8.64%, N: 15.85%. FT-IR (cm–1, selected bands): 3029, 2960, 2921, 1590, 1541, 1492, 1433, 1345, 1296, 1287, 1228, 1110, 1031, 944, 727. Crystals suitable for X-ray structure determination were obtained by layering Et2O on a solution of complex [LNiOH]2– in DMF (see additional details in the SI).
Cyclic Voltammetry of [LNiOH]2–
A total of 3 mL of a DMF solution of [LNiOH]2– (1 mM) containing 0.1 M NBu4PF6 was prepared in the glovebox and transferred via a syringe to an argon-purged electrochemical cell outside. A glassy carbon working electrode, CHI112 Ag/AgNO3 (0.1 M) reference electrode with a porous Teflon tip, and Pt wire counter electrode were used. The potentials were measured with respect to the Ag/AgNO3 reference electrode and converted to Fc0/+ after running a cyclic voltammogram of Fc under the same conditions. The cyclic voltammogram was recorded at a scan rate of 100 mV/s, carried out under an argon atmosphere.
UV–Vis Characterization of [LNiOH]2–, [LNiOH]−, and [LNiOH]
A total of 3 mL solutions of [LNiOH]2– was prepared in DMF (0.25 mM) inside a N2-filled glovebox. The addition of stoichiometric amounts of oxidant (FcPF6) solution generated the respective high-valent oxidant states [LNiOH]− and [LNiOH]. They can also be reduced back to [LNiOH]2– by the addition of stoichiometric amounts of a reductant (CoCp2) solution. The UV–vis spectra were recorded using a Schlenk quartz cuvette with a rubber septum and magnetic stirring under an argon flow at room temperature (see further details in the SI).
EPR Characterization of [LNiOH]2–, [LNiOH]−, and [LNiOH]
In the glovebox, a stock solution of [LNiOH]2– (1 mM) in DMF was prepared along with a stock solution of FcPF6 (1 mM). Differing ratios of complex and oxidant were added such that all of the final solutions consisted of 0.3 mL of a 1 mM solution of [LNiOH]2–, [LNiOH]−, and [LNiOH] and were thoroughly mixed. Each solution was injected into an EPR tube and immediately frozen in liquid N2. Note that [LNiOH]2– and [LNiOH] had no observable signal in the standard perpendicular mode (see further details in the SI).
NMR Characterization of [LNiOH]2–, [LNiOH]−, and [LNiOH]
In the glovebox, solutions of [LNiOH]2–, [LNiOH]−, and [LNiOH] were prepared in DMF-d7 (10 mM) with requisite amounts of FcPF6 oxidant and transferred into an NMR tube. All samples were recorded at room temperature (see the SI for additional details).
Evans Method Measurements
A DMF-d7 solution containing DCM (∼100 mM) and [LNiOH]n− (10 mM) was placed in an NMR tube. A coaxial inner tube containing the same solvent (DMF-d7 with DCM) was placed inside the standard NMR tube, and NMR spectra were taken at room temperature (298 K). The difference in the internal standard resonances was based on the shift of DCM peaks in the presence of a paramagnetic material (see the SI for additional details).
PCET Reactivity
Analysis of the Stoichiometry
UV–vis experiments: In a typical experiment, 3 mL of a [LMOH]n− solution (0.25 mM) in DMF was placed in a 10-mm-path-length quartz cell with a stir bar, capped with a rubber septum. All reactivity experiments were performed at room temperature (25 °C). Differing amounts of a solution of FcPF6 (0.25 mM or 0.5 mM) were injected into the complex solution to generate the corresponding “high-valent” species [LMOH]− and [LMOH]. DMF solutions of substrate (5 mM for TEMPOH or 25 mM for PhNHNHPh) or DMF·CF3SO3H (0.25 mM to 0.5 mM) were stored in 500 μL gastight syringes and injected into the quartz cell correspondingly. EPR experiments: In the glovebox, 300 μL solutions of [LMOH]n− (1 mM), requisite amounts of FcPF6 (1 or 2 mM), requisite amounts of DMF·CF3SO3H (0–2 mM), and TEMPOH (20 mM) were mixed, and the mixture was allowed to incubate for 60 min. They were then transferred to EPR tubes and rapidly transferred outside the glovebox to be frozen in liquid N2. NMR experiments: In the glovebox, 1.0 mL of a DMF-d7 solution of [LNiOH]2– (10 mM), FcPF6 (10 mM or 20 mM), hexamethylbenzene (10 mM, internal standard), and substrate (40–50 mM) were combined, and the mixture was transferred to a 7-in., 5-mm-o.d. NMR tube, which was capped and sealed before NMR spectra were taken (see additional details in the SI).
Analysis of the Kinetics
UV–vis experiments: In a typical reaction, 2.7 mL of a DMF solution of [LMOH]n− (0.25 mM) was transferred to a 10-mm-path-length quartz cell with a stir bar and a rubber septum. Differing amounts of a solution of FcPF6 (0.25 mM or 0.5 mM) were injected into the complex solution to generate the corresponding “high-valent” species [LMOH]− and [LMOH]. DMF solutions of substrate (5 mM for TEMPOH) were stored in 500 μL gastight syringes and injected into the quartz cell correspondingly. All reactions were run under an Ar flow and at −40 °C, with the exception of [LCuOH]−, which was performed at room temperature. The decays of the [LMOH]n− bands were monitored by UV–vis, which were fitted to the following exponential function.
DFT Calculations
All DFT calculations were performed with the Amsterdam Density Functional (ADF)39,40 and QUILD(41) programs, and were performed using the unrestricted Kohn-Sham scheme. Molecular orbitals were expanded in an uncontracted set of Slater type orbitals (STOs) of triple-ζ quality with double polarization functions (TZ2P), or the TDZP basis set which consists of triple-ζ quality on the metal and double-ζ quality on all other atoms, in both cases including one polarization function.41,42 Core electrons were not treated explicitly during the geometry optimizations (frozen core approximation40). An auxiliary set of s, p, d, f, and g STOs was used to fit the molecular density and to represent the coulomb and exchange potentials accurately for each SCF cycle.
Geometries of all possible spin states were optimized with the QUILD(41) program using adapted delocalized coordinates until the maximum gradient component was less than 10–4 a.u. Energies, gradients and Hessians43 (for vibrational frequencies) were calculated using S12g,44,45 in all cases by including solvation effects through the COSMO dielectric continuum model with appropriate parameters for the solvents.46,47 For computing Gibbs free energies, all small frequencies were raised to 100 cm–1 in order to compensate for the breakdown of the harmonic oscillator model.48,49 Scalar relativistic corrections have been included self-consistently in all calculations by using the zerothorder regular approximation (ZORA50). The geometry optimizations (with the TZ2P basis set) have been performed with S12g with a Becke grid of VeryGood quality. See additional details in the Supporting Information.
Acknowledgments
The National Science Foundation (Grant 1941220 to I.G.-B.), AEI/MCIU (PID2020-114548GB-I00 and PID2023-152415NB-I00 to M.S.) and GenCat (Grant 2021SGR00487 to M.S.) are gratefully acknowledged for financial support. We thank the Center for Information Technology of the University of Groningen for their support and for providing access to the Hábrók high-performance computing cluster. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM137914 (to I.G.B.) and Award Number R35GM141948 (to M.P.H.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.4c03370.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Special Issue
Published as part of Inorganic Chemistryspecial issue “Proton-Coupled Electron Transfer in Coordination Chemistry”.
Supplementary Material
References
- Huang D.; Makhlynets O. V.; Tan L. L.; Lee S. C.; Rybak-Akimova E. V.; Holm R. H. Kinetics and mechanistic analysis of an extremely rapid carbon dioxide fixation reaction. Proc. Nat. Acad. Sci. 2011, 108 (4), 1222–1227. 10.1073/pnas.1017430108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D.; Holm R. H. Reactions of the Terminal NiII-OH Group in Substitution and Electrophilic Reactions with Carbon Dioxide and Other Substrates: Structural Definition of Binding Modes in an Intramolecular NiII¬∑¬∑¬∑FeII Bridged Site. J. Am. Chem. Soc. 2010, 132 (13), 4693–4701. 10.1021/ja1003125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day C. S.; Rentería-Gómez Á.; Ton S. J.; Gogoi A. R.; Gutierrez O.; Martin R. Elucidating electron-transfer events in polypyridine nickel complexes for reductive coupling reactions. Nature Catalysis 2023, 6 (3), 244–253. 10.1038/s41929-023-00925-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavun A.; Niess R.; Scheibel L. A.; Seidl M.; Hohloch S. A mesoionic carbene stabilized nickel(ii) hydroxide complex: a facile precursor for C–H activation chemistry. Dalton Trans. 2024, 53 (6), 2749–2761. 10.1039/D3DT03746K. [DOI] [PubMed] [Google Scholar]
- Cámpora J.; Palma P.; del Río D.; Conejo M. M.; Álvarez E. Synthesis and Reactivity of a Mononuclear Parent Amido Nickel Complex. Structures of Ni[C6H3–2,6-(CH2PiPr2)2](NH2) and Ni[C6H3–2,6-(CH2PiPr2)2](OMe). Organometallics 2004, 23 (24), 5653–5655. 10.1021/om049312r. [DOI] [Google Scholar]
- Powell-Jia D.; Ziller J. W.; DiPasquale A. G.; Rheingold A. L.; Borovik A. S. A structure and reactivity analysis of monomeric Ni(ii)–hydroxo complexes prepared from water. Dalton Trans. 2009, (16), 2986–2992. 10.1039/b820209e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikari D.; Mossin S.; Basuli F.; Dible B. R.; Chipara M.; Fan H.; Huffman J. C.; Meyer K.; Mindiola D. J. A Dinuclear Ni(I) System Having a Diradical Ni2N2 Diamond Core Resting State: Synthetic, Structural, Spectroscopic Elucidation, and Reductive Bond Splitting Reactions. Inorg. Chem. 2008, 47 (22), 10479–10490. 10.1021/ic801137p. [DOI] [PubMed] [Google Scholar]
- Martínez-Prieto L. M.; Palma P.; Álvarez E.; Cámpora J. Nickel Pincer Complexes with Frequent Aliphatic Alkoxo Ligands [(iPrPCP)Ni-OR] (R = Et, nBu, iPr, 2-hydroxyethyl). An Assessment of the Hydrolytic Stability of Nickel and Palladium Alkoxides. Inorg. Chem. 2017, 56 (21), 13086–13099. 10.1021/acs.inorgchem.7b01868. [DOI] [PubMed] [Google Scholar]
- Donoghue P. J.; Tehranchi J.; Cramer C. J.; Sarangi R.; Solomon E. I.; Tolman W. B. Rapid C–H Bond Activation by a Monocopper(III)–Hydroxide Complex. J. Am. Chem. Soc. 2011, 133 (44), 17602–17605. 10.1021/ja207882h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.; MacMillan S. N.; Rajabimoghadam K.; Siegler M. A.; Lancaster K. M.; Garcia-Bosch I. Structure, Spectroscopy, and Reactivity of a Mononuclear Copper Hydroxide Complex in Three Molecular Oxidation States. J. Am. Chem. Soc. 2020, 142 (28), 12265–12276. 10.1021/jacs.0c03867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazzolotto D.; Bogart J. A.; Ross D. L.; Ziller J. W.; Borovik A. S. Stabilizing a NiII-aqua complex via intramolecular hydrogen bonds: Synthesis, structure, and redox properties. Inorg. Chim. Acta 2019, 495, 118960 10.1016/j.ica.2019.118960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal R. G.; Coste S. C.; Groff B. D.; Heuer A. M.; Noh H.; Parada G. A.; Wise C. F.; Nichols E. M.; Warren J. J.; Mayer J. M. Free Energies of Proton-Coupled Electron Transfer Reagents and Their Applications. Chem. Rev. 2022, 122 (1), 1–49. 10.1021/acs.chemrev.1c00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darcy J. W.; Koronkiewicz B.; Parada G. A.; Mayer J. M. A Continuum of Proton-Coupled Electron Transfer Reactivity. Acc. Chem. Res. 2018, 51 (10), 2391–2399. 10.1021/acs.accounts.8b00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado-Domínguez M.; Srnec M. H-Atom Abstraction Reactivity through the Lens of Asynchronicity and Frustration with Their Counteracting Effects on Barriers. Inorg. Chem. 2022, 61 (47), 18811–18822. 10.1021/acs.inorgchem.2c03269. [DOI] [PubMed] [Google Scholar]
- Bím D.; Maldonado-Domínguez M.; Rulíšek L.; Srnec M. Beyond the classical thermodynamic contributions to hydrogen atom abstraction reactivity. Proc. Natl. Acad. Sci. U.S.A. 2018, 115 (44), E10287–E10294. 10.1073/pnas.1806399115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barman S. K.; Yang M.-Y.; Parsell T. H.; Green M. T.; Borovik A. S. Semiempirical method for examining asynchronicity in metal–oxido-mediated C–H bond activation. Proc. Natl. Acad. Sci. U.S.A. 2021, 118 (36), e2108648118 10.1073/pnas.2108648118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz M. K.; Anderson J. S. Experimental Evidence for pKa-Driven Asynchronicity in C–H Activation by a Terminal Co(III)–Oxo Complex. J. Am. Chem. Soc. 2019, 141 (9), 4051–4062. 10.1021/jacs.8b13490. [DOI] [PubMed] [Google Scholar]
- Zhao X.; Yang J.-D.; Cheng J.-P. Revisiting the Electrochemistry of TEMPOH Analogues in Acetonitrile. J. Org. Chem. 2023, 88 (1), 540–547. 10.1021/acs.joc.2c02537. [DOI] [PubMed] [Google Scholar]
- Warren J. J.; Tronic T. A.; Mayer J. M. Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Chem. Rev. 2010, 110 (12), 6961–7001. 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorabacher D. B. Electron transfer by copper centers. Chem. Rev. 2004, 104 (2), 651–697. 10.1021/cr020630e. [DOI] [PubMed] [Google Scholar]
- Munhá R. F.; Zarkesh R. A.; Heyduk A. F. Tuning the Electronic and Steric Parameters of a Redox-Active Tris(amido) Ligand. Inorg. Chem. 2013, 52 (19), 11244–11255. 10.1021/ic401496w. [DOI] [PubMed] [Google Scholar]
- Kaim W.; Schwederski B. Non-innocent ligands in bioinorganic chemistry--An overview. Coord. Chem. Rev. 2010, 254 (13–14), 1580–1588. 10.1016/j.ccr.2010.01.009. [DOI] [Google Scholar]
- Luca O. R.; Crabtree R. H. Redox-active ligands in catalysis. Chem. Soc. Rev. 2013, 42 (4), 1440–1459. 10.1039/C2CS35228A. [DOI] [PubMed] [Google Scholar]
- Moutet J.; Philouze C.; du Moulinet d’Hardemare A.; Leconte N.; Thomas F. Ni(II) Complexes of the Redox-Active Bis(2-aminophenyl)dipyrrin: Structural, Spectroscopic, and Theoretical Characterization of Three Members of an Electron Transfer Series. Inorg. Chem. 2017, 56 (11), 6380–6392. 10.1021/acs.inorgchem.7b00433. [DOI] [PubMed] [Google Scholar]
- Mondal P.; Lovisari M.; Twamley B.; McDonald A. R. Fast Hydrocarbon Oxidation by a High-Valent Nickel–Fluoride Complex. Angew. Chem., Int. Ed. 2020, 59 (31), 13044–13050. 10.1002/anie.202004639. [DOI] [PubMed] [Google Scholar]
- Leach I. F.; Klein J. E. M. N. Oxidation States: Intrinsically Ambiguous?. ACS Central Science 2024, 10 (7), 1406–1414. 10.1021/acscentsci.4c00825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignolet L. H.; Horrocks W. D. Jr.; Holm R. H. Thermodynamics and kinetics of the planar-tetrahedral interconversion of dihalobis(diarylmethylphosphine)nickel(II) complexes. J. Am. Chem. Soc. 1970, 92 (7), 1855–1863. 10.1021/ja00710a011. [DOI] [Google Scholar]
- Qamar O. A.; Cong C.; Ma H. Solid state mononuclear divalent nickel spin crossover complexes. Dalton Trans. 2020, 49 (47), 17106–17114. 10.1039/D0DT03421E. [DOI] [PubMed] [Google Scholar]
- Hill E. A.; Weitz A. C.; Onderko E.; Romero-Rivera A.; Guo Y.; Swart M.; Bominaar E. L.; Green M. T.; Hendrich M. P.; Lacy D. C.; et al. Reactivity of an FeIV-Oxo Complex with Protons and Oxidants. J. Am. Chem. Soc. 2016, 138 (40), 13143–13146. 10.1021/jacs.6b07633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.; Puri A.; Qiu Y. L.; Ye D.; Sarma R.; Wang Y.; Kowalewski T.; Siegler M. A.; Swart M.; Garcia-Bosch I. Tuning the Thermochemistry and Reactivity of a Series of Cu-Based 4H+/4e– Electron-Coupled-Proton Buffers. Inorg. Chem. 2024, 63 (20), 9014–9025. 10.1021/acs.inorgchem.4c00835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.; Rajabimoghadam K.; Puri A.; Hebert D. D.; Qiu Y. L.; Eichelberger S.; Siegler M. A.; Swart M.; Hendrich M. P.; Garcia-Bosch I. A. 4H+/4e– Electron-Coupled-Proton Buffer Based on a Mononuclear Cu Complex. J. Am. Chem. Soc. 2022, 144 (37), 16905–16915. 10.1021/jacs.2c05454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. Y.; Peterson R. L.; Ohkubo K.; Garcia-Bosch I.; Himes R. A.; Woertink J.; Moore C. D.; Solomon E. I.; Fukuzumi S.; Karlin K. D. Mechanistic Insights into the Oxidation of Substituted Phenols via Hydrogen Atom Abstraction by a Cupric–Superoxo Complex. J. Am. Chem. Soc. 2014, 136, 9925–9937. 10.1021/ja503105b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bosch I.; Company A.; Cady C. W.; Styring S.; Browne W. R.; Ribas X.; Costas M. Evidence for a Precursor Complex in C–H Hydrogen Atom Transfer Reactions Mediated by a Manganese(IV) Oxo Complex. Angew. Chem., Int. Ed. 2011, 50 (25), 5648–5653. 10.1002/anie.201100907. [DOI] [PubMed] [Google Scholar]
- Schneider J. E.; Goetz M. K.; Anderson J. S. Variable temperature kinetic isotope effects demonstrate extensive tunnelling in the C–H activation reactivity of a transition metal-oxo complex. Chem. Commun. 2023, 59 (55), 8584–8587. 10.1039/D3CC02130K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsell T. H.; Yang M.-Y.; Borovik A. S. C–H Bond Cleavage with Reductants: Re-Investigating the Reactivity of Monomeric MnIII/IV–Oxo Complexes and the Role of Oxo Ligand Basicity. J. Am. Chem. Soc. 2009, 131 (8), 2762–2763. 10.1021/ja8100825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barman S. K.; Jones J. R.; Sun C.; Hill E. A.; Ziller J. W.; Borovik A. S. Regulating the Basicity of Metal–Oxido Complexes with a Single Hydrogen Bond and Its Effect on C–H Bond Cleavage. J. Am. Chem. Soc. 2019, 141 (28), 11142–11150. 10.1021/jacs.9b03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoyama Y.; Kojima T. Metal–Oxyl Species and Their Possible Roles in Chemical Oxidations.. Inorg. Chem. 2019, 58 (15), 9517–9542. 10.1021/acs.inorgchem.8b03459. [DOI] [PubMed] [Google Scholar]
- Dong Y.; Clarke R. M.; Porter G. J.; Betley T. A. Efficient C–H Amination Catalysis Using Nickel-Dipyrrin Complexes. J. Am. Chem. Soc. 2020, 142 (25), 10996–11005. 10.1021/jacs.0c02126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerends E. J.; Ziegler T.; Autschbach J.; Bashford D.; Berger A.; Bérces A.; Bickelhaupt F. M.; Bo C.; de Boeij P. L.; Boerrigter P. M.; Borini S.; Bulo R. E.; Cavallo L.; Chong D. P.; Deng L.; Dickson R. M.; van Duin A. C. T.; Ellis D. E.; van Faassen M.; Fan L.; Fischer T. H.; Fonseca Guerra C.; Ghysels A.; Giammona A.; van Gisbergen S. J. A.; Götz A. W.; Groeneveld J. A.; Gritsenko O. V.; Grüning M.; Gusarov S.; Harris F. E.; Heine T.; van den Hoek P.; Jacob C. R.; Jacobsen H.; Jensen L.; Kadantsev E. S.; Kaminski J. W.; van Kessel G.; Klooster R.; Kootstra F.; Kovalenko A.; Krykunov M. V.; van Lenthe E.; Louwen J. N.; McCormack D. A.; McGarrity E.; Michalak A.; Mitoraj M.; Neugebauer J.; Nicu V. P.; Noodleman L.; Osinga V. P.; Patchkovskii S.; Philipsen P. H. T.; Post D.; Pye C. C.; Ravenek W.; Rodríguez J. I.; Romaniello P.; Ros P.; Schipper P. R. T.; Schreckenbach G.; Seldenthuis J. S.; Seth M.; Skachkov D. G.; Snijders J. G.; Solà M.; Swart M.; Swerhone D.; te Velde G.; Vernooijs P.; Versluis L.; Visscher L.; Visser O.; Wang F.; Wesolowski T. A.; van Wezenbeek E. M.; Wiesenekker G.; Wolff S. K.; Woo T. K.; Yakovlev A. L.. ADF2012.01; SCM, Theoretical Chemistry, Vrije Universiteit: Amsterdam, The Netherlands, 2012. [Google Scholar]
- te Velde G.; Bickelhaupt F. M.; Baerends E. J.; Fonseca Guerra C.; van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. Chemistry with ADF. J. Comput. Chem. 2001, 22 (9), 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]
- Swart M.; Bickelhaupt F. M. QUILD: QUantum-Regions Interconnected by Local Descriptions. J. Comput. Chem. 2008, 29 (5), 724–734. 10.1002/jcc.20834. [DOI] [PubMed] [Google Scholar]
- Van Lenthe E.; Baerends E. J. Optimized Slater-Type Basis Sets for the Elements 1–118. J. Comput. Chem. 2003, 24 (9), 1142–1156. 10.1002/jcc.10255. [DOI] [PubMed] [Google Scholar]
- Wolff S. K. Analytical Second Derivatives in the Amsterdam Density Functional Package. Int. J. Quantum Chem. 2005, 104 (5), 645–659. 10.1002/qua.20653. [DOI] [Google Scholar]
- Swart M. A New Family Of Hybrid Density Functionals. Chem. Phys. Lett. 2013, 580, 166–171. 10.1016/j.cplett.2013.06.045. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132 (15), 154104 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Klamt A.; Schüürmann G. COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. Journal of the Chemical Society, Perkin Transactions 2 1993, 0 (5), 799–805. 10.1039/P29930000799. [DOI] [Google Scholar]
- Swart M.; Rösler E.; Bickelhaupt F. M. Proton Affinities in Water of Maingroup-element Hydrides – Effects of Hydration and Methyl Substitution. Eur. J. Inorg. Chem. 2007, 2007 (23), 3646–3654. 10.1002/ejic.200700228. [DOI] [Google Scholar]
- Averkiev B. B.; Truhlar D. G. Free Energy of Reaction by Density Functional Theory: Oxidative Addition of Ammonia by an Iridium Complex with PCP Pincer Ligands. Catalysis Science & Technology 2011, 1 (8), 1526–1529. 10.1039/c1cy00227a. [DOI] [Google Scholar]
- Klein J. E. M. N.; Dereli B.; Que L.; Cramer C. J. Why Metal-Oxos React with Dihydroanthracene and Cyclohexadiene at Comparable Rates, despite Having Different C-H Bond Strengths. A Computational Study. Chem. Commun. (Camb.) 2016, 52 (69), 10509–10512. 10.1039/C6CC05395E. [DOI] [PubMed] [Google Scholar]
- van Lenthe E.; Baerends E. J.; Snijders J. G. Relativistic Regular Two-Component Hamiltonians. J. Chem. Phys. 1993, 99 (6), 4597–4610. 10.1063/1.466059. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.